

Abstract
Background
Vectors are widely used to drive gene expression using a promoter. However, not all promoters are able to drive ectopic gene expression efficiently, including CMV promoter. Here, we report our data using CMV promoter for high-level gene expression in a B lymphoma cell line DG75.
Material/Methods
A plasmid (pcDNA3.1(+)) containing the CD21 gene driven under CMV promoter was constructed. The plasmid was stably transfected into a human B lymphoma cell line DG75 for cellular surface CD21 expression, and flow cytometry was used to monitor CD21 expression. CD21+ cells in the stable cell line were purified using anti-CD21 antibody-coupled Dynabeads for CD21-mediated antigen presentation experiment.
Results
The percentage of CD21+ cells in newly generated stable DG75pcDNA3.1(+)CD21 cells was only 6.5% as determined by flow cytometry, which was unexpected and did not fit the requirements for further experiments. However, CD21+ cells could be purified to 100% using antiCD21 antibody-coupled beads. The percentage of CD21+ cells in purified cells can be kept at 95%, 82%, 42%, 15%, and 42% at 7 d, 14 d, 34 d, and 42 d after purification, respectively. Specific T cell response against CD21mediated antigen presentation can be activated successfully only when surface CD21 expression remains high.
Conclusions
A commonly down-regulated CMV promoter can be used to drive ectopic gene expression at a high-level in stable cell lines. Our results should facilitate future experimental design using other down-regulated promoters containing vectors such as SV40 and PGK1.
MeSH Keywords: Burkitt Lymphoma; Cell Line; Down-Regulation; Gene Expression; Promoter Regions, Genetic
Background
Vectors are widely used to deliver ectopic genes into cells in vitro and in vivo, and the ectopic gene expression efficiency varies depending on cell types and utilized vectors [1–3]. Many attempts had been made to improve ectopic gene expression efficiency, such as constructing vectors using different transcriptional regulatory elements [4,5], or using different vectors such as viral or nonviral vectors [6]; however, no vector has been found to be able to achieve efficient ectopic gene expression in all situations, and some commonly used promoters in the vector were found to be frequently suppressed in the transfected cells [7]. Thus, vectors with different promoters may have to be evaluated in different settings when designing scientific experiments.
Human cytomegalovirus (CMV) is a Herpes virus that can infect various human cell types such as epithelial cells, endothelial cells, fibroblasts, smooth muscle cells, connective tissue cells, macrophages, dendritic cells, and lymphocytes [8,9]. During productive infection, CMV genes are expressed from immediate early (IE) genes to early genes and then to late genes in a coordinated order, with gene expression kicking off by CMV IE promoter with assistance from its proximal and distal enhancer [10].
Thus, CMV IE promoter together with enhancer is widely used as a constitutive promoter (often abbreviated as CMV promoter) to drive gene expression in a variety of cell types [8,11,12]. However, the strength of the target gene expression driven by CMV promoter varies depending on cell types; for example, CMV promoter driven-green fluorescence protein (GFP) signal was strong in human embryonic kidney cells (293T) and human fibrosarcoma cells (HT1080), while it was weak in fibroblasts (MRC5) [1] and inactivated in mouse embryonic stem cells (D3 and J1) [2]. Xia et al. showed that the reporter gene was shut off in more than 95% of targeted cells under CMV promoter in human embryonic stem cells, and which no experiment could be performed using such a gene expression system [7].
Here, we report our data using a CMV promoter-driving ectopic gene expression system in a cell line derived from human B lymphoma cells. The ectopic gene was cloned into a pcDNA3.1(+) plasmid under CMV promoter, and the new plasmid was transfected into the target DG75 cells for stable cell line generation under antibiotic selection. Finally, stably transfected DG75 cells were able to be purified and monitored using anti-ectopic gene antibody as the ectopic gene product as a cellular surface molecule. By using the steps mentioned above, a timeline for high-level ectopic gene expression was be established using CMV promoter; therefore, the experiment can be performed during this conditional timeline when the expression level of the ectopic gene remains high. This method could be used in similar experimental settings to improve ectopic surface molecule or selectable intracellular molecule expression.
Material and Methods
Cell culture
Cell cultures were maintained in a HERA cell 150 incubator (Thermo Scientific) with constant 5% CO2 and 37°C temperature under humidified conditions. Cell handling was done in a Herasafe KS12 Safety Cabinet (Thermo Electron Corporation) laminar flow workstation. Cell lines including DG75 [13] and HEK293 cells [11] were maintained in RPMI medium supplemented with 10% fetal bovine serum (FBS) and were split 1: 10 twice per week. CD4+ T cells were maintained as described before [14], and stable cell lines were maintained as described below in the stable transfection section.
CD21 cloning
To stably transfect a CD21 expression plasmid into the DG75 cell line, a plasmid expressing CD21 was generated. The CD21 gene was cloned into the pcDNA3.1(+) plasmid under the control of a CMV promoter using the neomycin resistance gene as a selection marker. The CD21 gene (together with a signal peptide) was cut out using the EcoRI cutting site from a CD21 expression plasmid, generated from the laboratory as described previously [15]. This was done by partial digestion with EcoRI (Fermentas) (because there is also 1 EcoRI cutting site within the CD21 gene itself) at 37°C for 4 min (5 μg DNA in 5 reaction tubes were loaded with 10, 3, 1, 0.3, and 0.1 U EcoRI, respectively). The insert was cut out of the agarose gel after gel electrophoresis (intact CD21 gene is 3165 bp compared to other completely digested fragments of 4927, 1721 and 1444 bp) and purified by using phenol and butanol. The pcDNA3.1(+) vector was cut by the EcoRI endonuclease restriction enzyme. The obtained insert and the vector were ligated by T4 ligase and transformed into a competent E. coli DH5α strain. The plasmid DNA prepared by the boiling Mini-preparation from the transformed DH5a bacteria was analyzed by using BamHI and EcoRI endonuclease restriction enzymes and thus the CD21-containing clones were identified. The new CD21-containing plasmid contained a neomycin resistance gene, which was used as a selection marker following stable transfection. The plasmid was tested in 293 cells by transient transfection for CD21 expression, which was further confirmed by immunostaining.
Stable transfection
Stable transfection was performed to introduce CD21 into DG75 cells using electroporation. Briefly, 20 μg pcDNA3.1(+)-CD21 was combined with 3×106 DG75 cells in RPMI 1640/10% FBS on ice, and the mixture was electroporated (260 Voltage, 1050 μF) for 1 min using gene pulser II (Bio-Rad). At 3 days after transfection, the culture medium was exchanged for the selection medium, which was the same medium but containing 800 μg/ml G418 (Geneticin; Gibco), which was subsequently changed every 3–4 d. The concentration of G418 in the selection medium was reduced to 400 μg/ml after establishment of the stable cell lines was confirmed. The pcDNA3.1(+) empty control plasmid was transfected into DG75 cells in parallel as negative control.
CD21+ DG75 cells selection
To obtain a pure population of CD21+ DG75 cells and perform T cell recognition assays, CD21+ DG75 cells were selected 1 week prior to the assay. Selection was performed by incubating DG75-pcDNA3.1(+)-CD21 cells with mouse anti-human CD21 antibody (Santa Cruz, #sc-18857) at 1 μg per 1×106 cells and subsequently extracting the bound cells using anti-mouse IgG Dynabeads (Invitrogen). The selected CD21+ cells were cultured for an additional 3–5 days to amplify the CD21+ population, and subsequently the Dynabead-bound cells were removed by negative selection under a Dynal single-tube MPC 1 magnetic particle concentrator (Invitrogen) and the unbound cells were washed and collected for further culture. The purity of the selected CD21+ DG75 cells was confirmed by FACS (fluorescence-activated cell sorting, BD Biosciences) analysis.
Immunostaining
To determine whether CD21 gene was successfully cloned into the pcDNA3.1(+) plasmid, immunostaining was performed. Briefly, cells (approx. 1–2×106) were harvested and resuspended in 200 μl PBS, and 20 μl cells were dropped onto each well of an 8-well microscope glass slide (Medco). After 5–10 s, excess liquid was removed and cells on each well were air-dried. Slides were fixed in Acetone (Sigma-Aldrich) for 20 min followed by 30-min incubation at 37°C with 20 μl primary antibody (1.3 μg/ml, undiluted or antibody transfections diluted in RPMI 1640+10% FBS medium) per slide. After washing the slide 2–5 min with PBS, a secondary antibody conjugated to an immunofluorescent dye (Goat anti-Mouse IgG-Cy3, Dianova), which was diluted 1: 300 in PBS previously, was added to the slide for 30 min at 37°C. The slide was then washed 2–5 min with PBS again. A glass coverslip was mounted with 90% glycerol. The slide was observed under a Leica DM5000B fluorescent microscope and stored at 4°C.
Flow cytometry assay
To assess the expression of CD21 on the cell surface, a flow cytometry assay was performed. Cells were handled in a 1.5-ml Eppendorf tube. All the procedures were done on ice. Briefly, 1×106 cells/samples were washed in PBS. For the detection of CD21, cells were incubated with primary (unconjugated) anti-CD21 antibody (1.3 μg/ml) for 20 min, washed twice with PBS, then incubated with PE-labeled secondary antibody (anti-mouse IgG-PE, Jackson Immunoresearch #115–116-068, diluted 1: 100 in PBS) for 30 min. Cells were then washed and filtered into polystyrene round-bottom 12×75 mm BD Falcon tubes (the filter was provided with the BD Falcon tube), and, finally, cells were counted on a BD FACSCalibur flow cytometer. BD CellQuest Pro software was used to analyze the cell populations (BD Biosciences).
Results
Generation of CD21 expression plasmid pcDNA3.1(+)-CD21 and its functional testing
To generate the CD21 expression plasmid, the DNA fragment of CD21 from a plasmid expressing CD21, which was generated previously [15], was cloned into a pcDNA3.1(+) plasmid under a CMV promoter with a neomycin gene under a SV40 (Simian virus 40) promoter (Figure 1A). The CD21 gene-containing plasmid was analyzed and selected by using BamHI (Figure 1B). The obtained pcDNA3.1(+)-CD21 plasmid was then tested in 293 cells for CD21 expression by transient transfection, which showed a strong positive signal by immunostaining at day 3 after transfection (Figure 2A).
Figure 1.
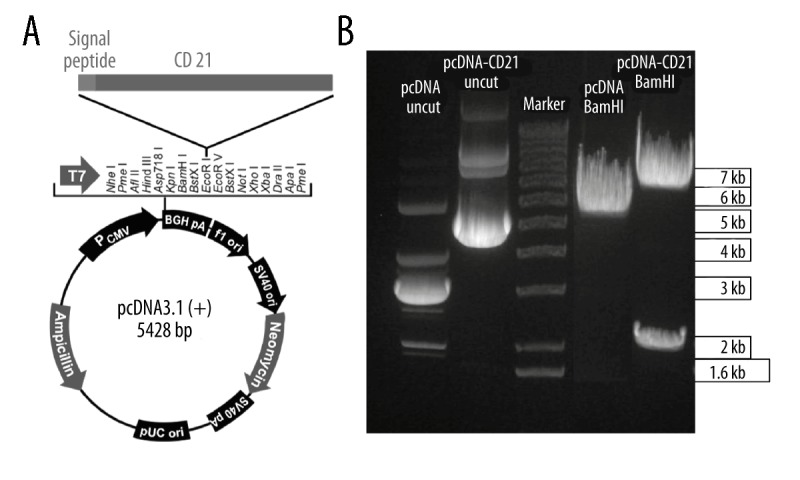
Constructing a CD21-expressing plasmid under CMV promoter. (A) The human CD21 gene, together with a signal peptide, was cloned into the pcDNA3.1(+) (short as pcDNA) expression vector under a CMV promoter. Picture was modified from the plasmid map of Invitrogen. (B) The generated CD21 expression plasmid pcDNA3.1(+)-CD21(abbreviated as pcDNA-CD21) was confirmed by endonuclease restriction enzyme BamHI, and the uncut pcDNA and pcDNA-CD21 plasmids were used as control, the pcDNA and pcDNA-CD21 were cut at 37°C for 1 h using BamHI, and corresponding predicted bands were 5428 bp for pcDNA and 6596 bp+2001 bp for pcDNA-CD21, which were shown in the gel.
Figure 2.

Expression of CD21 on DG75 cells using generated pcDNA3.1(+)-CD21. (A) The generated CD21 expression plasmid (DG75-pcDNA3.1(+)-CD21) was tested by transient expression of 293 cells for 3 d, which was confirmed by immunostaining. LCLs and DG75 cells were used as positive and negative controls for CD21 expression, respectively. Untransfected 293 cells were also compared in parallel as a negative control. Both immunofluorescent and phase-contrast pictures were taken, and scale bars represent 100 μm. (B) Stable transfection of DG75 cells with pcDNA3.1(+)-CD21 plasmid was performed by electroporation. FACS analysis of untransfected cells (DG75) and cells transfected with pcDNA3.1(+)-CD21 plasmid (DG75-pcDNA3.1(+)-CD21) or empty vector (DG75-pcDNA3.1(+)), the expression of CD21 was tested after establishment of the stable cell line, and the selected CD21+ DG75 was confirmed by FACS analysis. The grey shading represents unstained samples alone, the black line represents samples stained with secondary fluorescent antibody alone, and the light dark line represents samples stained with anti-CD21 antibody coupled with secondary fluorescent antibody. pcDNA3.1(+) is shortened to pcDNA for convenience.
Stable transfection of pcDNA3.1(+)-CD21 into DG75 cell line
To stably express CD21 in DG75 cells, pcDNA3.1(+)-CD21 plasmid was transfected into DG75 by using electroporation, and a pcDNA3.1(+)-CD21-containing stable DG75 cell line was generated under antibiotic G418 selection. CD21 expression of the newly generated DG75-pcDNA3.1(+)-CD21 cell line was analyzed using anti-CD21 antibody by flow cytometry. Surprisingly, the percentage of CD21+ population in the selected pcDNA3.1(+)-CD21-containing DG75 cells was only 6.5% after a stable cell line was generated (Figure 2B).
Down-regulation of CD21 expression driven by CMV promoter in DG75 cells
Because the proportion of CD21-positive cells in the stable transfected DG75 cells only reached 6.5% after establishment of the stable cell line, questions arise regarding the dynamics of CD21 expression in these cells. To further evaluate CD21 expression, CD21+ cells were purified using mouse anti-human CD21 antibody-coupled with anti-mouse IgG-conjugated Dynabeads (Figure 2B). The Dynabead-bound cells were removed with a magnet (negative selection) after 3–5 d of growing. FACS analysis was done to assess CD21 expression on these selected cells. The results clearly showed that CD21 expression on the selected CD21+ DG75 cells decreases over time (Figure 3). In fact, the half-life of CD21 expressed on these selected cells is only around 30 d.
Figure 3.

CD21 expression on stable DG75 cells driven by CMV promoter is down-regulated over time. DG75 cells were stably transfected with the pcDNA3.1(+)-CD21 plasmid, and 30 d following transfection, CD21+ cells were selected by binding to anti-human CD21 IgG antibody followed by purification with anti-mouse IgG Dynabeads. The purified CD21+ DG75 cells were allowed to grow for 3–5 d and then negative selection was performed to remove the bead-bound cells. The proportion of CD21+ DG75 cells was examined by FACS staining at 7, 14, 34, and 42 d after selection, and is marked on each chart. The black line represents cells stained with anti-CD21 antibody coupled with the secondary fluorescent antibody and the light grey shading shows unstained cells. The experiment was performed in duplicate. The secondary only antibody control for DG75 cells was ignored as it was shown to be no background on DG75 cells.
High-level surface CD21 on DG75 cells restores CD21-mediated antigen presentation
Because there is no CD21 expressed on DG75 cells [13], and antibody-mediated antigen delivery to antigen presenting cells could activate CD4+ T cells [16], we tried to test whether restoring surface CD21 would restore cognate CD4+ T cell response by B cell lymphoma DG75 cells. Our results clearly showed that T cell response could be activated by CD21+ DG75 cells targeted by anti-CD21 antibody-mediated antigen delivery in the first week after CD21+ selection (Figure 4). Interestingly, the same T cell response failed to be activated by using selected CD21+ DG75 cells 3 weeks after CD21+ selection (data not shown), indicating it is critical to perform the experiment during a timeline when expression level of ectopic genes is high.
Figure 4.

High CD21 expression on DG75 cells driven by CMV promoter restores CD21-mediated antigen presentation. (A) FACS staining was performed on untransfected DG75, selected stable transfected DG75-pcDNA-CD21, and stable transfected DG75-pcDNA for CD21 expression by using αCD21 antibody. DG75-pcDNA-CD21 was taken at 14 d after CD21+ selection. The black line represents cells stained with anti-CD21 antibody-coupled with the secondary fluorescent antibody and the light grey shading shows unstained cells. The secondary only antibody control for DG75 cells was ignored as it was shown to have no background on DG75 cells. pcDNA refers to pcDNA3.1(+). (B) The T cell recognition assay was performed on untransfected DG75, stable transfected DG75-pcDNA-CD21 (within 7 d after CD21+ selection), and stable transfected DG75-pcDNA by using 10 ng αCD21-3BB9 antibody, corresponding 1μg 3BB9 peptide, and medium only (peptide and medium as controls). The cognate CD4+ T cell activation was determined by IFN-γ release by ELISA and IFN-γ release from αCD21-3BB9 antibody treatment (shorted to A in figure) was divided by that of peptide treatment (shorted to P in figure) in the same cell type. Untransfected DG75, DG75-pcDNA-CD21, and DG75-pcDNA were incubated with respective medium or 1 μg 3BB9 peptide or 10 ng αCD21-3BB9 for 24 h before co-culturing with cognate T cells for 18 h. A: P ratio represents a ratio value: IFN-γ released from antigen coupled antibody group divided by IFN-γ released from peptide group. Experiments were performed in triplicate and repeated at least 2 times. The black shading shows medium only group, the black line shows peptide treatment group, and the grey line shows CD21-3BB9 antibody treatment group. pcDNA refers to pcDNA3.1(+) for convenience.
Workflow of the method
The workflow of this method includes: 1. Transfect vector into cells for target gene expression and positive selection marker; 2. Generate a stable cell line; 3. Check whether there is insufficient target gene expression; and 4. Antibody-based target gene purification (Figure 5).
Figure 5.
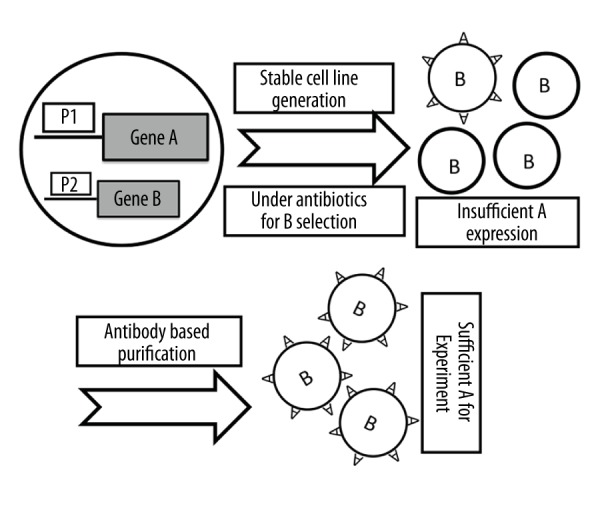
Schematic view of the method workflow. The original cells without target gene A are transfected with vector containing target gene A and gene B for positive selection. In many cases, target gene A under promoter 1 (P1) is expressed insufficiently. Following target gene A specific antibody-based purification, sufficient target gene A expression can be achieved for further experiments and this protocol could be repeated if necessary.
Discussion
Ectopic gene expression in targeted cells is an important method with which to investigate functions of genes. Unfortunately, down-regulation of ectopic gene expression in targeted cells is quite common [7,17–19]. We report here a method to recover a down-regulated cellular surface molecule expressed on a B lymphoma cell line based on antibody-based selection from a CMV promoter-containing vector (in our case, recovery of CD21 expression driven by CMV promoter in DG75 cells); therefore, the selected cells can be enriched for further experiments.
The CD21 gene generated in the lab previously [15] was cloned into a pcDNA3.1(+) plasmid using respective cutting sites, which was compatible with eukaryotic cells to express CD21 in DG75 cells. The plasmid was proved to strongly express CD21 by transient transfection (Figure 2A), confirming correct cloning of CD21 gene in the plasmid when low CD21+ cells occurred after establishment of a stable cell line (Figure 2B). The percentage of CD21+ cells in stable DG75-pcDNA3.1(+)-CD21 cells generated based on standard protocol was very low; therefore, we focused on CMV promoter activity of the plasmid after considering the problem, as there were reports on suppression of CMV promoter activity [1,7]. As CD21 detection was done by FACS analysis on the pools of transfected cells, as it is proposed that loss of CD21 expression is caused by CMV promoter inactivation in the minority of cells initially and outgrowth of such cells afterwards.
Since low CD21 expression on stable DG75-pcDNA3.1(+)-CD21 cells does not meet the requirement of following the CD21-mediated antigen presentation experiment, which requires high CD21 expression, so that CD21+ purification was attempted to restore CD21+ cell purity based on antibody-based selection which included an anti-CD21 antibody for binding to cellular surface CD21 molecule and a secondary antibody for purification. Purification was done successfully (Figure 2B). However, high CD21 expression on these purified CD21+ DG75 cells does not keep long and its expression level was decreased continuously (Figure 3). The stable cell line was generated under antibiotic G418 selection; therefore, loss of CD21 expression is not expected to be simply due to loss of the plasmid from DG75 cells; it may be caused by methylation, deacetylation, or inhibitors of DNA methylation and histone deacetylation, thus leading to CD21 down-regulation [7,18–20], but this needs to be further investigated. Intriguingly, in this experiment we could restore CD21 expression by using anti-CD21 antibody-based positive selection without additional inhibitor treatment of the cells, which is useful and efficient enough for further experiments.
The advantage of this method is applicable for long-timeline experiments such as stable transfection experiments that normally last for several weeks or months, as promoter activity is often down-regulated, such as CMV promoter during such a long period [1]. However, for short experiments such as transient transfection experiments that last for only several days, purification is not necessary, as expression of ectopic gene usually does not change much for a promoter activity such as CMV. For example, in this study, the “half-life” of CD21 driven by CMV promoter is estimated to be around 30 d; therefore, there is still enough time left to do purification after establishment of the stable cell line (it takes around 30 d); on the other hand, down-regulation of CD21 will be faint for transient transfection that normally takes 3–5d, so CD21+ purification will not be useful. Thus, we recommend the ectopic gene purification method for long-term experiments. This method could be potentially used in cells that need a sustained high level of ectopic surface molecules or selectable intracellular molecules, and the normal expression level is lower than that required. For example, cell surface molecule or GFP driven by CMV promoter lost during culture in embryonic stem cell lines could be restored to high-level expression by this method (GFP-positive cells could be enriched by FACS sorting).
Conclusions
We described a novel method to recover down-regulated target gene expression driven by a CMV promoter using antibody-based selection, so that ectopic molecule-positive cells could be enriched for further experiments. This study provides an example of recovery of a down-regulated molecule using antibody-based selection and should facilitate future experimental design when using CMV promoter-containing vectors.
Acknowledgements
We are grateful to Professor Henri Jacques Delecluse for his support for the experiments.
Footnotes
Conflicts of interests
None.
Source of support: The First Affiliated Hospital of Anhui Medical University funded this research (2016KJ10)
References
- 1.Qin JY, Zhang L, Clift KL, et al. Systematic comparison of constitutive promoters and the doxycycline-inducible promoter. PLoS One. 2010;5:e10611. doi: 10.1371/journal.pone.0010611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chung S, Andersson T, Sonntag KC, et al. Analysis of different promoter systems for efficient transgene expression in mouse embryonic stem cell lines. Stem Cells. 2002;20:139–45. doi: 10.1634/stemcells.20-2-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou L, Bu Y, Liang Y, et al. Epstein-Barr Virus (EBV)-BamHI-A rightward transcript (BART)-6 and cellular microRNA-142 synergistically compromise immune defense of host cells in EBV-positive burkitt lymphoma. Med Sci Monit. 2016;22:4114–20. doi: 10.12659/MSM.897306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Addison CL, Hitt M, Kunsken D, Graham FL. Comparison of the human versus murine cytomegalovirus immediate early gene promoters for transgene expression by adenoviral vectors. J Gen Virol. 1997;78(Pt 7):1653–61. doi: 10.1099/0022-1317-78-7-1653. [DOI] [PubMed] [Google Scholar]
- 5.Damdindorj L, Karnan S, Ota A, et al. A comparative analysis of constitutive promoters located in adeno-associated viral vectors. PLoS One. 2014;9:e106472. doi: 10.1371/journal.pone.0106472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McMahon JM, Conroy S, Lyons M, et al. Gene transfer into rat mesenchymal stem cells: A comparative study of viral and nonviral vectors. Stem Cells Dev. 2006;15:87–96. doi: 10.1089/scd.2006.15.87. [DOI] [PubMed] [Google Scholar]
- 7.Xia X, Zhang Y, Zieth CR, Zhang SC. Transgenes delivered by lentiviral vector are suppressed in human embryonic stem cells in a promoter-dependent manner. Stem Cells Dev. 2007;16:167–76. doi: 10.1089/scd.2006.0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rice GP, Schrier RD, Oldstone MB. Cytomegalovirus infects human lymphocytes and monocytes: virus expression is restricted to immediate-early gene products. Proc Natl Acad Sci USA. 1984;81:6134–38. doi: 10.1073/pnas.81.19.6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zawilinska B, Kopec J, Szostek S, et al. Lymphotropic herpesvirus DNA detection in patients with active CMV infection – a possible role in the course of CMV infection after hematopoietic stem cell transplantation. Med Sci Monit. 2011;17:CR432–41. doi: 10.12659/MSM.881904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Isomura H, Tsurumi T, Stinski MF. Role of the proximal enhancer of the major immediate-early promoter in human cytomegalovirus replication. J Virol. 2004;78:12788–99. doi: 10.1128/JVI.78.23.12788-12799.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graham FL, Smiley J, Russell WC, Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol. 1977;36:59–74. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- 12.Moritz B, Woltering L, Becker PB, Gopfert U. High levels of histone H3 acetylation at the CMV promoter are predictive of stable expression in Chinese hamster ovary cells. Biotechnol Prog. 2016;32:776–86. doi: 10.1002/btpr.2271. [DOI] [PubMed] [Google Scholar]
- 13.Ben-Bassat H, Goldblum N, Mitrani S, et al. Establishment in continuous culture of a new type of lymphocyte from a “Burkitt like” malignant lymphoma (line D.G.-75) Int J Cancer. 1977;19:27–33. doi: 10.1002/ijc.2910190105. [DOI] [PubMed] [Google Scholar]
- 14.Adhikary D, Behrends U, Moosmann A, et al. Control of Epstein-Barr virus infection in vitro by T helper cells specific for virion glycoproteins. J Exp Med. 2006;203:995–1006. doi: 10.1084/jem.20051287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Busse C, Feederle R, Schnolzer M, et al. Epstein-Barr viruses that express a CD21 antibody provide evidence that gp350’s functions extend beyond B-cell surface binding. J Virol. 2010;84:1139–47. doi: 10.1128/JVI.01953-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schjetne KW, Gundersen HT, Iversen JG, et al. Antibody-mediated delivery of antigen to chemokine receptors on antigen-presenting cells results in enhanced CD4+ T cell responses. Eur J Immunol. 2003;33:3101–8. doi: 10.1002/eji.200324299. [DOI] [PubMed] [Google Scholar]
- 17.Rodova M, Jayini R, Singasani R, et al. CMV promoter is repressed by p53 and activated by JNK pathway. Plasmid. 2013;69:223–30. doi: 10.1016/j.plasmid.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brooks AR, Harkins RN, Wang P, et al. Transcriptional silencing is associated with extensive methylation of the CMV promoter following adenoviral gene delivery to muscle. J Gene Med. 2004;6:395–404. doi: 10.1002/jgm.516. [DOI] [PubMed] [Google Scholar]
- 19.Nuo MT, Yuan JL, Yang WL, et al. Promoter methylation and histone modifications affect the expression of the exogenous DsRed gene in transgenic goats. Genet Mol Res. 2016;15(3) doi: 10.4238/gmr.15038560. [DOI] [PubMed] [Google Scholar]
- 20.Grassi G, Maccaroni P, Meyer R, et al. Inhibitors of DNA methylation and histone deacetylation activate cytomegalovirus promoter-controlled reporter gene expression in human glioblastoma cell line U87. Carcinogenesis. 2003;24:1625–35. doi: 10.1093/carcin/bgg118. [DOI] [PubMed] [Google Scholar]