

Abstract
Many pathogens such as Shigella and other bacteria assemble the type III secretion system (T3SS) nanoinjector to inject virulence proteins into their target cells to cause infectious diseases in humans. The rise of drug resistance among pathogens that rely on the T3SS for infectivity, plus the dearth of new antibiotics require alternative strategies in developing new antibiotics. The Shigella T3SS tip protein IpaD is an attractive target for developing anti-infectives because of its essential role in virulence and its exposure on the bacterial surface. Currently, the only known small molecules that bind to IpaD are bile salts sterols. Here, we identified four new small molecule scaffolds that bind to IpaD based on the methylquinoline, pyrrolidin-aniline, hydroxyindole, and morpholinoaniline scaffolds. NMR mapping revealed potential hotspots in IpaD for binding small molecules. These scaffolds can be used as building blocks in developing small molecule inhibitors of IpaD that could lead to new anti-infectives.
Keywords: type III secretion system, IpaD, NMR, small molecules, surface plasmon resonance
Introduction
Shigella is endemic in many countries and infects over 90 million people worldwide annually[1] causing an estimated 100,000 deaths per year.[2] Like many Gram-negative pathogens such as Pseudomonas, Salmonella, Chlamydia, and Yersinia sp. that cause infectious diseases in humans, Shigella deploys the type III secretion system (T3SS) to inject virulence proteins directly into its host cells to establish infection.[3] The T3SS is essential in the pathogenesis of these pathogens, and defects in the proper assembly of the T3SS render these pathogens non-infective. The structural component of the T3SS is a needle apparatus that functions like a nanoscale injector of bacterial virulence proteins directly into eukaryotic cells.
The T3SS needle apparatus is comprised of a basal body that spans the two bacterial membranes, an extracellular needle, a tip complex, which in Shigella, is formed by the tip protein IpaD (37 kDa), and two membrane proteins IpaB (62 kDa) and IpaC (42 kDa). IpaB and IpaC are membrane proteins that insert into the host cell membrane, forming a translocon pore, to allow the passage of bacterial effector proteins into the host cell. IpaD is expected to form a pentameric ring complex at the tip of the needle and functions as a platform for the assembly of the translocon.[4] IpaD has a dumbbell-like structure with a long central coiled-coil attached to an N-terminal α-helical hairpin domain, and a C-terminal globular domain of mixed a-helices and b-sheets at the other end.[5] Results of mutagenesis showed that the coiled-coil helices are responsible for the proper assembly of the needle apparatus, while the distal domains are involved in signal transduction.[5–6]
The increase in antibiotic resistance among pathogens, including multidrug-resistant strains of Shigella, is a serious public health problem.[7] Therefore, there is a need to identify novel targets for developing new antibiotics.[7b, 7d, 8] Because the T3SS is essential for virulence, it is an attractive target for developing anti-infectives or drugs that prevent infection but not necessarily destroy pathogens.[9] IpaD in particular is an attractive target for several reasons: (i) it is essential for infectivity;[10] (ii) it is exposed on the bacterial surface; and (iii) it is conserved in other bacteria. Developing new anti-infectives that target IpaD require the identification of small molecules that can bind and inhibit the function of IpaD. There are currently no known small-molecule inhibitors of IpaD or any of its homologs. Further, the only known small molecules that bind to IpaD are sterol-like compounds like bile salts that are present in the digestive tract and trigger the final steps in the assembly of the translocon.[11] We have previously identified small molecule scaffolds that bind to the homolog of IpaD in Salmonella, SipD.[12] Nevertheless, despite 56% sequence similarity between IpaD and SipD, they bind to small molecules differently. Here, we used fragment-based approach by SPR screening to identify four new small molecule scaffolds that bind to IpaD. Our NMR characterization of the IpaD-small molecule interaction identified potential hotspots in IpaD for binding a variety of small molecules.
Results
SPR screening
Our SPR-based screening of a commercial library of 288 fragments from Zenobia identified 4 small molecule scaffolds (Figure 1) that bind to IpaD (Figure 2). These compounds are based on the quinoline [4-amino-2-methylquinoline, compound 1, Figure 1], aniline [4-(2-(Pyrrolidin-1-yl)ethyl)aniline, compound 2; and 4-morpholinoaniline, compound 4], and hydroxyindole [5-hydroxyindole, compound 3] scaffolds. Analogues of these four compounds (1a–d, 2a–b, 3a–c, 4a–b, Figure 1) present in the library that did not bind to IpaD enabled the identification of chemical groups that are important in binding IpaD. In compound 1, the amino and methyl groups are important for binding IpaD as removal of the amino group (in 1a and 1d, Figure 1), removal of the methyl group (in 1c or 1d), or bromination (in 1c or 1d) abolished binding to IpaD. In compound 2, the pyrrolidine group is important for binding, which when substituted by either piperidine, piperazine, or amine group; or altering the spacing between the two rings (in 2a or 2b) resulted in loss of binding to IpaD. In compound 3, removal of the hydroxyl group (in 3b) or addition of acetyl (in 3a) or carboxylic group (in 3c) abrogated binding to IpaD. Finally, in compound 4, removal of the amine group (in 4a) and introduction of a hydroxyl group (in 4a) or methylene group in between the two rings (in 4b) resulted in loss of binding to IpaD. This information is useful in further derivatization of compounds 1, 2, 3, and 4 for optimal binding to IpaD.
Figure 1.
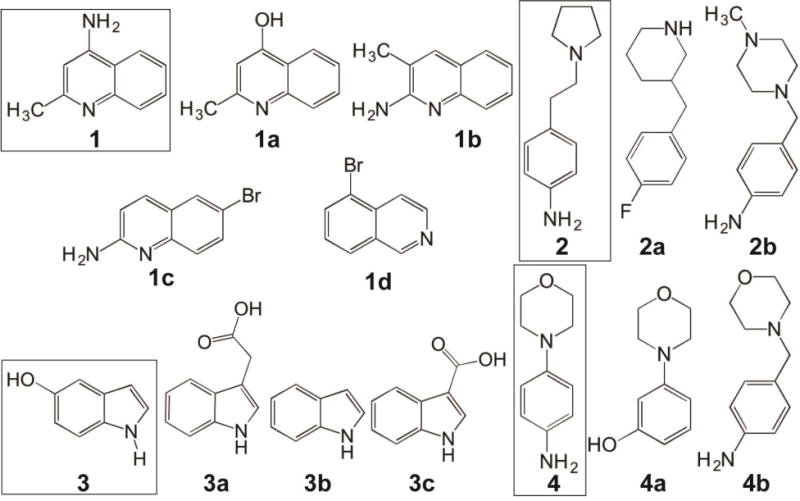
The four scaffolds that bind to IpaD (boxed) are 4-amino-2-methylquinoline (1), 4-[2-(pyrrolidin-1-yl) ethyl] aniline (2), 5-hydroxyindole (3), and 4-morpholinoaniline (4). Analogs of these four scaffolds that did not bind to IpaD identified which chemical moieties are important for binding to IpaD. The analogs are 4-hydroxy-2-methylquinoline (1a), 2-amino-3-methylquinoline (1b), 2-amino-6-bromoquinoline (1c), 5-bromoisoquinoline (1d), 3-(4-fluorobenzyl)-piperidine (2a), 4-(4-methyl-piperazin-1-ylmethyl)-phenylamine (2b), 3-indole acetic acid (3a), indole (3b), indole-3-carboxylic acid (3c), 3-morpholinophenol (4a) and 4-morpholin-4-ylmethyl-phenylamine (4b).
Figure 2.
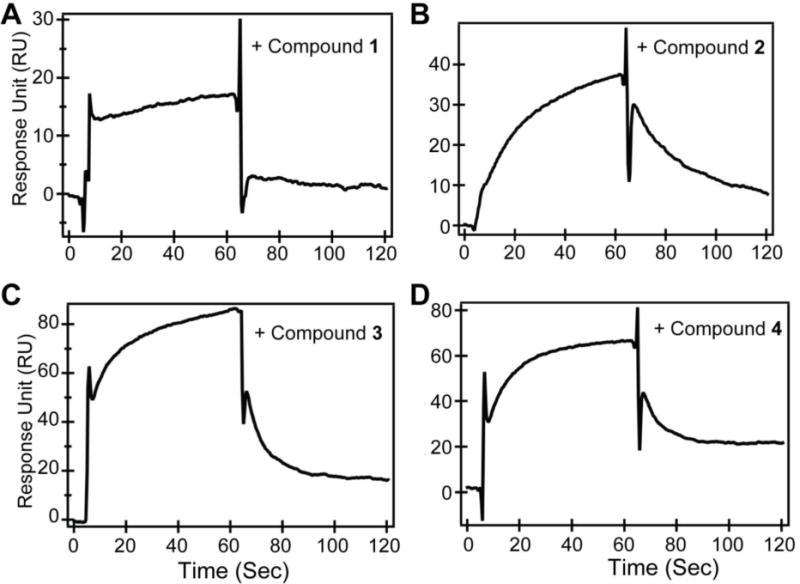
Surface plasmon resonance sensorgrams of IpaD with compounds A) 1, B) 2, C) 3 and D) 4.
STD NMR
We used saturation transfer difference (STD) NMR,[13] to determine how the small molecules interact with the protein. In STD NMR, the proton resonances of the protein are saturated with a selective pulse that is turned on or off, and two spectra are acquired. On the on-resonance spectrum, where the protein is saturated by the selective pulse, magnetization is transferred to the small molecule and detected. The off-resonance spectrum (top panels, Figure 3) show the NMR peaks of the small molecule and the protein. The STD spectrum (bottom panels, Figure 3) is the difference between the off-resonance and the on-resonance spectra, and shows protons of the small molecule that are in contact with the protein. Small molecules that do not interact with the protein will not show peaks in the STD spectra. Results of STD NMR showed that all the protons of the four small molecules tested showed STD peaks (Figure 3) indicating close proximity of these protons to the protein. In compound 1, both the quinoline and methyl groups showed STD peaks (Figure 3A) indicating these group are in contact with the protein. Additionally, the relatively stronger STD peak from the methyl group compared to the quinoline ring (Figure 3A), suggested the importance of hydrophobic interaction of 1 with the protein and corroborated the results from the SPR screen (Figure 1) showing that removal of the methyl group in (1) eliminated binding to IpaD. Upon binding of compound 2 to IpaD, there were relatively stronger STD peaks for the ethyl group (marked b/h, Figure 3B) and the pyrrolidine ring (marked a/j, Figure 3B) compared to that of the aniline group. The STD peaks for compounds 3 and 4 (Figure 3C, Figure 3D) showed relatively equal intensities suggesting that every part of 3 and 4 was equally important in binding to IpaD. Overall, the results of STD NMR suggested that the four small molecules were essentially embedded in IpaD.
Figure 3.
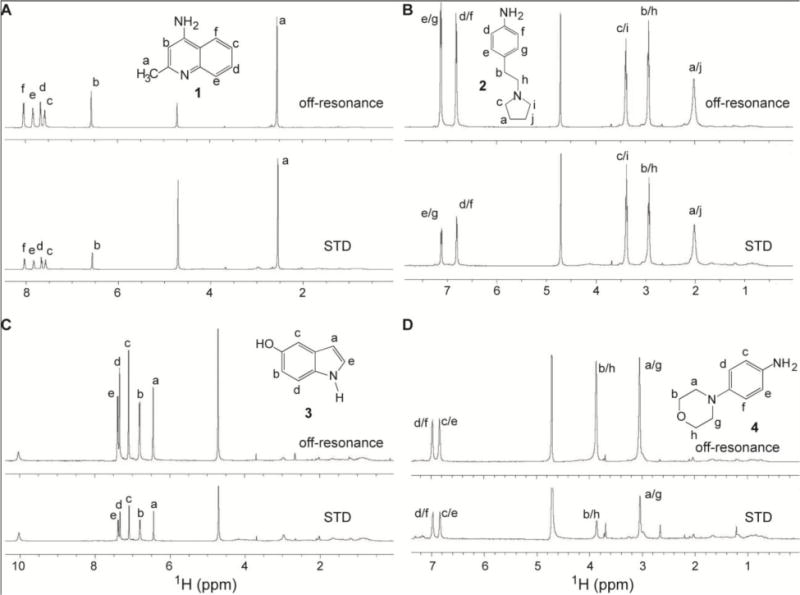
STD NMR of IpaD with compounds A) 1, B) 2, C) 3 and D) 4. Top panel shows the off-resonance spectra and the assignment of protons for each scaffold. Lower panel shows the STD spectra, after the on-resonance spectra (not shown) were subtracted from their corresponding off-resonance spectra, indicating which protons of the scaffolds are in contact with the protein.
ILV assignments of IpaD
NMR studies of protein-ligand interaction commonly use 15N-labeled proteins. However, results of STD NMR above suggested the importance of hydrophobic interaction of small molecules with IpaD, thus, in the NMR titrations of IpaD, we also used the hydrophobic ILV probes – where specific methyl groups of Isoleucines, Leucines and Valines in IpaD are 13C-labeled. The ILV 2D 1H-13C HSQC spectra of IpaD (Supporting Information Figure S1) show 117 peaks consisting of 13Cδ methyl resonances of 15 isoleucines, 13Cδ paired resonances of 35 leucines and 13Cγ paired resonances of 16 valines. Site directed mutagenesis of Ile to Leu assisted in assigning the isoleucine 13Cδ methyl resonances. Additionally, 1H-1H nuclear Overhauser effects (NOEs) observed through-space using 3D HMQC-NOESY of perdeuterated ILV IpaD and (Cδ/γ-Cδ/γ) distance calculations from the crystal structure of IpaD [PDB ID: 2J0O],[5] helped in assigning all the 16 valines and 32 out of 35 leucines in IpaD.
NMR titrations of 15N and ILV labeled IpaD
We used NMR methods to identify which surfaces of IpaD are involved in binding the small molecules. IpaD labeled simultaneously with 15N and ILV was titrated with increasing concentrations of the compounds 1, 2, 3, or 4; and the titrations were monitored by acquiring 2D 1H-15N TROSY and 2D 1H-13C HSQC spectra. We used the 111 ILV assignments of IpaD described above in addition to the 201 15N backbone amides that were previously assigned by others[11b] to determine which residues of IpaD were affected by the small molecules.
Titration of IpaD with the compounds (1, 2, 3, 4) showed chemical shift perturbations of both 15N amide and ILV peaks in a concentration dependent manner as shown in 2D 1H-15N TROSY and 2D 1H-13C ILV HSQC spectra (Figure 4, Supporting Information Figures S2, S3, S4, S5). The IpaD residues that were affected showed significant changes in their peak positions upon titration of increasing amounts of compounds, indicating protein-ligand interaction in fast exchange NMR time scale (Figure 4). Plots of the weighted chemical shift deviations for each of the compound showed which 15N and ILV peaks were strongly affected relative to the rest of the IpaD residues (Figure 5). Further, mapping the chemical shift deviations (Figure 5) on the surface of the crystal structure of IpaD, identified the binding pockets in IpaD for the small molecules (Figure 6). The results of the ILV titrations complemented and added additional information from the results of the more traditional 15N-based NMR titrations. For example, when compound 1 binds to IpaD, the most strongly affected 15N residues are Q148 and Y149 (Figure 5A), and for ILV residues, I145 and V152 (Figure 5B). All these residues (I145, Q148, Y149, and V152) cluster on the same surface of IpaD (Figure 6A), identifying the binding pocket in IpaD for compound 1. The functional groups of compounds 1, 2, 3, and 4, and the IpaD residues affected upon binding suggested that to bind IpaD, the compounds rely on polar contacts mediated by hydrogen bonding and ionic contacts, as well as hydrophobic interaction via aromatic and aliphatic rings present in the functional groups of compounds.
Figure 4.
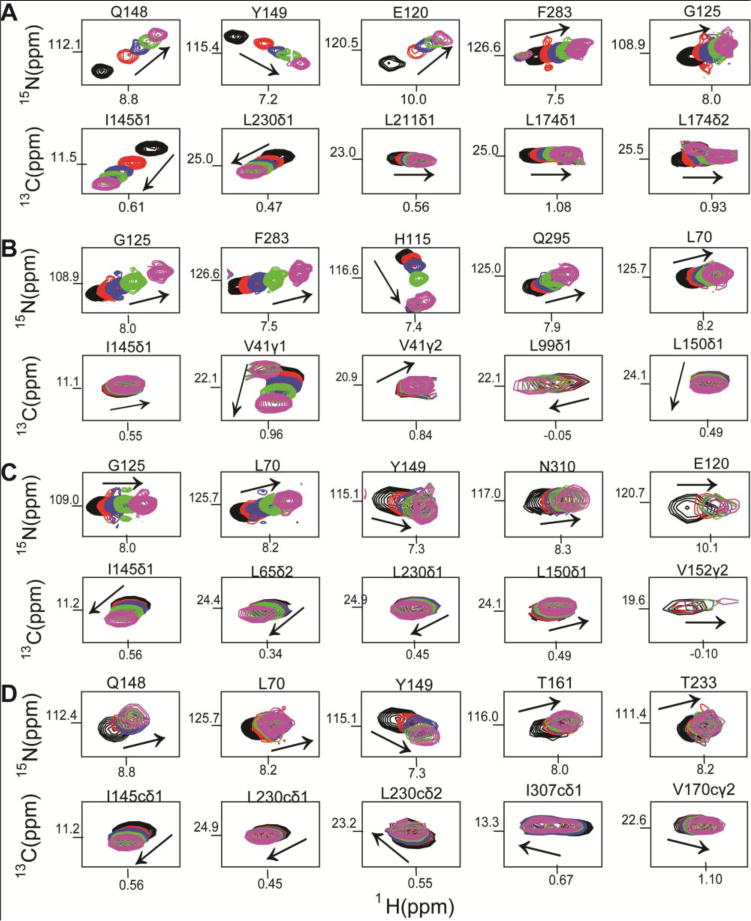
Selected regions from 2D 1H 15N HSQC spectra of IpaD titrated with compounds A) 1, B) 2, C) 3 and D) 4. Only IpaD residues that showed changes in peak positions are shown. Arrows indicate the movement of peaks upon titration of IpaD with increasing molar ratios of each compound. Peaks are colored (black, red, blue, green, magenta) according to increasing molar ratios of IpaD:scaffold. The full HSQC spectra, including the molar ratios used for each titration, are in the Supplementary.
Figure 5.
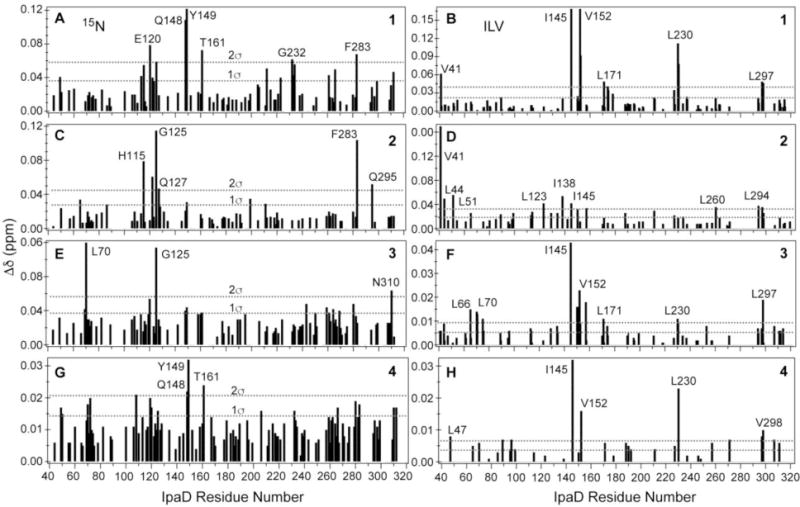
Plots of the weighted chemical shift deviations (Δδ) of IpaD with compounds A,B) 1, C,D) 2, E,F) 3 and G,H) 4. The results of the 15N titrations are on the left panels (A, C, E, G) and the results of the ILV-titrations are on the right panels (B, D, F, H).
Figure 6.
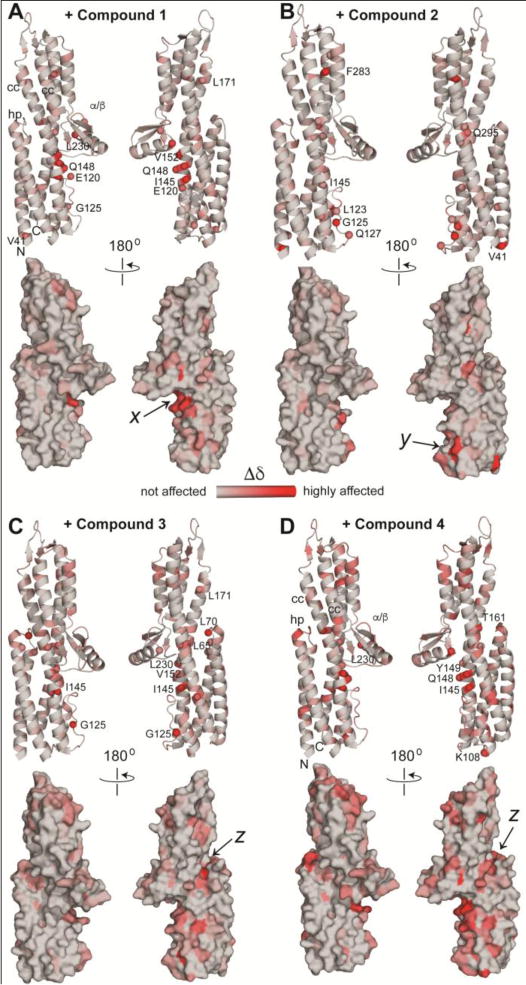
The chemical shift deviation (Δδ) of compounds A) 1, B) 2, C) 3 and D) 4 is shown on the ribbon and surface structures of IpaD, and colored according to the value of (Δδ), with the least affected residues colored gray, to the highly affected residues colored red. The binding pockets x, y, and z are indicated. The different parts of IpaD are indicated as hairpin (hp), coiled-coil (cc), the mixed α/β domain (α/β) as well as the amino (N) and the carboxy (C) termini.
Compound 1 binds in a pocket – herein designated as pocket x – formed at the interface of the mixed α/β domain and the long central coiled-coil (Figure 6). Compound 1 affected residues Y149, I145, and V152 (Figure 5) that form pocket x. On the other hand, compound 2 binds in a pocket formed by the long central coiled-coil, here referred to as pocket y (Figure 6B). The residues most perturbed by Compounds 3 and 4 are located the interface of the hairpin and the coiled-coil, and herein designated as pocket z. Additionally, compounds 3 and 4 showed chemical shift perturbations of residues near binding pockets x and y, suggesting perhaps, non-specific interactions at other sites. Currently, the only known small molecules that bind to IpaD are bile salts,[11] and the bile salt deoxycholate, a sterol-based scaffold, binds in pocket y based on the co-crystal structure of IpaD-deoxycholate.[11a] Thus, there are hotspots in IpaD for small molecule scaffolds – pocket x for scaffolds similar to compounds 1 and pocket y for compound 2 and sterols. Pockets x and y are approximately 22 Å apart and close enough that small molecule binders in both pockets could be linked together in designing the next generation of small molecule binders for IpaD. The binding pockets (x, y, and z) identified by NMR (Figure 6) were also similar to druggable sites identified by two prediction servers, DoGSiteScorer[14] and PockDrug-Server[15] (Supporting Information Figure S6). Likewise, the SwissDock server[16] predicted the docking of Compounds 1, 3, and 4 to IpaD (Supporting Information Figure S7), in binding the pockets x (for Compound 1) and z (for Compounds 3 and 4) identified by NMR.
We previously reported that compound 3 (5-hydroxyindole) and compound 4 (4-morpholinoaniline) bind to SipD, the IpaD-homolog in Salmonella.[12] IpaD and SipD share 38% sequence identity and 56% sequence similarity, as well as structural homology,[5, 17] however they differ in their binding pockets for compounds 3 and 4. Compounds 3 and 4 binds in pocket x in IpaD (Figure 6C), however, they bind in SipD in a pocket that is roughly similar to the IpaD pocket y (Figure 6B) suggesting that surface residues that are not conserved between SipD and IpaD are likely important in forming the pockets for binding small molecules.
Discussion
The rise of antibiotic resistance among bacterial pathogens coupled with the dearth of new antibiotics is a serious public health problem that necessitates the development of novel antibiotics. Because of its critical role in virulence among pathogens, the T3SS is an attractive target for developing anti-infectives. There is a growing number of small molecules that have been reported to inhibit the T3SS, however, the specific targets within the T3SS for many of those inhibitors remain unknown.[9, 18] Likewise, the number of known small molecules that bind directly to T3SS proteins are limited.[9] Currently, the only known small molecules that interact with IpaD, a protein that plays a critical role in the T3SS and pathogenesis of Shigella, are the bile salt sterols deoxycholate,[11a, 11b, 19] cholate, chenodeoxycholate, and taurodeoxycholate.[19]
We sought to identify small molecules by fragment based approach to increase the known chemical space of IpaD with the long term goal of developing T3SS inhibitors. Fragment based approach is a method that can identify new small molecule binders without a priori knowledge of their potential binding sites or their mechanism of action on the target protein. We screened a library of 288 drug-like fragments (Zenobia library 2.0) by SPR (Figure 2), which identified 4 compounds (1, 2, 3, & 4; Figure 1), that bound to IpaD. The results of SPR were confirmed and further validated by NMR methods to determine how the compounds bound to IpaD (Figure 3, Figure 4, Figure 5). Our results identified three possible binding pockets as hotspots in IpaD for binding small molecules (Figure 6).
Our NMR results identified three potential binding pockets for various scaffolds, which we have arbitrarily designated herein as pockets x, y, and z (Figure 6). These binding pockets are also used in the protein-protein interactions of IpaD. Previous studies have identified IpaD residues near pocket x to be involved in protein-protein interaction of IpaD with its cognate translocon protein IpaB.[11c, 12, 20] Likewise, previous studies have reported that residues near the IpaD pocket x and pocket y are involved in protein-protein interaction with the needle protein MxiH.[4a, 21] Pocket z has also been shown to be the primary binding site of sterol-like compounds as bile salts,[11b] and binding of bile salts trigger conformational change in IpaD that allows IpaD to interact with IpaB during the assembly of the translocon.[11c]
There is a good match between the binding pockets x, y, and z (Figure 6) identified by NMR and the predicted druggable sites identified by the computational prediction servers DoGSiteScorer[14] and PockDrug-Server[15] (Supporting Information Figure S6). These computational methods can be used to identify potential druggable sites in other T3SS tip proteins. With respect to docking, SwissDock was able to generate models of Compounds 1, 3 and 4 docked to their respective pockets – pocket x for Compound 1; and pocket z for Compounds 3 and 4, but not for Compounds 2 (Figure 6 and Supporting Figure S7). Future work using NMR data as constraints in computational docking should enable the generation of structural models that will aid in designing the next generation of small molecules that can bind, and potentially, inhibit the function of IpaD.
Enquist et al.[22] reported that a compound, INP1750 (Supporting Information Figure S8), inhibits the T3SS of Yersinia pseudotuberculosis and Chlamydia trachomatis. The specific target of INP1750 in type III secretion is unknown. INP1750 is based on the quinolone scaffold and has some similar structural features with Compound 1. Our results suggest that INP1750 should be investigated for binding/inhibiting the activity of the tip proteins of Yersinia pseudotuberculosis and Chlamydia trachomatis.
Conclusions
To summarize, we report four new small molecules that bind to IpaD. These molecules are based on the quinoline, pyrrolidin aniline, hydroxyindole, and morpholinoaniline scaffolds. These scaffolds, together with the bile sterols cholate, deoxycholate, taurodeoxycholate, and chenodeoxycholte, are currently the only known small molecules that interact with IpaD. Our NMR mapping identified three binding pockets in IpaD for the four scaffolds, suggesting three potential hotspots in IpaD for binding small molecules. This new knowledge is needed in designing small molecule inhibitors of IpaD to develop new anti-infectives against drug resistant bacteria.
Experimental Section
Protein Expression and Purification
The cloning, expression and purification of recombinant IpaD (residues 38–332 C322S) have been described previously.[12] The plasmid harboring IpaD was transformed into E. coli BL21(DE3) for protein expression. To obtain unlabeled IpaD, 1 L LB was inoculated with 10 mL LB starter culture and cells were grown at 37°C. Protein expression was induced with 1 mM IPTG at OD600 of 0.6–0.8 and cell growth was continued overnight at 15°C before harvest. For NMR studies, IpaD was simultaneously labeled with 15N and ILV (where the methyl groups of isoleucine, leucine, and valine are 13C-labeled). Cells were grown in M9 minimal medium supplemented with 1 g/L 15N-ammonium chloride (Sigma) at 37°C. When OD600 reached ~0.4, the growth medium was supplemented with 60 mg/L 2-ketobutyric acid-4-13C (Sigma #571342) to label the isoleucine Cδ1 methyl group and 100 mg/L 2-keto-3-(methyl-13C)-butyric-4-13C acid sodium salt (Sigma #571334) to label the leucine Cδ and valine Cγ geminal methyl groups. Perdeuterated 15N/ILV-labeled IpaD used for assigning the ILV resonances was obtained by growing cells in M9 minimal media in 1 liter of D2O and 2 g/L deuterated D-glucose-1,2,3,4,5,6,6-d7 (Cambridge Isotope Laboratories #CLM-2062) and 1 g/L 15N-ammonium chloride. The ILV precursors were added at OD600 ~0.4 as described above. At OD600 0.6–0.8, protein expression was induced with 1 mM IPTG, and growth temperature was dropped to 15°C; cells were grown overnight prior to harvest. Recombinant IpaD was purified by nickel affinity chromatography and the His tag was cleaved by digestion with TEV protease as described.[12] Recombinant proteins were concentrated by Amicon Ultra 3K centrifugal filter (Millipore) and protein concentration was determined by absorbance at 280 nm.
SPR screening
Biacore 3000 Surface Plasmon Resonance (GE Healthcare) was used for SPR screening of 288 fragments of drug like molecules in the Zenobia Fragment Library 2 (Zenobia Therapeutics, San Diego, CA, USA). IpaD was dialyzed in PBS buffer (137mM NaCl, 2,7mM KCl, 10mM Na2HPO4, 2mM KH2PO4, pH 7.4) and immobilized covalently on a CM5 sensor chip (#BR-1003-99 GE healthcare) by standard amine coupling (#BR-1106-33 GE healthcare) technique using 1.05× PBS as running buffer. The CM5 chip was activated for 7 min with a mixture of N-hydroxysuccinimide (NHS) and 1-ethyl-3- (3 dimethylaminopropyl) carbodiimide (EDC) at 1:1 ratio and 15 μL/min flow rate at 25°C. IpaD was diluted to 50 μg/μL in 10 mm sodium acetate buffer (pH 4.3) and injected for 7 min until a response unit (RU) of 10700 was achieved. The flow cells were later treated with 1M ethanolamine (pH 8.0) for 7 min to remove unbound protein or unreacted esters of NHS/EDC from the flow cells. Another flow cell in the CM5 chip was kept as reference cell without any immobilized protein. Each fragment in the library as received was reconstituted with 100 μL DMSO to form 100 mM stock solutions, and diluted to a final concentration of 1 mM in 5% DMSO and 1.05 × PBS buffer. The compounds were injected at a flow rate of 60 μL/min over the CM5 chip for 60 sec and dissociation was allowed for additional 60 sec. Unbound fragments from the flow system was washed with a 1:1 DMSO:water, and running buffer was injected in between fragment runs to eliminate carryover effects. The calibration curve for DMSO was obtained by sequentially injecting eight varying concentrations of DMSO (from 4 to 6%) at the beginning and end of the screening at 60 μL/min flow rate.
Saturation transfer difference (STD) NMR
Saturation transfer difference (STD) NMR data was acquired as previously described[12] using a Bruker Avance 600 MHz NMR spectrometer equipped with a TXI-RT probe, the pulseprogram stddiffesgp.3,[13a] and processed using Topspin. One-dimensional 1H STD NMR was obtained at 30°C from samples containing protein and compound in 1:100 molar ratio (40 μM protein, 4000 μM compound, 10% D2O, 0.4% [D6]DMSO). The protein selective saturation pulse used was a 50 ms Gaussian, and applied for 2 s. The center of the selective pulse was varied from −0.5 to 0.1 ppm to optimize the STD signals, while the off-resonance center was kept at 40 ppm. Other acquisition parameters were typically 256 scans, 16 ppm 1H sweep width centered at 4.701 ppm, and 2 s recycle delay.[12]
NMR Spectroscopy
NMR data was acquired using a Bruker Avance 800 MHz spectrometer equipped with a cryogenic triple resonance probe, processed with NMRPipe[23] and analyzed with NMRView.[24] Two dimensional 1H-15N TROSY and 1H-13C HSQC spectra were collected at 25°C using 0.4 mM 15N/ILV-labeled IpaD in NMR buffer (20 mm NaCl, 20 mm sodium phosphate, pH 6.8 and 10%D2O). The isoleucines in the ILV 2D 1H-13C HSQC spectra of IpaD were assigned by point mutagenesis of isoleucine into leucine. These mutants were individually expressed in M9 media supplemented with 2-ketobutyric acid-4-13C as described above to label the isoleucine 13Cδ1 methyl group, and acquiring 2D 1H-13C HSQC spectra to identify the missing isoleucine 13Cδ1 peak in comparison with wild type 2D 1H-13C HSQC spectra. Completion of the ILV assignments of IpaD was done following the method of Xiao et al.[25] After dialysis in NMR buffer, 500 μL of 0.4 mM perdeuterated 15N/ILV-labeled IpaD was lyophilized and resuspended in 100% D2O. A 3D 1H-13C-13C HMQC-NOESY-HMQC dataset was acquired using 8 scans with 2048 complex points (1H), 80 complex points (13C) and 100 complex points (NOE 13C) with a 300 ms mixing time and a recycle delay of 2 s. Sweep widths were 10 ppm for 1H centered at 4.69 ppm and 20 ppm for 13C centered at 19 ppm. The leucine and valine 13C methyl peaks were assigned by analysis of the 3D 1H-13C-13C HMQC-NOESY-HMQC dataset with distance information from the crystal structure of IpaD (PDB ID: 2J0O).[5]
NMR titrations
For NMR titrations, the small molecules were dissolved in [D6]DMSO (Cambridge Isotope Laboratories, Andover, MA, USA) and titrated into 15N/ILV-labeled IpaD. Typically, ~500 mg of stock compounds were obtained from Zenobia, and requisite amounts were dissolved in ~250 μL 100% [D6]DMSO to form a 1 to 2 M stock solution, and titrated into 440 μL of 0.2–0.3 mM 15N/ILV-labeled IpaD. Five titration points were obtained with increasing molar ratio of compound:protein ranging from 12 for Compound 1 to 100 for Compound 4. All samples used in the NMR titrations were dissolved in a final buffer condition of 2% (v/v) [D6]DMSO in NMR buffer. For 15N-titrations monitored by acquiring 2D 1H-15N TROSY spectra, typical acquisition parameters were 16 scans at 30 ppm 15N sweep width centered at 118 ppm. For ILV-titrations monitored by acquiring 2D 1H–13C HSQC spectra, typical acquisition parameters were 32 scans, 18 ppm 13C sweep width centered at 18 ppm and 10 ppm 1H sweep width centered at 4.69 ppm. The weighted chemical shift deviation (Δδ) were calculated using the equation ΔδHN = ½ [(ΔδH)2 + (ΔδN/5)2][26] for backbone amides and ΔδILV = ½ [(ΔδH)2 + (ΔδC/2)2] for ILV.
Druggable sites and molecular docking
The druggable sites in IpaD (PDB 2J0O)[5] were predicted using the servers DoGSiteScorer[14] and PockDrug-Server[15]. Models of molecular docking of the small molecules to IpaD were generated using the SwissDock server[16].
Supplementary Material
Acknowledgments
We are grateful to Andrew McShan for assistance in assignment and Kawaljit Kaur for helpful discussions. This work was supported by US National Institutes of Health grants AI074856 (R.N.D.), P30-GM110761 (University of Kansas Biomolecular NMR Core Facility) and University of Kansas Strategic Initiative Grant #INS72410 (R.N.D.).
Abbreviations
- NMR
nuclear magnetic resonance
- CSD
chemical shift deviations
- SPR
surface plasmon resonance
- STD
saturation transfer difference
- T3SS
type III secretion system
References
- 1.Agaisse H. Lancet Infect Dis. 2015;15:867–868. doi: 10.1016/S1473-3099(15)00003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, Wu Y, Sow SO, Sur D, Breiman RF, Faruque AS, Zaidi AK, Saha D, Alonso PL, Tamboura B, Sanogo D, Onwuchekwa U, Manna B, Ramamurthy T, Kanungo S, Ochieng JB, Omore R, Oundo JO, Hossain A, Das SK, Ahmed S, Qureshi S, Quadri F, Adegbola RA, Antonio M, Hossain MJ, Akinsola A, Mandomando I, Nhampossa T, Acacio S, Biswas K, O’Reilly CE, Mintz ED, Berkeley LY, Muhsen K, Sommerfelt H, Robins-Browne RM, Levine MM. Lancet. 2013;382:209–222. doi: 10.1016/S0140-6736(13)60844-2. [DOI] [PubMed] [Google Scholar]
- 3.Galan JE, Wolf-Watz H. Nature. 2006;444:567–573. doi: 10.1038/nature05272. [DOI] [PubMed] [Google Scholar]
- 4.a) Epler CR, Dickenson NE, Bullitt E, Picking WL. J Mol Biol. 2012;420:29–39. doi: 10.1016/j.jmb.2012.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Espina M, Olive AJ, Kenjale R, Moore DS, Ausar SF, Kaminski RW, Oaks EV, Middaugh CR, Picking WD, Picking WL. Infect Immun. 2006;74:4391–4400. doi: 10.1128/IAI.00440-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Roehrich AD, Guillossou E, Blocker AJ, Martinez-Argudo I. Mol Microbiol. 2013;87:690–706. doi: 10.1111/mmi.12124. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Meghraoui A, Schiavolin L, Allaoui A. Microbes Infect. 2014;16:532–539. doi: 10.1016/j.micinf.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 5.Johnson S, Roversi P, Espina M, Olive A, Deane JE, Birket S, Field T, Picking WD, Blocker AJ, Galyov EE, Picking WL, Lea SM. J Biol Chem. 2007;282:4035–4044. doi: 10.1074/jbc.M607945200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schiavolin L, Meghraoui A, Cherradi Y, Biskri L, Botteaux A, Allaoui A. Mol Microbiol. 2013;88:268–282. doi: 10.1111/mmi.12185. [DOI] [PubMed] [Google Scholar]
- 7.a) Spellberg B, Guidos R, Gilbert D, Bradley J, Boucher HW, Scheld WM, Bartlett JG, Edwards J., Jr Clin Infect Dis. 2008;46:155–164. doi: 10.1086/524891. [DOI] [PubMed] [Google Scholar]; b) Folster JP, Pecic G, Bowen A, Rickert R, Carattoli A, Whichard JM. Antimicrob Agents Chemother. 2011;55:1758–1760. doi: 10.1128/AAC.01463-10. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Le Hello S, Hendriksen RS, Doublet B, Fisher I, Nielsen EM, Whichard JM, Bouchrif B, Fashae K, Granier SA, Jourdan-Da Silva N, Cloeckaert A, Threlfall EJ, Angulo FJ, Aarestrup FM, Wain J, Weill FX. J Infect Dis. 2011;204:675–684. doi: 10.1093/infdis/jir409. [DOI] [PubMed] [Google Scholar]; d) Gu B, Cao Y, Pan S, Zhuang L, Yu R, Peng Z, Qian H, Wei Y, Zhao L, Liu G, Tong M. Int J Antimicrob Agents. 2012;40:9–17. doi: 10.1016/j.ijantimicag.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 8.a) Wong MR, Reddy V, Hanson H, Johnson KM, Tsoi B, Cokes C, Gallagher L, Lee L, Plentsova A, Dang T, Krueger A, Joyce K, Balter S. Microb Drug Resist. 2010;16:155–161. doi: 10.1089/mdr.2009.0130. [DOI] [PubMed] [Google Scholar]; b) Ahmed AM, Shimamoto T. Int J Food Microbiol. 2015;194:78–82. doi: 10.1016/j.ijfoodmicro.2014.11.013. [DOI] [PubMed] [Google Scholar]
- 9.McShan AC, De Guzman RN. Chem Biol Drug Des. 2015;85:30–42. doi: 10.1111/cbdd.12422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Picking WL, Nishioka H, Hearn PD, Baxter MA, Harrington AT, Blocker A, Picking WD. Infect Immun. 2005;73:1432–1440. doi: 10.1128/IAI.73.3.1432-1440.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Barta ML, Guragain M, Adam P, Dickenson NE, Patil M, Geisbrecht BV, Picking WL, Picking WD. Proteins. 2012;80:935–945. doi: 10.1002/prot.23251. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dickenson NE, Zhang L, Epler CR, Adam PR, Picking WL, Picking WD. Biochemistry. 2011;50:172–180. doi: 10.1021/bi101365f. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Dickenson NE, Arizmendi O, Patil MK, Toth RTt, Middaugh CR, Picking WD, Picking WL. Biochemistry. 2013;52:8790–8799. doi: 10.1021/bi400755f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McShan AC, Anbanandam A, Patnaik S, De Guzman RN. Chem Med Chem. 2016;11:963–971. doi: 10.1002/cmdc.201600065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Mayer M, Meyer B. Angew Chem Int Ed. 1999;38:1784–1788. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1784::AID-ANIE1784>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]; b) Meyer B, Peters T. Angew Chem Int Ed Engl. 2003;42:864–890. doi: 10.1002/anie.200390233. [DOI] [PubMed] [Google Scholar]
- 14.Volkamer A, Kuhn D, Grombacher T, Rippmann F, Rarey M. J Chem Inf Model. 2012;52:360–372. doi: 10.1021/ci200454v. [DOI] [PubMed] [Google Scholar]
- 15.Hussein HA, Borrel A, Geneix C, Petitjean M, Regad L, Camproux AC. Nucleic Acids Res. 2015;43:W436–442. doi: 10.1093/nar/gkv462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grosdidier A, Zoete V, Michielin O. Nucleic Acids Res. 2011;39:W270–277. doi: 10.1093/nar/gkr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Lunelli M, Hurwitz R, Lambers J, Kolbe M. PLoS Pathog. 2011;7:e1002163. doi: 10.1371/journal.ppat.1002163. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chatterjee S, Zhong D, Nordhues BA, Battaile KP, Lovell SW, De Guzman RN. Protein Sci. 2011;20:75–86. doi: 10.1002/pro.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Veenendaal AK, Sundin C, Blocker AJ. J Bacteriol. 2009;191:563–570. doi: 10.1128/JB.01004-08. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gu L, Zhou S, Zhu L, Liang C, Chen X. Molecules. 2015;20:17659–17674. doi: 10.3390/molecules200917659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stensrud KF, Adam PR, La Mar CD, Olive AJ, Lushington GH, Sudharsan R, Shelton NL, Givens RS, Picking WL, Picking WD. J Biol Chem. 2008;283:18646–18654. doi: 10.1074/jbc.M802799200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaur K, Chatterjee S, De Guzman RN. Chembiochem. 2016;17:745–752. doi: 10.1002/cbic.201500556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang L, Wang Y, Olive AJ, Smith ND, Picking WD, De Guzman RN, Picking WL. J Biol Chem. 2007;282:32144–32151. doi: 10.1074/jbc.M703403200. [DOI] [PubMed] [Google Scholar]
- 22.Enquist PA, Gylfe A, Hagglund U, Lindstom P, Norberg-Scherman H, Sundin C, Elofsson M. Bioorg Med Chem Lett. 2012;22:3550–3553. doi: 10.1016/j.bmcl.2012.03.096. [DOI] [PubMed] [Google Scholar]
- 23.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 24.Johnson BA. Methods Mol Biol. 2004;278:313–352. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- 25.Xiao Y, Lee T, Latham MP, Warner LR, Tanimoto A, Pardi A, Ahn NG. Proc Natl Acad Sci U S A. 2014;111:2506–2511. doi: 10.1073/pnas.1318899111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grzesiek S, Stahl SJ, Wingfield PT, Bax A. Biochemistry. 1996;35:10256–10261. doi: 10.1021/bi9611164. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.