

Abstract
Chromatin accessibility, a crucial component of genome regulation, has primarily been studied in homogeneous and simple systems, such as isolated cell populations or early-development models. Whether chromatin accessibility can be assessed in complex, dynamic systems in vivo with high sensitivity remains largely unexplored. In this study, we use ATAC-seq to identify chromatin accessibility changes in a whole animal, the model organism Caenorhabditis elegans, from embryogenesis to adulthood. Chromatin accessibility changes between developmental stages are highly reproducible, recapitulate histone modification changes, and reveal key regulatory aspects of the epigenomic landscape throughout organismal development. We find that over 5000 distal noncoding regions exhibit dynamic changes in chromatin accessibility between developmental stages and could thereby represent putative enhancers. When tested in vivo, several of these putative enhancers indeed drive novel cell-type- and temporal-specific patterns of expression. Finally, by integrating transcription factor binding motifs in a machine learning framework, we identify EOR-1 as a unique transcription factor that may regulate chromatin dynamics during development. Our study provides a unique resource for C. elegans, a system in which the prevalence and importance of enhancers remains poorly characterized, and demonstrates the power of using whole organism chromatin accessibility to identify novel regulatory regions in complex systems.
Chromatin accessibility represents an essential level of genome regulation and plays a pivotal role in many biological and pathological processes, including development, tissue regeneration, aging, and cancer (Stergachis et al. 2013; Simon et al. 2014; Tsompana and Buck 2014). However, most genome-wide chromatin accessibility studies have been in relatively simple systems to date, including cultured or purified cells as well as early embryos (Thomas et al. 2011; Wang et al. 2012; Lara-Astiaso et al. 2014; West et al. 2014; Zhu et al. 2015). Assessing chromatin accessibility directly in complex systems composed of multiple cell types could allow for high-throughput discovery of regulatory regions whose activities are restricted to rare or undefined subpopulations of cells. This is particularly relevant for enhancers, which are thought to be highly cell-type- and temporally specific (Ren and Yue 2015).
The primary limitation for studying chromatin accessibility in complex systems is that most assays lack the sensitivity and precision to detect regions active only in rare subpopulations of cells or require so many cells that precise temporal synchronization of samples is impractical. However, the Assay for Transposase Accessible Chromatin using sequencing (ATAC-seq) has been shown to assess native chromatin accessibility with high sensitivity and base pair resolution while requiring orders of magnitude less starting material than other assays (Buenrostro et al. 2013). This approach has been used successfully in cultured or purified cells even down to single cells, though such low input relies heavily on existing knowledge (Buenrostro et al. 2015; Cusanovich et al. 2015). Rather than purifying specific cell types, we wondered whether ATAC-seq could be sensitive enough to detect subtle changes in chromatin accessibility in complex mixtures of tissues and, in so doing, uncover novel biological insights that would have otherwise been obscured.
The nematode Caenorhabditis elegans is a particularly powerful model to study chromatin accessibility in a complex system and potentially identify novel regulatory regions. C. elegans has highly synchronous life stages, as well as consistent and well-characterized cellular composition throughout each stage of development (Sulston et al. 1983). Rapid transgenesis and transparency (Mello and Fire 1995) also make C. elegans an ideal system to efficiently validate genomic regions of functional importance and visualize tissue- or cell-specificity (Jantsch-Plunger and Fire 1994; Harfe et al. 1998; Lei et al. 2009). While there exist some preliminary reports on chromatin states in C. elegans (Valouev et al. 2008; Shi et al. 2009; Gerstein et al. 2010; Liu et al. 2011b; Hsu et al. 2015; Evans et al. 2016), high-resolution, genome-wide chromatin accessibility maps throughout development have not yet been reported. In this study, we show that studying high-resolution chromatin accessibility dynamics in synchronized C. elegans populations allows us to characterize highly reproducible changes in chromatin accessibility between developmental stages and to identify functional temporal- and tissue-specific novel enhancers in vivo. Our study provides a unique resource for defining C. elegans regulatory regions as well as a guide for the interpretation of chromatin structure in complex multitissue systems in vivo.
Results
High-resolution chromatin accessibility profiles from three C. elegans life stages
To sensitively measure high-resolution chromatin accessibility at different life stages in C. elegans, we used the Assay for Transposase Accessible Chromatin using sequencing. We optimized the ATAC-seq protocol for C. elegans by including a step of native nuclei isolation by mechanical homogenization before the transposition step (see Methods; Supplemental Extended Protocol). The low input requirements of ATAC-seq (several orders of magnitude less than standard ChIP-seq [Furey 2012]) allowed us to grow C. elegans in standard conditions (i.e., plates, whereas most high-throughput assays in C. elegans require growth in liquid) and to generate three independent biological replicates that are tightly synchronized at three key life stages—early embryo, larval stage 3 (L3), and young adults—thereby limiting variation within stages (see Methods; Fig. 1A). We generated and sequenced ATAC-seq libraries, as well as an input control, to a median depth of over 17 million unique, high-quality mapping reads per sample (Supplemental Table S1). The insert size distribution of each C. elegans ATAC-seq library displays a stereotypical 147-bp periodicity that is consistent with the expected nucleosome occupancy of chromatin (Supplemental Fig. S1A), indicative of ATAC-seq library quality (Buenrostro et al. 2013). We designed a computational framework to integrate the input control (Supplemental Fig. S1B) and emphasize single base pair resolution (Fig. 1B), resulting in the identification of 13,000–27,000 high-confidence, accessible peaks per developmental stage and more than 30,000 consensus ATAC-seq peaks found in at least one of the three stages (Supplemental Tables S2, S3; see Methods). The high correlation of ATAC-seq signal between each of the three biological replicates (Spearman's ρ > 0.837) demonstrates the high reproducibility of this approach (Fig. 1C). The ability to cluster samples by their developmental stage also shows that chromatin accessibility is strikingly different between these three life stages (Fig. 1C). These differences between developmental stages are likely due to both changes in accessibility within cells, as well as the organisms’ changing cellular composition throughout development. Finally, ATAC-seq signal is enriched at transcription start sites (TSSs) (Fig. 2B), another indication of the quality of ATAC-seq data (Buenrostro et al. 2013). Together, these results indicate that reproducible high-resolution chromatin accessibility can be obtained from low amounts (at least an order of magnitude less than standard histone ChIP-seq or DNase-seq) of complex, multitissue samples.
Figure 1.

ATAC-seq in whole C. elegans captures chromatin accessibility dynamics across three life-stages. (A) Three independent biological replicates each consisting of tightly temporally synchronized C. elegans were used for ATAC-seq. Hours post-egg lay are at 20°C. (B) C. elegans were flash frozen and nuclei were isolated before assaying accessible chromatin using transposons loaded with next-generation sequencing adaptors, allowing paired-end sequencing. A custom analysis pipeline emphasizing high-resolution signal and consistent peaks, as well as accommodating input control, was developed to generate stage-specific and consensus (i.e., across stages) ATAC-seq peaks. (C) ATAC-seq signal within consensus ATAC-seq peaks was compared between all samples using Spearman's ρ to cluster samples. Replicate batches are noted as letters following the stage. (D,E) Comparison of ATAC-seq signal (normalized by total sequencing depth) between all three stages at a region that decreases (D) or increases (E) in accessibility during development. (F,G) Genes that lose accessibility between embryo and larval stage 3 (L3) are enriched for early development functions (F), while genes that gain accessibility are enriched for larval-related functions (G); all calculations and genes lists are from GOrilla (Eden et al. 2009), and the number of genes enriched in each term are listed in parentheses.
Figure 2.

ATAC-seq as a single assay describes the epigenome. (A) Enrichment of stage-specific ATAC-seq peaks in ChromHMM-predicted chromatin states relative to values expected by chance; significance derived via 10,000 bootstrapping iterations, and error bars reflect 95% confidence intervals (P ≤ 1 × 10−4, except for early embryo transcribed gene body [P = 0.633]). The decrease in enrichment of ATAC-seq peaks in H3K27me3-repressed regions in L3 compared to young adult and early embryo is likely due to the low sequencing depth of the L3 H3K27me3 ChIP-seq. (B) Larval stage 3 (L3) ATAC-seq signal at 19,899 previously defined transcription start sites (TSS ± 0.5 kb) is correlated with active histone modifications (H3K4me3) and anti-correlated with heterochromatin (H3K9me3) around the TSS. (C,D) Regions that increase in accessibility in ATAC-seq peaks are enriched for transitions from inactive chromatin states to active regulatory states (C), while regions that decrease in accessibility are enriched for transitions from active regulatory states to inactive chromatin states (D). (E) An example of an increase in chromatin accessibility overlapping with a transition from heterochromatin to a predicted active enhancer chromatin state. Multiple TSSs have been noted for kin-1, but only the 5′-most is shown here for ease.
To investigate the changes in chromatin accessibility between life stages, we identified and characterized the ATAC-seq peaks that significantly changed accessibility between early embryo and L3 (12,193 peaks) (Supplemental Fig. S1C) and between L3 and young adult (783 peaks; FDR < 0.05) (Supplemental Fig. S1D; see Methods). The larger number of differentially accessible peaks (both decreased and increased) observed during the transition from early embryo to L3 versus L3 to young adult could not simply be explained by differences in sequencing depth (see Methods) and is likely due to the massive changes in cell number and tissue composition that occur during this transition (Byerly et al. 1976).
An example of a decrease in chromatin accessibility from embryo to L3 can be seen in the promoter region of the cav-1 gene, which is expressed during embryogenesis but not larval development (Fig. 1D; Parker and Baylis 2009). Conversely, several ATAC-seq peaks drastically increase from embryo to L3 in the promoter and regions upstream of the daf-12 gene, which is a key regulator of stage-specific developmental programs, particularly at L3 (Fig. 1E; Antebi et al. 1998, 2000). Confirming these specific examples, the most enriched gene ontology (GO) terms for genes with decreased chromatin accessibility from embryo to L3 include early-development terms such as embryonic morphogenesis and cell fate specification (Fig. 1F; Supplemental Table S4), while the most enriched terms for genes with increased chromatin accessibility from embryo to L3 include larval development and locomotion (Fig. 1G; Supplemental Table S5). Similarly, we observe strong enrichments of GO terms reflecting the major phenotypic changes occurring between L3 and adult, including terms like larval development and reproduction (Supplemental Fig. S1E,F; Supplemental Tables S6, S7).
Together, these results indicate that ATAC-seq in whole organisms can identify changes in DNA accessibility that represent key biological differences between stages, regardless of whether these changes are due to activation/repression of specific regions within a cell type or to changes in cell-type composition.
ATAC-seq as a single assay describes the epigenome
Accessible chromatin encompasses several key features of the epigenome, including active and poised regulatory regions. To verify that our ATAC-seq data correctly identify regulatory regions throughout the epigenome, we used multiple histone modification ChIP-seq data sets from modENCODE (Li et al. 2009; Ho et al. 2014) and ChromHMM (Ernst and Kellis 2012) to build predictive models of the epigenome. ChromHMM is a hidden Markov model that classifies regions of the genome into chromatin states (e.g., heterochromatin) using the co-occurrence of multiple histone modifications from each life stage—in our case, ChIP-seq data sets characterizing eight distinct histone modifications across all three stages (Supplemental Fig. S3A–E; Supplemental Tables S8–S11; see Methods). ATAC-seq peaks from all three stages were significantly enriched in active and poised regulatory chromatin states (e.g., promoter), as defined by this ChromHMM model, and significantly depleted in heterochromatic states (Fig. 2A; Supplemental Fig. S3B,C). ATAC-seq peaks were also enriched in H3K27me3-repressed regions, which is likely due to H3K27me3 marking inactive, yet accessible poised sites (Rada-Iglesias et al. 2011; Zentner et al. 2011; Zhang et al. 2012; Lei et al. 2015; Lorzadeh et al. 2016; Sen et al. 2016). ATAC-seq signal was correlated with individual active histone modifications at TSSs (Fig. 2B) and genome-wide (Supplemental Fig. S3F). Thus, ATAC-seq correctly identifies poised and active regulatory regions at both specific loci and genome-wide.
To further assess the relevance of whole organism chromatin accessibility, we compared our ATAC-seq data to publicly available gene expression data (GRO-seq, and RNA-seq) (Hillier et al. 2009; Gerstein et al. 2010, 2014; Kruesi et al. 2013). Both GRO-seq (Supplemental Fig. S2A,B) and RNA-seq (Supplemental Fig. S2C,D) were positively correlated with ATAC-seq signal near the TSS. These observations support the relationship between chromatin accessibility at the TSS and gene expression, despite the numerous other layers of gene regulation (e.g., RNA stability).
Beyond simply identifying regulatory regions important for individual life stages, chromatin accessibility dynamics should highlight regulatory regions critical for transitions from embryo to larval stages, and from larval stages to adulthood. We examined whether genomic regions that showed accessibility changes from one life stage to another were enriched for specific chromatin state transitions. Regions that lost chromatin accessibility from embryo to L3 or from L3 to young adult were enriched for transitions from active regulatory chromatin states (especially predicted enhancers) to repressed or heterochromatic states (Fig. 2D; Supplemental Fig. S3G). Conversely, the regions that gained accessibility during development were enriched for transitions from inactive chromatin states to active regulatory states (again, especially predicted enhancers) (Fig. 2C,E; Supplemental Fig. S3H). Collectively these results show that ATAC-seq performed on an entire organism, even a complex multitissue adult, is sensitive enough to detect important global changes in chromatin structure that are consistent with predicted chromatin state transitions. Thus, ATAC-seq as a single assay constitutes an attractive alternative to performing multiple histone modification ChIP-seq experiments for identifying key regulatory regions, especially considering that ATAC-seq requires orders of magnitude less input than a single ChIP-seq.
ATAC-seq identifies new distal regulatory regions that serve as tissue- and stage-specific enhancers in C. elegans
Enhancers are key regulators of temporal- and tissue-specific gene expression that play important and conserved functions during development (Ren and Yue 2015; Sun et al. 2015). However, the identification and characterization of novel enhancers remains a challenge because they can be specifically active in rare cell populations, some of which may not have even been characterized (Heinz et al. 2015). The extent and functional importance of distal regulatory regions in the C. elegans genome has been particularly underexplored (Reinke et al. 2013). While several methods to identify potential enhancers genome-wide have recently been developed, these methods suffer from notable drawbacks (Giresi et al. 2007; Mito et al. 2007; Wang et al. 2008; Visel et al. 2009; He et al. 2010; Arnold et al. 2013; Chen et al. 2013a; Zhu et al. 2013, 2015). For example, the co-occurrence of multiple histone modifications (e.g., H3K27ac, H3K4me1) or RNA polymerase II (PolII) through ChIP-seq experiments lacks the sensitivity to detect enhancers active only in rare subpopulations of cells and the resolution to precisely identify the active enhancer region (Furey 2012). We therefore investigated whether whole-organism ATAC-seq could overcome these challenges and facilitate the identification of tissue- and stage-specific enhancers. To this end, we examined distal noncoding ATAC-seq peaks (defined as ATAC-seq peaks at least 1 kb upstream of or 0.5 kb downstream from a TSS and not in an exon). Distal noncoding ATAC-seq peaks in all three stages were highly and significantly enriched for active and repressed enhancers (Supplemental Fig S4A), as defined by the ChromHMM model and distinguished by the presence of H3K27ac (active enhancers) or H3K27me3 (repressed enhancers) (Supplemental Fig. S3A). Furthermore, these distal noncoding ATAC-seq peaks were significantly more conserved than expected by chance (Supplemental Fig. S4B,C), a defining feature of enhancers (Pennacchio et al. 2006). Finally, distal noncoding ATAC-seq peaks also exhibited significant overlap with regions previously predicted to be enhancers by overlapping short capped RNA, transcription factor binding, and a H3K4me3 dearth (P < 1 × 10−323, one-sided Fisher's exact test) (Chen et al. 2013a). These analyses support the notion that distal noncoding ATAC-seq peaks include enhancers.
In C. elegans, a small number of functional enhancers have been experimentally mapped, including four enhancers in the upstream regulatory region of hlh-1, the C. elegans MyoD ortholog that regulates muscle development (Lei et al. 2009). ATAC-seq peaks overlap three of these four regions, and despite its lack of statistical significance, the fourth region still exhibits noticeable ATAC-seq signal in embryos (Supplemental Fig. S4D). Given that hlh-1 is exclusively expressed in muscle (Krause et al. 1994), a tissue comprising <10% of C. elegans’ cellular composition (Altun and Hall 2009), these observations indicate that ATAC-seq performed on a whole organism is sensitive enough to identify functional tissue-specific enhancers.
We next sought to determine whether ATAC-seq dynamics could be leveraged to identify novel functional enhancers. Previous work has demonstrated that enhancers are precisely activated/inactivated at very specific times in development (Bonn et al. 2012; Kvon 2015; Sun et al. 2015). We hypothesized that the temporal resolution of our ATAC-seq data (due to the precise synchronization of C. elegans populations) would enable us to capture distal regulatory dynamics of development. To experimentally test the functional enhancers predicted by our ATAC-seq data, we selected 13 distal ATAC-seq peaks (i.e., at least 1 kb away from the nearest TSS, defined on the UCSC Genome Browser) that exhibited the largest fold changes in accessibility between any two stages. We generated multiple transgenic C. elegans strains with these 13 putative enhancer regions upstream of a minimal promoter (pes-10) driving expression of green fluorescent protein (GFP) containing a nucleolar localization signal (NoLS) (Fig. 3A; Table 1; Supplemental Tables S12, S13). To control for specificity, we also tested regions flanking 10 of the 13 putative enhancer regions (Table 1; Supplemental Tables S12, S13). We created numerous (8–11) independent genetic strains for each validated regulatory site to ensure that no artifact of transgenesis (e.g., hybridization of the rol-6 marker in the extrachromosomal array) could be driving spurious GFP expression (Table 1). By fluorescence microscopy, we examined the spatiotemporal GFP pattern in these transgenic strains to assess the temporal- and tissue-specificity of the putative enhancers and control regions. Using stringent criteria for defining enhancer activity (see Methods), we found that six of the 13 putative enhancer regions (identified by dynamic distal noncoding ATAC-seq peaks) led to specific and consistent spatiotemporal GFP pattern and, for all but one of these, this pattern was observed regardless of the genomic orientation of the region—an important characteristic of enhancers (Table 1; Fig. 3B–G; Supplemental Fig. S5A–E; Ren and Yue 2015). In contrast, zero of the 10 flanking regions led to such a GFP pattern, indicating enrichment for functional enhancers in distal noncoding ATAC-seq peaks that change between stages (P = 0.038, one-sided Fisher's exact test) (Table 1). Thus, dynamic chromatin accessibility on its own can mark functional enhancers and can be used to successfully identify novel distal regulatory regions.
Figure 3.

Dynamic ATAC-seq peaks identify functional enhancers with unique spatiotemporal specificity during C. elegans development. (A) Functional enhancer constructs used to generate C. elegans transgenic lines. Putative regulatory regions, or corresponding flanking regions for negative controls, are inserted upstream of a minimal promoter (pes-10) driving green fluorescent protein (GFP) localized to the nucleolus. (B,D,F) Distal (>1 kb from a TSS) noncoding ATAC-seq peaks with the largest fold change in ATAC-seq signal between any two stages were screened for potential enhancer activity. The approximate regions tested near C54G6.2 (B), nhr-25 (D), and swip-10 (F) are boxed in red. Note that for swip-10 (F), the plot orientation is reversed for consistency with other plots. (C,E,G) Specific patterns of spatiotemporal enhancer activity in transgenic lines. Representative images of GFP expression in staged C. elegans transgenic lines are presented with a 50-µm scale bar. All images were straightened with ImageJ and are grayscale images with florescence overlaid.
Table 1.
Transgenic reporter lines generated for validation of ATAC-seq peaks as functional enhancers
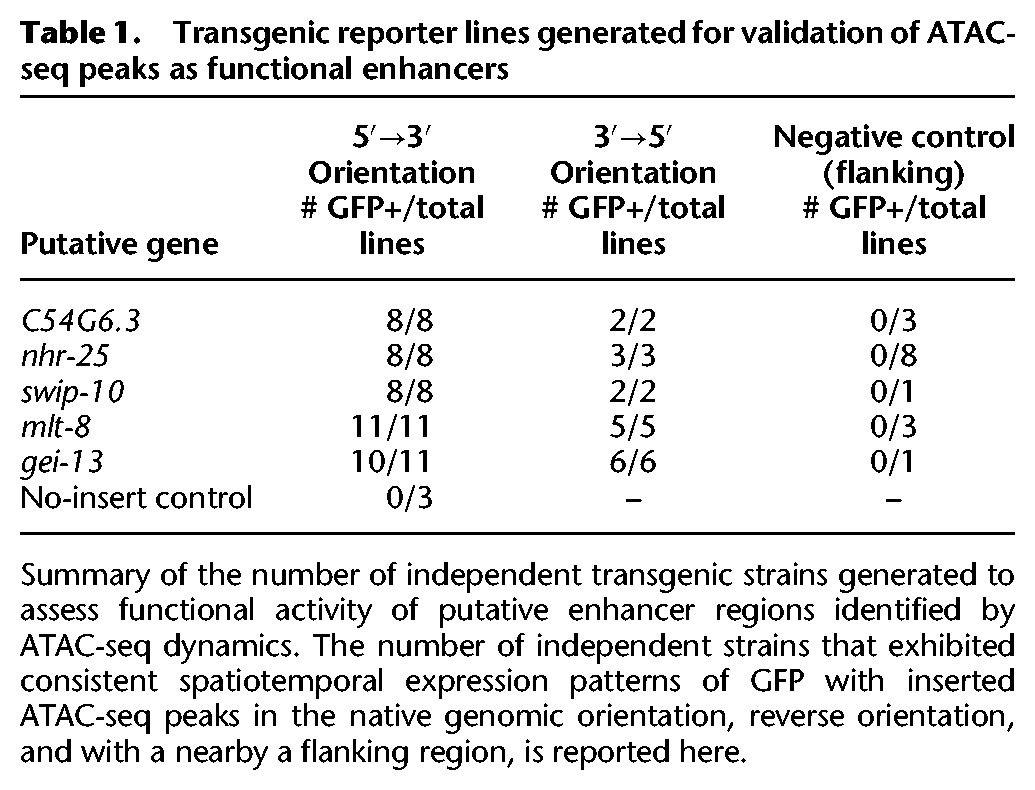
Importantly, the enhancers we experimentally identified display diverse spatiotemporal activity patterns. Three of the enhancer regions (putatively associated with gei-13, mlt-8, and nhr-25) are active in the head or tail hypodermis during development, while others are active specifically in the pharynx (C54G6.3) or along the flank of the worm (swip-10). In addition, the enhancers are located at a wide diversity of genomic positions relative to their putatively associated gene: upstream of the TSS (at distances varying from 1 to 9 kb), within introns, as well as downstream from the coding sequence. An interesting example is the nhr-25-associated enhancer, which is downstream from the 3′ UTR, more than 5 kb downstream from the TSS (as defined on the UCSC Genome Browser; this enhancer is also downstream from npr-10 3′ UTR on the other side, about 10 kb from the TSS) (Fig. 3D). nhr-25 is a conserved nuclear receptor primarily expressed in the hypodermis (and somatic gonad) during larval development (Gissendanner and Sluder 2000). The 3′ nhr-25 enhancer specifically drives GFP expression in approximately 20 hypodermal cells in the head and tail of the worm during larval development (Fig. 3E). The limited expression pattern of the 3′ nhr-25 enhancer (as well as the other enhancers we identified) indicates that ATAC-seq performed in whole worms is sensitive enough to identify regulatory regions active only in specific cell types, though we cannot rule out the possibility that these regions are also accessible but their activity repressed (e.g., by H3K27me3) in other cells.
Collectively, these findings demonstrate the power of using ATAC-seq dynamics as an unbiased approach to identify functional and conserved enhancers active in a small subset of cells. When applied to whole organisms or complex samples composed of diverse cellular populations, this approach could capture regulatory regions that may have been missed in studies of isolated cell populations.
Specific motifs for transcription factors predict changes in chromatin accessibility
To explore the regulatory underpinnings of chromatin accessibility dynamics, including that of enhancers, we examined the occurrence of experimentally defined C. elegans transcription factor (TF) binding motifs (Narasimhan et al. 2015) in the dynamic chromatin accessibility regions identified by ATAC-seq. In regions that change chromatin accessibility between early embryo and L3 or between L3 and young adult, we observe significant enrichment of motifs associated with TF homologs and orthologs that have previously been connected to chromatin accessibility dynamics (Fig. 4A; Supplemental Fig. S6A). For example, the motif for BLMP-1, the C. elegans ortholog of human BLIMP-1/PRDM1, a TF that recruits chromatin-remodeling complexes in human B cells (Minnich et al. 2016), is enriched in peaks that are more accessible in L3 when compared to embryos or adults. Furthermore, the motif for ELT-3, a GATA TF, is enriched in peaks more accessible in L3 than embryos (the GATA family was recently shown to be an important regulator of chromatin accessibility during human hematopoiesis [Corces et al. 2016]).
Figure 4.

Motifs associated with increases in chromatin accessibility during development reveal key transcription factors with unique binding loci. (A) ATAC-seq peaks which decreased (left) or increased (right) accessibility between early embryo and L3 are enriched for previously identified transcription factor binding motifs; P-values are Benjamini-Hochberg-corrected for multiple hypothesis testing. (B) The number of instances of previously identified as well as de novo motifs (see Supplemental Fig. S6C) in each consensus ATAC-seq peak were used as features in a machine learning model to predict how each ATAC-seq peak changed between early embryo and L3 (increasing, decreasing, or no change). A training set (70% of all ATAC-seq peaks) was used to build the model, while the remaining held-out testing set was used to assess model quality (see Supplemental Fig. S6D). (C) The relative influence of every motif from the machine learning model in Figure 4B was quantified. Solid bars are previously defined motifs, while hashed bars are de novo identified motifs in dynamic ATAC-seq peaks. (D) The median L3 ATAC-seq signal and fragment length at the midpoint (±50 bp) of L3 ChIP-seq peaks; box plots of the same data are in Supplemental Figure S7C,D. (E) Histograms of L3 ATAC-seq fragment size at the midpoint (±50 bp) of L3 ChIP-seq peaks were calculated and normalized to percentages. Canonical TFs and chromatin factors were then aggregated and plotted. (F) The change in H3 nucleosome occupancy between early embryo and larval stage 3 at the midpoint of each L3 transcription factor ChIP-seq peak was calculated using DANPOS (Chen et al. 2013b) and publicly available H3 ChIP-seq.
The DNA binding motif for EOR-1, which resembles a dimeric version of the canonical GAGA motif, was significantly enriched in distal noncoding ATAC-seq peaks (Supplemental Fig. S6B) and in ATAC-seq peaks that gained in accessibility in L3 versus embryo (Fig. 4A). This GAGA/EOR-1 motif was also present in two of the five functional enhancers that we experimentally validated, including the nhr-25-associated enhancer described above. TFs binding GAGA motifs have been identified as modulators of chromatin structure dynamics from plants to humans (Lund et al. 2013; Srivastava et al. 2013; Hecker et al. 2015), and the GAGA motif itself is required for full functionality in two previously defined C. elegans enhancers (Jantsch-Plunger and Fire 1994; Harfe et al. 1998). Finally, GAGA/EOR-1 factors might play an important role in the regulation of chromatin accessibility as EOR-1 genetically interacts with components of two nucleosome remodeling complexes, SWI/SNF and RSC (Lehner et al. 2006), and a dimeric GAGA motif nearly identical to the EOR-1 motif is highly enriched in ChIP-seq peaks for two separate SWI/SNF components in C. elegans (Riedel et al. 2013).
To independently verify the importance of the GAGA/EOR-1 motif, we developed a machine learning model to identify the TF binding motifs that are predictive of regions that gained or lost accessibility between early embryo and larval stage 3. This machine learning method is unbiased and allowed for the integration of the largest available set of previously defined C. elegans TF binding motifs (107 high-confidence motifs from 98 TFs out of more than 750 TFs in the C. elegans genome [Narasimhan et al. 2015]) as well as 59 motifs discovered de novo in regions that changed in accessibility between early embryo and L3 (Fig. 4B; Supplemental Fig. S6C; see Methods). Using this machine learning framework, we identified the motifs that are the most predictive of ATAC-seq dynamics; the top three most informative motifs in our model are the GAGA motif that we identified above and motifs for known chromatin regulators (BLMP-1 and ELT-3) (Fig. 4C). Thus, machine learning independently supports the GAGA/EOR-1 motif as a potential important regulator of chromatin accessibility.
We next investigated whether EOR-1 may play a role in chromatin accessibility changes. An EOR-1 ChIP-seq data set at L3 (Araya et al. 2014) revealed enrichment for a dimeric GAGA motif (Supplemental Fig. S7A) and was significantly enriched for regions that overlapped ATAC-seq peaks that gain accessibility in L3 compared to early embryo (Supplemental Fig. S7B), indicating that EOR-1 is indeed bound to the GAGA motif and that EOR-1 may be involved in regulating chromatin accessibility. To examine if EOR-1 is found at closed chromatin, we quantified not only ATAC-seq signal but also ATAC-seq fragment size, as larger ATAC-seq fragments correlate with higher nucleosome occupancy and less accessible chromatin regions (Buenrostro et al. 2013; Bao et al. 2015; Schep et al. 2015). EOR-1 ChIP-seq peak summits at L3 have significantly larger fragment sizes (indicative of less accessible chromatin regions) than all 24 canonical TFs and the histone deacetylase HDA-1 ChIP-seq peak summits (FDR < 0.05) (Fig. 4D; Supplemental Fig. S7C,D; Supplemental Table S14). In fact, the only factor with larger ATAC-seq insert sizes was the heterochromatin-associated protein HPL-2. The larger ATAC-seq insert sizes at EOR-1 binding sites are unlikely to be due to the binding of other TFs hindering the ability of transposons to interact with the genomic DNA because EOR-1 ChIP-seq peaks did not show unusual overlap with the other TFs assessed (Supplemental Fig. S7E). These observations are consistent with the possibility that EOR-1 is uniquely present at regions with less accessible chromatin compared to other TFs.
To more closely investigate the chromatin accessibility landscape of EOR-1 peaks at the L3 stage, we generated aggregated histograms of the median ATAC-seq fragment sizes for the ChIP-seq peak summits of EOR-1, all canonical TFs, and the chromatin-associated factors HPL-1 and HDA-1 (Fig. 4E; Supplemental Fig. S7F). As expected, canonical TFs are predominantly found at regions with short ATAC-seq insert sizes (<147 bp, the length of DNA wrapped around a nucleosome), whereas chromatin factors, especially HPL-2, are also present at regions with larger ATAC-seq fragment sizes (with many >147 bp). Strikingly, EOR-1 was found both at regions with short ATAC-seq insert sizes as well as at regions with larger ATAC-seq fragment sizes (>147 bp). Together, these results suggest that EOR-1 may, in some cases, bind to or at least be immediately adjacent to nucleosome-occupied DNA.
We next quantified nucleosome occupancy dynamics in the summits of ChIP-seq peaks using publicly available histone H3 ChIP-seq data (Gerstein et al. 2010; Contrino et al. 2012). EOR-1 peak summits exhibited a large decrease in nucleosome occupancy from embryo to L3, and this was distinct from the canonical TFs assessed (Fig. 4F). In contrast, chromatin factors such as HPL-2 and HDA-1 exhibited an increase in nucleosome occupancy. These results suggest that EOR-1 binding sites exhibit changes in chromatin accessibility during the transition from early embryos to L3 that are unique from the other factors analyzed.
Taking these observations collectively, we propose four potential models to explain the unique binding profile of EOR-1: (1) EOR-1 is capable of binding to both open and closed chromatin (depending on the loci or the tissue); for example, EOR-1 could bind open sites in one tissue but closed sites in another tissue (Fig. 5A); (2) EOR-1 binds immediately adjacent to nucleosomes (resulting in larger ATAC-seq fragment sizes), including those nucleosomes that shift location between early embryo and L3 (explaining the change in accessibility and nucleosome signal) (Fig. 5B); (3) EOR-1 binds open chromatin as part of a larger complex, including factors not assayed here (e.g., EOR-2 [Howell et al. 2010]), thereby hindering the ability of transposons to interact with the genomic DNA (Fig. 5C); and (4) EOR-1 binds closed chromatin as a pioneer factor and contributes to its opening (Fig. 5D). Future experiments, such as nucleosome binding assays and chromatin profiling of eor-1 mutants, will be needed to distinguish between these models and to fully elucidate the mechanism underlying the unusual binding profile of EOR-1. Collectively, these analyses suggest that ATAC-seq on a complex sample can identify factors with unusual chromatin binding patterns that could regulate chromatin dynamics during development.
Figure 5.
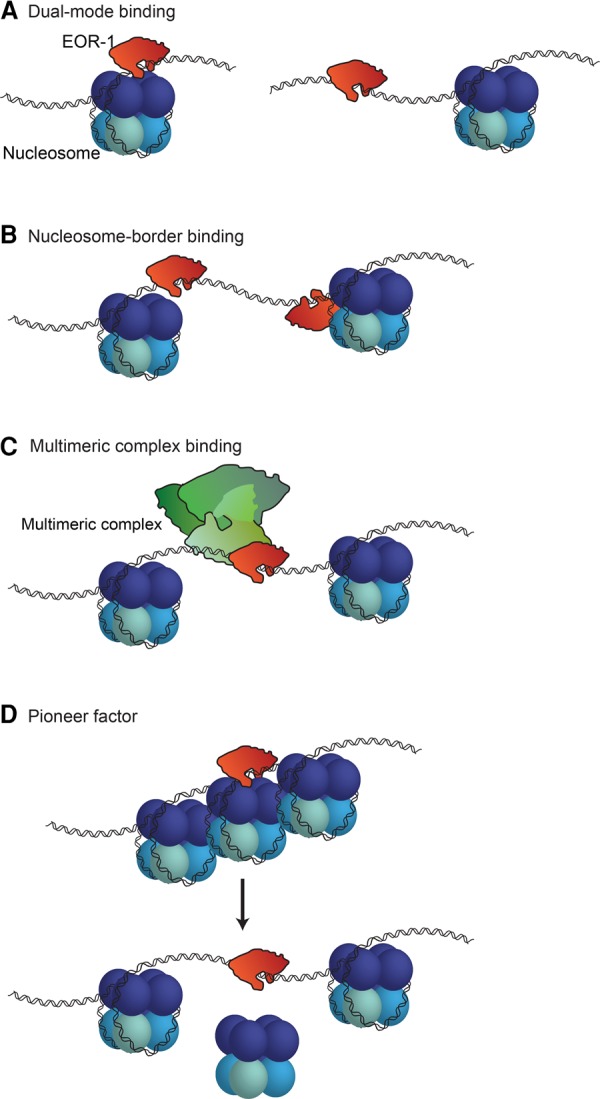
Possible models to explain EOR-1 binding characteristics. EOR-1 could either (A) bind both open and closed chromatin, depending on the genomic loci or the tissues; for example, EOR-1 could bind open sites in one tissue but closed sites in another tissue, (B) bind immediately adjacent to nucleosomes, (C) act as part of a large complex, or (D) act as a pioneer factor by binding and contributing to opening closed chromatin.
Discussion
Here, we show for the first time that sensitively measuring chromatin accessibility in a whole metazoan can detect dynamic changes throughout development and even identify novel functional enhancers active in only a small subset of the whole organism. Among the three developmental stages surveyed, we identified over 30,000 accessible sites which could serve as a catalog to facilitate the discovery of previously unknown distal regulatory loci (Supplemental Table S3) such as insulators and enhancers. This approach, developed here for C. elegans, should be readily applicable to other complex samples in vivo such as whole mammalian organs or tumor samples.
In C. elegans, enhancer identification has historically been limited, with most previous studies employing a single gene approach and focusing on promoter-proximal regions (Jantsch-Plunger and Fire 1994; Harfe et al. 1998; Lei et al. 2009). Other groups have identified enhancers genome-wide but have not functionally validated predicted enhancer activity (Vavouri et al. 2007; Chen et al. 2013a). In this study, we identify and functionally characterize distal ATAC-seq peaks as novel, active enhancers. These enhancers have a range of spatiotemporal activity patterns that are orientation-independent and are found at a diversity of genomic locations, suggesting that enhancers may be more prevalent in C. elegans gene regulation than previously appreciated. We note that some of these regions could also be promoters, especially when they are closer to the TSS, since promoters could also function in a bidirectional manner (Core et al. 2008; Jin et al. 2017). Indeed, two of the five regions we identified as enhancers (gei-13 and mlt-8) have previously been suggested to be promoters (despite their ChromHMM enhancer annotation) (Chen et al. 2013a). Thus, further experimental work will be required to verify the exact nature of the regulatory regions defined by ATAC-seq.
An interesting example is the enhancer downstream from the conserved transcription factor nhr-25. This putative nhr-25 enhancer is located 5 kb downstream from the nhr-25 TSS and acts in an orientation-independent manner. Thus, this enhancer might be looping to the nhr-25 TSS to enhance transcription, although the limited spatial resolution of current Hi-C data in C. elegans (Crane et al. 2015) does not provide information on the three-dimensional chromatin structure of the nhr-25 locus. This enhancer model contrasts with the promoter-proximal model most often thought to regulate gene expression in C. elegans but agrees with the recent Hi-C study which identified insulator-like loci throughout the C. elegans genome (Crane et al. 2015). Insulators are important regulators of three-dimensional chromatin architecture (Schoborg and Labrador 2014). Interestingly, nhr-25 orthologs in fly (Ftz-F1), mouse (Nr5a1), and human (NR5A1) exhibit a consistent signature of three-dimensional chromatin architecture downstream from the 3′ UTR (insulator class I in flies and CTCF-binding sites in mice and humans [Sharov et al. 2006; Rosenbloom et al. 2013; Attrill et al. 2016]). While C. elegans does not possess a CTCF ortholog, our results raise the intriguing possibility that the nhr-25 enhancer we have identified is evolutionarily conserved.
Furthermore, we have uncovered a potential role for a likely GAGA factor in C. elegans, EOR-1. The EOR-1 motif we initially detected closely resembles a dimeric version of the canonical GAGA motif bound by Trl/GAGA-Associated Factor (GAF) in Drosophila. GAF is a multifaceted transcription factor that can associate with heterochromatin (Raff et al. 1994), remodel chromatin in concert with nucleosome remodelers (Okada and Hirose 1998), and act as a transcriptional activator, in part due to its ability to increase chromatin accessibility (Adkins et al. 2006). Like Drosophila GAF, C. elegans EOR-1 is a transcriptional activator in the Ras/ERK signaling pathway that has been suggested to also repress gene expression (Liu et al. 2011a). GAF and EOR-1 are also similar in that both proteins have a BTB/POZ domain on their N terminal as well as C2H2 zinc-fingers and polyQ domains on their C terminals (Howard and Sundaram 2002). This dual action (gene activation and repression) is particularly interesting considering the bimodal binding pattern we found when examining ATAC-seq fragment size in EOR-1 binding sites.
In C. elegans, EOR-1 genetically interacts with at least two chromatin remodeling complexes (SWI/SNF and RSC) (Lehner et al. 2006). This is noteworthy given our findings that EOR-1 is found in less accessible and potentially even nucleosome-occupied regions of the genome and that the EOR-1 motif is predictive of increased accessibility in development. GAGA-factors are conserved regulators of gene expression (Ohtsuki and Levine 1998; Mahmoudi et al. 2002; Petrascheck et al. 2005; Srivastava et al. 2013), and a GAGA motif has been found to be necessary for enhancer functionality in C. elegans (Harfe et al. 1998). Indeed, two out of five of the novel validated enhancers identified in this study (nhr-25 and swip-10) contain an EOR-1/GAGA motif. Collectively, these data suggest that EOR-1 might regulate chromatin accessibility at enhancers in C. elegans and potentially other species.
Using C. elegans as a paradigm, we have shown that ATAC-seq performed on complex, heterogeneous samples can reveal novel, spatiotemporally specific genetic regulators and that measuring chromatin accessibility across a developmental time course can identify important dynamic regions. We have highlighted important applications of this approach: discovering functional distal regulatory regions active in only a small subset of the total sample and identifying candidate regulators of genome-wide chromatin dynamics. This data set, which represents an initial atlas of genome-wide chromatin accessibility and candidate distal regulatory sites in C. elegans, should be a valuable resource for the community. The fact that a genome-wide chromatin accessibility assay performed in whole organisms can sensitively identify previously undiscovered functional enhancers in vivo raises the exciting possibility that distal regulation plays a more important role than previously believed in the nematode.
Methods
Brief methods can be found below, but in all cases more details can be found in the Supplemental Methods.
C. elegans ATAC-seq
Three sets of completely independent biological replicates were prepared by harvesting and then flash-freezing tightly temporally synchronized samples at three different stages: early embryo (in utero), larval stage 3 (36 h post-egg lay), and young adult (57 h post-egg lay). Native nuclei were purified from frozen samples using mechanical homogenization as previously described (Haenni et al. 2012). The purified nuclei were immediately used for the ATAC-seq protocol (Buenrostro et al. 2013). An input control was also generated by using 10 ng of genomic DNA. Sequencing was performed using 101-bp paired-end sequencing on an Illumina HiSeq 2000.
ATAC-seq alignment and peak calling
The ATAC-seq libraries were sequenced to a median depth of over 17 million unique, high-quality mapping reads per sample (Supplemental Table S1). Prior to mapping, standard next-generation sequencing quality control steps, as well as ATAC-seq-specific quality control steps, were performed. Individual replicate ATAC-seq peaks were called using a custom pipeline which used MACS (v2.1) (Zhang et al. 2008) to call the peaks. To identify the most high-confidence set of peaks, we employed a strategy that emphasized peaks which were consistently observed across replicates. All peaks were masked for regions with significant signal in the input control and for previously identified regions known to give spurious results in next generation sequencing assays (Boyle et al. 2014). The result was a set of 30,832 consensus ATAC-seq peaks.
Selection of putative enhancers and generation of enhancer reporter constructs
For the enhancer screen, each putative regulatory region was cloned in the pL4051 plasmid (a gift from Andrew Fire) upstream of a minimal promoter (pes-10) driving expression of a C. elegans intron- and photo-stability-optimized GFP containing an N-terminal nucleolar localization signal. Putative regulatory regions were chosen by selecting ATAC-seq peaks that exhibited the largest differential accessibility between two stages and that were at least 1 kb from a transcription start site (as defined on the UCSC Genome Browser). Flanking negative control regions were chosen by selecting regions within 2 kb of the putative regulatory regions that were not in peaks of accessibility. Primers were designed to amplify each region as well as 50–500 bp flanking either side (Supplemental Table S13).
Enhancer screen in C. elegans
Stable extrachromosomal transgenic lines for putative enhancer regions (in both orientations), negative control regions, and the no-insert controls were generated. Multiple independent lines were generated per construct (Table 1). For each line, mixed-staged worms were screened for GFP signal distinct from the no-insert control background signal, which is 1–2 nuclei near the pharynx in all larval and adult stages (Supplemental Fig. S5E). To quantify the consistency of GFP expression pattern, all lines (including the negative control lines) (Table 1) were scored for GFP expression pattern in a blinded manner.
Machine learning models to predict accessibility changes with motifs
To predict changes in accessibility between early embryo and larval stage 3, the number of each mapped C. elegans motif from cisBP (v1.02) (Weirauch et al. 2014) and each de novo discovered motif found in the dynamics ATAC-seq peaks between early embryo and L3 (59 in total) was counted to create a matrix of 166 motif-counts and 30,832 ATAC-seq peaks. The ATAC-seq peaks were split into a training set (70%) and a testing set and several classification models evaluated. We found that a generalized boosting model (GBM) (https://CRAN.R-project.org/package=gbm) had the best accuracy while still allowing for interpretation of which motifs were the most informative. Given the unbalanced classification problem, we used balanced accuracy as our primary metric of classification success (Supplemental Fig. S6D).
Software availability
All analysis source code is freely available at https://github.com/brunetlab/CelegansATACseq, as well as in the Supplemental Material.
Data access
The raw data as well as stage-specific peaks for all three stages and the input control have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE89608.
Supplementary Material
Acknowledgments
We thank Dr. Elizabeth Noblin and Max Lenail for their help imaging enhancer reporter strains, and Dr. Eric Greer for assistance in developing the nuclei isolation protocol. We thank Dr. Elizabeth Noblin, Dr. Lauren Booth, and Dr. Bérénice Benayoun for critically reading the manuscript. We thank Dr. Bérénice Benayoun for feedback on computational pipeline and analysis, Daniel Kim for use of his ‘smartMerge’ script, and Dr. Andy Fire and Dr. Joanna Wysocka for suggestions on the project. This work is supported by the National Institutes of Health DP1 AG044848 (A.B.), a seed grant from the Discovery Innovation Fund (A.B.), and NSF graduate fellowship (A.C.D.). J.D.B. is a member of the Harvard Society of Fellows.
Author contributions: A.C.D. and A.B. planned the study. A.C.D. performed the ATAC-seq experiments, designed the analytical pipeline, analyzed and interpreted the data, and wrote the manuscript with help from A.B. R.W.Y. generated the transgenic enhancer reporter lines, imaged and analyzed GFP expression, and led revisions to the manuscript. A.K. provided intellectual guidance with ATAC-seq analysis and machine learning. J.D.B. and W.J.G. provided early access to ATAC-seq protocols and feedback on the design. All authors discussed the results and commented on the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.226233.117.
Freely available online through the Genome Research Open Access option.
References
- Adkins NL, Hagerman TA, Georgel P. 2006. GAGA protein: a multi-faceted transcription factor. Biochem Cell Biol 84: 559–567. [DOI] [PubMed] [Google Scholar]
- Altun Z, Hall D. 2009. Muscle system, somatic muscle. In WormAtlas. 10.3908/wormatlas.1.7. [DOI] [Google Scholar]
- Antebi A, Culotti JG, Hedgecock EM. 1998. daf-12 regulates developmental age and the dauer alternative in Caenorhabditis elegans. Development 125: 1191–1205. [DOI] [PubMed] [Google Scholar]
- Antebi A, Yeh WH, Tait D, Hedgecock EM, Riddle DL. 2000. daf-12 encodes a nuclear receptor that regulates the dauer diapause and developmental age in C. elegans. Genes Dev 14: 1512–1527. [PMC free article] [PubMed] [Google Scholar]
- Araya CL, Kawli T, Kundaje A, Jiang L, Wu B, Vafeados D, Terrell R, Weissdepp P, Gevirtzman L, Mace D, et al. 2014. Regulatory analysis of the C. elegans genome with spatiotemporal resolution. Nature 512: 400–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold CD, Gerlach D, Stelzer C, Boryn LM, Rath M, Stark A. 2013. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 339: 1074–1077. [DOI] [PubMed] [Google Scholar]
- Attrill H, Falls K, Goodman JL, Millburn GH, Antonazzo G, Rey AJ, Marygold SJ, FlyBase C. 2016. FlyBase: establishing a Gene Group resource for Drosophila melanogaster. Nucleic Acids Res 44: D786–D792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X, Rubin AJ, Qu K, Zhang J, Giresi PG, Chang HY, Khavari PA. 2015. A novel ATAC-seq approach reveals lineage-specific reinforcement of the open chromatin landscape via cooperation between BAF and p63. Genome Biol 16: 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonn S, Zinzen RP, Girardot C, Gustafson EH, Perez-Gonzalez A, Delhomme N, Ghavi-Helm Y, Wilczynski B, Riddell A, Furlong EE. 2012. Tissue-specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nat Genet 44: 148–156. [DOI] [PubMed] [Google Scholar]
- Boyle AP, Araya CL, Brdlik C, Cayting P, Cheng C, Cheng Y, Gardner K, Hillier LW, Janette J, Jiang L, et al. 2014. Comparative analysis of regulatory information and circuits across distant species. Nature 512: 453–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, Greenleaf WJ. 2015. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523: 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byerly L, Cassada RC, Russell RL. 1976. The life cycle of the nematode Caenorhabditis elegans. I. Wild-type growth and reproduction. Dev Biol 51: 23–33. [DOI] [PubMed] [Google Scholar]
- Chen RA, Down TA, Stempor P, Chen QB, Egelhofer TA, Hillier LW, Jeffers TE, Ahringer J. 2013a. The landscape of RNA polymerase II transcription initiation in C. elegans reveals promoter and enhancer architectures. Genome Res 23: 1339–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Xi Y, Pan X, Li Z, Kaestner K, Tyler J, Dent S, He X, Li W. 2013b. DANPOS: Dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res 23: 341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contrino S, Smith RN, Butano D, Carr A, Hu F, Lyne R, Rutherford K, Kalderimis A, Sullivan J, Carbon S, et al. 2012. modMine: flexible access to modENCODE data. Nucleic Acids Res 40: D1082–D1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, Koenig JL, Snyder MP, Pritchard JK, Kundaje A, Greenleaf WJ, et al. 2016. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet 48: 1193–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, Lis JT. 2008. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322: 1845–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane E, Bian Q, McCord RP, Lajoie BR, Wheeler BS, Ralston EJ, Uzawa S, Dekker J, Meyer BJ. 2015. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature 523: 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusanovich DA, Daza R, Adey A, Pliner HA, Christiansen L, Gunderson KL, Steemers FJ, Trapnell C, Shendure J. 2015. Epigenetics. Multiplex single-cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348: 910–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. 2009. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kellis M. 2012. ChromHMM: automating chromatin-state discovery and characterization. Nat Methods 9: 215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans KJ, Huang N, Stempor P, Chesney MA, Down TA, Ahringer J. 2016. Stable Caenorhabditis elegans chromatin domains separate broadly expressed and developmentally regulated genes. Proc Natl Acad Sci 113: E7020–E7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furey TS. 2012. ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nat Rev Genet 13: 840–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A, Ikegami K, et al. 2010. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science 330: 1775–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein MB, Rozowsky J, Yan KK, Wang D, Cheng C, Brown JB, Davis CA, Hillier L, Sisu C, Li JJ, et al. 2014. Comparative analysis of the transcriptome across distant species. Nature 512: 445–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD. 2007. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res 17: 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gissendanner CR, Sluder AE. 2000. nhr-25, the Caenorhabditis elegans ortholog of ftz-f1, is required for epidermal and somatic gonad development. Dev Biol 221: 259–272. [DOI] [PubMed] [Google Scholar]
- Haenni S, Ji Z, Hoque M, Rust N, Sharpe H, Eberhard R, Browne C, Hengartner MO, Mellor J, Tian B, et al. 2012. Analysis of C. elegans intestinal gene expression and polyadenylation by fluorescence-activated nuclei sorting and 3′-end-seq. Nucleic Acids Res 40: 6304–6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harfe BD, Vaz Gomes A, Kenyon C, Liu J, Krause M, Fire A. 1998. Analysis of a Caenorhabditis elegans Twist homolog identifies conserved and divergent aspects of mesodermal patterning. Genes Dev 12: 2623–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He HH, Meyer CA, Shin H, Bailey ST, Wei G, Wang Q, Zhang Y, Xu K, Ni M, Lupien M, et al. 2010. Nucleosome dynamics define transcriptional enhancers. Nat Genet 42: 343–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecker A, Brand LH, Peter S, Simoncello N, Kilian J, Harter K, Gaudin V, Wanke D. 2015. The Arabidopsis GAGA-binding factor BASIC PENTACYSTEINE6 recruits the POLYCOMB-REPRESSIVE COMPLEX1 component LIKE HETEROCHROMATIN PROTEIN1 to GAGA DNA motifs. Plant Physiol 168: 1013–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Romanoski CE, Benner C, Glass CK. 2015. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol 16: 144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier LW, Reinke V, Green P, Hirst M, Marra MA, Waterston RH. 2009. Massively parallel sequencing of the polyadenylated transcriptome of C. elegans. Genome Res 19: 657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho JW, Jung YL, Liu T, Alver BH, Lee S, Ikegami K, Sohn KA, Minoda A, Tolstorukov MY, Appert A, et al. 2014. Comparative analysis of metazoan chromatin organization. Nature 512: 449–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard RM, Sundaram MV. 2002. C. elegans EOR-1/PLZF and EOR-2 positively regulate Ras and Wnt signaling and function redundantly with LIN-25 and the SUR-2 Mediator component. Genes Dev 16: 1815–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell K, Arur S, Schedl T, Sundaram MV. 2010. EOR-2 is an obligate binding partner of the BTB–zinc finger protein EOR-1 in Caenorhabditis elegans. Genetics 184: 899–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu HT, Chen HM, Yang Z, Wang J, Lee NK, Burger A, Zaret K, Liu T, Levine E, Mango SE. 2015. TRANSCRIPTION. Recruitment of RNA polymerase II by the pioneer transcription factor PHA-4. Science 348: 1372–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jantsch-Plunger V, Fire A. 1994. Combinatorial structure of a body muscle-specific transcriptional enhancer in Caenorhabditis elegans. J Biol Chem 269: 27021–27028. [PubMed] [Google Scholar]
- Jin Y, Eser U, Struhl K, Churchman LS. 2017. The ground state and evolution of promoter region directionality. Cell 170: 889–898.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause M, Harrison SW, Xu SQ, Chen L, Fire A. 1994. Elements regulating cell- and stage-specific expression of the C. elegans MyoD family homolog hlh-1. Dev Biol 166: 133–148. [DOI] [PubMed] [Google Scholar]
- Kruesi WS, Core LJ, Waters CT, Lis JT, Meyer BJ. 2013. Condensin controls recruitment of RNA polymerase II to achieve nematode X-chromosome dosage compensation. eLife 2: e00808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvon EZ. 2015. Using transgenic reporter assays to functionally characterize enhancers in animals. Genomics 106: 185–192. [DOI] [PubMed] [Google Scholar]
- Lara-Astiaso D, Weiner A, Lorenzo-Vivas E, Zaretsky I, Jaitin DA, David E, Keren-Shaul H, Mildner A, Winter D, Jung S, et al. 2014. Immunogenetics. Chromatin state dynamics during blood formation. Science 345: 943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner B, Crombie C, Tischler J, Fortunato A, Fraser AG. 2006. Systematic mapping of genetic interactions in Caenorhabditis elegans identifies common modifiers of diverse signaling pathways. Nat Genet 38: 896–903. [DOI] [PubMed] [Google Scholar]
- Lei H, Liu J, Fukushige T, Fire A, Krause M. 2009. Caudal-like PAL-1 directly activates the bodywall muscle module regulator hlh-1 in C. elegans to initiate the embryonic muscle gene regulatory network. Development 136: 1241–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei I, West J, Yan Z, Gao X, Fang P, Dennis JH, Gnatovskiy L, Wang W, Kingston RE, Wang Z. 2015. BAF250a protein regulates nucleosome occupancy and histone modifications in priming embryonic stem cell differentiation. J Biol Chem 290: 19343–19352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Rogers J, Murphy CT, Rongo C. 2011a. EGF signalling activates the ubiquitin proteasome system to modulate C. elegans lifespan. EMBO J 30: 2990–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Rechtsteiner A, Egelhofer TA, Vielle A, Latorre I, Cheung MS, Ercan S, Ikegami K, Jensen M, Kolasinska-Zwierz P, et al. 2011b. Broad chromosomal domains of histone modification patterns in C. elegans. Genome Res 21: 227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorzadeh A, Bilenky M, Hammond C, Knapp DJ, Li L, Miller PH, Carles A, Heravi-Moussavi A, Gakkhar S, Moksa M, et al. 2016. Nucleosome density ChIP-seq identifies distinct chromatin modification signatures associated with MNase accessibility. Cell Rep 17: 2112–2124. [DOI] [PubMed] [Google Scholar]
- Lund E, Oldenburg AR, Delbarre E, Freberg CT, Duband-Goulet I, Eskeland R, Buendia B, Collas P. 2013. Lamin A/C-promoter interactions specify chromatin state–dependent transcription outcomes. Genome Res 23: 1580–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoudi T, Katsani KR, Verrijzer CP. 2002. GAGA can mediate enhancer function in trans by linking two separate DNA molecules. EMBO J 21: 1775–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello C, Fire A. 1995. DNA transformation. Methods Cell Biol 48: 451–482. [PubMed] [Google Scholar]
- Minnich M, Tagoh H, Bonelt P, Axelsson E, Fischer M, Cebolla B, Tarakhovsky A, Nutt SL, Jaritz M, Busslinger M. 2016. Multifunctional role of the transcription factor Blimp-1 in coordinating plasma cell differentiation. Nat Immunol 17: 331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mito Y, Henikoff JG, Henikoff S. 2007. Histone replacement marks the boundaries of cis-regulatory domains. Science 315: 1408–1411. [DOI] [PubMed] [Google Scholar]
- Narasimhan K, Lambert SA, Yang AW, Riddell J, Mnaimneh S, Zheng H, Albu M, Najafabadi HS, Reece-Hoyes JS, Fuxman Bass JI, et al. 2015. Mapping and analysis of Caenorhabditis elegans transcription factor sequence specificities. eLife 4 10.7554/eLife.06967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuki S, Levine M. 1998. GAGA mediates the enhancer blocking activity of the eve promoter in the Drosophila embryo. Genes Dev 12: 3325–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada M, Hirose S. 1998. Chromatin remodeling mediated by Drosophila GAGA factor and ISWI activates fushi tarazu gene transcription in vitro. Mol Cell Biol 18: 2455–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker S, Baylis HA. 2009. Overexpression of caveolins in Caenorhabditis elegans induces changes in egg-laying and fecundity. Commun Integr Biol 2: 382–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennacchio LA, Ahituv N, Moses AM, Prabhakar S, Nobrega MA, Shoukry M, Minovitsky S, Dubchak I, Holt A, Lewis KD, et al. 2006. In vivo enhancer analysis of human conserved non-coding sequences. Nature 444: 499–502. [DOI] [PubMed] [Google Scholar]
- Petrascheck M, Escher D, Mahmoudi T, Verrijzer CP, Schaffner W, Barberis A. 2005. DNA looping induced by a transcriptional enhancer in vivo. Nucleic Acids Res 33: 3743–3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. 2011. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470: 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff JW, Kellum R, Alberts B. 1994. The Drosophila GAGA transcription factor is associated with specific regions of heterochromatin throughout the cell cycle. EMBO J 13: 5977–5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke V, Krause M, Okkema P. 2013. Transcriptional regulation of gene expression in C. elegans. WormBook. 10.1895/wormbook.1.45.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, Yue F. 2015. Transcriptional enhancers: bridging the genome and phenome. Cold Spring Harb Symp Quant Biol 80: 17–26. [DOI] [PubMed] [Google Scholar]
- Riedel CG, Dowen RH, Lourenco GF, Kirienko NV, Heimbucher T, West JA, Bowman SK, Kingston RE, Dillin A, Asara JM, et al. 2013. DAF-16 employs the chromatin remodeller SWI/SNF to promote stress resistance and longevity. Nat Cell Biol 15: 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM, Wong MC, Maddren M, Fang R, Heitner SG, et al. 2013. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res 41: D56–D63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep AN, Buenrostro JD, Denny SK, Schwartz K, Sherlock G, Greenleaf WJ. 2015. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Res 25: 1757–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoborg T, Labrador M. 2014. Expanding the roles of chromatin insulators in nuclear architecture, chromatin organization and genome function. Cell Mol Life Sci 71: 4089–4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen S, Block KF, Pasini A, Baylin SB, Easwaran H. 2016. Genome-wide positioning of bivalent mononucleosomes. BMC Med Genomics 9: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharov AA, Dudekula DB, Ko MS. 2006. CisView: a browser and database of cis-regulatory modules predicted in the mouse genome. DNA Res 13: 123–134. [DOI] [PubMed] [Google Scholar]
- Shi B, Guo X, Wu T, Sheng S, Wang J, Skogerbo G, Zhu X, Chen R. 2009. Genome-scale identification of Caenorhabditis elegans regulatory elements by tiling-array mapping of DNase I hypersensitive sites. BMC Genomics 10: 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JM, Hacker KE, Singh D, Brannon AR, Parker JS, Weiser M, Ho TH, Kuan PF, Jonasch E, Furey TS, et al. 2014. Variation in chromatin accessibility in human kidney cancer links H3K36 methyltransferase loss with widespread RNA processing defects. Genome Res 24: 241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Puri D, Garapati HS, Dhawan J, Mishra RK. 2013. Vertebrate GAGA factor associated insulator elements demarcate homeotic genes in the HOX clusters. Epigenetics Chromatin 6: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stergachis AB, Neph S, Reynolds A, Humbert R, Miller B, Paige SL, Vernot B, Cheng JB, Thurman RE, Sandstrom R, et al. 2013. Developmental fate and cellular maturity encoded in human regulatory DNA landscapes. Cell 154: 888–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston JE, Schierenberg E, White JG, Thomson JN. 1983. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol 100: 64–119. [DOI] [PubMed] [Google Scholar]
- Sun Y, Nien CY, Chen K, Liu HY, Johnston J, Zeitlinger J, Rushlow C. 2015. Zelda overcomes the high intrinsic nucleosome barrier at enhancers during Drosophila zygotic genome activation. Genome Res 25: 1703–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S, Li XY, Sabo PJ, Sandstrom R, Thurman RE, Canfield TK, Giste E, Fisher W, Hammonds A, Celniker SE, et al. 2011. Dynamic reprogramming of chromatin accessibility during Drosophila embryo development. Genome Biol 12: R43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsompana M, Buck MJ. 2014. Chromatin accessibility: a window into the genome. Epigenetics Chromatin 7: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valouev A, Ichikawa J, Tonthat T, Stuart J, Ranade S, Peckham H, Zeng K, Malek JA, Costa G, McKernan K, et al. 2008. A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning. Genome Res 18: 1051–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vavouri T, Walter K, Gilks WR, Lehner B, Elgar G. 2007. Parallel evolution of conserved non-coding elements that target a common set of developmental regulatory genes from worms to humans. Genome Biol 8: R15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Rubin EM, Pennacchio LA. 2009. Genomic views of distant-acting enhancers. Nature 461: 199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, et al. 2008. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 40: 897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YM, Zhou P, Wang LY, Li ZH, Zhang YN, Zhang YX. 2012. Correlation between DNase I hypersensitive site distribution and gene expression in HeLa S3 cells. PLoS One 7: e42414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K, et al. 2014. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158: 1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JA, Cook A, Alver BH, Stadtfeld M, Deaton AM, Hochedlinger K, Park PJ, Tolstorukov MY, Kingston RE. 2014. Nucleosomal occupancy changes locally over key regulatory regions during cell differentiation and reprogramming. Nat Commun 5: 4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, Tesar PJ, Scacheri PC. 2011. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res 21: 1273–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based Analysis of ChIP-Seq (MACS). Genome Biol 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wu Y, Schnable JC, Zeng Z, Freeling M, Crawford GE, Jiang J. 2012. High-resolution mapping of open chromatin in the rice genome. Genome Res 22: 151–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Sun L, Chen Z, Whitaker JW, Wang T, Wang W. 2013. Predicting enhancer transcription and activity from chromatin modifications. Nucleic Acids Res 41: 10032–10043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B, Zhang W, Zhang T, Liu B, Jiang J. 2015. Genome-wide prediction and validation of intergenic enhancers in Arabidopsis using open chromatin signatures. Plant Cell 27: 2415–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.