

Abstract
Trichloroethylene (TCE), a prevalent environmental contaminant, is a potent renal and hepatic toxicant through metabolites such as S-(1, 2-dichlorovinyl)-L-cysteine (DCVC). However, effects of TCE on other target organs such as the placenta have been minimally explored. Because elevated apoptosis and lipid peroxidation in placenta have been observed in pregnancy morbidities involving poor placentation, we evaluated the effects of DCVC exposure on apoptosis and lipid peroxidation in a human extravillous trophoblast cell line, HTR-8/SVneo. We exposed the cells in vitro to 10–100 μM DCVC for various time points up to 24 h. Following exposure, we measured apoptosis using flow cytometry, caspase activity using luminescence assays, gene expression using qRT-PCR, and lipid peroxidation using a malondialdehyde quantification assay. DCVC significantly increased apoptosis in time- and concentration-dependent manners (p<0.05). DCVC also significantly stimulated caspase 3, 7, 8 and 9 activities after 12 h (p<0.05), suggesting that DCVC stimulates the activation of both the intrinsic and extrinsic apoptotic signaling pathways simultaneously. Pre-treatment with the tBID inhibitor Bl-6C9 partially reduced DCVC-stimulated caspase 3 and 7 activity, signifying crosstalk between the two pathways. Additionally, DCVC treatment increased lipid peroxidation in a concentration-dependent manner. Co-treatment with the antioxidant peroxyl radical scavenger (±)-α-tocopherol attenuated caspase 3 and 7 activity, suggesting that lipid peroxidation mediates DCVC-induced apoptosis in extravillous trophoblasts. Our findings suggest that DCVC-induced apoptosis and lipid peroxidation in extravillous trophoblasts could contribute to poor placentation if similar effects occur in vivo in response to TCE exposure, indicating that further studies into this mechanism are warranted.
Keywords: S-(1,2-dichlorovinyl)-L-cysteine [DCVC]; trichloroethylene [TCE]; apoptosis; lipid peroxidation; extravillous trophoblasts; placenta
Graphical Abstract
Introduction
Trichloroethylene (TCE) is a chlorinated volatile organic solvent most commonly used in chemical production and as an industrial metal degreaser (Waters et al., 1977; Chiu et al., 2013; NTP, 2015). Ranked as number sixteen on the U.S. Agency for Toxic Substances and Disease Registry’s Priority List of Hazardous Substances, TCE is a common environmental contaminant found in approximately 800 Environmental Protection Agency-designated Superfund sites (Chiu et al., 2013; ATSDR, 2015). Despite being classified as a “known human carcinogen,” (Guha et al., 2012; NTP, 2015), approximately 1.9 million pounds of TCE were released into the environment in 2015 (EPA, 2017). Because of its continued use and widespread persistent environmental contamination, TCE exposure continues to pose a threat to human health through ingestion of contaminated drinking water and inhalation of the volatilized chemical.
Although TCE is most commonly recognized as a renal and liver toxicant (Chiu et al., 2013), its effects during pregnancy are not well understood. TCE-induced fetal cardiotoxicity is particularly controversial: for example, some laboratory animal studies reported TCE effects (Das and Scott, 1994; Johnson et al., 2003; Rufer et al., 2010) and other studies reported no cardiac defects (Fisher et al., 2001; Carney et al., 2006). Furthermore, two systematic literature reviews concluded there was insufficient evidence to support an association between TCE exposure and congenital heart abnormalities (Watson et al., 2006; Bukowski, 2014). With regard to other pregnancy outcomes, an earlier study found no association between maternal TCE exposure and low birth weight (Lagakos et al., 1986), but more recent epidemiology studies report positive associations of TCE exposure during pregnancy with decreased fetal weight and preterm birth (Forand et al., 2012) (Ruckart et al., 2014).
Placental toxicity could potentially mediate adverse birth outcomes such as preterm birth and decreased birth weight (Ilekis et al., 2016). In particular, emerging recent studies implicate placental insufficiency, defined as inadequate maternal-fetal nutrient and waste exchange resulting from placental abnormalities, as a potential cause of premature labor (Morgan, 2014; Morgan, 2016) Furthermore, a recent epidemiology study found a significant association between pre-eclampsia and preterm birth (Davies et al., 2016). Because it is highly perfused, the placenta is readily exposed to circulating TCE and its metabolites, and may be a target for TCE toxicity (Laham, 1970). Moreover, the placenta is capable of metabolizing compounds, which puts it at risk for tissue generation of toxic TCE metabolites (Burton and Fowden, 2015).
Previous studies have shown that TCE exerts its toxic effects primarily through its metabolites (Lash et al., 2014). For example, exposure to the glutathione conjugation pathway metabolite S-(1, 2-dichlorovinyl)-L-cysteine (DCVC) is toxic in vitro to renal proximal tubular cells, the main putative target in the kidney, of rats, mice and humans (Lash and Anders, 1986; Darnerud et al., 1989; Chen et al., 2001; Lash et al., 2001; Xu et al., 2008). Furthermore, numerous studies demonstrated that DCVC induces mitochondrial dysfunction, excessive reactive oxygen species generation and subsequent lipid peroxidation in kidney cells (Lash and Anders, 1986; Chen et al., 1990; van de Water et al., 1994; van de Water et al., 1995; Chen et al., 2001; Lash et al., 2003; Xu et al., 2008). Similarly, our lab recently showed that DCVC induces a loss of mitochondrial membrane potential and increases ROS generation in the first trimester extravillous trophoblast HTR-8/SVneo cell line (Hassan et al., 2016). Taken together, the evidence indicates that mitochondrial dysfunction and aberrant ROS-generating lipid peroxidation play central roles in DCVC-mediated TCE cytotoxicity (Lash and Anders, 1986).
Mitochondria and lipid peroxidation are involved in the regulation of apoptotic pathways (Simon et al., 2000; Ayala et al., 2014), and multiple studies have demonstrated that DCVC initiates apoptosis in proximal tubular cells of humans and rodents (Van de Water et al., 1996; Chen et al., 2001; Lash et al., 2001; Xu et al., 2008). Apoptosis is a highly organized form of cell death that is tightly regulated by two primary signaling pathways depending upon the stimulus: the mitochondrial-dependent or intrinsic pathway and the cell surface death receptor-mediated or extrinsic pathway. Apoptosis is especially critical during pregnancy (Straszewski-Chavez et al., 2005). During placental development, apoptosis plays an important role in removing damaged cells without injuring surrounding tissues (Smith et al., 1997b; Straszewski-Chavez et al., 2005; Sharp et al., 2010). Despite its role in normal placentation, evidence suggests that an abnormal increase in apoptosis of extravillous trophoblasts contributes to multiple placental pathologies including intrauterine growth restriction and pre-eclampsia (Smith et al., 1997a; DiFederico et al., 1999; Genbacev et al., 1999). The present study investigated the effects of DCVC on the two primary apoptosis signaling pathways and lipid peroxidation in the human extravillous trophoblast cell line HTR-8/SVneo.
Materials and Methods
Chemicals and reagents
The trichloroethylene metabolite S-(1, 2-dichlorovinyl)-L-cysteine (DCVC) was synthesized by the University of Michigan Medicinal Chemistry Core according to procedures described by McKinney et al. (McKinney et al., 1959). Purity (98.7%) was determined by HPLC analysis. A stock solution was prepared in PBS and identity was confirmed by proton nuclear magnetic resonance spectroscopy performed at the University of Michigan Biochemical Nuclear Magnetic Resonance Core. Phosphate buffered saline (PBS) and 0.25% trypsin were purchased from Invitrog en Life Technologies (Carlsbad, CA, USA). BI-6C9 tBID inhibitor, tert-butyl hydroperoxide (TBHP) and (±)-α-tocopherol were purchased from Sigma-Aldrich (St. Louis, MO). Camptothecin was purchased from Cayman Chemical (Ann Arbor, MI). RPMI 1640 culture medium with L-glutamine and without phenol red, 10,000 U/mL penicillin/10,000 μg/mL streptomycin (P/S) solution, and fetal bovine serum (FBS) were purchased from Thermo Fisher Scientific (Waltham, MA, USA).
Cell culture and treatment
The HTR-8/SVneo cell line, a gift from Dr. Charles H. Graham (Queen’s University, Kingston, Ontario, Canada), models first-trimester extravillous cytotrophoblasts in vitro (Graham et al., 1993). HTR-8/SVneo cells were originally derived from first trimester female human cytotrophoblast cells and immortalized with simian virus 40 large T antigen (Graham et al., 1993). HTR-8/SVneo cells were cultured as previously described (Tetz et al., 2013; Hassan et al., 2016). Briefly, cells were cultured between passages 71–87 in RPMI 1640 medium supplemented with 10% FBS and 1% P/S at 37°C in a 5% CO2 humidified incubator. Cells were maintained in RPMI 1640 growth medium with 10% FBS and 1% P/S prior to and during experiments to ensure optimal cell growth (Graham et al., 1993). Cells were grown to 70–90% confluence at least 24 h after subculture before starting any experiment. A stock solution of 1 mM DCVC was prepared in PBS and stored in 1-ml aliquots at −20°C to minimize freeze/thaw cycles. Prior to each experiment, a DCVC stock solution aliquot was quickly thawed in a 37°C water bath and then diluted in RPMI 1640 medium with 10% FBS and 1% P/S to final exposure concentrations of 10–100 μM DCVC. The DCVC concentrations were selected for the current study to include the mean peak blood concentration of 13.4 μM S-(1,2-dichlorovinyl)glutathione, the stable precursor of DCVC, measured in female volunteers exposed to 100 ppm of TCE by inhalation for 4 h (Lash et al., 1999), with higher concentrations consistent with DCVC-induced cytotoxicity in human placental cells and human proximal tubular cells in vitro (Xu et al., 2008; Hassan et al., 2016).
Cell line validation
HTR-8/SVneo cells were seeded at a density of 400,000 cells per well in a 6-well cell culture plate and allowed to adhere for 24 h. Cells were treated with RPMI 1640 medium alone for 24 h. Following exposure, DNA was extracted using QIAamp® DNA Mini Kit (Qiagen; Hilden, Germany). RNA samples were frozen at −20°C and transported to the University of Michigan DNA Sequencing Core for completion of the cell line validation process using microsatellite genotyping. At the core, AmpFLSTR Identifiler Plus PCR Amplification Kit run on an 3730XL Genetic Analyzer purchased from Applied Biosystems (Waltham, MA) was utilized to identify human genomic DNA for 8 tetranucleotide repeat loci and the Amelogenin gender determination marker. The short tandem repeat profile generated for our cells was compared to the short tandem repeat profile for HTR-8/SVneo (ATCC® CRL-3271™) published by American Type Culture Collection (Manassas, VA) (ATCC, 2015). The short tandem repeat profile was an exact match: Amelogenin gender determination marker: X, CSF1PO: 12, D13S317: 9,12, D16S539: 13D5S818: 12, D7S820: 12, TH01: 6,9.3, vWA: 13,18, TPOX: 8 (ATCC, 2015).
Apoptosis assessment with flow cytometry
HTR-8/SVneo cells were seeded at a density of 50,000 cells per well in a 24-well plate and allowed to adhere for 24 h prior to treatment. Cells were treated with medium alone (control), DCVC (10, 20, 50 and 100 μM), or 4 μM camptothecin (positive control) in triplicate. After 12 or 24 h of exposure, viable, early apoptotic, late-apoptotic, and necrotic cells were measured using the Annexin V FITC Assay Kit (Cayman Chemical; Ann Arbor, MI). This assay uses two staining solutions simultaneously: annexin V, which exclusively binds to phosphatidylserines externalized on the plasma membrane during apoptosis, and propidium iodide (PI), which enters the cell when the plasma membrane is compromised in late -apoptotic and necrotic cells. The assay was performed according to the manufacturer’s protocol with modifications. Following exposure, cells were treated with 0.25% trypsin-EDTA and incubated for 2 min to detach adherent cells. Following incubation, trypsin was deactivated with cell culture medium and triplicate wells were pooled into one 5-ml round-bottom fluorescence-activated cell sorting tube per treatment group. Cells were washed with 1 ml Annexin V Binding Buffer and resuspended with 500 μl Annexin V FITC/Propidium Iodide Staining Solution diluted in Binding Buffer, incubated for 10 min at room temperature and transported to the University of Michigan Flow Cytometry Core for analysis. Early apoptotic, late apoptotic and necrotic cells were quantified using a FACSAria II Flow Cytometer (BD Biosciences; San Jose, CA). Quantification and dye-based visualization were performed using BD FACSDiva Software (BD Sciences; San Jose, CA) for PC. Cell aggregates and cellular debris were excluded using the forward scatter and side scatter modes and the analysis was performed using 10,000 events per sample. Annexin V-FITC was detected using the 488 nm laser, while PI was detecting using the 561 nm laser.
Measurement of caspase 3, 7, 8 and 9 activity
HTR-8/SVneo cells were seeded at a density of 10,000 cells per well in a 96-well white, clear-bottom plate and allowed to adhere for 24 h prior to treatment. Cells were treated in quadruplicate with RPMI 1640 medium alone (control) or DCVC (20 or 100 μM). Following 3, 6, 12, or 24 h treatments, caspase activity was measured using luminescence-based Caspase-Glo 3/7, 8 and 9 Assays (Promega; Madison, WI) following the manufacturer’s recommended protocols. Caspase assays utilize luminogenically engineered caspase substrates that are specific to each type of caspase being measured based on specific amino acid sequences embedded within the substrates: DEVD (Asp -Glu-Val-Asp) for caspase 3 and 7, LETD (Leu-Glu-Thr-Asp) for caspase 8, and LEHD (Leu-Glu-His-Asp) for caspase 9. Following cleavage of the substrates, the product generated, aminoluciferin, reacts with luciferase to produce light proportional to the amount of caspase activity (Niles et al., 2008). Briefly, for Caspase-Glo 3/7, 8 and 9 assays, respectively, 100 μL Caspase-Glo substrate in Caspase-Glo buffer was added to cell cultures and incubated at room temperature for 1 h following DCVC treatment. In order to reduce non-specific background signal, MG-132, a proteasome inhibitor, was added to caspase 8 and 9 cell cultures only. Following incubation, luminescence signal was measured on Glomax Multi Plus Detection System (Promega; Madison, WI).
Apoptosis PCR array
Because DCVC stimulated apoptosis as shown by annexin V-FITC/PI staining, we evaluated changes in the gene expression of 84 genes specifically involved in apoptosis using the commercially available Apoptosis PCR Array manufactured by SABiosciences (Valencia, CA). HTR-8/SVneo cells were seeded at a density of 400,000 cells per well in a 6-well cell culture plate and allowed to adhere for 24 h. Following the adjustment period, the cells were treated with RPMI 1640 medium alone (control) or DCVC (20, 50 and 100 μM) and exposed for 24 h. The concentrations of 20, 50 and 100 μM were chosen to span a range of concentrations from no effect to effective concentrations that induced apoptosis in our earlier experiment. Following exposure, cell lysates were homogenized using QIA shredder (Qiagen; Hilden, Germany) and RNA was extracted using the RNeasy Plus Mini Kit (Qiagen) following the manufacture’s recommended protocol. RNA samples were frozen at −20°C and transported to the University of Michigan DNA Sequencing Core for completion of the Apoptosis PCR Array. At the core, cDNA was synthesized using the RT2 First Strand Kit (SABiosciences; Valencia, CA) following the manufacturer’s recommended protocol. For the array, cDNA from the culture medium-only control, 20 μM DCVC, 50 μM DCVC and 100 μM DCVC treatment groups was analyzed using the Applied Biosystems 7900HT Sequence Detection System following the SABiosciences recommended protocol. Fold changes were calculated from ΔCT values (gene of interest CT value – average of all housekeeping gene CT values) using the ΔΔCT method. Mean ΔCT values were compared between groups using paired t-tests from the Limma package of Bioconductor (Smyth, 2004). The resulting P-values were adjusted for multiplicity using the Benjamini and Hochberg false discovery rate method (Benjamini and Hochberg, 1995).
Real-time quantitative PCR (qRT-PCR) validation
We validated the findings of the PCR-based array using qRT-PCR for genes with significant differential mRNA expression at least 2 fold higher or 0.5 fold lower than control following 100 μM DCVC treatment for 24 h. These genes included: Harakiri, BCl-2 interaction protein (HRK), BCL2-related protein A1 (BCL2A1), growth arrest and DNA-damage-inducible, alpha (GADD45α), DNA fragmentation factor alpha polypeptide (c), Receptor-interacting serine-threonine kinase 2 (RIPK2), nuclear factor kappa B subunit 1 (NFKB1), caspase 1 (CASP1) caspase 4 (CASP4), BCL2-antagonist/killer 1 (BAK1), and TNF receptor-associated factor 3 (TRAF3). We also tested differential expression of the tumor suppressor protein p53 (TP53) and TNF receptor superfamily, member 6 (FAS) genes because they showed a trend towards upregulation in the PCR-based array. Primary sequences are shown in Supplemental Table 1. Primary sequences for BCL2A1, DFFA, RIPK2, TRAF3, NFKB1, TP53, CASP1, BAK1, FAS AND CASP4 were obtained from the online primer sequence database PrimerBank (Spandidos et al., 2010). HRK primer sequence was obtained from Zaker et al. (Zaker et al., 2016). GADD45α primer sequence was obtained from Zhang et al. (Zhang et al., 2011). qRT-PCR was performed on the above genes using samples from cells treated with RPMI 1640 medium alone (control), or DCVC (20 and 100 μM) for 24 h. qRT-PCR reactions were prepared with SYBR Green Mastermix (SABiosciences) and custom synthesized primers (Integrated DNA Technologies; Coralville, IA), and run on a Bio-Rad (Hercules, CA) CFX96 Real Time C1000 thermal cycler following the manufacturer’s recommended protocols. mRNA levels of each gene of interest were normalized to β-2-microglobulin (B2M) mRNA levels.
Visualization of intrinsic and extrinsic apoptosis pathways
We used PathVisio software (version 3.2.4) to visualize DCVC-induced gene expression changes in the intrinsic and extrinsic apoptotic pathways. Pathways were constructed from WikiPathways (Apoptosis-Homo Sapiens) (Kelder et al., 2012; Zambon et al., 2017) with modifications from Kegg Pathway Database (Apoptosis [map04210] and NOD-like receptor [map04621]-Homo sapiens signaling pathways) (Kanehisa et al., 2017) and other sources from the literature (Inohara et al., 1997; McCarthy et al., 1998; Gao and Abu Kwaik, 2000; Haupt et al., 2003; Roth et al., 2003; Perfettini et al., 2004; Amaral et al., 2010; Graupner et al., 2011; Nishizaki et al., 2014). Genes found to be differentially expressed in either the 84-gene qRT-PCR array experiments or in qRT-PCR experiments were included in the visualization (except CD70 and IGR1F).
ROS lipid peroxidation assessment
Because our evidence indicated that DCVC exposure activated the mitochondria-dependent intrinsic apoptosis pathway, we investigated the effect of DCVC treatment on the degradation of membrane lipids by ROS by measuring a lipid peroxidation by-product, malondialdehyde (MDA). HTR-8/SVneo cells were seeded at a density of 150,000 cells per well in 12-well plates and allowed to adhere for 24 h prior to treatment. Cells were treated with medium alone (control), DCVC (10, 20, and 50 μM), or 20 μM tert-butyl hydroperoxide (TBHP) (positive control) for 12 h. MDA was measured using a Lipid Peroxidation (MDA) Assay Kit (Sigma-Aldrich; St Louis, MO) according to the manufacturer’s protocol. The assay uses the reaction of MDA with thiobarbituric acid (TBA) to form an MDA-TBA adduct product proportional to the amount of MDA present in the samples. Briefly, cells were lysed using MDA lysis buffer containing butylated hydroxytoluene. Insoluble material was removed by centrifugation and lysates were transferred to microcentrifuge tubes. TBA solution was added to the lysate and incubated at 95°C for 1 h. Lysate was cooled to room temperature with an ice bath for 10 min and plated on a 96-well plate for analysis. MDA concentration was measured colorimetrically (OD = 532 nm) using a SpectraMax M2e Multi-Mode Microplate Reader (Molecular Devices; Sunnyvale, CA).
Modulation of DCVC-stimulated caspase 3 and 7 activity
In order to investigate the role of aberrant ROS generation on DCVC-stimulated caspase 3 and 7 activity, the antioxidant peroxyl radical scavenger (±)-α-tocopherol was used to block lipid peroxidation. HTR-8/SVneo cells were seeded at a density of 10,000 cells per well in a 96-well white, clear-bottom plate and allowed to adhere for 24 h prior to treatment. Cells were treated in quadruplicate with medium alone (control) or DCVC (20 μM) plus (±)-α-tocopherol (50 μM). The cells were pre-treated for 15 min with (±)-α-tocopherol prior to co-treatment with DCVC for 12 h. We used a concentration of 50 μM (±)-α-tocopherol because it was previously shown by our lab to attenuate DCVC-stimulated IL-6 release in HTR-8/SVneo cells (Hassan et al., 2016). In addition, it showed a protective role in TCE-induced human epidermal keratinocytes at a concentration of one order of magnitude higher (50 mM) (Zhu et al., 2005). Caspase 3 and 7 activity was measured with Caspase-Glo 3/7 Assay as previously described.
In order to gain more insight into the roles that the intrinsic and extrinsic apoptosis pathways play in DCVC-stimulated caspase 3 and 7 activity, we explored the involvement of the truncated BID protein, tBID, as a source of cross talk between the two pathways. HTR-8/SVneo cells were seeded and cultured as previously described. Cells were treated in triplicate with medium alone (control), DCVC (20 μM) or DCVC plus tBID inhibitor BI-6C9 (10 μM). We used a concentration of 10 μM BI-6C9 because it was shown in a previous study to moderately inhibit BID-induced apoptosis without cytotoxicity (Becattini et al., 2004). The cells were pre-treated for 1 h prior with BI-6C9, followed by treatment with DCVC for 12 h. Caspase 3 and 7 activity was measured with Caspase-Glo 3/7 Assays as previously described.
Statistical Analysis
All experiments were performed independently in at least triplicate and repeated at least three times. The technical replicates were averaged within each experiment, and these values were analyzed using student’s t-test, one-way or two-way analysis of variance (ANOVA), followed by Tukey’s or Dunnett’s post-hoc test for comparison of means using GraphPad Prism software (GraphPad Software Inc., San Diego, CA, USA). Data are expressed as means ± SEM. N=number of independent experiments. P < 0.05 was considered statistically significant.
Results
Effect of DCVC treatment on apoptosis
Because our laboratory previously demonstrated that DCVC treatment impacts cell viability, we investigated the effects of DCVC exposure on a specific form of cell death, apoptosis. Early apoptosis, late apoptosis, and necrosis were quantified in HTR-8/SVneo cells with annexin V-FITC and propidium iodide fluorescence indicators using flow cytometry (Fig. 1). Statistically significant concentration-dependent increases of total apoptosis were observed after 24 h but not 12 h of DCVC exposure (ANOVA interaction effect, P=0.001, Fig. 1). No statistically significant treatment-related differences were observed with 12 h of exposure, but treatment with 50 and 100 μM DCVC increased total apoptosis in a concentration-dependent manner at 24 h. Following 24-h treatment, total apoptosis increased from 6.38% (control) to 14.73% and 23.48% with 50 and 100 μM DCVC treatment, respectively (P<0.001). The 100 μM DCVC treatment increased apoptosis 2.3 fold at 24 h compared with 12 h (P<0.0001).
Figure 1. Effects of DCVC on apoptosis in HTR-8/SVneo cells.

Cells were treated for 12 h or 24 h with 0 (no treatment control), 10, 20, 50, or 100 μM DCVC. A) Annexin V-FITC and propidium iodide fluorescence indicators were used to quantify apoptosis using flow cytometry. The dot plots are representative of each experiment at 24 h. B) Graphical representation of total apoptosis (early+late apoptosis). Bars represent means±SEM. Data were analyzed by two -way ANOVA (interaction between time and treatment, P=0.001) with posthoc Tukey multiple comparisons. #Significantly different compared to all 12 h time points (P<0.0022). &Significantly different compared to control and 10 μM DCVC at 24 h time point (P<0.0088). +Significantly different compared to control, 10 and 20 μM DCVC at 24 h time point (P<0.0001). *Significantly different compared to 50 μM DCVC at 24 h time (P=0.005). N=4 independent experiments for 12 h and N=5 independent experiments for 24 h. All experiments were performed in triplicate. Camptothecin (4 μM) was included as a positive control and increased total apoptosis 32.89% +/−15.23% at 12 h and 39.11% +/− 6.36 at 24 h.
Effect of DCVC treatment on caspase activity
We measured the effects of DCVC on caspase activity to evaluate the involvement of the intrinsic and extrinsic apoptotic pathways in DCVC-induced apoptosis (Fig. 2). Caspases 3 and 7, which are downstream executor caspases activated by both the intrinsic and extrinsic pathways, showed time - and concentration-dependent increases in activity following DCVC exposure for 12 and 24 h (ANOVA time and treatment interaction, P<0.0001, Fig. 2A). Caspases 3 and 7 measured after 3 and 6 h of DCVC treatment showed no significant changes in activity. In contrast, treatment with 20 and 100 μM DCVC increased caspase 3 and 7 activity by 86.2% (P=0.0015) and 187.5% (P<0.001) at 12 h, and 88.6% (P<0.001) and 155.5% (P<0.001) at 24 h, respectively, compared with time -matched controls. Likewise, increases of caspase 3 and 7 activity were observed at 12 h and 24 h compared to earlier time points, ranging from 2.4 to 3.5 fold within each DCVC treatment (P<0.0001).
Figure 2. DCVC effects on caspase activity in HTR-8/SVneo cells.

Cells were treated for 3, 6, 12 or 24 h with 0 (control), 20 or 100 μM DCVC. Caspase activity was measured using the Caspase Glo® luminescence assays. A) Time course of caspase 3 and 7 activity. B) Caspase 9 activity. C) Caspase 8 activity. Symbols represent means±SEM. N=3 independent experiments. Data were analyzed by two-way ANOVA (interaction between time and treatment, P<0.0001) with posthoc Tukey multiple comparisons. Given the very large number of significant mean comparisons, only significant comparisons within a treatment across time points and within a time point across treatments are indicated. Asterisks indicate significant differences with all lower concentrations within a single time point across treatments: * P<0.05, ** P<0.005, *** P<0.0001. Pound signs indicate significantly different compared with all earlier time points for same treatment: # P<0.05, ### P<0.0001. N=3 independent experiments. All experiments were performed in quadruplicate.
Caspase 9, an initiator caspase specific to the mitochondria-mediated intrinsic pathway, showed time- and concentration-dependent increases in activity following DCVC exposure (ANOVA time and treatment interaction, P<0.0001, Fig. 2B). Caspase 9 activity measured after 3 and 6 h of DCVC treatment showed no changes in activity level compared to time-matched controls. However, caspase 9 activity measured at 12 h showed a 56.3% increase in activity compared to time-matched control for 100 μM DCVC, whereas activity measured at 24 h showed 41.4% and 69.6% increases in activity compared to time-matched controls (P<0.0001). The 20 μM DCVC treatment increased caspase 9 activity 1.32 fold at 12 h compared to 6 h (P=0.0228) and 1.42 fold at 24 h compared to 12 h (P<0.0001). Similarly, 100 μM DCVC treatment increased caspase 9 activity 1.66 fold at 12 h compared to 6 h and 1.29 fold at 24 h compared to 12 h (P<0.0001).
Caspase 8, an initiator caspase specific to the cell surface receptor-mediated extrinsic pathway, showed time- and concentration-dependent increases in activity following DCVC exposure (ANOVA time and treatment interaction, P<0.0001, Fig. 2C). Caspase 8 activity measured after 3 and 6 h post-DCVC treatment showed no significant differences in activity compared to time-matched controls. However, caspase 8 activity measured at 12 h showed 36.5% (P=0.029) and 72.0% (P<0.0001) increases in activity compared to time-matched control for 20 and 100 μM DCVC, respectively, and activity measured at 24 h showed 30.6% (P=0.0002) and 70.2% (P<0.0001) increases in activity compared to time-matched control. The 20 μM DCVC treatment increased caspase 8 activity 1.86 fold at 12 h compared to 6 h and 1.39 fold at 24 h compared to 12 h (P<0.0001). The 100 μM DCVC treatment increased caspase 8 activity 2.39 fold at 12 h compared to 6 h and 1.43 fold at 24 h compared to 12 h (P<0.0001).
Inhibition of DCVC-stimulated caspase 3 and 7 by tBID protein inhibitor
In order to ascertain whether the DCVC mechanism of cytotoxicity involves interaction between the intrinsic and extrinsic apoptosis pathways, we used BI-6C9, which inhibits tBID-mediated cross talk between the two pathways. HTR-8/SVneo cells were pre-treated for 1 h with 10 μM BI-6C9 followed by treatment with 20 μM DCVC for 12 h. BI-6C9 pre-treatment attenuated DCVC-induced caspase 3 and 7 activity by 60.77% compared to cells treated with DCVC without the inhibitor (ANOVA interaction between DCVC and BI-6C9 treatments, P=0.028; Fig 3).
Figure 3. Effect of tBID inhibitor pre-treatment on DCVC-stimulated caspase 3/7 activity.

Cells were exposed to 20 μM DCVC for 12 h without pretreatment or after pretreatment with the tBID inhibitor BI-6C9 (10 μM for 1 h). Controls were not exposed to DCVC. Bars represent means±SEM. Data were analyzed by two-way ANOVA (interaction between DCVC and BI-6C9 treatments, P=0.028), followed by Tukey’s multiple comparison of means. #Significantly different compared to control with no inhibitor (P=0.001). *Significantly different compared to 20 μM DCVC with no inhibitor (P<0.010).
Apoptosis PCR array and qRT-PCR Validation
Because there are potentially hundreds of genes involved in the activation of apoptosis in cells, we used a commercially available PCR-based apoptosis array to screen 84 apoptosis-related genes for DCVC-induced changes in mRNA expression following treatment with 100 μM DCVC for 24 h (Supplement Table 2; P<0.05). Using this array, we identified ten genes that were significantly upregulated at least 2 fold or significantly downregulated at least 0.5 fold compared to control, and two other genes that showed a trend towards upregulation (Fig 4; P<0.05). We used qRT-PCR to follow up on these array results. In agreement with the PCR-based array results, 20 and 100 μM DCVC increased mRNA expression of the BCL-2 family genes HRK, BAK1 and BCL2A1 by 13.1 and 19.4 fold, 1.3 and 1.4 fold, and 7.6 and 8.0 fold, respectively, compared to control (P<0.05). Tumor suppressor gene TP53 showed 1.7 and 1.3 fold increases in mRNA expression following 24-h treatment with 20 and 100 μM DCVC, respectively (P<0.05). Cell cycle arrest gene GADD45α expression increased 9.8 and 9.7 fold, while FAS death receptor gene increased 1.8 fold with 20 and 100 μM DCVC treatments, respectively, compared to control (P<0.05). Inflammatory response genes NFKB1, RIPK2, and CASP4 were upregulated 2.0 and 1.8 fold, 2.0 and 1.6 fold, and 1.7 and 2.5 fold, respectively, while CASP1 was downregulated 0.5 and 0.2 fold, respectively, compared to control (P<0.05). Contrary to the array data, TRAF3 and DFFA did not yield a significant increase in expression, although DFFA showed a trend towards upregulation of 1.5 fold compared to control with 100 μM DCVC treatment (P=0.0582). Relevant genes significantly up or down regulated in the apoptosis array and/or qRT-PCR are shown in Figure 5.
Figure 4. DCVC effects on HTR-8/SVneo cell mRNA expression of genes implicated in apoptosis signaling.

Based on the results of an apoptosis PCR array, we identified ten genes that were significantly upregulated by at least 2 fold or downregulated at least 0.5 fold compared to control, following treatment with 100 μM DCVC for 24 h. The PCR array data results were confirmed using qRT-PCR. Two other genes were also included in the qRT-PCR analysis because they showed a trend of upregulation. Cells were exposed to 0 (control), 20 μM DCVC or 100 μM DCVC for 24 h followed by qRT-PCR analysis. A) The mRNA expression of TP53, FAS, DFFA and BAK1. B) The mRNA expression of NFKB1, RIPK2, TRAF3 and CASP1. B) The mRNA expression of HRK, GADD45α, BCL2A1 and CASP4. Bars represent means±SEM. * Significantly different compared to control (P<0.05). ** Significantly different compared to control (P<0.01). *** Significantly different compared to control (P<0.001). Data were analyzed by one -way ANOVA with posthoc Dunnett’s multiple comparison. N=3–6 independent experiments. All experiments were performed in triplicate.
Figure 5. Visualization of gene expression changes in intrinsic and extrinsic apoptosis pathways in HTR-8/SVneo cells placental after exposure to DCVC.

In order to elucidate the gene expression patterns that regulate the apoptosis observed in cells after treatment with DCVC, we mapped gene expression changes for extrinsic and intrinsic apoptosis pathways using Pathvisio software. Gene expression changes (log2 fold-change/control) are shown with red=upregulation, green=downregulation, grey=gene not evaluated. All gene expression data were obtained from an 84-gene qRT-PCR array except that genes indicated with an asterisk (*) had expression data obtained from separate qRT-PCR experiments. Two genes, IGF1R and CD70, were differentially expressed in the array but are not included in the diagram. This figure was constructed in part from Wikipathways: Apoptosis (Homo sapiens) (Zambon et al., 2017).
Effect of DCVC treatment on ROS lipid peroxidation
In order to further investigate the mechanism of DCVC-induced cytotoxicity, we evaluated the effects of DCVC on lipid peroxidation in HTR-8/SVneo cells by measuring cellular malondialdehyde concentrations, a byproduct and proxy measurement for lipid peroxidation. We detected a significant concentration-dependent increase in cellular malondialdehyde concentration following 24-h exposure to 10, 20 and 50 μM DCVC compared to control (Fig 6; P<0.05).
Figure 6. Effects of DCVC on lipid peroxidation in HTR-8/SVneo cells.
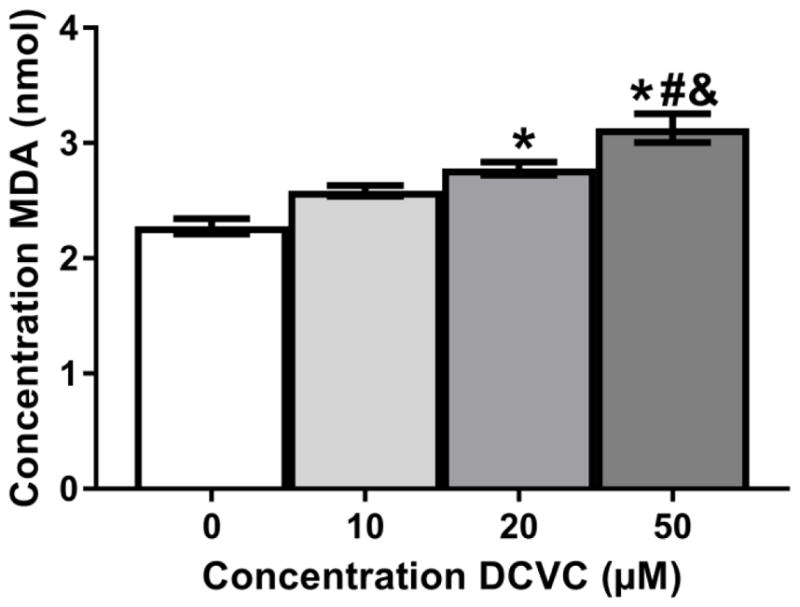
Cells were treated for 24 h with 0 (control), 10, 20, and 50 μM DCVC or 20 μM tert-butyl hydroperoxide (positive control). As a proxy measure for lipid peroxidation, malondialdehyde (MDA) concentrations were quantified using thiobarbituric acid (TBA), which generates a MDA-TBA adduct when in contact with MDA. The MDA concentrations were quantified colorimetrically (OD = 532). Bars represent means±SEM. *Significantly different compared to control (P<0.004). #Significantly different compared to 10 μM DCVC (P=0.0020). &Significantly different compared 20 μM DCVC (P= 0.0389). Data were analyzed by one -way ANOVA with posthoc Tukey multiple comparisons. N=4 independent experiments. All experiments were performed in triplicate. TBHP (20 μM) was used as a positive control (2.87 nmol +/− 0.167 nmol).
Attenuation of DCVC-stimulated caspase 3 and 7 activity by an antioxidant
To test the hypothesis that lipid peroxidation mediates DCVC-stimulated caspase 3 and 7 activity and apoptosis, we co-treated HTR-8/SVneo cells with either 20 μM DCVC only or 20 μM DCVC plus (±)-α-tocopherol (50 μM), an antioxidant peroxyl radical scavenger. Co-treatment with (±)-α-tocopherol significantly attenuated DCVC-stimulated caspase 3 and 7 activity by 80% compared to cells treated only with 20 μM DCVC for 12 h (ANOVA interaction between DCVC and α-tocopherol treatments, P=0.0103; Fig Discussion
Trichloroethylene continues to be a pervasive environmental contaminant in soil and groundwater despite reduction of improper disposal of the parent compound in recent decades. When TCE is ingested or inhaled, it is metabolized by two primary pathways into harmful metabolites, one of which is S-(1, 2-dichlorovinyl)-L-cysteine (DCVC), the focus of the current study. Although DCVC has been studied and established as a renal toxicant and carcinogen (Lash et al., 2014), epidemiological and animal studies linking TCE to pregnancy complications and adverse birth outcomes thus far remain inconclusive (Chiu et al., 2013), prompting our laboratory to examine DCVC as a placental toxicant. The general objectives of the current study were to further test the biological plausibility of DCVC as a placental toxicant and to examine its possible mechanism of action. In this study, we demonstrated that DCVC induced lipid peroxidation-associated apoptosis in placental cells through activation of both the intrinsic and extrinsic signaling pathways, providing new insights into the biological plausibility of DCVC-induced placental injury and the cytotoxic mechanism of action of DCVC.
Our laboratory previously reported that DCVC decreased cell viability and increased cytotoxicity in HTR-8/SVneo, an immortalized cell line that retains key molecular and functional characteristics of extravillous trophoblasts and which serves as an in vitro model for these cells (Kilburn et al., 2000; Hannan et al., 2010; Khan et al., 2011; Hassan et al., 2016). The present study extends those findings with evidences that DCVC directly induced aberrant apoptosis, a specific form of cell death, in the same HTR-8/SVneo cell type. These findings are consistent with previous reports that DCVC exposure induces apoptosis in kidney proximal tubular cells of humans and rodents (Van de Water et al., 1996; Chen et al., 2001; Lash et al., 2001; Xu et al., 2008). In addition, aberrant and widespread apoptosis of extravillous trophoblasts has been observed in several pregnancy-related morbidities specifically characterized by poor placentation such as pre-eclampsia and intrauterine growth restriction (DiFederico et al., 1999; Genbacev et al., 1999; Reister et al., 2001). Aberrant placental apoptosis is linked to major pathological features of these pregnancy morbidities including insufficient extravillous trophoblast invasion into the maternal decidua and inadequate remodeling of the spiral arteries needed to accommodate increased maternal blood flow to the placenta (Brosens et al., 1972; Meekins et al., 1994; Caniggia et al., 1999; Aardema et al., 2001; Kadyrov et al., 2006; Pennington et al., 2012). Because a growing body of evidence ties excessive apoptosis to impaired trophoblast invasion and inadequate remodeling of the spiral arties commonly observed with pregnancy-related morbidities, our results suggest that DCVC-induced apoptosis in extravillous trophoblasts could be a mechanism by which TCE contributes to poor placentation and early pregnancy morbidities if it occurs in vivo.
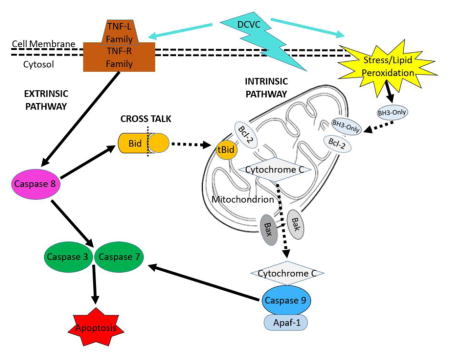
In order to gain further insight into the cytotoxic mechanism of action, we evaluated specific signaling pathways involved in DCVC-stimulated apoptosis. Depending upon the stimulus, there are two well-characterized pathways that are capable of inducing apoptosis: the extrinsic and intrinsic pathways, as illustrated in Figure 8 (Ashkenazi, 2008; Beesoo et al., 2014). The intrinsic pathway is activated by non-receptor stress stimuli such as hypoxia, damaged DNA and ROS (Elmore, 2007). These stimuli induce apoptosis through interactions with the BCL-2 family of mitochondrial proteins including BAX and BAK, which control mitochondrial membrane permeability. Upon activation, the pro-apoptotic BCL-2 family proteins dimerize to increase the permeability of the mitochondrial membrane, allowing cytochrome c to leak into the cytoplasm and prompting activation of the intrinsic pathway-specific initiator caspase 9 (Elmore, 2007). In contrast, the extrinsic pathway is stimulated when specific tumor necrosis factor (TNF) superfamily cytokines bind to TNF cell surface death receptors, prompting cytoplasmic adapter proteins to form complexes that activate extrinsic pathway-specific initiator caspase 8 (Straszewski-Chavez et al., 2005; Elmore, 2007). Following activation of caspase 8 and 9, both apoptosis pathways converge in a common pathway when caspases 8 and 9 activate executioner caspases 3 and 7, ultimately leading to the cleavage of cellular proteins and destruction of the cell (Levy and Nelson, 2000; Elmore, 2007). In the current study, we showed that DCVC exposure simultaneously activated both major apoptosis pathways, as indicated by caspase 8 and 9 activity results, in time- and concentration-dependent manners. We chose to measure caspase enzymatic activity as an endpoint that reflects caspase zymogen activation as well as enzyme abundance (Niles et al., 2008). Although western blot analysis would provide specific information on protein expression, a study that compared the sensitivity and specificity of Caspase Glo assays to western blot analysis demonstrated a strong correlation between activity levels measured by Caspase Glo 3/7, 8, and 9 assays and western blot detection of active caspase enzymes (Alvero et al., 2008).
Figure 8. Intrinsic and extrinsic apoptosis pathways.

The mitochondria-mediated intrinsic pathway is activated in response to stress stimuli. BCL-2 family proteins with the BH3-only domain such as HRK translocate into mitochondria and dimerize with anti-apoptotic BCL-2, releasing pro-apoptotic BAK and BAX, which increase mitochondrial membrane permeability. In response, mitochondrial components such as cytochrome c leak out and bind to caspase 9 and APAF-1 to form the apoptosome. This complex activates caspases 3 and 7, which execute apoptosis. The extrinsic pathway is activated when TNF family ligands bind to receptors and activate caspase 8, which then activates caspases 3 and 7. Cross talk occurs when extrinsic pathway protein caspase 8 cleaves BID into truncated tBID. tBID translocates into the mitochondria and dimerizes with BCL-2 family proteins, engaging the intrinsic pathway.
To our knowledge, our study is the first to demonstrate that DCVC is capable of activating both the intrinsic and extrinsic apoptosis pathways simultaneously, as indicated by the caspase results, as part of its cytotoxicity mechanism. Interestingly, these results only partially agree with a previous study that reported involvement of the mitochondrial-mediated intrinsic but not extrinsic pathway in DCVC-induced apoptosis (Xu et al., 2008). The differences between the study findings may be attributed to the different cell types and/or exposure concentrations examined. For example, contrary to our study, Xu et al. investigated the effects of DCVC in concentrations up to 300 μM in primary human proximal tubular cells.
Because our results clearly showed that DCVC exposure activated both intrinsic and extrinsic apoptosis signaling pathways in HTR-8/SVneo cells, we examined potential interactions between the two pathways by evaluating cross talk between the pathways. The best characterized source of cross talk between the two pathways involves the truncated protein tBID. The pathway interaction typically occurs when extrinsic pathway-specific caspase 8 cleaves the cytosolic BCL-2 protein family member BID into its truncated form, tBID. tBID then translocates across the mitochondrial membrane and engages the intrinsic pathway by dimerizing with BCL-2 family proteins and increasing mitochondrial membrane permeability (Li et al., 1998; Wei et al., 2000). Our results indicated that pre-treatment with the tBID inhibitor Bl-6C9 partially attenuated DCVC-induced downstream activation of caspases 3 and 7, demonstrating that cross talk between the two pathways contributes to the DCVC mechanism of stimulating apoptosis in placental cells.
In agreement with these results, we further observed that DCVC treatment stimulated an increase in caspase 3 and 7 activity that was more than double the increase in activity observed in either caspase 8 or 9 when compared to control. As reported in previous studies, the latter results strongly suggest that the cross talk occurring between the two pathways amplifies the effect on the common terminal pathway, which may also explain the magnitude of the DCVC-induced apoptotic response (Kuwana et al., 1998; Chou et al., 1999). Although multiple studies have established that first-trimester extravillous trophoblasts are capable of undergoing apoptosis via either the intrinsic or extrinsic pathways (Belkacemi et al., 2009; Belkacemi et al., 2011; Huang et al., 2014), to our knowledge, our study is the first to demonstrate a simultaneous pathway activation involving tBID-mediated cross talk in response to exposure to an exogenous chemical stimulant.
To elucidate the specific signaling mechanism of DCVC-induced apoptosis in placental cells, we evaluated expression of apoptosis-related genes using a targeted PCR-based array and qRT-PCR. The DCVC-induced gene expression changes for the extrinsic and intrinsic apoptotic pathways are summarized in Figure 6. We detected a notable DCVC-stimulated increase in TP53 (p53) tumor suppressor gene expression consistent with previous studies (Chen et al., 2002; Rehman et al., 2013). The p53 protein activates apoptosis through multiple transcription-dependent and independent mechanisms involving both apoptosis pathways (Haupt et al., 2003; Amaral et al., 2010). In fact, the effect of DCVC on p53 may explain, at least in part, why both pathways are activated in DCVC-induced apoptosis. For example, we observed a significant upregulation of the p53 transcriptional target FAS, a gene encoding a TNF family death receptor, indicating involvement of p53 in extrinsic pathway regulation. On the other hand, we also observed a significant gene upregulation of BAK1, another p53 transcriptional target which codes for a BCL-2 family protein, indicating simultaneous involvement of p53 in intrinsic pathway regulation (Perfettini et al., 2004; Graupner et al., 2011). Additionally, p53 regulates the intrinsic pathway in a non-transcriptional manner by directly translocating into the mitochondria following stress-induced upregulation and cytosolic accumulation of the protein (Haupt et al., 2003; Amaral et al., 2010). Once inside the mitochondria, p53 forms an antagonist complex with anti-apoptotic Bcl-XL, releasing pro-apoptotic BAX and BAK1 to dimerize and increase mitochondrial membrane permeability (Perfettini et al., 2004; Amaral et al., 2010).
In addition to p53-associated apoptosis, we observed that DCVC treatment substantially increased the expression of another p53 transcriptionally regulated gene, GADD45α, which induces cell cycle arrest in response to stress stimuli (Papathanasiou et al., 1991; Hollander et al., 1993). Although previous studies failed to reach a consensus on the role of GADD45α in apoptosis signaling (Sheikh et al., 2000), GADD45α-mediated gene expression has been implicated in physiological irregularities and insufficient trophoblast invasion in pre-eclamptic placentas (Xiong et al., 2009; Xiong et al., 2013; Liu et al., 2014; Liu et al., 2016b). Because DCVC exposure increased GADD45α gene expression by a magnitude of nearly ten-fold, our findings suggest that this gene may play a pivotal role in DCVC-induced placental cell cytotoxicity: however, further investigation is needed to clarify this role.
Several other salient apoptosis-related genes demonstrated differential DCVC-stimulated expression in the current study. For example, DNA fragmentation factor gene, DFFA, a common pathway caspase 3 cleavage target that promotes DNA fragmentation, showed a trend in upregulation following DCVC exposure. In addition, CASP4, the gene coding for caspase 4, was significantly upregulated, while CASP1 was significantly downregulated. Caspases 1 and 4 are part of a family of caspases that are strongly implicated in inflammatory processes and responses to pathogens (McIlwain et al., 2015). Although their role in the induction of apoptosis is not fully understood (Denes et al., 2012), several studies have suggested that caspase 4 participates in endoplasm-reticulum stress-induced apoptosis in neuronal cells (Hitomi et al., 2004; Yamamuro et al., 2011; Li et al., 2013). More research in needed to clarify the function of caspases 1 and 4 expression in placental cells. Lastly, HRK, which showed a large 19-fold increase in expression, belongs to a BCL-2 subfamily of genes called BH3-only because they only contain one domain in common with other BCL-2 family genes. This subfamily of genes plays a pivotal role in the induction of intrinsic apoptosis because they travel freely in the cytoplasm until they are activated by receptor-free stimuli. Upon activation, they translocate into the mitochondria and interact with other mitochondrial-sequestered BCL-2 proteins like BAK1, directly stimulating the mitochondrial components of the intrinsic pathway (Inohara et al., 1997). Interestingly, a previous study also reported an increase HRK mRNA expression in abnormally fragmented pre-implantation embryos (Jurisicova et al., 2003). These results may indicate that HRK plays an important role in regulating both normal and pathological cell death during early pregnancy.
Because our prior study showed that DCVC increases ROS generation in HTR-8/SVneo cells, we tested the hypothesis that aberrant ROS generation contributes to DCVC-induced apoptosis. We found that DCVC treatment increased malondialdehydes in HTR-8/SVneo cells, a byproduct and proxy measure for lipid peroxidation. These results are consistent with previous studies that indicate DCVC cause s lipid peroxidation in kidney tubular cells of humans and rodents (Beuter et al., 1989; Chen et al., 1990; Groves et al., 1991). Lipid peroxidation occurs when reactive oxygen species, including free radicals, attack carbon double bonds in lipids resulting in lipid peroxyl radicals and hydroperoxides (Ayala et al., 2014). This process damages lipid membranes within the cell and affects their ability to function properly, especially the plasma and mitochondrial membranes. Membrane damage caused by lipid peroxidation is capable of inducing a loss of mitochondrial membrane potential because it destroys the selective barrier and ion transport properties of the membrane (Stark, 2005). Our finding that DCVC induced lipid peroxidation offers a possible mechanistic explanation for a DCVC-induced loss of mitochondrial membrane potential in HTR-8/SVneo cells, previously reported by our lab (Hassan et al., 2016).
Consistent with our finding of DCVC-induced lipid peroxidation, we observed a significant increase in NFKB1 gene expression in placental cells, as well as its upstream regulator RIPK2 (McCarthy et al., 1998). NFKB1 (nuclear factor kappa-light-chain-enhancer of activated B cells) is a transcription factor primarily involved in inflammatory responses to a variety of stimuli, most notably, reactive oxygen species (ROS) (Schreck et al., 1991). NFKB1 directly regulates the transcription levels of many cytokines such as the pro-inflammatory cytokine interleuken-6 (IL-6) (Libermann and Baltimore, 1990). Our result indicating a DCVC-induced increase in NFKB1 gene expression is consistent with our previous study that showed DCVC caused an ROS-mediated increase in IL-6 production (Hassan et al., 2016). Our NFKB1 gene expression results are particularly significant because increased NFKB1 activity and concomitant aberrant ROS generation have been observed both in early pre-eclamptic pregnancies in vivo and in trophoblast-like cells in vitro (Luppi et al., 2006; Vaughan and Walsh, 2012). Additionally, we observed the upregulation of another NFKB1 transcriptional target BCL2A1, an anti-apoptotic BCL-2 family gene. The exact nature of the upregulation of this gene remains unclear but it is likely related to a stress response -induced biological feedback system.
Consistent with lipid peroxidation, we demonstrated that the antioxidant peroxyl radical scavenger (±)-α-tocopherol attenuated DCVC-induced increase caspase 3 and 7 activity. These results suggest that DCVC-stimulated apoptotic activity is dependent on increased ROS generation and lipid peroxidation. These data agree with prior findings that increased ROS generation is capable of inducing apoptosis (Smith et al., 1999; Simon et al., 2000; Myatt and Cui, 2004). Lipid peroxidation activity has been observed in pregnancy complications that involve poor placentation. For example, multiple studies reported that pre-eclamptic pregnancies have increased placental lipid peroxidation and serum circulating lipid peroxidation byproducts compared to normal pregnancies (Madazli et al., 2002; Atamer et al., 2005; Gupta et al., 2005). Studies have also implicated lipid peroxidation as a pathological feature of intrauterine growth restriction (Biri et al., 2007; Karowicz-Bilinska et al., 2007). The evidence presented in the current study suggests that increased ROS generation and lipid peroxidation are involved in the mechanism by which DCVC expo sure placental toxicity.
Multiple studies have reported that HTR-8/SVneo cells retain key functional characteristics of extravillous trophoblasts such as invasive and proliferative capacities which change in response to stress and oxygen availability, expression of adhesion molecules and a mesenchymal proteomic phenotype (Kilburn et al., 2000; Hannan et al., 2010; Liu et al., 2016a; Szklanna et al., 2017). In addition, several studies have also shown that HTR-8/SVneo cells express specific markers that uniquely identify extravillous trophoblasts including cytokeratin 7 (CK7) and α5β1 integrin, and when grown on matrigel, histocompatibility antigen, class I, G (HLA-G) (Irving et al., 1995; Kilburn et al., 2000; Khan et al., 2011; Takao et al., 2011). Overall, HTR-8/SVneo cells have proven useful in modeling first-trimester extravillous trophoblasts because primary first-trimester placental tissue tends to have limited availability for research purposes. Regardless, in vitro cultured cells lack cell signaling and tissue interactions that would otherwise be present in in vivo models. Thus, in vivo studies are needed to further validate our results. In addition, HTR-8/SVneo is an immortalized cell line. Even though HTR-8/SVneo were originally derived from first-trimester non-cancerous placental cells, the process of immortalization changes cells to allow them to be cultured for extended periods of time (Graham et al., 1993). Moreover, cell culture conditions may also be responsible for reported changes in the genetic and epigenetic profiles of HTR-8/SVneo cells (Bilban et al., 2010; Novakovic et al., 2011). For these reasons, the mechanisms of cytotoxicity of DCVC should be further investigated in primary first-trimester extravillous trophoblasts.
The current study includes DCVC concentrations selected for relevance to human exposure. Although the EPA’s maximum contaminant level (MCL) for drinking water is 5 μg/L or parts-per-billion of TCE (NTP, 2015), some people may regularly be exposed to TCE levels in drinking water that exceed the EPA MCL (ATSDR, 2016). Moreover, the Occupational Safety and Health Administration (OSHA) Permissible Exposure Limit (PEL) is 100 ppm averaged over an 8-hour work day (ATSDR, 2016). We suggest that the 10 μM and 20 μM DCVC concentrations used in our study are relevant to occupational exposures because the average peak concentration in blood of female volunteers exposed to 100 ppm TCE by inhalation for 4 h was 13.4 μM for the metabolic precursor to DCVC, S-(1,2-dichlorovinyl)glutathione (Lash et al., 1999). Moreover, the most recent human physiologically based pharmacokinetic models are in good agreement with human data of the latter study, though the need for further study was noted (EPA, 2011). Adding support that the concentrations used in our study are plausible for some occupational exposures, a recent study detected concentrations up to 229 ppm using personal aerosolized TCE exposure of 80 workers (29% women) (Walker et al., 2016). We measured a number of significant effects on HTR-8/SVneo cells at the 20 μM DCVC treatment level, including increased caspase 3, 7, 8 and 9 activation, increased malondialdehyde formation, and increased expression of multiple apoptosis and inflammatory-related genes, suggesting that DCVC stimulated apoptosis, lipid peroxidation, and pro -inflammatory responses at a concentration relevant for human exposure. Additionally, we included higher DCVC concentrations in our study that allowed us to characterize DCVC-induced cell death more completely. The concentrations of 50 μM and 100 μM DCVC are still within an order of magnitude of occupationally relevant concentrations, and similar to (Hassan et al., 2016) or less than those used in previously published in vitro studies, with multiple studies using concentrations up to 500 μM and 1 mM DCVC (van de Water et al., 1995; Chen et al., 2001; Lash et al., 2001; Lash et al., 2003; Xu et al., 2008).
In summary, we detected significant DCVC-induced cytotoxic effects in placental cells utilizing concentrations that encompass those detected of the DCVC precursor in human serum samples (Lash et al., 1999). We demonstrated that the TCE metabolite DCVC stimulated activation of both the intrinsic and extrinsic apoptotic signaling pathways, culminating in aberrant apoptosis in a first-trimester extravillous trophoblast cell line. In addition, we demonstrated that DCVC-induced lipid peroxidation-associated apoptosis. To our knowledge, our study is the first to report that DCVC induces apoptosis via two signaling pathways simultaneously and that cross talk occurs between the two pathways. Our findings contribute to the biological plausibility of DCVC-induced placental toxicity, indicating that further studies into matter are warranted.
Supplementary Material
Figure 7. Effect of antioxidant co-treatment on DCVC-stimulated caspase 3/7 activity.

Cells were untreated (media only control) for 12 h or exposed to 20 μM DCVC without or with 50 μM (±)α-tocopherol pre-treatment for 15 min followed by co -treatment for 12 h. Bars represent means±SEM. Data were analyzed by two-way ANOVA (interaction between DCVC and α-tocopherol treatments, P=0.0103), followed by Tukey’s multiple comparison of means. #Significantly different compared to control with no (±)α-tocopherol treatment (P=0.0016). *Significantly different compared to 20 μM DCVC with no (±)α-tocopherol treatment (P<0.0075). N=3 independent experiments. All experiments were performed in quadruplicate.
Highlights.
DCVC induced apoptosis in a placental cell line in a concentration-dependent manner.
DCVC activated both intrinsic and extrinsic apoptotic pathways via tBID cross talk.
DCVC stimulated differential intrinsic and extrinsic pathway gene expression.
Cellular lipid peroxidation byproduct malondyaldehyde levels were increased by DCVC.
Antioxidant (±)-α-tocopherol attenuated DCVC-induced caspase activity.
Acknowledgments
This work was supported by the National Institute of Environmental Health Sciences, National Institutes of Health (grant numbers P42 ES0171982, P30 ES017885, and T32 ES007062); and The University of Michigan. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIEHS or NIH. We thank the University of Michigan’s DNA Sequencing Core and the Omics and Bioinformatics Core of the Michigan Center on Lifestage Environmental Exposures and Disease for assistance with the Apoptosis PCR Array, and the University of Michigan’s Flow Cytometry Core for assistance with the Annexin-FITC/PI Staining Assay. We gratefully acknowledge Dr. Kelly A. Hogan, Anthony Su and Faith Bjork for assistance with learning new laboratory techniques and for helpful scientific discussions, and Gloria Choi for assistance with qRT-PCR.
Abbreviations
- BAK1
BCL2-antagonist/killer 1
- BAX
BCL2-associated X protein
- BCL-2
B-cell CLL/lymphoma 2
- BCL2A1
BCL2-related protein A1
- BID
BH3 interacting domain death agonist
- CASP1
Caspase 1, apoptosis-related cysteine peptidase (interleukin 1, beta, convertase)
- CASP3
Caspase 3, apoptosis-related cysteine peptidase
- CASP4
Caspase 4, apoptosis-related cysteine peptidase
- CASP7
Caspase 7, apoptosis-related cysteine peptidase
- CASP8
Caspase 8, apoptosis-related cysteine peptidase
- CASP9
Caspase 9, apoptosis-related cysteine peptidase
- DCVC
S-(1, 2-dichlorovinyl)-L-cysteine
- DEVD
Asp-Glu-Val-Asp
- DFFA
DNA fragmentation factor
- 45kDa
alpha polypeptide
- FAS
Fas (TNF receptor superfamily, member 6)
- FBS
fetal bovine serum
- FITC
fluorescein isothiocyanate
- GADD45A
Growth arrest and DNA-damage-inducible, alpha
- HRK
Harakiri
- BCL2
interacting protein (contains only BH3 domain)
- LEHD
Leu-Glu-His-Asp
- LETD
Leu-Glu-Thr-Asp
- IGF1R
Insulin-like growth factor 1 receptor
- MCL
maximum contaminant level
- MDA
malondialdehyde
- NFKB1
Nuclear factor of kappa light polypeptide gene enhancer in B-cells 1
- RIPK2
Receptor-interacting serine-threonine kinase 2
- ROS
reactive oxygen species
- TBA
thiobarbituric
- TBHP
tert-butyl hydroperoxide
- tBID
truncated BH3 interacting domain death agonist
- TCE
trichloroethylene
- TP53
tumor protein p53
- TRAF3
TNF receptor-associated factor 3
Appendix A. Supplementary data
Supplementary data to this article can be found online at:
Footnotes
Conflict of interest
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- Aardema MW, Oosterhof H, Timmer A, van Rooy I, Aarnoudse JG. Uterine artery Doppler flow and uteroplacental vascular pathology in normal pregnancies and pregnancies complicated by pre-eclampsia and small for gestational age fetuses. Placenta. 2001;22:405–411. doi: 10.1053/plac.2001.0676. [DOI] [PubMed] [Google Scholar]
- Alvero AB, Montagna MK, Mor G. Correlation of caspase activity and in vitro chemo -response in epithelial ovarian cancer cell lines. Methods Mol Biol. 2008;414:79–82. doi: 10.1007/978-1-59745-339-4_7. [DOI] [PubMed] [Google Scholar]
- Amaral JD, Xavier JM, Steer CJ, Rodrigues CM. The role of p53 in apoptosis. Discov Med. 2010;9:145–152. [PubMed] [Google Scholar]
- Atamer Y, Kocyigit Y, Yokus B, Atamer A, Erden AC. Lipid peroxidation, antioxidant defense, status of trace metals and leptin levels in preeclampsia. Eur J Obstet Gynecol Reprod Biol. 2005;119:60–66. doi: 10.1016/j.ejogrb.2004.06.033. [DOI] [PubMed] [Google Scholar]
- ATCC. ATCC Product Sheet: HTR8/SVneo (ATCC® CRL3271™) American Type Culture Collection; 2015. [Google Scholar]
- ATSDR. 2015 Priority List of Hazardous Substances. Agency for Toxic Substances and Disease Registry; Atlanta, GA: 2015. [Google Scholar]
- ATSDR. Public Health Statement on Trichloroethylene. Agency for Toxic Substances and Disease Registry; 2016. [Google Scholar]
- Ayala A, Munoz MF, Arguelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev. 2014;2014:360438. doi: 10.1155/2014/360438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becattini B, Sareth S, Zhai D, Crowell KJ, Leone M, Reed JC, Pellecchia M. Targeting apoptosis via chemical design: inhibition of bid -induced cell death by small organic molecules. Chem Biol. 2004;11:1107–1117. doi: 10.1016/j.chembiol.2004.05.022. [DOI] [PubMed] [Google Scholar]
- Belkacemi L, Chen CH, Ross MG, Desai M. Increased placental apoptosis in maternal food restricted gestations: role of the Fas pathway. Placenta. 2009;30:739–751. doi: 10.1016/j.placenta.2009.06.003. [DOI] [PubMed] [Google Scholar]
- Belkacemi L, Desai M, Nelson DM, Ross MG. Altered mitochondrial apoptotic pathway in placentas from undernourished rat gestations. Am J Physiol Regul Integr Comp Physiol. 2011;301:R1599–1615. doi: 10.1152/ajpregu.00100.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B (Methodological) 1995;57:289–300. [Google Scholar]
- Beuter W, Cojocel C, Muller W, Donaubauer HH, Mayer D. Peroxidative damage and nephrotoxicity of dichlorovinylcysteine in mice. J Appl Toxicol. 1989;9:181–186. doi: 10.1002/jat.2550090308. [DOI] [PubMed] [Google Scholar]
- Bilban M, Tauber S, Haslinger P, Pollheimer J, Saleh L, Pehamberger H, Wagner O, Knofler M. Trophoblast invasion: assessment of cellular models using gene expression signatures. Placenta. 2010;31:989–996. doi: 10.1016/j.placenta.2010.08.011. [DOI] [PubMed] [Google Scholar]
- Biri A, Bozkurt N, Turp A, Kavutcu M, Himmetoglu O, Durak I. Role of oxidative stress in intrauterine growth restriction. Gynecol Obstet Invest. 2007;64:187–192. doi: 10.1159/000106488. [DOI] [PubMed] [Google Scholar]
- Brosens IA, Robertson WB, Dixon HG. The role of the spiral arteries in the pathogenesis of preeclampsia. Obstet Gynecol Annu. 1972;1:177–191. [PubMed] [Google Scholar]
- Bukowski J. Critical review of the epidemiologic literature regarding the association between congenital heart defects and exposure to trichloroethylene. Critical reviews in toxicology. 2014;44:581–589. doi: 10.3109/10408444.2014.910755. [DOI] [PubMed] [Google Scholar]
- Burton GJ, Fowden AL. The placenta: a multifaceted, transient organ. Philos Trans R Soc Lond B Biol Sci. 2015;370:20140066. doi: 10.1098/rstb.2014.0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caniggia I, Grisaru-Gravnosky S, Kuliszewsky M, Post M, Lye SJ. Inhibition of TGF-beta 3 restores the invasive capability of extravillous trophoblasts in preeclamptic pregnancies. J Clin Invest. 1999;103:1641–1650. doi: 10.1172/JCI6380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney EW, Thorsrud BA, Dugard PH, Zablotny CL. Developmental toxicity studies in Crl:CD (SD) rats following inhalation exposure to trichloroethylene and perchloroethylene. Birth Defects Res B Dev Reprod Toxicol. 2006;77:405–412. doi: 10.1002/bdrb.20091. [DOI] [PubMed] [Google Scholar]
- Chen Q, Jones TW, Brown PC, Stevens JL. The mechanism of cysteine conjugate cytotoxicity in renal epithelial cells. Covalent binding leads to thiol depletion and lipid peroxidation. J Biol Chem. 1990;265:21603–21611. [PubMed] [Google Scholar]
- Chen SJ, Wang JL, Chen JH, Huang RN. Possible involvement of glutathione and p53 in trichloroethylene- and perchloroethylene-induced lipid peroxidation and apoptosis in human lung cancer cells. Free Radic Biol Med. 2002;33:464–472. doi: 10.1016/s0891-5849(02)00817-1. [DOI] [PubMed] [Google Scholar]
- Chen Y, Cai J, Anders MW, Stevens JL, Jones DP. Role of mitochondrial dysfunction in S-(1,2-dichlorovinyl)-l-cysteine-induced apoptosis. Toxicol Appl Pharmacol. 2001;170:172–180. doi: 10.1006/taap.2000.9107. [DOI] [PubMed] [Google Scholar]
- Chiu WA, Jinot J, Scott CS, Makris SL, Cooper GS, Dzubow RC, Bale AS, Evans MV, Guyton KZ, Keshava N, Lipscomb JC, Barone S, Jr, Fox JF, Gwinn MR, Schaum J, Caldwell JC. Human health effects of trichloroethylene: key findings and scientific issues. Environ Health Perspect. 2013;121:303–311. doi: 10.1289/ehp.1205879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou JJ, Li H, Salvesen GS, Yuan J, Wagner G. Solution structure of BID, an intracellular amplifier of apoptotic signaling. Cell. 1999;96:615–624. doi: 10.1016/s0092-8674(00)80572-3. [DOI] [PubMed] [Google Scholar]
- Darnerud PO, Brandt I, Feil VJ, Bakke JE. Dichlorovinyl cysteine (DCVC) in the mouse kidney: tissue-binding and toxicity after glutathione depletion and probenecid treatment. Arch Toxicol. 1989;63:345–350. doi: 10.1007/BF00303121. [DOI] [PubMed] [Google Scholar]
- Das RM, Scott JE. Trichloroethylene -induced pneumotoxicity in fetal and neonatal mice. Toxicol Lett. 1994;73:227–239. doi: 10.1016/0378-4274(94)90062-0. [DOI] [PubMed] [Google Scholar]
- Davies EL, Bell JS, Bhattacharya S. Preeclampsia and pre term delivery: A population-based case-control study. Hypertens Pregnancy. 2016;35:510–519. doi: 10.1080/10641955.2016.1190846. [DOI] [PubMed] [Google Scholar]
- Denes A, Lopez-Castejon G, Brough D. Caspase-1: is IL-1 just the tip of the ICEberg? Cell Death Dis. 2012;3:e338. doi: 10.1038/cddis.2012.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFederico E, Genbacev O, Fisher SJ. Preeclampsia is associated with widespread apoptosis of placental cytotrophoblasts within the uterine wall. Am J Pathol. 1999;155:293–301. doi: 10.1016/S0002-9440(10)65123-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EPA. Toxicological Review of Trichloroethylene (CAS No. 79-01-6) National Center for Environmental Assessment; Washtingon D.C: 2011. [Google Scholar]
- EPA. TRI Explorer: Release Trends Report. Environmental Protection Agency; 2017. [Google Scholar]
- Fisher JW, Channel SR, Eggers JS, Johnson PD, MacMahon KL, Goodyear CD, Sudberry GL, Warren DA, Latendresse JR, Graeter LJ. Trichloroethylene, trichloroacetic acid, and dichloroacetic acid: do they affect fetal rat heart development? Int J Toxicol. 2001;20:257–267. doi: 10.1080/109158101753252992. [DOI] [PubMed] [Google Scholar]
- Forand SP, Lewis-Michl EL, Gomez MI. Adverse birth outcomes and maternal exposure to trichloroethylene and tetrachloroethylene through soil vapor intrusion in New York State. Environ Health Perspect. 2012;120:616–621. doi: 10.1289/ehp.1103884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Abu Kwaik Y. Hijacking of apoptotic pathways by bacterial pathogens. Microbes Infect. 2000;2:1705–1719. doi: 10.1016/s1286-4579(00)01326-5. [DOI] [PubMed] [Google Scholar]
- Genbacev O, DiFederico E, McMaster M, Fisher SJ. Invasive cytotrophoblast apoptosis in pre -eclampsia. Hum Reprod. 1999;14(Suppl 2):59–66. doi: 10.1093/humrep/14.suppl_2.59. [DOI] [PubMed] [Google Scholar]
- Graham CH, Hawley TS, Hawley RG, MacDougall JR, Kerbel RS, Khoo N, Lala PK. Establishment and characterization of first trimester human trophoblast cells with extended lifespan. Experimental cell research. 1993;206:204–211. doi: 10.1006/excr.1993.1139. [DOI] [PubMed] [Google Scholar]
- Graupner V, Alexander E, Overkamp T, Rothfuss O, De Laurenzi V, Gillissen BF, Daniel PT, Schulze-Osthoff K, Essmann F. Differential regulation of the proapoptotic multidomain protein Bak by p53 and p73 at the promoter level. Cell Death Differ. 2011;18:1130–1139. doi: 10.1038/cdd.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves CE, Lock EA, Schnellmann RG. Role of lipid peroxidation in renal proximal tubule cell death induced by haloalkene cysteine conjugates. Toxicol Appl Pharmacol. 1991;107:54–62. doi: 10.1016/0041-008x(91)90330-h. [DOI] [PubMed] [Google Scholar]
- Guha N, Loomis D, Grosse Y, Lauby-Secretan B, El Ghissassi F, Bouvard V, Benbrahim-Tallaa L, Baan R, Mattock H, Straif K International Agency for Research on Cancer Monograph Working G. Carcinogenicity of trichloroethylene, tetrachloroethylene, some other chlorinated solvents, and their metabolites. Lancet Oncol. 2012;13:1192–1193. doi: 10.1016/s1470-2045(12)70485-0. [DOI] [PubMed] [Google Scholar]
- Gupta S, Agarwal A, Sharma RK. The role of placental oxidative stress and lipid peroxidation in preeclampsia. Obstet Gynecol Surv. 2005;60:807–816. doi: 10.1097/01.ogx.0000193879.79268.59. [DOI] [PubMed] [Google Scholar]
- Hannan NJ, Paiva P, Dimitriadis E, Salamonsen LA. Models for study of human embryo implantation: choice of cell lines? Biology of reproduction. 2010;82:235–245. doi: 10.1095/biolreprod.109.077800. [DOI] [PubMed] [Google Scholar]
- Hassan I, Kumar AM, Park HR, Lash LH, Loch-Caruso R. Reactive Oxygen Stimulation of Interleukin-6 Release in the Human Trophoblast Cell Line HTR-8/SVneo by the Trichlorethylene Metabolite S-(1,2-Dichloro)-l-Cysteine. Biology of reproduction. 2016;95:66. doi: 10.1095/biolreprod.116.139261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis - the p53 network. J Cell Sci. 2003;116:4077–4085. doi: 10.1242/jcs.00739. [DOI] [PubMed] [Google Scholar]
- Hitomi J, Katayama T, Eguchi Y, Kudo T, Taniguchi M, Koyama Y, Manabe T, Yamagishi S, Bando Y, Imaizumi K, Tsujimoto Y, Tohyama M. Involvement of caspase -4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J Cell Biol. 2004;165:347–356. doi: 10.1083/jcb.200310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollander MC, Alamo I, Jackman J, Wang MG, McBride OW, Fornace AJ., Jr Analysis of the mammalian gadd45 gene and its response to DNA damage. J Biol Chem. 1993;268:24385–24393. [PubMed] [Google Scholar]
- Huang Y, Dong F, Du Q, Zhang H, Luo X, Song X, Zhao X, Zhang W, Tong D. Swainsonine induces apoptosis through mitochondrial pathway and caspase activation in goat trophoblasts. Int J Biol Sci. 2014;10:789–797. doi: 10.7150/ijbs.9168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilekis JV, Tsilou E, Fisher S, Abrahams VM, Soares MJ, Cross JC, Zamudio S, Illsley NP, Myatt L, Colvis C, Costantine MM, Haas DM, Sadovsky Y, Weiner C, Rytting E, Bidwell G. Placental origins of adverse pregnancy outcomes: potential molecular targets: an Executive Workshop Summary of the Eunice Kennedy Shriver National Institute of Child Health and Human Development. Am J Obstet Gynecol. 2016;215:S1–S46. doi: 10.1016/j.ajog.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inohara N, Ding L, Chen S, Nunez G. harakiri, a novel regulator of cell death, encodes a protein that activates apoptosis and interacts selectively with survival-promoting proteins Bcl-2 and Bcl-X(L) EMBO J. 1997;16:1686–1694. doi: 10.1093/emboj/16.7.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irving JA, Lysiak JJ, Graham CH, Hearn S, Han VK, Lala PK. Characteristics of trophoblast cells migrating from first trimester chorionic villus explants and propagated in culture. Placenta. 1995;16:413–433. doi: 10.1016/0143-4004(95)90100-0. [DOI] [PubMed] [Google Scholar]
- Johnson PD, Goldberg SJ, Mays MZ, Dawson BV. Threshold of trichloroethylene contamination in maternal drinking waters affecting fetal heart development in the rat. Environ Health Perspect. 2003;111:289–292. doi: 10.1289/ehp.5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurisicova A, Antenos M, Varmuza S, Tilly JL, Casper RF. Expression of apoptosis -related genes during human preimplantation embryo development: potential roles for the Harakiri gene product and Caspase-3 in blastomere fragmentation. Molecular human reproduction. 2003;9:133–141. doi: 10.1093/molehr/gag016. [DOI] [PubMed] [Google Scholar]
- Kadyrov M, Kingdom JC, Huppertz B. Divergent trophoblast invasion and apoptosis in placental bed spiral arteries from pregnancies complicated by maternal anemia and early -onset preeclampsia/intrauterine growth restriction. Am J Obstet Gynecol. 2006;194:557–563. doi: 10.1016/j.ajog.2005.07.035. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karowicz-Bilinska A, Kedziora-Kornatowska K, Bartosz G. Indices of oxidative stress in pregnancy with fetal growth restriction. Free Radic Res. 2007;41:870–873. doi: 10.1080/10715760701291647. [DOI] [PubMed] [Google Scholar]
- Kelder T, van Iersel MP, Hanspers K, Kutmon M, Conklin BR, Evelo CT, Pico AR. WikiPathways: building research communities on biological pathways. Nucleic Acids Res. 2012;40:D1301–1307. doi: 10.1093/nar/gkr1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan GA, Girish GV, Lala N, Di Guglielmo GM, Lala PK. Decorin is a novel VEGFR-2-binding antagonist for the human extravillous trophoblast. Mol Endocrinol. 2011;25:1431–1443. doi: 10.1210/me.2010-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilburn BA, Wang J, Duniec-Dmuchowski ZM, Leach RE, Romero R, Armant DR. Extracellular matrix composition and hypoxia regulate the expression of HLA -G and integrins in a human trophoblast cell line. Biology of reproduction. 2000;62:739–747. doi: 10.1095/biolreprod62.3.739. [DOI] [PubMed] [Google Scholar]
- Kuwana T, Smith JJ, Muzio M, Dixit V, Newmeyer DD, Kornbluth S. Apoptosis induction by caspase-8 is amplified through the mitochondrial release of cytochrome c. J Biol Chem. 1998;273:16589–16594. doi: 10.1074/jbc.273.26.16589. [DOI] [PubMed] [Google Scholar]
- Lagakos S, Wessen B, Zelen M. An analysis of contaminated well water and health effects in Woburn, Massachusetts. Journal of the American Statistical Association. 1986;81:583–596. [Google Scholar]
- Laham S. Studies on placental transfer. Trichlorethylene. IMS Ind Med Surg. 1970;39:46–49. [PubMed] [Google Scholar]
- Lash LH, Anders MW. Cytotoxicity of S-(1,2-dichlorovinyl)glutathione and S-(1,2-dichlorovinyl)-L-cysteine in isolated rat kidney cells. J Biol Chem. 1986;261:13076–13081. [PubMed] [Google Scholar]
- Lash LH, Chiu WA, Guyton KZ, Rusyn I. Trichloroethylene biotransformation and its role in mutagenicity, carcinogenicity and target organ toxicity. Mutat Res Rev Mutat Res. 2014;762:22–36. doi: 10.1016/j.mrrev.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lash LH, Hueni SE, Putt DA. Apoptosis, necrosis, and cell proliferation induced by S -(1,2-dichlorovinyl)-L-cysteine in primary cultures of human proximal tubular cells. Toxicol Appl Pharmacol. 2001;177:1–16. doi: 10.1006/taap.2001.9295. [DOI] [PubMed] [Google Scholar]
- Lash LH, Putt DA, Brashear WT, Abbas R, Parker JC, Fisher JW. Identification of S -(1,2-dichlorovinyl)glutathione in the blood of human volunteers exposed to trichloroethylene. J Toxicol Environ Health A. 1999;56:1–21. doi: 10.1080/009841099158204. [DOI] [PubMed] [Google Scholar]
- Lash LH, Putt DA, Hueni SE, Krause RJ, Elfarra AA. Roles of necrosis, Apoptosis, and mitochondrial dysfunction in S-(1,2-dichlorovinyl)-L-cysteine sulfoxide-induced cytotoxicity in primary cultures of human renal proximal tubular cells. J Pharmacol Exp Ther. 2003;305:1163–1172. doi: 10.1124/jpet.102.046185. [DOI] [PubMed] [Google Scholar]
- Levy R, Nelson DM. To be, or not to be, that is the question. Apoptosis in human trophoblast. Placenta. 2000;21:1–13. doi: 10.1053/plac.1999.0450. [DOI] [PubMed] [Google Scholar]
- Li C, Wei J, Li Y, He X, Zhou Q, Yan J, Zhang J, Liu Y, Liu Y, Shu HB. Transmembrane Protein 214 (TMEM214) mediates endoplasmic reticulum stress -induced caspase 4 enzyme activation and apoptosis. J Biol Chem. 2013;288:17908–17917. doi: 10.1074/jbc.M113.458836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- Libermann TA, Baltimore D. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol Cell Biol. 1990;10:2327–2334. doi: 10.1128/mcb.10.5.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Wang Y, Shen C, He J, Liu X, Ding Y, Gao R, Chen X. Benzo(a)pyrene inhibits migration and invasion of extravillous trophoblast HTR-8/SVneo cells via activation of the ERK and JNK pathway. J Appl Toxicol. 2016a;36:946–955. doi: 10.1002/jat.3227. [DOI] [PubMed] [Google Scholar]
- Liu X, Deng Q, Luo X, Chen Y, Shan N, Qi H. Oxidative stress -induced Gadd45alpha inhibits trophoblast invasion and increases sFlt1/sEng secretions via p38 MAPK involving in the pathology of pre-eclampsia. J Matern Fetal Neonatal Med. 2016b;29:3776–3785. doi: 10.3109/14767058.2016.1144744. [DOI] [PubMed] [Google Scholar]
- Liu X, Mu H, Luo X, Xiao X, Ding Y, Yin N, Deng Q, Qi H. Expression of Gadd45alpha in human early placenta and its role in trophoblast invasion. Placenta. 2014;35:370–377. doi: 10.1016/j.placenta.2014.03.020. [DOI] [PubMed] [Google Scholar]
- Luppi P, Tse H, Lain KY, Markovic N, Piganelli JD, DeLoia JA. Preeclampsia activates circulating immune cells with engagement of the NF-kappaB pathway. Am J Reprod Immunol. 2006;56:135–144. doi: 10.1111/j.1600-0897.2006.00386.x. [DOI] [PubMed] [Google Scholar]
- Madazli R, Benian A, Aydin S, Uzun H, Tolun N. The plasma and placental levels of malondialdehyde, glutathione and superoxide dismutase in pre -eclampsia. J Obstet Gynaecol. 2002;22:477–480. doi: 10.1080/0144361021000003573. [DOI] [PubMed] [Google Scholar]
- McCarthy JV, Ni J, Dixit VM. RIP2 is a novel NF-kappaB-activating and cell death-inducing kinase. J Biol Chem. 1998;273:16968–16975. doi: 10.1074/jbc.273.27.16968. [DOI] [PubMed] [Google Scholar]
- McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2015:7. doi: 10.1101/cshperspect.a026716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney LL, Picken JC, Jr, Weakley FB, Eldridge AC, Campbell RE, Cowan JC, Biester HE. Possible Toxic Factor of Trichloroethylene -extracted Soybean Oil Meal3. Journal of the American Chemical Society. 1959;81:909–915. [Google Scholar]
- Meekins JW, Pijnenborg R, Hanssens M, McFadyen IR, van Asshe A. A study of placental bed spiral arteries and trophoblast invasion in normal and severe pre -eclamptic pregnancies. Br J Obstet Gynaecol. 1994;101:669–674. doi: 10.1111/j.1471-0528.1994.tb13182.x. [DOI] [PubMed] [Google Scholar]
- Morgan T. Placental Insufficiency Is a Leading Cause of Preterm Labor. NewReviews. 2014;15:5618–e5525. [Google Scholar]
- Morgan TK. Role of the Placenta in Preterm Birth: A Review. Am J Perinatol. 2016;33:258–266. doi: 10.1055/s-0035-1570379. [DOI] [PubMed] [Google Scholar]
- Myatt L, Cui X. Oxidative stress in the placenta. Histochem Cell Biol. 2004;122:369–382. doi: 10.1007/s00418-004-0677-x. [DOI] [PubMed] [Google Scholar]
- Niles AL, Moravec RA, Riss TL. Caspase activity assays. Methods Mol Biol. 2008;414:137–150. doi: 10.1007/978-1-59745-339-4_11. [DOI] [PubMed] [Google Scholar]
- Nishizaki T, Kanno T, Tsuchiya A, Kaku Y, Shimizu T, Tanaka A. 1-[2-(2-Methoxyphenylamino)ethylamino]-3-(naphthalene-1- yloxy)propan-2-ol may be a promising anticancer drug. Molecules. 2014;19:21462–21472. doi: 10.3390/molecules191221462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novakovic B, Gordon L, Wong NC, Moffett A, Manuelpillai U, Craig JM, Sharkey A, Saffery R. Wide-ranging DNA methylation differences of primary trophoblast cell populations and derived cell lines: implications and opportunities for understanding trophoblast function. Molecular human reproduction. 2011;17:344–353. doi: 10.1093/molehr/gar005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NTP. National Toxicology Progam. 2015. Monograph on Trichloroethylene Reports on Carcinogens. [PubMed] [Google Scholar]
- Papathanasiou MA, Kerr NC, Robbins JH, McBride OW, Alamo I, Jr, Barrett SF, Hickson ID, Fornace AJ., Jr Induction by ionizing radiation of the gadd45 gene in cultured human cells: lack of mediation by protein kinase C. Mol Cell Biol. 1991;11:1009–1016. doi: 10.1128/mcb.11.2.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennington KA, Schlitt JM, Jackson DL, Schulz LC, Schust DJ. Preeclampsia: multiple approaches for a multifactorial disease. Dis Model Mech. 2012;5:9–18. doi: 10.1242/dmm.008516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perfettini JL, Kroemer RT, Kroemer G. Fatal liaisons of p53 with Bax and Bak. Nat Cell Biol. 2004;6:386–388. doi: 10.1038/ncb0504-386. [DOI] [PubMed] [Google Scholar]
- Rehman MU, Tahir M, Quaiyoom Khan A, Khan R, Lateef A, Hamiza OO, Ali F, Sultana S. Diosmin protects against trichloroethylene -induced renal injury in Wistarrats: plausible role of p53, Bax and caspases. Br J Nutr. 2013;110:699–710. doi: 10.1017/S0007114512005752. [DOI] [PubMed] [Google Scholar]
- Reister F, Frank HG, Kingdom JC, Heyl W, Kaufmann P, Rath W, Huppertz B. Macrophage-induced apoptosis limits endovascular trophoblast invasion in the uterine wall of preeclamptic women. Lab Invest. 2001;81:1143–1152. doi: 10.1038/labinvest.3780326. [DOI] [PubMed] [Google Scholar]
- Roth W, Kermer P, Krajewska M, Welsh K, Davis S, Krajewski S, Reed JC. Bifunctional apoptosis inhibitor (BAR) protects neurons from diverse cell death pathways. Cell Death Differ. 2003;10:1178–1187. doi: 10.1038/sj.cdd.4401287. [DOI] [PubMed] [Google Scholar]
- Ruckart PZ, Bove FJ, Maslia M. Evaluation of contaminated drinking water and preterm birth, small for gestational age, and birth weight at Marine Corps Base Camp Lejeune, North Carolina: a cross-sectional study. Environ Health. 2014;13:99. doi: 10.1186/1476-069X-13-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rufer ES, Hacker TA, Flentke GR, Drake VJ, Brody MJ, Lough J, Smith SM. Altered cardiac function and ventricular septal defect in avian embryos exposed to low-dose trichloroethylene. Toxicol Sci. 2010;113:444–452. doi: 10.1093/toxsci/kfp269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreck R, Rieber P, Baeuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991;10:2247–2258. doi: 10.1002/j.1460-2075.1991.tb07761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AN, Heazell AE, Crocker IP, Mor G. Placental apoptosis in health and disease. Am J Reprod Immunol. 2010;64:159–169. doi: 10.1111/j.1600-0897.2010.00837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh MS, Hollander MC, Fornance AJ., Jr Role of Gadd45 in apoptosis. Biochem Pharmacol. 2000;59:43–45. doi: 10.1016/s0006-2952(99)00291-9. [DOI] [PubMed] [Google Scholar]
- Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis: an international journal on programmed cell death. 2000;5:415–418. doi: 10.1023/a:1009616228304. [DOI] [PubMed] [Google Scholar]
- Smith SC, Baker PN, Symonds EM. Increased placental apoptosis in intrauterine growth restriction. Am J Obstet Gynecol. 1997a;177:1395–1401. doi: 10.1016/s0002-9378(97)70081-4. [DOI] [PubMed] [Google Scholar]
- Smith SC, Baker PN, Symonds EM. Placental apoptosis in normal human pregnancy. Am J Obstet Gynecol. 1997b;177:57–65. doi: 10.1016/s0002-9378(97)70438-1. [DOI] [PubMed] [Google Scholar]
- Smith SC, Guilbert LJ, Yui J, Baker PN, Davidge ST. The role of reactive nitrogen/oxygen intermediates in cytokine-induced trophoblast apoptosis. Placenta. 1999;20:309–315. doi: 10.1053/plac.1998.0383. [DOI] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Statistical applications in genetics and molecular biology. 2004;3 doi: 10.2202/1544-6115.1027. Article3. [DOI] [PubMed] [Google Scholar]
- Spandidos A, Wang X, Wang H, Seed B. PrimerBank: a resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic Acids Res. 2010;38:D792–799. doi: 10.1093/nar/gkp1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark G. Functional consequences of oxidative membrane damage. J Membr Biol. 2005;205:1–16. doi: 10.1007/s00232-005-0753-8. [DOI] [PubMed] [Google Scholar]
- Straszewski-Chavez SL, Abrahams VM, Mor G. The role of apoptosis in the regulation of trophoblast survival and differentiation during pregnancy. Endocr Rev. 2005;26:877–897. doi: 10.1210/er.2005-0003. [DOI] [PubMed] [Google Scholar]
- Szklanna PB, Wynne K, Nolan M, Egan K, Ainle FN, Maguire PB. Comparative proteomic analysis of trophoblast cell models reveals their differential phenotypes, potential uses and limitations. Proteomics. 2017 doi: 10.1002/pmic.201700037. [DOI] [PubMed] [Google Scholar]
- Takao T, Asanoma K, Kato K, Fukushima K, Tsunematsu R, Hirakawa T, Matsumura S, Seki H, Takeda S, Wake N. Isolation and characterization of human trophoblast side -population (SP) cells in primary villous cytotrophoblasts and HTR-8/SVneo cell line. PloS one. 2011;6:e21990. doi: 10.1371/journal.pone.0021990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetz LM, Cheng AA, Korte CS, Giese RW, Wang P, Harris C, Meeker JD, Loch-Caruso R. Mono-2-ethylhexyl phthalate induces oxidative stress responses in human placental cells in vitro. Toxicol Appl Pharmacol. 2013;268:47–54. doi: 10.1016/j.taap.2013.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Water B, Kruidering M, Nagelkerke JF. F-actin disorganization in apoptotic cell death of cultured rat renal proximal tubular cells. Am J Physiol. 1996;270:F593–603. doi: 10.1152/ajprenal.1996.270.4.F593. [DOI] [PubMed] [Google Scholar]
- van de Water B, Zoeteweij JP, de Bont HJ, Mulder GJ, Nagelkerke JF. Role of mitochondrial Ca2+ in the oxidative stress-induced dissipation of the mitochondrial membrane potential. Studies in isolated proximal tubular cells using the nephrotoxin 1,2-dichlorovinyl-L-cysteine. J Biol Chem. 1994;269:14546–14552. [PubMed] [Google Scholar]
- van de Water B, Zoeteweij JP, de Bont HJ, Nagelkerke JF. Inhibition of succinate:ubiquinone reductase and decrease of ubiquinol in nephrotoxic cysteine S-conjugate-induced oxidative cell injury. Molecular pharmacology. 1995;48:928–937. [PubMed] [Google Scholar]
- Vaughan JE, Walsh SW. Activation of NF-kappaB in placentas of women with preeclampsia. Hypertens Pregnancy. 2012;31:243–251. doi: 10.3109/10641955.2011.642436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DI, Uppal K, Zhang L, Vermeulen R, Smith M, Hu W, Purdue MP, Tang X, Reiss B, Kim S, Li L, Huang H, Pennell KD, Jones DP, Rothman N, Lan Q. High-resolution metabolomics of occupational exposure to trichloroethylene. Int J Epidemiol. 2016;45:1517–1527. doi: 10.1093/ije/dyw218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters EM, Gerstner HB, Huff JE. Trichloroethylene. I. An overview. J Toxicol Environ Health. 1977;2:671–707. doi: 10.1080/15287397709529469. [DOI] [PubMed] [Google Scholar]
- Watson RE, Jacobson CF, Williams AL, Howard WB, DeSesso JM. Trichloroethylene -contaminated drinking water and congenital heart defects: a critical analysis of the literature. Reprod Toxicol. 2006;21:117–147. doi: 10.1016/j.reprotox.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, Thompson CB, Korsmeyer SJ. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000;14:2060–2071. [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Liebermann DA, Holtzman EJ, Jeronis S, Hoffman B, Geifman-Holtzman O. Preeclampsia-associated stresses activate Gadd45a signaling and sFlt-1 in placental explants. J Cell Physiol. 2013;228:362–370. doi: 10.1002/jcp.24139. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Liebermann DA, Tront JS, Holtzman EJ, Huang Y, Hoffman B, Geifman-Holtzman O. Gadd45a stress signaling regulates sFlt-1 expression in preeclampsia. J Cell Physiol. 2009;220:632–639. doi: 10.1002/jcp.21800. [DOI] [PubMed] [Google Scholar]
- Xu F, Papanayotou I, Putt DA, Wang J, Lash LH. Role of mitochondrial dysfunction in cellular responses to S-(1,2-dichlorovinyl)-L-cysteine in primary cultures of human proximal tubular cells. Biochem Pharmacol. 2008;76:552–567. doi: 10.1016/j.bcp.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamuro A, Kishino T, Ohshima Y, Yoshioka Y, Kimura T, Kasai A, Maeda S. Caspase -4 directly activates caspase-9 in endoplasmic reticulum stress-induced apoptosis in SH-SY5Y cells. J Pharmacol Sci. 2011;115:239–243. doi: 10.1254/jphs.10217SC. [DOI] [PubMed] [Google Scholar]
- Zaker F, Amirizadeh N, Nasiri N, Razavi SM, Teimoori-Toolabi L, Yaghmaie M, Mehrasa R. Gene Expression and Methylation Pattern in HRK Apoptotic Gene in Myelodysplastic Syndrome. Int J Mol Cell Med. 2016;5:90–99. [PMC free article] [PubMed] [Google Scholar]
- Zambon A, Hanspers K, Pico A, Kalafati M, van Iersel MP, nuno Riutta AMK, Angelika Lawlor B, Willighagen E, Bouwman J, Josip, Gillespie M, Coort S, Kelder T. Wikipathways: Apoptosis (Homo sapiens) Wikipathways 2017 [Google Scholar]
- Zhang XY, Qu X, Wang CQ, Zhou CJ, Liu GX, Wei FC, Sun SZ. Over-expression of Gadd45a enhances radiotherapy efficacy in human Tca8113 cell line. Acta Pharmacol Sin. 2011;32:253–258. doi: 10.1038/aps.2010.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu QX, Shen T, Ding R, Liang ZZ, Zhang XJ. Cytotoxicity of trichloroethylene and perchloroethylene on normal human epidermal keratinocytes and protective role of vitamin E. Toxicology. 2005;209:55–67. doi: 10.1016/j.tox.2004.12.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.