

Abstract
Background
Fish skin mucosal surfaces (SMS) are quite similar in composition and function to some mammalian MS and, in consequence, could constitute an adequate niche for the evolution of mucosal aquatic pathogens in natural environments. We aimed to test this hypothesis by searching for metagenomic and genomic evidences in the SMS-microbiome of a model fish species (Anguilla Anguilla or eel), from different ecosystems (four natural environments of different water salinity and one eel farm) as well as the water microbiome (W-microbiome) surrounding the host.
Results
Remarkably, potentially pathogenic Vibrio monopolized wild eel SMS-microbiome from natural ecosystems, Vibrio anguillarum/Vibrio vulnificus and Vibrio cholerae/Vibrio metoecus being the most abundant ones in SMS from estuary and lake, respectively. Functions encoded in the SMS-microbiome differed significantly from those in the W-microbiome and allowed us to predict that successful mucus colonizers should have specific genes for (i) attachment (mainly by forming biofilms), (ii) bacterial competence and communication, and (iii) resistance to mucosal innate immunity, predators (amoeba), and heavy metals/drugs. In addition, we found several mobile genetic elements (mainly integrative conjugative elements) as well as a series of evidences suggesting that bacteria exchange DNA in SMS. Further, we isolated and sequenced a V. metoecus strain from SMS. This isolate shares pathogenicity islands with V. cholerae O1 from intestinal infections that are absent in the rest of sequenced V. metoecus strains, all of them from water and extra-intestinal infections.
Conclusions
We have obtained metagenomic and genomic evidence in favor of the hypothesis on the role of fish mucosal surfaces as a specialized habitat selecting microbes capable of colonizing and persisting on other comparable mucosal surfaces, e.g., the human intestine.
Electronic supplementary material
The online version of this article (10.1186/s40168-017-0376-1) contains supplementary material, which is available to authorized users.
Keywords: Vibrio, Skin mucus, Microbiome, Metagenomics, Attached microbiota
Background
Accidental pathogens are bacteria that live in the environment and occasionally infect humans. In the particular case of the aquatic environment, accidental pathogens usually present a life cycle in which they shift between free and sessile life styles, the latter by forming biofilms on abiotic or biotic surfaces. Free-living aquatic pathogens use active motility to swim towards nutrients (positive chemotaxis) that are more abundant on water/surface interfaces. Once on the surface, they may lose motility, attach to, multiply, and colonize the surface by forming biofilms.
One of the most nutrient-rich surfaces available to aquatic pathogens is mucosal surfaces of fish, in particular, the skin mucous surface (SMS). It has been suggested before that teleost SMS share many characteristics with type I mucosal surfaces of mammalian intestines, respiratory tract, and uterus [1]. Similar to these type I mucosal surfaces, teleost SMS is also formed by mucus-secreting cells arranged with epithelial cells and a diffuse skin-associated lymphoid-tissue containing macrophages, mast cells, dendritic cells, and lymphocytes [2, 3]. Furthermore, fish mucosal secretions also contain a wide variety of innate immune molecules including complement proteins, lysozyme, proteases, esterases, lactoferrin (an iron chelator), and anti-microbial peptides [2, 3].
Most studies performed in aquatic accidental pathogens have been focused on disease mechanisms or environmental survival. For instance, in the case of Vibrio cholerae, most environmental isolates have been demonstrated to be non-pathogenic, as they lack specific virulence genes located in mobile genetic elements (MGE), mainly phages and pathogenicity islands [4, 5]. Consequently, it has been proposed that pathogenic clones responsible for cholera epidemics have emerged by acquisition of such virulence genes by horizontal gene transfer (HGT) events, either in the natural environment or in the human gut [6].
We hypothesize that the fish SMS could be one of the natural niches for aquatic mucosal pathogen evolution. Accordingly, fish SMS could act as an intermediate niche between water and human mucus (i.e., intestinal) pre-selecting bacteria best adapted to survive and multiply in mucus and favoring mucus-fitness by DNA exchange. As a first approach to test this hypothesis, we selected the European eel (henceforth “eel”) and sequenced the SMS-metagenome from eels captured in different ecosystems, four natural (two estuaries, one lake, and a river) and one artificial (an intensive eel farm) located close to the Mediterranean Sea or the Atlantic coast of Spain as well as the metagenome of the eel-surrounding water (W-metagenome) from one of the natural ecosystems [7]. We selected eels as the best fish candidate because (i) they lack macroscopic scales and are surrounded by a thick layer of mucus similar in function to mammal mucus [8], which can be easily sampled without significant manipulation; (ii) wild eel SMS can contain accidental or opportunistic human pathogens like Vibrio vulnificus [9]; and (iii) eels are eurihaline teleostei that inhabit multiple natural environments (lakes, oceans, ponds…) along its life cycle, thus being in contact with a great variety of microorganisms [7]. In fact, adult eels spawn in the Sargasso Sea, migrate as young larvae towards Europe with the Gulf Stream, arriving 1 to 3 years later. Subsequently, they undergo three metamorphoses while colonizing ponds, lagoons, or lakes before reaching sexual maturity and, finally, migrate back to the Sargasso Sea (more than 6000 km against the Gulf Stream) to spawn, closing their life cycle [7]. In addition, the European eel is a species of commercial interest that is cultured (or grown since reproduction in captivity has not been achieved) in intensive farms. Interestingly, a preliminary analysis of the eel-SMS metagenomes suggested that the superficial mucus harbors a microbiota that is quite distinct from that of the surrounding environment [8]. In that work, no analysis of these metagenomes in terms of composition in bacterial species as well as evaluation of potential pathogenicity or functionality of the microbiome was performed (i.e., distinction between potentially virulent vs non-virulent bacteria).
Taken all together, the main objective of this work was to analyze in depth these metagenomic data under the hypothesis that it might be possible (1) to detect genomic features of microbes best adapted to survive and multiply in mucus, (2) examine if potentially pathogenic genomic elements are detectable in fish SMS vs natural environment, and (3) assess the potential for horizontal gene exchange events in the evolution of aquatic mucosal pathogens (i.e., intestinal human pathogens). In the process, we also sought to obtain a deeper insight into the natural SMS-microbiome that travels with eels in natural and artificial ecosystems.
Methods
Sampling and DNA isolation
The sampled habitats, their description, and positional coordinates as well as the main physico-chemical parameters of the water samples are presented in Additional file 1: Figure S1 and Table 1. Nets were strategically placed at different locations in the Nature Parks and trapped wild eels were recovered after 24 h (Additional file 1: Figure S2). We also sampled 20 eels (around 250 g) grown in an intensive eel farm (farmed eels) close to Prat de Cabanes-Torreblanca as well as about 10,000 wild eels (glass eels; average weight per individual, 0.33 g) from various rivers of the Atlantic coast of Spain (Galicia) that were fished and transported to the same farm. Wild and farmed eels were deposited in fishbowls of 100 L (6 or 1000 individuals per bowl, depending on the animal size) containing 10–50 mL of sterile PBS for 20 min, and the detached mucus was collected in sterile glass bottles that were stored at 4 °C (Additional file 1: Figure S2). Wild and farmed eels were returned without damage to their habitats or their respective tanks. In parallel to wild eel sampling in the Ebro Delta, water samples were taken directly with sterile glass bottles and were stored at 4 °C. Water- and mucus bottles were transported to the laboratory and were sequentially filtered through five, 1- and 0.22-μm-pore-size filters by using a peristaltic pump. Finally, the prokaryotic biomass recovered was treated with 1 mg/mL lysozyme and 0.2 mg/mL proteinase K (final concentrations). Nucleic acids were extracted with phenol/chloroform/isoamyl alcohol and chloroform/isoamyl alcohol, and DNA integrity was checked by agarose gel electrophoresis [9].
Table 1.
Sampling points, types of samples, and designation as well as main physico-chemical parameters of water of the sampled environments
| Sampling pointa | Sample type | Sample designationb | SRA accession | Salinity (g/l) | pH | T (°C) |
|---|---|---|---|---|---|---|
| Fish farm close to the Mediterranean sea | Skin mucus from eels | FE4 5.3 | SRX702744 | 4 | 5.3 | 24 |
| Rivers (North Atlantic coast: Galicia) | WE≤1 7 | SRX702748 | ≤ 1c | 7 | 7 | |
| Natural Park: Albufera Lake | WE1 9.5 | SRX703656 | 1 | 9.5 | 19 | |
| Natural Park: Prado Cabanes | WE7 8 | SRX703655 | 7 | 8 | 20 | |
| Natural Park: Ebro Delta | WE3 8 | SRX702749 | 3 | 8 | 19 | |
| WE10 8 | SRX710670 | 10 | 8 | 20 | ||
| Water | WE10 8W | SRX710703 | 10 | 8 | 20 |
aCoordinates of each sampling point are indicated in Additional file 2: Table S1
bSuper- and sub-indexes indicate pH and salinity values, respectively
cThe salinity of the rivers where eels were fished was ≤ 1 g/l
Sequencing and assembly
DNA samples were sequenced either by using a FLX sequencer (454 Roche) with Titanium chemistry (Centro Superior de Investigación en Salud Pública [CSISP, Valencia, Spain]) or an Illumina HiSeq 2000 sequencer with pair-end technology (Macrogen [Seoul, Korea]). To this end, 500 bp and 100 pair-end libraries were prepared for Roche and Illumina, respectively. Default values of dynamictrim were run (phred cutoff 13) to quality filter reads [10]. Assembly of 454 metagenomic reads was performed using Geneious Pro 5.4 with a minimum overlap of 50 nucleotides, 95% identity, and allowing 1% of mismatches per read, while Illumina reads were assembled using Velvet (k-mer 51) [11]. All the metagenomes have been previously deposited in NCBI SRA with the following accession codes: SRX702744, SRX702748, SRX702749, SRX703649, SRX703655, SRX703656, SRX710670, and SRX710703 [8, 12]. Only assembled contigs bigger than 1 kb were considered for the following analysis.
Sequence analysis and annotation of the assembled contigs
The %GC of the metagenomes was determined using the “geecee” program from EMBOSS package [13]. BioEdit software was used to manipulate the sequences [14]. The assembled contigs were annotated using Prodigal [11, 15] and the MG-RAST pipeline [16]. The KEGG and SEED databases were used to analyze metabolic pathways and functional classification of the proteins [17]. To allow the interactive visualization of genomic fragment comparisons, Artemis Comparison Tool ACTv.8 [18] was used. Annotation was refined manually using HHpred [19].
Community structure using all reads and ribosomal RNA
For taxonomy, the entire datasets were compared using BLASTX or BLASTP [20] from the NCBI NR database (cut off expectation e-value 10−5, minimum length 50 bp, and minimum similarity of 95%) and analyzed using MEGAN with the “Percent Identity Filter” active [21]. Ribosomal RNA (rRNA) genes were identified by comparing the data sets against the RDP database [22]. All reads that matched an rRNA sequence with an identity 95% and an alignment length of 100 bases against either the RDP or the LSU database were extracted. The best hit with a taxonomic affiliation was considered a reasonable closest attempt to classify the rRNA sequences.
Functional classification of reads in SEED categories
In order to categorize the reads obtained, we used the pipeline from MG-RAST. This program automatically annotates reads and then classifies them in SEED categories. We downloaded the results in .xls format and compared abundance using Microsoft Office Excel manually.
Vibrio isolation, identification, and genome sequencing
A volume of 1 mL of water or mucus from each one of the samples was inoculated into 4 mL of the Vibrio enrichment medium, Alkaline Peptone Water (APA; 1% peptone extract supplemented with 1% [wt/vol] NaCl at pH 8.6) and incubated for 12 h at 28 °C with agitation (150 rpm). Then, volumes of 0.1 mL of a 1:10,000 dilution were spread on plates containing the Vibrio selective media, TCBS (thiosulfate-citrate-bile salts-sucrose) (Conda 1074), and VVM (Vibrio vulnificus medium) [23] agar and on the general medium TSA-1 (trypticase soy agar supplemented with 1% [wt/vol] NaCl). Plates were incubated 24 h at 28 °C. Suspected colonies were purified on TSA-1, isolated and lyophilized at − 80 °C in LB-1 (Luria Bertani broth1% [wt/vol] NaCl) supplemented with 20% (vol/vol) glycerol. The isolates were phenotypically identified with API 20E kit (bioMerieux) according to manufacturer’s instructions. Bacterial suspensions in PBS were used as inocula. Examination of the strips was conducted after 24 h using the API Database https://apiweb.biomerieux.com/. In parallel, genomic DNA was extracted from suspected colonies and amplified using PCR targeting rDNA by using universal primers 699R (5′- RGGGTTGCGCTCGTT-3′) and 616V (5′- AGAGTTTGATYMTGGCTCAG-3′) to identify bacteria. The amplicons were sequenced and identified in the Genomic section of the SCSIE (Servicios Centrales de Soporte a la Investigación Experimental) from the Universidad of Valencia (Spain). To identify suspected V. vulnificus, primers vvhA-F (5′- CGCCACCCACTTTCGGGCC-3′) and vvhA-R (5′-CC GCGGTACAG GTTGGCGC-3′) were used to amplify the hemolysin gene corresponding exclusively to V. vulnificus [24].
Selected strains were sequenced using Illumina HiSeq2000 (Macrogen [Seoul, Korea]), genomic DNA was extracted using the Wizard Genomic DNA extraction kit (Promega), and assembly and annotation was followed as described before.
The contigs assembled from sequencing the strain M12v were compared with the deposited Vibrio metoecus and Vibrio cholerae O1 El Tor N16961 using GCviewer Comparison tool [25]. Default values were used for BLAST atlas comparison.
Mobile genetic elements detection
Multiple approaches were used to detect MGE in contigs > 10 kb: on one hand to detect intra-contig variations in GC content, taxonomical annotation, and hexanucleotide usage pattern (HUP) and on the other to look for MGE-signature genes (sgMGE) by using BLASTP. BLASTN and ISfinder and ISbrowser were used to identify plasmids and pathogenic islands deposited in databases [26, 27]. We considered as sgMGE: integrases, transposases, conjugative elements, and phage or viral proteins. HUP values were calculated by using compseq program from EMBOSS package [13]. Microsoft Excel tools helped us differentiate genes annotated to the same taxon and visualize the GC variation in the contigs. The workflow diagram is shown in Additional file 1: Figure S3. Contigs that contained genes for ribosomal proteins or for lipopolysaccharide biosynthesis and clustered with contigs identified as putative MGE (cpMGE) but did not contain sgMGE were considered false positives. Intra-contig changes in the taxonomic annotation plus presence of an integrase located next to tRNA or several sgMGE were considered to establish the presence of a MGE in a cpMGE.
Three metagenomes generated in the present work (WE3 8, WE10 8, and WE10 8W), and six metagenomes from various origins that were downloaded from EBI metagenomics or MG-RAST were tested for the protocol (Additional file 1: Table S1) [16, 28].
Abundance of bacteria in the metagenomes
In order to compare the abundance of bacteria in our datasets, we counted the number of reads recruited to bacterial concatenated contigs from the sequenced metagenomes. The number of reads was calculated using BLASTN, considering a minimum identity of 95% and a maximum e-value of 10−3 for filtering the results. The number of reads recruited per kb of the genome per Gb of the metagenomic dataset (RPKG) was used as a normalized value for comparing abundances.
Results
Some summary statistics for each metagenome are shown in Additional file 1: Table S2. Assuming that a contig’s length is inversely proportional to bacterial diversity, the results of the assembly suggested that SMS is a not a very diverse niche. This observation was also confirmed by calculating alpha diversity indexes (Additional file 1: Table S2).
Skin-mucus microbiome of wild European eel
GC profiles of the SMS-metagenomes from wild eels are shown in Additional file 1: Figure S4. The profiles vary according to waterbody salinity rather than to the eel specific habitat. Thus, wild eel SMS from river (water salinity below 1 g/L) was unimodal with a peak around 65% GC, that from estuarine waters of intermediate salinity (1–3 g/L) was bimodal, with an additional peak around 50% GC, and that from estuarine and wetland waters of higher salinity (7–10 g/L) was again unimodal but with a peak around 45% GC (Additional file 1: Figure S4).
We analyzed the bacterial taxa present in the SMS-metagenomes by using 16S reads (Fig. 1). We found that the SMS-microbiome was dominated by Gammaproteobacteria, whose proportion in the metagenomes ranged from 30% (wild eels from river) to 95% (wild eels from estuarine water of 7 g/L). Flavobacteria (1.6–30%), Betaproteobacteria (2.6–26%), and Alphaproteobacteria (5–24%) were other phyla dominant in the SMS-microbiome (Fig. 1a).
Fig. 1.

Main bacterial classes and genera detected in eel SMS-metagenomes. a Bacterial classes determined by 16S rRNA gene fragments classification in the metagenomics dataset. b Bacterial genera determined by taxonomic classification of all the reads in the metagenomes. Salinity of each habitat is shown at the bottom
The main bacterial genera in the SMS-metagenomes are shown in Fig. 1b and Additional file 1: Figure S5. Results from gene annotation (Fig. 1b) and 16S reads (Additional file 1: Figure S5) were very similar. Remarkably, Vibrio was the dominant genus in the wild eel SMS-microbiome from estuary and wetland (46–93.5%) but decreased significantly in lake and river samples (9.8 and 0.2%, respectively). In these last samples, Pseudomonas (22–38%), Stenotrophomonas (10–14%), and Achromobacter (2–5%) were the most abundant genera (Fig. 1b).
Genome abundance based on metagenomic fragment recruitment showed differences in species composition related to the origin of the sampled eels (waterbody salinity) (Fig. 2). Thus, Vibrio anguillarum, V. metoecus (a recently described species closely related to V. cholerae) [29], and V. cholerae were the most abundant Vibrio species in mucus from waterbodies of 7–10 g/L, 3 g/L, and 1 g/L, respectively (Fig. 2). Vibrio fischeri (currently Aliivibrio fischeri), Vibrio furnissii, and V. vulnificus were also associated to eel mucus in the Nature Parks (Fig. 2). Abundance values for V. vulnificus were very low in all the metagenomes with the exception of WE10 8 (Fig. 2). All the mentioned species, except the symbiotic species Al. fischeri, are well-known human and/or fish pathogens. Among them, V. anguillarum and V. vulnificus are pathogenic for a wide range of teleosts, including eels [30, 31]. In both species, the virulence in fish relies on the virulence plasmids pJM1 and pVvBt2, respectively [32, 33]. However, we did not find any evidence of the presence of any of these plasmids (or their genes) in our metagenomes.
Fig. 2.

Main bacterial species detected in wild eel SMS metagenomes. The abundance of different species in the microbiomes is shown. The result was normalized dividing by the size of the genome (kb) and the dataset (Gb) (RPKG). Aliivibrio (Vibrio) fischeri was considered inside the Vibrio
In parallel to the metagenomic study, we also isolated V. metoecus from SMS of wild eels captured in Ebro-Delta (salinity 3 g/L). We selected the major yellow colony on TCBS agar, which was green on VVM agar (isolate M12v), and sequenced its genome. The average nucleotide identity (ANI) between M12v and V. metoecus RC341 was 98.24%, suggesting that isolate M12v belonged to the species V. metoecus. The comparison of M12v contigs to published genomes of V. metoecus (five strains isolated from a brackish coastal pond on the US east coast [34], as well as four clinical strains) and with that of V. cholerae O1 biovar El Tor N16961, highlighted that M12v shared with V. cholerae O1 but not with V. metoecus (i) two phage genes, zot (Zonula Occludens toxin), belonging to CTXphi and rtsA (encoding a phage protein), belonging to RS1 [35, 36]; (ii) most of the genes present in the pathogenicity island VPI-1, including the loci for the toxin co-regulated pilus (TCP) and the regulator ToxT; (iii) the loci for El Tor-RTX (Repeat in Toxin) biosynthesis, modification, and transport; and finally, (iv) most genes for vibriobactin biosynthesis and transport, including its outer membrane receptor ViuA (Fig. 3). Other genes present in M12v and V. cholerae El Tor but absent in strains of V. metoecus were genes for a nickel transport system, a phosphotransferase system, and a monovalent cation/proton antiporter (Additional file 1: Figure S6). Remarkably, downstream of the TCP locus we found a Type 6 secretion system (T6SS) absent in both V. metoecus strains and V. cholerae El Tor but present in V. cholerae strain 1421-77, a no-O1/no-O139 clinical isolate previously sequenced. Finally, M12v lacked the pathogenicity islands VSP-1 and -2 as the rest of V. metoecus strains and presented some genes of the VPI-2 in common with most of V. metoecus strains (Fig. 3; Additional file 1: Figure S7).
Fig. 3.
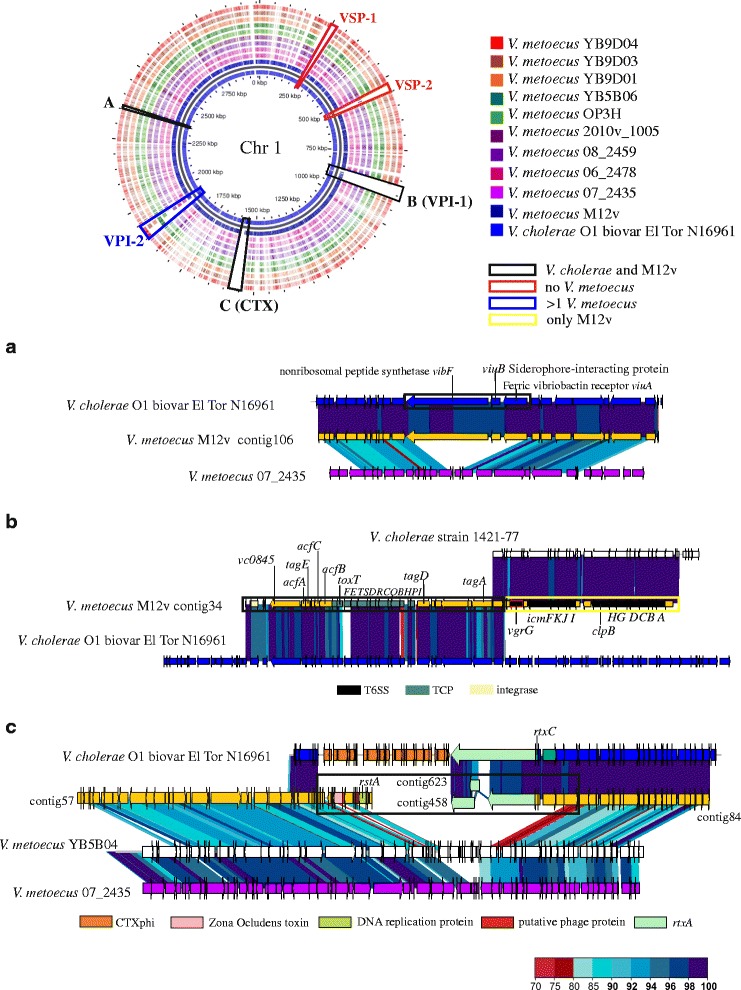
V. metoecus M12v BLAST atlas. Chromosome I of V. cholerae O1 biovar ElTor N16961 (reference) was compared with all available V. metoecus genomes and the strain M12v (sequenced in this study). Each ring represents a single color coded strain. Genomic islands are highlighted. Black boxes represent islands shared by V. cholerae and V. metoecus M12v while blue and red are used when ≥ 1 and 0 V. metoecus strains had the island, respectively. Islands found only in M12v (black box a, b, and c) were plotted using BLASTX against reference and the most similar V. metoecus strain
Other accidental pathogens identified in the SMS-microbiome from wild eels were Pseudomonas aeruginosa, Stenotrophomonas maltophilia, Achromobacter xylosoxidans, and Aeromonas veronii, the last being considered as potential eel pathogen [37].
Mucus-attached vs free-living bacteria
The community composition of the attached mucus microbiome to that of the surrounding water (W) in the case of Ebro Delta ecosystem (water salinity 10 g/L) was quite different in spite of samples being taken at the same time (Fig. 4). Such differences have been previously shown by us in SMS-microbiome of eels from Lake Albufera as well [8]. While Actinobacteria (20%) and Gammaproteobacteria (17%) were the dominant taxa in the W-microbiome, however, the genus compositions of Gammaproteobacteria were quite different (Additional file 1: Figure S8). Only Pseudomonas was present in the same proportions in both metagenomes (around 2%) while Vibrio was remarkably overrepresented in SMS (32 vs 1% in water). The percentage of 16S reads that could not be assigned to any known genera was higher in W-metagenome (82%) than in SMS-metagenome (55%) (Additional file 1: Figure S8).
Fig. 4.

Main bacterial classes detected in eel SMS- and W-metagenomes. High level taxa determined by 16S rRNA gene fragment classification of the two metagenomics datasets. WE10 8 (eel SMS); WE10 8W (water)
We analyzed by culture the waterbody where V. anguillarum was not detected by sequencing in spite of being the dominant species in eel SMS. The most abundant colony recovered on TCBS plates corresponded to V. vulnificus (identification by API20E plus PCR against vvhA). The isolated colonies were identified as belonging to biotypes 1 and 2 because they were yellow on VVM agar [38]. One of the isolates was sequenced to examine presence/absence of virulence genes. We detected an rtxA1 gene that appears to be a new type emerged after hybridization between the two main rtxA1 genes described in V. vulnificus rtxA1 1, present in the most virulent biotype 1 strains and rtxA1 3, and present in biotype 2 strains [39] (Additional file 1: Figure S9).
Functional differences between mucus-attached and free-living microbiome
We analyzed and compared the functional capabilities of SMS- and W-metagenomes by classifying metagenomic reads in functional categories [40] (Additional file 2: Table S1). At the highest classification level, cell-wall/capsule, membrane-transport, virulence/disease/defense, regulation/cell-signaling, iron-acquisition/metabolism, N/S/K-metabolism, and motility/chemotaxis were the over-represented categories in SMS-metagenome (Fig. 5). On the other hand, amino-acids/derivatives, protein metabolism, nucleosides/nucleotides, phages/prophages/transposable-elements/plasmids, and photosynthesis were the over-represented ones in W-metagenome.
Fig. 5.

Differences in functional capacities detected in eel SMS- and W-metagenomes. The presence of each category is presented as percentage of reads assigned from the whole annotated metagenome. WE10 8 (eel SMS); WE10 8W (water).*Category visually over-represented in water; **Category visually over-represented in mucus
Specific searches in the classification for cholera toxin yielded no results in either metagenome. However, Ace (accessory cholera toxin) and ZOT (zonula occludens toxin), both present in CTXphi phage of V. cholerae [41] were found in the SMS-microbiome. Importantly, all types of bacterial secretion systems were overrepresented in mucus samples, e.g., T6SS which is known to be advantageous for bacterial competition [42, 43] (Additional file 1: Figures S10 and S11).
We analyzed in depth the SMS-metagenomes assembled contigs to look for other toxin genes. We found a paralog for rtxA gene in Ali. fischeri. We also found two genes for toxins in two Pseudomonas contigs: ExoU and ToxA which were 100 and 99% identical to their respective copies in P. aeruginosa WH-SGI-V-07317.
Finally, we looked for differences in antibiotic resistance genes between SMS- and W-metagenomes (Fig. 6). Genes for cobalt(Co)-zinc(Zn)-cadmium(Cd), multidrug efflux pumps, copper (Cu), aminoglycoside adenylyltransferases, chromium compounds, fosfomycin resistance, lysozyme inhibitors, MAR (Multiple Antibiotic Resistance) locus, and bile hydrolysis were duplicated in SMS-metagenome (Fig. 6). On the contrary, resistance to fluoroquinolones was clearly dominant in W-metagenome.
Fig. 6.

Abundance of gene categories involved in resistance to antibiotics and toxic compounds in wild eel SMS and W-metagenomes. An asterisk at the top indicates at least two times more abundance in SMS-metagenome than in W-metagenome; two asterisks indicate at least two times more abundance in W-metagenome than in SMS-metagenome; and three asterisks indicate at least two times more abundant in farmed eel SMS than in wild eel SMS
SMS-microbiome from farmed vs wild eels
In order to find differences between the SMS-microbiomes of farmed vs wild eels, we used SMS-metagenome from wild (WE3 8) and farmed eels (FE4 5.3) as they had similar salinities (Table 1). At high taxonomic levels, Alphaproteobacteria was present in higher levels in mucus from farmed eels (Fig. 1a). However, at genus level, Vibrio, Aeromonas, and Xanthomonas were not present in farmed eel, microbiome while Comamonas, Citrobacter, and Chryseobacterium were significantly abundant in farmed eels (Fig. 1b; Additional file 1: Figure S5). Curiously, farmed eels maintained some of the genera found in wild eels from estuary and wetlands, such as Pseudomonas, Acinetobacter, Stenotrophomonas, and Sphingobium in significant proportions, (Additional file 1: Figure S5). This suggests these genera could be part of the eel SMS-resident microbiome.
Moreover, farmed eels microbiome presented less antimicrobial-related genes than wild metagenomes, with only one exception, resistance to Zn and Cd (Fig. 6). In fact, methicillin- and vancomycin-resistance clearly were overrepresented in SMS from wild eels.
MGE in the attached microbiome: the flexible metagenome
We looked for MGE in metagenomes WE3 8 and WE10 8 as well as in nine previously published metagenomes (Additional file 1: Table S1) by using the workflow presented in Additional file 1: Figure S3. PCA analysis from HUP values showed contigs assigned to the same bacterial species but with and without pMGE as clearly distinguishable clusters (Additional file 1: Figures S12-S14). Regarding GC variation, the interchange of DNA material between GC-rich populations was easily detected since GC content of the genera varied from 50 to 70%. For example, Pseudomonas and Sphingobium apparently exchanged long fragments of DNA (> 35 kb) (Fig. 7). However, we could not find inserted sequences looking at the GC content in the samples mainly composed by Vibrio.
Fig. 7.

Horizontal gene transfer of an ICE between different genera. a The contig was compared against the genomes of the most similar strains in NCBI. b The pMGE (putative MGE) was re-annotated and classified as an ICE. Minimum identity of 80 was used to filter the BLASTN results
We found 118 and 12 cpMGE in WE3 8 and WE10 8, respectively, and none in the rest of SMS-metagenomes probably because very few contigs larger than 10 kb were assembled. The protocol was useful to highlight the presence of lytic phages and prophages previously described in these metagenomes (Additional file 1: Figures S12-S14) [12]. Once we identified the cpMGE in the metagenome, we re-annotated the genes inside using HHpred and, then, classified them according to gene content. Additional file 1: Table S3 shows the confirmed contigs and the type of pMGE that contained in each case. Only 34 of the pMGE could be clearly classified: integrative and conjugative elements (ICE), integrons, CRISPR, prophages (described in 43), and transposons. These MGE were detected in the contigs assigned to the dominant genus Vibrio, Pseudomonas, Sphingobium, Achromobacter, Enterobacterium, and Aeromonas (Additional file 1: Table S3 and Figure S15). Most of them encoded genes for colonization and resistance to drugs and heavy metals.
We also identified various MGE in Pseudomonas contigs, which had been previously related to pathogenicity islands encoding two toxins associated with resistance to innate immune system (phagocytosis), the toxins ToxA and ExoU [44] (Additional file 1: Figure S16). The genetic context of both genes in our metagenomes was also compatible with a pathogenicity island.
Finally, we found evidences of exchange of long DNA fragments among bacteria. For example, we found sequences from different genera inserted within Sphingobium and Achromobacter contigs. We focused our attention on one contig, which contained a putative ICE (WE3 8C14) of significant size (47 kb) with a 100% identity and coverage to a contig of Ps. aeruginosa strain AZPAE14707 and AZPAE14724 (Additional file 1: Figure S17). These strains were isolated from the respiratory tract of a patient in Greece and from an intra-abdominal tract infection in Italy, respectively. Moreover, this ICE was highly similar to other Pseudomonas sequences from different ecosystems (Additional file 1: Figure S17). Three genes encoding a multidrug efflux pump were inserted within this ICE. Other interesting contigs were WE3 8C164 and WE3 8C158 that were part of the same ICE in Achromobacter (Additional file 1: Figure S18). Surprisingly, resistance to three different components (mercuric, arsenic, and multidrug efflux pump) were encoded in this MGE. The putative ICE (> 48 kb) separated in these contigs also hit Pseudomonas genomes in the databases including one that also matched the previously mentioned one (WE3 8C11). Pseudomonas, Sphingobium, and Achromobacter have similar GC content, and their abundance in our metagenomes was also similar.
Discussion
The hypothesis underlying this work is that the fish SMS could constitute an appropriate environment for evolution and emergence of new mucosal pathogens of aquatic origin. These mucosal surfaces are exposed to water and are similar in composition, structure, and defense mechanisms to the human intestinal mucosa [45]. As a first approach to test this hypothesis, we have analyzed the microbiome of a euryhaline fish, the European eel, whose surface is covered by a thick layer of mucus [46]. This species inhabits distinct environments, from the open sea to rivers and lakes, and even can be grown in farms.
The eel natural SMS-microbiome was dominated by Gammaproteobacteria while the W-microbiome did not present any clear dominant class being rich in unknown bacteria and, according to [12] also by phages. Further, the natural SMS-microbiome varied in bacterial composition, both qualitative and quantitatively, according to the salinity of the waterbody where eels were captured. Thus, the genus Vibrio was dominant in natural SMS-microbiome from estuarine eels at salinities between 7 and 10 g/L while a mixture of genera predominated in estuarine, lake, and river eels at salinities ≤ 3 g/L. We identified V. anguillarum, V. vulnificus, V. fischeri, V. furnissii, V. metoecus, and V. cholerae as the dominant vibrios in all the eel SMS samples, which suggests that they are an important part of the resident natural microbiome. In general terms, each one of these species of resident vibrios increased its proportion in SMS-microbiome at the salinity values closest to its optimal value for growth [47–49]. Thus, the dominant Vibrio species in natural SMS-microbiome was V. anguillarum at salinities between 7 and 10 g/L, V. metoecus at salinity of 3 g/L, and V. cholerae at salinity of 1 g/L. Interestingly, while V. anguillarum was dominant in the SMS-microbiome, it was not detected in water, suggesting that bacterial species composition in fish SMS is not a simple reflection of that of surrounding water. Instead, it appears that SMS attracts and selectively concentrates specific members of the aquatic microbiome, among which there are multiple vibrios. We hypothesized that such “specialized” bacteria, although could be present in low concentration in the water, because of these “specialized capabilities” would attach to and multiply in the fish SMS, a part of them being finally established as resident microbiota. Supporting this hypothesis, we found that alpha diversity indices decreased four times in the attached microbiome in comparison to the water microbiome.
In parallel, we analyzed by culture the waterbody negative for V. anguillarum contigs in spite of this bacterium being the dominant one in eel SMS-microbiome. The recovered bacteria belonged to V. vulnificus. The genome of the selected isolate lacked the plasmid pVvBt2, essential to cause fish vibriosis [38, 50, 51] and was positive for cellobiose fermentation (biotype 3 strains are negative), and therefore, it corresponded to a biotype 1 strain. The most striking feature of this genome was that it contained a new type of rtxA1. V. vulnificus produces seven types of RTX toxins, all of them classified within the subfamily MARTX (multifunctional autoprocessing repeats-in-toxin) [52]. MARTX present two common external modules, containing the repeated sequences, together with a specific internal module, and containing a unique combination of effector domains responsible for the toxic activity of the protein (nine specific domains have been described) [52]. The new type of MARTXVv (MARTX type VIII) seems to be a hybrid from types I (produced by the most human virulent biotype 1 strains) and type III (biotype 2 strains), both involved in resistance to phagocytosis by immune cells and, at least type III, in resistance to amoeba, one of the main bacterial predators in the environment [53]. It has been proposed that rtxA1 3 emerged by recombination between a new variant of rtxA1, arriving with the plasmid pVvBt2, and the resident chromosomal gene [54]. We proposed that rtxA1 8 could have emerged in the environment by recombination between rtxA1 1 and rtxA1 3 following an HGT event. In any case, this finding strongly supports that new variants of these modular toxins are continuously emerging in the environment, in this case in water.
V. vulnificus biotype 2 is highly virulent for eels and has caused the closure of many eel farms due to massive mortality [55]. To control eel vibriosis, some farmers decided to use freshwater instead of brackish water in farm facilities, but this measure resulted in the emergence of new serovars [56]. Subsequently, most of the intensive eel farms closed and the remaining ones combined low salinity with low pH to eradicate vibriosis. This seems to be quite effective since no Vibrio, including V. vulnificus, was detected in the SMS-microbiome from farmed eels (artificial SMS-microbiome). In fact, the artificial SMS-microbiome was dominated by bacterial species resistant to acid pH such as members of Comamonas, Citrobacter, and Chryseobacterium and, in particular, by Comamonas testosteroni, a bacterium resistant to low pH and antibiotics [57, 58]. This result is quite interesting, because it confirms that the outcome of infectious diseases in aquatic animals in captivity can be modified by a change in a selected water physicochemical parameter(s) (such as pH) that results in a major change in the composition of fish SMS-microbiome.
We also analyzed the differences in heavy metals and drug resistance genes between the artificial and natural SMS-microbiome. Resistance to Co, Zn, and Cd were significantly overrepresented in the artificial SMS-metagenome. Fish farmers frequently use antibiotics and metal containing products to prevent fouling, to feed, and to treat fish in order to limit the spread of infections [59]. The overrepresentation of these resistance genes in mucus from farmed eels stresses the importance of substituting these practices by methods such as immunostimulants in diet or vaccination to prevent not only the disease but also the spread of resistance genes to the environment.
Among the rest of Vibrio pathogenic species found in the wild eel SMS-microbiome, V. metoecus is perhaps the least studied. Originally, V. metoecus was described as a non-virulent clone isolated from Chesapeake Bay (USA) [60]. This species is the closest to V. cholerae. In fact, both species share the aquatic habitat and have been co-isolated from Oyster Ponds in the USA [34]. In this work, we found both species together with other vibrios and when eels were fished in water of 0.3 g/L salinity; V. metoecus was the dominant Vibrio species. This finding suggests that fish SMS constitutes a new environment from which V. metoecus could be co-isolated with V. cholerae, the first species dominating at 0.3 g/L water salinity.
V. metoecus was successfully isolated from SMS-samples, and one strain was sequenced, M12v. The M12v genome showed strong evidences of HGT from V. cholerae O1 ElTor since it contained (i) the VPI-1, an island that encodes the ability to colonize human intestine [61], (ii) some CTXphi and RS1 phage genes [35, 36], (iii) an RTX toxin cluster [62], and (iv) a locus for vibriobactin biosynthesis and transport [63, 64]. In all these traits, our eel mucus isolate was different from the rest of V. metoecus strains, all of them from water and extra-intestinal infections. The presence of all these V. cholerae genes, especially those involved in intestinal colonization (siderophore and TCP), strongly suggests that M12v would be better adapted to mucus than the rest of V. metoecus strains sequenced so far. In consequence, V. metoecus avirulent clones could become virulent by the oral route after acquiring V. cholerae virulence genes when both species co-exist in the same environment and are under selective pressure.
To find out what forces were acting to select the best adapted bacteria or to favor HGT events lending expansion of the best mucus-adapted recombinants, we compared the functionalities of SMS- and W-metagenomes. The genes that were particularly enriched in the SMS allow us to predict the following functional categories as essential for successful mucus colonizers: (i) Biofilm (exopolysaccharide production, sigma-dependent biofilm formation, VieSAB signal transduction system, etc.), (ii) bacterial communication (quorum sensing, autoinducer-2-transport/processing etc.), (iii) bacterial competition (bacteriocin-like peptides, ABC transporter peptide and type T6SS, etc.), (iv) adherence (colonization-factor-antigen-1, curli, accessory-colonization-factors, Campylobacter-adhesion etc.), (v) resistance to humoral innate immunity (including nutritional immunity) (lysozyme resistance, bile hydrolysis, multidrug efflux pumps, siderophore biosynthesis and transport, hemin-uptake, etc.), (vi) resistance to phagocytosis: (RTX toxins, two lytic toxins of Pseudomonas and for types II, III, and IV secretion systems), (vii) resistance to lysis by phages: (CRISPR systems), and (viii) resistance to heavy metals and drugs: Co-Zn-Cd, Cu, multidrug efflux pumps, antibiotic resistance. Remarkably, most of the genes found in each category could be classified within the SMS-virulome and SMS-resistome, respectively.
Regarding the SMS-resistome, this would contain the genes encoding resistance to the heavy metals Co, Zn, Cd, and Cu, which were clearly over-represented in SMS-metagenomes in comparison to water. Although the sampled habitats are protected from human pollution, there are numerous rice fields in the surrounding areas [65, 66], and in consequence, this contamination would have probably derived from agricultural activities. Remarkably, some of these resistances were detected to be encoded in pMGE, mostly ICEs, in the analyzed metagenomes. It has been reported that the mucin of the mucus covering the fish epidermis can bind heavy metals by electrostatic forces and concentrate them on the fish surface [67], which could favor the exchange of these pMGEs by HGT events. Finally, these genes were overrepresented in artificial vs natural SMS-metagenome, confirming the relationship between its frequency in a sample and its degree of contamination.
Curiously, we also found drug-resistance genes that were overrepresented in W-metagenome from Nature Park and that corresponded to resistance to fluoroquinolones. Fluoroquinolones can be excreted by humans and animals into hospital or municipal sewage [68], resist the wastewater treatment plants, and remain for decades in the environment [69–71]. Two large hospitals are located in the surroundings of the Ebro Delta, which was declared protected area in August 1983, what could explain the prevalence of fluoroquinolone resistance genes in the W-metagenome from this location.
With regard to SMS-virulome, it appears to contain all the genes found in our metagenomes for tissue colonization, from genes for bacterial adhesins to genes for resistance to mucosal innate immunity such as those involved in iron uptake. Among them, there were genes encoding cytolytic toxins that could be putatively involved in resistance to phagocytosis as it is MARTXVv type III [53]: (i) an rtxA gene found in Ali. fischeri contigs; (ii) ace (accessory cholera enterotoxin) and zot genes found in V. cholerae contigs, originally described as part of the phage CTXphi; and finally, (iii) exoU and toxA genes found in Pseudomonas contigs, practically identical to their paralogs in P. aeruginosa WH-SGI-V-07317. All these toxins are secreted by different secretion systems, i.e., ToxA and ExoU are injected by T3SS [72]. In accordance, we found that all types of secretion systems were overrepresented in mucus samples, including T6SS, which is known to be essential for bacterial competition in the environment [42, 43]. Interestingly, some of these genes were found to be associated to pMGE suggesting that HGT events occur in mucus under the selective pressure of innate immunity and, probably, natural predators.
When the genes presumptively acquired by our SMS V. metoecus isolate from V. cholerae were compared with the functional categories over-represented in mucus, we found that the cluster of genes for the T6SS, the TCP pilus biogenesis, the vibriobactin biosynthesis and uptake, and the RTX toxin and its transport could be classified in the categories, iii (Bacterial competition), iv (Adherence), v (Resistance to mucosal innate immunity), and vi (Resistance to phagocytosis), respectively. All these findings suggest that fish SMS selects phenotypic traits that favor fitness in mucus and that these traits could be advantageous for these bacteria if they accidentally arrive to the human intestine.
Conclusions
In conclusion, we have obtained multiple evidences from metagenomic, genomic, and culture in favor of the hypothesis on the role of fish SMS as an important niche for mucosal pathogen evolution in nature. First, SMS concentrates bacteria present in water, in particular vibrios; second, selects those bacteria with particular abilities to attach, resist innate immunity, and compete with other bacteria; and third, favors the exchange of genes encoding these functions. Particularly interesting is that we have isolated a new variant of Vibrio with intermediate properties between V. metoecus and V. cholerae O1 El Tor, abilities that are encoded in pathogenicity islands and phages. It appears that genetic exchange takes place primarily in the fish SMS and that this niche, conceivably, provides the selective pressures for acquisition and maintenance of the colonization and competition associated virulence phenotypes, phenotypes that could collaterally mediate bacterial fitness in the human intestine.
Additional files
Metagenomes used to detect MGE. Table S2. General data for each metagenome and alpha diversity. Table S3. Contigs with MGE detected using the methodology described in Fig. 1. Figure S1. Sampling points, location and description. Figure S2. From nature to the laboratory: skin mucus sampling from wild eels and DNA extraction. Figure S3. Mobile genetic elements (MGE) detection workflow diagram. Figure S4. %GC content profiles of the eel’s SMS- and W-metagenomes. Figure S5. Wild eel’s versus farmed eel’s SMS metagenomes. Figure S6. V. metoecus M12v BLAST atlas. Figure S7. Schematic representation of VPI-2 in M12v. Figure S8. Main bacterial genera detected in eel’s SMS- and W-metagenomes. Figure S9. rtxA1 gene comparison. Figure S10. Differences in functional capacities between SMS-associated and water microbiomes. Figure S11. Differences in membrane transport functional categories between SMS-associated and water microbiomes. Figure S12. PCA analysis of hexanucleotide usage pattern (HUP) of water metagenomes. Figure S13. PCA analysis of hexanucleotide usage pattern (HUP) of metagenomes associated to different hosts. Figure S14. Hexanucleotide usage pattern (HUP) distribution of the attached microbiome to epidermal mucus of European eels in WE3 8. Figure S15. Contigs with pMGE. Genes of interest are colored differently. Figure S16. MGE in a contig of Pseudomonas. Figure S17. Distribution of an ICE identified in contigWE3 8C14 between Pseudomonas strains. Figure S18. Exchange of long DNA stretches between genera with similar %GC of the genome. (DOCX 4303 kb)
Abundance and comparison of functions in water and skin-mucus surface microbiomes. (XLSX 646 kb)
Acknowledgements
We would like to express our sincere gratitude to Dr. Karla Satchell for helping in the analysis of rtxA1 gene.
Funding
This work has been financed by grants AGL2014-58933-P and AGL2017-87723-P (both cofunded with FEDER funds) and Programa Consolider-Ingenio 2010 CSD2009-00006 from MICINN. We thank the Servicio de Vida Silvestre (DG de Medio Natural, Conselleria de Infrestructuras, Territorio y Medio Ambiente) for helping in sampling of wild eels. M. Carda-Diéguez thanks MICINN for the Fellowship FPI (BES-2012-052361). RG was supported by the research grant 17-04828S from the Grant Agency of the Czech Republic. FR-V was supported by grant “VIREVO” CGL2016-76273-P [AEI/FEDER, EU], (cofunded with FEDER funds) Acciones de dinamización “REDES DE EXCELENCIA” CONSOLIDER- CGL2015-71523-REDC from the Spanish Ministerio de Economía y Competitividad and PROMETEO II/2014/012 “AQUAMET” from Generalitat Valenciana.
Availability of data and materials
The datasets analyzed during the current study are available at NCBI SRA database with the following accession codes: SRX702744, SRX702748, SRX702749, SRX703649, SRX703655, SRX703656, SRX710670, and SRX710703 (https://www.ncbi.nlm.nih.gov/bioproject/261319).
Abbreviations
- cpMGE
Contigs identified as putative mobile genetic elements
- HGT
Horizontal gene transfer
- HUP
Hexanucleotide usage pattern
- ICE
Integrative and conjugative element
- MARTX
Multifunctional autoprocessing repeats-in-toxin
- MGE
Mobile genetic elements
- PBS
Phosphate buffer saline
- PCA
Principal component analysis
- RPKG
Reads recruited per kb of the genome per Gb of the metagenomic dataset
- sgMGE
Signature genes for mobile genetic elements
- SMS
Skin-mucosal-surface
- T6SS
Type 6 secretion system
- TCBS
Thiosulfate-citrate-bile salts-sucrose
- TCP
Toxin co-regulated pilus
- VVM
Vibrio vulnificus medium
- W
Water
Authors’ contributions
CA and FRV planned the study. MCD was responsible of the sampling. MCD and RG analyzed the sequencing results. CA, FRV, RG, and FRV interpreted the data. MCD drafted the manuscript. RG, CA, and FRV revised the manuscript critically, and CA was in charge of the final preparation and submission. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s40168-017-0376-1) contains supplementary material, which is available to authorized users.
References
- 1.Gomez D, Sunyer JO, Salinas I. The mucosal immune system of fish: the evolution of tolerating commensals while fighting pathogens. Fish Shellfish Immunol. 2013;35:1729–1739. doi: 10.1016/j.fsi.2013.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pérez T, Balcázar JL, Ruiz-Zarzuela I, Halaihel N, Vendrell D, de Blas I, et al. Host-microbiota interactions within the fish intestinal ecosystem. Mucosal Immunol. 2010;3:355–360. doi: 10.1038/mi.2010.12. [DOI] [PubMed] [Google Scholar]
- 3.Gómez GD, Balcázar JL. A review on the interactions between gut microbiota and innate immunity of fish. FEMS Immunol Med Microbiol. 2008;52:145–154. doi: 10.1111/j.1574-695X.2007.00343.x. [DOI] [PubMed] [Google Scholar]
- 4.Shapiro BJ, Levade I, Kovacikova G, Taylor RK, Almagro-Moreno S. Origins of pandemic Vibrio cholerae from environmental gene pools. Nat Microbiol. 2016;2:16240. doi: 10.1038/nmicrobiol.2016.240. [DOI] [PubMed] [Google Scholar]
- 5.Jackson RW, Vinatzer B, Arnold DL, Dorus S, Murillo J. The influence of the accessory genome on bacterial pathogen evolution. Mob Genet Elements. 2011;1:55–65. doi: 10.4161/mge.1.1.16432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ochman H, Moran NA. Genes lost and genes found: evolution of bacterial pathogenesis and symbiosis. Science. 2001;292:1096–1099. doi: 10.1126/science.1058543. [DOI] [PubMed] [Google Scholar]
- 7.Tsukamoto K. Oceanic biology: spawning of eels near a seamount. Nature. 2006;439:929. doi: 10.1038/439929a. [DOI] [PubMed] [Google Scholar]
- 8.Carda-Dieguez M, Ghai R, Rodriguez-Valera F, Amaro C. Metagenomics of the mucosal microbiota of European eels. Genome Announc. 2014;2:e01132–e01114. doi: 10.1128/genomeA.01132-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghai R, Hernandez CM, Picazo A, Mizuno CM, Ininbergs K, Díez B, et al. Metagenomes of Mediterranean coastal lagoons. Sci Rep. 2012 [cited 2013 Dec 11];2:490. A. [DOI] [PMC free article] [PubMed]
- 10.Cox MP, Peterson DA, Biggs PJ. SolexaQA: at-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics. 2010;11:485. doi: 10.1186/1471-2105-11-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008 [cited 2014 Jan 9];18:821–9. [DOI] [PMC free article] [PubMed]
- 12.Carda-Diéguez M, Mizuno CM, Ghai R, Rodriguez-Valera F, Amaro C. Replicating phages in the epidermal mucosa of the eel (Anguilla anguilla) Front Microbiol. 2015;6:3. doi: 10.3389/fmicb.2015.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rice P, Longden I, Bleasby A. EMBOSS: the European molecular biology open software suite. Trends Genet. 2000 [cited 2013 Dec 27];16:2–3. [DOI] [PubMed]
- 14.Hall T. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999 [cited 2013 Dec 27];
- 15.Hyatt D, Chen G-L, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meyer F, Paarmann D, D’Souza M, Olson R, Glass EM, Kubal M, et al. The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 2008 [cited 2014 Jan 21];9:386. [DOI] [PMC free article] [PubMed]
- 17.Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004 [cited 2013 Dec 16];32:D277–80. [DOI] [PMC free article] [PubMed]
- 18.Carver TJ, Rutherford KM, Berriman M, Rajandream M-A, Barrell BG, Parkhill J. ACT: the Artemis comparison tool. Bioinformatics 2005 [cited 2013 Dec 13];21:3422–3. [DOI] [PubMed]
- 19.Söding J. Protein homology detection by HMM-HMM comparison. Bioinformatics 2005 [cited 2014 Jul 17];21:951–60. [DOI] [PubMed]
- 20.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huson DH, Mitra S, Ruscheweyh H-J, Weber N, Schuster SC. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011 [cited 2014 Jan 21];21:1552–60. [DOI] [PMC free article] [PubMed]
- 22.Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009 [cited 2013 Dec 15];37:D141–5. [DOI] [PMC free article] [PubMed]
- 23.Cerdà-Cuéllar M, Jofre J, Blanch AR. A selective medium and a specific probe for detection of Vibrio vulnificus. Appl Environ Microbiol. 2000;66:855–859. doi: 10.1128/AEM.66.2.855-859.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanjuan E, Amaro C. Multiplex PCR assay for detection of Vibrio vulnificus biotype 2 and simultaneous discrimination of Serovar E strains. Appl Environ Microbiol. 2007;73:2029–2032. doi: 10.1128/AEM.02320-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grant JR, Arantes AS, Stothard P. Comparing thousands of circular genomes using the CGView Comparison Tool. BMC Genomics. 2012;13:202. doi: 10.1186/1471-2164-13-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siguier P. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006;34:D32–D36. doi: 10.1093/nar/gkj014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kichenaradja P, Siguier P, Perochon J, Chandler M. ISbrowser: an extension of ISfinder for visualizing insertion sequences in prokaryotic genomes. Nucl Acids Res. 2010;38:D62–D68. doi: 10.1093/nar/gkp947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hunter S, Corbett M, Denise H, Fraser M, Gonzalez-Beltran A, Hunter C, et al. EBI metagenomics—a new resource for the analysis and archiving of metagenomic data. Nucleic Acids Res. 2014;42:D600–D606. doi: 10.1093/nar/gkt961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirchberger PC, Turnsek M, Hunt DE, Haley BJ, Colwell RR, Polz MF, et al. Vibrio metoecus sp. nov., a close relative of Vibrio cholerae isolated from coastal brackish ponds and clinical specimens. Int J Syst Evol Microbiol. 2014;64:3208–14. [DOI] [PubMed]
- 30.Amaro C, Biosca EG. Vibrio vulnificus biotype 2, pathogenic for eels, is also an opportunistic pathogen for humans. Appl Environ Microbiol. 1996;62:1454–1457. doi: 10.1128/aem.62.4.1454-1457.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frans I, Michiels CW, Bossier P, Willems KA, Lievens B, Rediers H. Vibrio anguillarum as a fish pathogen: virulence factors, diagnosis and prevention. J Fish Dis. 2011;34:643–661. doi: 10.1111/j.1365-2761.2011.01279.x. [DOI] [PubMed] [Google Scholar]
- 32.Valiente E, Lee C-T, Hor L-I, Fouz B, Amaro C. Role of the metalloprotease Vvp and the virulence plasmid pR99 of Vibrio vulnificus serovar E in surface colonization and fish virulence. Environ Microbiol. 2008 [cited 2014 Nov 26];10:328–38. [DOI] [PubMed]
- 33.Naka H, Dias GM, Thompson CC, Dubay C, Thompson FL, Crosa JH. Complete genome sequence of the marine fish pathogen Vibrio anguillarum harboring the pJM1 virulence plasmid and genomic comparison with other virulent strains of V. anguillarum and V. ordalii. Infect Immun. 2011 [cited 2014 Oct 20];79:2889–900. [DOI] [PMC free article] [PubMed]
- 34.Orata FD, Kirchberger PC, Méheust R, Barlow EJ, Tarr CL, Boucher Y. The dynamics of genetic interactions between Vibrio metoecus and Vibrio cholerae, two close relatives co-occurring in the environment. Genome Biol. Evol. 2015;7:2941–2954. doi: 10.1093/gbe/evv193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Waldor MK, Mekalanos JJ. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science. 1996;272:1910–1914. doi: 10.1126/science.272.5270.1910. [DOI] [PubMed] [Google Scholar]
- 36.Faruque SM, Asadulghani, Kamruzzaman M, Nandi RK, Ghosh AN, Nair GB, et al. RS1 element of Vibrio cholerae can propagate horizontally as a filamentous phage exploiting the morphogenesis genes of CTXphi. Infect Immun. 2002;70:163–170. doi: 10.1128/IAI.70.1.163-170.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yi S-W, You M-J, Cho H-S, Lee C-S, Kwon J-K, Shin G-W. Molecular characterization of Aeromonas species isolated from farmed eels (Anguilla japonica). Vet Microbiol. 2013;164:195–200. [DOI] [PubMed]
- 38.Bisharat N, Agmon V, Finkelstein R, Raz R, Ben-Dror G, Lerner L, et al. Clinical, epidemiological, and microbiological features of Vibrio vulnificus biogroup 3 causing outbreaks of wound infection and bacteraemia in Israel. Israel Vibrio Study Group. Lancet. 1999;354:1421–1424. doi: 10.1016/S0140-6736(99)02471-X. [DOI] [PubMed] [Google Scholar]
- 39.Roig FJ, González-Candelas F, Amaro C. Domain organization and evolution of multifunctional autoprocessing repeats-in-toxin (MARTX) toxin in Vibrio vulnificus. Appl Environ Microbiol. 2011;77:657–668. doi: 10.1128/AEM.01806-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang H-Y, Cohoon M, et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005;33:5691–5702. doi: 10.1093/nar/gki866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dalsgaard A, Serichantalergs O, Forslund A, Lin W, Mekalanos J, Mintz E, et al. Clinical and environmental isolates of Vibrio cholerae serogroup O141 carry the CTX phage and the genes encoding the toxin-coregulated pili. J Clin Microbiol. 2001;39:4086–4092. doi: 10.1128/JCM.39.11.4086-4092.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma AT, Mekalanos JJ. In vivo actin cross-linking induced by Vibrio cholerae type VI secretion system is associated with intestinal inflammation. Proc Natl Acad Sci U S A. 2010 [cited 2013 Nov 14];107:4365–70. [DOI] [PMC free article] [PubMed]
- 43.Hachani A, Lossi NS, Hamilton A, Jones C, Bleves S, Albesa-Jové D, et al. Type VI secretion system in Pseudomonas aeruginosa: secretion and multimerization of VgrG proteins. J Biol Chem. 2011 [cited 2013 Nov 14];286:12317–27. [DOI] [PMC free article] [PubMed]
- 44.Sato H, Frank DW. Intoxication of host cells by the T3SS Phospholipase ExoU: PI(4,5)P2-associated, cytoskeletal collapse and late phase membrane blebbing. Samuel JE, editor. PLoS One. 2014;9:e103127. doi: 10.1371/journal.pone.0103127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ángeles EM. An overview of the immunological defenses in fish skin. ISRN Immunol. 2012;2012:1–29. doi: 10.5402/2012/853470. [DOI] [Google Scholar]
- 46.van Ginneken VJT, Maes GE. The European eel (Anguilla anguilla, Linnaeus), its lifecycle, evolution and reproduction: a literature review. Rev Fish Biol Fish. 2005;15:367–98.
- 47.Bordas MA, Balebona MC, Rodriguez-Maroto JM, Borrego JJ, Morinigo MA. Chemotaxis of pathogenic Vibrio strains towards mucus surfaces of gilt-head sea bream (Sparus aurata L.) Appl. Environ. Microbiol. 1998;64:1573–1575. doi: 10.1128/aem.64.4.1573-1575.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singleton FL, Attwell R, Jangi S, Colwell RR. Effects of temperature and salinity on Vibrio cholerae growth. Appl Environ Microbiol. 1982;44:1047–1058. doi: 10.1128/aem.44.5.1047-1058.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singleton FL, Attwell RW, Jangi MS, Colwell RR. Influence of salinity and organic nutrient concentration on survival and growth of Vibrio cholerae in aquatic microcosms. Appl Environ Microbiol. 1982;43:1080–1085. doi: 10.1128/aem.43.5.1080-1085.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Amaro C, Biosca EG, Fouz B, Alcaide E, Esteve C. Evidence that water transmits Vibrio vulnificus biotype 2 infections to eels. Appl Environ Microbiol. 1995;61:1133–1137. doi: 10.1128/aem.61.3.1133-1137.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tison DL, Nishibuchi M, Greenwood JD, Seidler RJ. Vibrio vulnificus biogroup 2: new biogroup pathogenic for eels. Appl Environ Microbiol. 1982;44:640–646. doi: 10.1128/aem.44.3.640-646.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Satchell KJF. Multifunctional-autoprocessing repeats-in-toxin (MARTX) Toxins of Vibrios. Microbiol Spectr. 2015;3. doi:10.1128/microbiolspec.VE-0002-2014. [DOI] [PMC free article] [PubMed]
- 53.Lee C-T, Pajuelo D, Llorens A, Chen Y-H, Leiro JM, Padrós F, et al. MARTX of Vibrio vulnificus biotype 2 is a virulence and survival factor. Environ Microbiol. 2013 [cited 2013 Oct 31];15:419–32. [DOI] [PubMed]
- 54.Kwak JS, Jeong H-G, Satchell KJF. Vibrio vulnificus rtxA1 gene recombination generates toxin variants with altered potency during intestinal infection. Proc Natl Acad Sci U S A. 2011;108:1645–1650. doi: 10.1073/pnas.1014339108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Amaro C, Sanjuán E, Fouz B, Pajuelo D, Lee C-T, Hor L-I, et al. The fish pathogen Vibrio vulnificus biotype 2: epidemiology, phylogeny, and virulence factors involved in warm-water vibriosis. Microbiol Spectr. 2015;3:1–23. [DOI] [PubMed]
- 56.Fouz B, Larsen JL, Amaro C. Vibrio vulnificus serovar A: an emerging pathogen in European anguilliculture. J Fish Dis. 2006;29:285–291. doi: 10.1111/j.1365-2761.2006.00719.x. [DOI] [PubMed] [Google Scholar]
- 57.Oppermann UC, Belai I, Maser E. Antibiotic resistance and enhanced insecticide catabolism as consequences of steroid induction in the gram-negative bacterium Comamonas testosteroni. J Steroid Biochem Mol Biol. 1996;58:217–223. doi: 10.1016/0960-0760(96)00021-0. [DOI] [PubMed] [Google Scholar]
- 58.Ma Y-F, Zhang Y, Zhang J-Y, Chen D-W, Zhu Y, Zheng H, et al. The complete genome of Comamonas testosteroni reveals its genetic adaptations to changing environments. Appl Environ Microbiol. 2009 [cited 2013 Dec 21];75:6812–9. [DOI] [PMC free article] [PubMed]
- 59.Romero J, Gloria C and Navarrete P. Antibiotics in Aquaculture – Use, Abuse and Alternatives, Health and Environment in Aquaculture, Dr. Edmir Carvalho (Ed.), InTech. 2012.
- 60.Haley BJ, Grim CJ, Hasan NA, Choi S, Chun J, Brettin TS, et al. Comparative genomic analysis reveals evidence of two novel Vibrio species closely related to V. cholerae. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Karaolis DK, Johnson JA, Bailey CC, Boedeker EC, Kaper JB, Reeves PR. A Vibrio cholerae pathogenicity island associated with epidemic and pandemic strains. Proc Natl Acad Sci U S A. 1998;95:3134–3139. doi: 10.1073/pnas.95.6.3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dolores J, Satchell KJF. Analysis of Vibrio cholerae genome sequences reveals unique rtxA variants in environmental strains and an rtxA-null mutation in recent altered El Tor isolates. MBio. 2013;4:e00624. doi: 10.1128/mBio.00624-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wyckoff EE, Stoebner JA, Reed KE, Payne SM. Cloning of a Vibrio cholerae vibriobactin gene cluster: identification of genes required for early steps in siderophore biosynthesis. J Bacteriol. 1997;179:7055–7062. doi: 10.1128/jb.179.22.7055-7062.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stoebner JA, Butterton JR, Calderwood SB, Payne SM. Identification of the vibriobactin receptor of Vibrio cholerae. J Bacteriol. 1992;174:3270–4. [DOI] [PMC free article] [PubMed]
- 65.Canet R, Chaves C, Pomares F, Albiach R. Agricultural use of sediments from the Albufera Lake (eastern Spain) Agric Ecosyst Environ. 2003;95:29–36. doi: 10.1016/S0167-8809(02)00171-8. [DOI] [Google Scholar]
- 66.Suárez-Serrano A, Alcaraz C, Ibáñez C, Trobajo R, Barata C. Procambarus clarkii as a bioindicator of heavy metal pollution sources in the lower Ebro River and Delta. Ecotoxicol Environ Saf. 2010;73:280–286. doi: 10.1016/j.ecoenv.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 67.Coello WF, Khan MA. Protection against heavy metal toxicity by mucus and scales in fish. Arch Environ Contam Toxicol. 1996;30:319–326. doi: 10.1007/BF00212289. [DOI] [PubMed] [Google Scholar]
- 68.Kaplan E, Ofek M, Jurkevitch E, Cytryn E. Characterization of fluoroquinolone resistance and qnr diversity in Enterobacteriaceae from municipal biosolids. Front Microbiol. 2013;4 [DOI] [PMC free article] [PubMed]
- 69.Weber W, Henkes H, Möller P, Bade K, Kühne D. Toxic spongiform leucoencephalopathy after inhaling heroin vapour. Eur Radiol. 1998;8:749–755. doi: 10.1007/s003300050467. [DOI] [PubMed] [Google Scholar]
- 70.Córdova-Kreylos AL, Scow KM. Effects of ciprofloxacin on salt marsh sediment microbial communities. ISME J. 2007;1:585–595. doi: 10.1038/ismej.2007.71. [DOI] [PubMed] [Google Scholar]
- 71.Kümmerer K. Antibiotics in the aquatic environment—a review—part I. Chemosphere. 2009;75:417–434. doi: 10.1016/j.chemosphere.2008.11.086. [DOI] [PubMed] [Google Scholar]
- 72.Michalska M, Wolf P. Pseudomonas exotoxin a: optimized by evolution for effective killing. Front Microbiol. 2015;6:963. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Metagenomes used to detect MGE. Table S2. General data for each metagenome and alpha diversity. Table S3. Contigs with MGE detected using the methodology described in Fig. 1. Figure S1. Sampling points, location and description. Figure S2. From nature to the laboratory: skin mucus sampling from wild eels and DNA extraction. Figure S3. Mobile genetic elements (MGE) detection workflow diagram. Figure S4. %GC content profiles of the eel’s SMS- and W-metagenomes. Figure S5. Wild eel’s versus farmed eel’s SMS metagenomes. Figure S6. V. metoecus M12v BLAST atlas. Figure S7. Schematic representation of VPI-2 in M12v. Figure S8. Main bacterial genera detected in eel’s SMS- and W-metagenomes. Figure S9. rtxA1 gene comparison. Figure S10. Differences in functional capacities between SMS-associated and water microbiomes. Figure S11. Differences in membrane transport functional categories between SMS-associated and water microbiomes. Figure S12. PCA analysis of hexanucleotide usage pattern (HUP) of water metagenomes. Figure S13. PCA analysis of hexanucleotide usage pattern (HUP) of metagenomes associated to different hosts. Figure S14. Hexanucleotide usage pattern (HUP) distribution of the attached microbiome to epidermal mucus of European eels in WE3 8. Figure S15. Contigs with pMGE. Genes of interest are colored differently. Figure S16. MGE in a contig of Pseudomonas. Figure S17. Distribution of an ICE identified in contigWE3 8C14 between Pseudomonas strains. Figure S18. Exchange of long DNA stretches between genera with similar %GC of the genome. (DOCX 4303 kb)
Abundance and comparison of functions in water and skin-mucus surface microbiomes. (XLSX 646 kb)
Data Availability Statement
The datasets analyzed during the current study are available at NCBI SRA database with the following accession codes: SRX702744, SRX702748, SRX702749, SRX703649, SRX703655, SRX703656, SRX710670, and SRX710703 (https://www.ncbi.nlm.nih.gov/bioproject/261319).