

Abstract
The cytokine interleukin-2 (IL-2) stimulates both activated CD4+ and CD8+ T cells to proliferate. On a molecular level, IL-2 signals through an identical receptor complex and promotes the same dose-dependent phosphorylation of the canonical transcription factor STAT5 in both cell types. Despite this, CD8+ T cells enter S-phase earlier and proliferate to a greater extent than CD4+ T cells in response to IL-2. Here, we identify distinct IL-2 signaling dynamics in CD4+ and CD8+ T-cell blasts. In CD8+ T cells, STAT5 phosphorylation increased rapidly and was sustained throughout a 6-hour time course. In contrast, CD4+ T cells had a biphasic response, with maxima at 15 minutes and 2–4 hours. Both cell types required vesicular trafficking, but only CD4+ T cells required new protein synthesis to maintain high phosphorylation of STAT5. Two subunits of the IL-2 receptor, IL-2Rβ and IL-2Rγ, were twice as abundant in CD8+ T cells compared to CD4+ T cells. Reduction of IL-2Rβ abundance by 50% was sufficient to convert CD8+ T cells to a CD4+-like signaling pattern and delay S-phase entry. These results suggest that the larger pool of IL-2Rβ chains found in CD8+ T-cells is required to sustain IL-2 signaling over time and contributes to the quantitatively greater IL-2 proliferative response relative to CD4+ T cells. This cell type–specific difference in IL-2Rβ abundance appears to tune responses, potentially preventing extensive, autoimmune expansion of CD4+ T cells while still enabling sufficient expansion of CD8+ T cells to control viral infections.
Introduction
The common γ-chain cytokine interleukin-2 (IL-2) plays an essential role in the development, expansion, and function of many lymphocyte subsets (1). IL-2 induces signaling by binding to a receptor complex minimally comprised of the IL-2Rβ and IL-2Rγ subunits. Binding induces a conformational change that activates the associated cytoplasmic kinases Janus kinase 1 (JAK1) and JAK3, which in turn phosphorylate three tyrosine residues on the IL-2Rβ chain. Phosphorylated Tyr392 and Tyr510 recruit the transcription factor signal transducer and activator of transcription 5 (STAT5), which is rapidly phosphorylated and alters transcription of IL-2–dependent genes (2). Phosphorylated Tyr338 recruits the adapter protein Shc, which can activate phosphoinosidie 3-kinase (PI3K) signaling and, in some cell types, mitogen-activated protein kinase (MAPK) signaling. Depending on the cell type and physiologic context, these signals combine to trigger proliferation, production of effector molecules, or differentiation into distinct T-cell subsets, such as regulatory (Treg) or memory T cells.
IL-2–driven cell fate changes require hours to days of sustained signaling. Differentiation into Tregs requires up to 3 days of IL-2 exposure (3), and S-phase entry by effector T cells requires 8–12 hours of continuous IL-2 stimulation (4, 5). Despite this, most studies of IL-2 signaling have focused on events immediately following the onset of stimulation. In our previous work in activated CD4+ T cells, we examined IL-2 signaling throughout this critical 8–12 hour period prior to S-phase entry and found that there are two waves of STAT5 phosphorylation (pSTAT5) following stimulation: an initial strong, rapid induction of pSTAT5 that peaks within 15 minutes and decays within an hour and a second wave, which appears between 2 and 12 hours after the onset of stimulation. Abolishing the second wave of pSTAT5 was sufficient to prevent S-phase entry (5). This unique signaling pattern, coupled with the importance of sustained IL-2 signaling, raises the question of how signaling dynamics vary by cell type or cytokine and how this impacts cell fate decisions.
At physiologic doses, IL-2 signals through the ‘high affinity’ (Kd ~10 pM) receptor complex composed of IL-2Rα (CD25), IL-2Rβ (CD122), and IL-2Rγ (CD132) (6). This trimeric architecture enables regulation of T-cell IL-2 responsiveness at multiple levels. Whereas all T cells express IL-2Rγ, resting naïve T cells do not express IL-2Rα and are unresponsive to physiologic doses of IL-2 (7). Upon activation of these cells through the T-cell receptor (TCR), IL-2Rα is induced to very high abundance, at least 10-fold higher than IL-2Rβ or IL-2Rγ (8–10), and cells become sensitive to IL-2. Additionally, on naïve CD4+ T cells, surface abundance of IL-2Rβ is at or below the limit of detection by fluorescence-activated cell sorting (FACS), rendering these cells minimally responsive to IL-2 until at least 24 hours after TCR activation (11). By contrast, naïve CD8+ T cells have a small, but detectable, quantity of IL-2Rβ chains and can respond to high doses of IL-2 through the ‘intermediate affinity’ (Kd~ 1 nM) receptor (6, 12). Upon IL-2 binding, receptor complexes are rapidly internalized (surface t1/2 ~15 minutes), and the majority of IL-2Rβ and IL-2Rγ subunits are degraded (t½~50–70 minutes) while IL-2Rα is recycled to the surface (t1/2 ≫6 hours) (13, 14).
Given the requirement for sustained IL-2 signaling and the short half-life of IL-2–bound receptor complexes (15), the precise number and subcellular localization of individual IL-2 receptor subunits is a potentially important regulatory mechanism. Fifty-fold overexpression of the epidermal growth factor receptor (EGFR) in a neuronal cell line converts a transient pulse of extracellular signal–regulated kinase (ERK) signaling into a sustained signal that triggers cell differentiation (16). A threshold for sustained TCR signaling allows 2–3-fold differences in receptor abundance to translate into binary outcomes (17) and provides a check on hyperresponsive T cells (18). However, in the IL-2R system, the relationship between receptor abundance, dynamics, and cell fate is not known.
Here, we compared IL-2 signaling dynamics in mouse CD4+ and CD8+ T cells. As others have reported (19), we found that IL-2 triggered a quantitatively stronger proliferative response in CD8+ T cells compared to CD4+ T cells. Unlike the biphasic response seen in CD4+ T cells, CD8+ T cells had a very different pattern: a strong and sustained induction of pSTAT5. We compared the abundance of IL-2R subunits and found that CD8+ T cells expressed approximately twice as much IL-2Rβ and IL-2Rγ as CD4+ T cells. By examining the dynamics of other cytokines that, like IL-2, signal through the common γ chain (γc, IL-2Rγc), we identified IL-2Rβ as a limiting receptor component in CD4+ T cells. A 50% reduction of IL-2Rβ abundance in CD8+ T cells converted their signaling dynamics to a CD4+-like phenotype and greatly blunted their proliferative response. These results suggest a model whereby IL-2Rβ abundance across a narrow range can dramatically alter a cell’s IL-2 responsiveness.
Results
CD8 T cells are hyper-proliferative in response to IL-2
Because naïve T cells do not express the ‘high affinity’ IL-2 receptor, we generated CD4+ and CD8+ T-cell blasts from primary murine T cells in order to compare IL-2 signaling in each cell type. Upon stimulation with a physiologic dose of IL-2 (50 u/mL), both resting CD4+ and CD8+ T-cell blasts enter S-phase and proliferate. However, CD8+ T-cell blasts entered S-phase sooner and more synchronously than did CD4+ T-cell blasts (Fig. 1A). Consequently, CD8+ T-cell blasts completed more rounds of cell division in 72 hours under continuous IL-2 stimulation (Fig. 1B). This increased proliferative response of CD8+ vs. CD4+ T cells to IL-2 has been reported previously (19). Furthermore, enhanced CD8+ vs. CD4+ proliferation has also been observed in response to simultaneous TCR and IL-2 stimulation in vitro and in vivo (20, 21).
Figure 1. Proliferative Responses of CD4 and CD8 T Cells to IL-2.
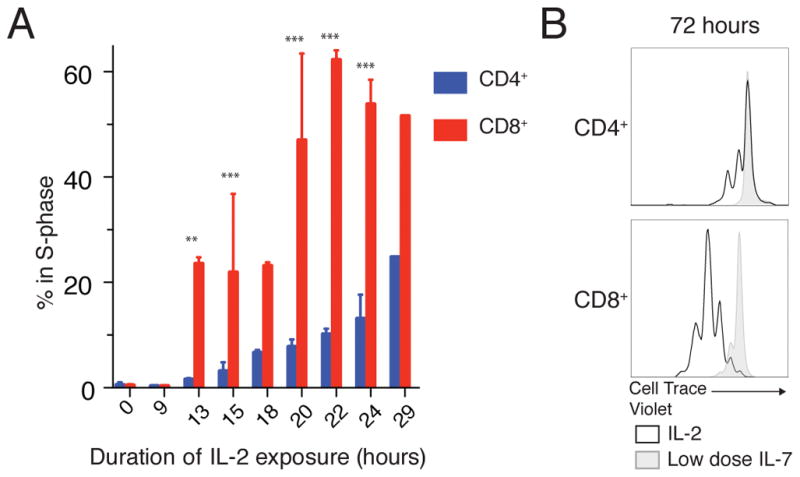
(A) S-phase entry of rested CD4+ and CD8+ T-cell blasts was assessed with a 1-hour pulse of ethynyl deoxyuridine (EdU) after the indicated duration of IL-2 stimulation. (B) Cell proliferation after 72 hours of IL-2 stimulation was assessed by dye dilution (Cell Trace Violet) compared to undivided cells sustained with low dose IL-7. Pooled results from 7 independent experiments plotted as mean +/− SEM (A) or representative of 3 independent experiments (B). Time course (A) compared by repeated measures ANOVA (cell type, time and interaction all p<0.0001) with Bonferroni multiple comparison test (significant p<xxx: ** p<0.01, *** p<0.001 for CD4s vs CD8s at a given time point).
CD4+ and CD8+ T cells have distinct IL-2 signaling dynamics
To understand the signaling basis for this enhanced response in CD8+ T-cell blasts, we first assessed the immediate (t=15 minutes) phosphorylation of STAT5, which is essential for IL-2–driven proliferation (2). In response to a wide titration of IL-2 concentrations, CD4+ and CD8+ T-cell blasts displayed nearly identical dose responses (EC50 0.35 u/mL and 0.28 u/mL respectively, Fig. 2, A and B). At high IL-2 concentrations, the magnitude of the response (mean fluorescence intensity of the responding population) was approximately 25% higher in CD8+ T cells compared to CD4+ T cells (fig. S1, A and B). Although it was tempting to attribute the enhanced proliferation of CD8+ T cells to this increase in maximal pSTAT5, the requirement for 8–12 hours of continuous IL-2 exposure led us to suspect a more complicated mechanism (4, 5).
Figure 2. Distinct IL-2 Signaling Dynamics in CD4 and CD8 T Cells.

(A) Rested CD4+ and CD8+ T cell blasts were stimulated with a 4-fold titration of IL-2 from 1000 to 0.001 units/mL, and phosphorylated STAT5 (pSTAT5) was assessed by phosphoflow cytometry. (B) Fraction of responding (pSTAT5+) cells at each concentration of IL-2. EC50 CD4 0.35 u/mL [0.29–0.42 95% confidence interval (CI)]; EC50 CD8 0.28 u/mL (0.24–0.32 95% CI). (C) Time course of pSTAT5 accumulation in CD4+ and CD8+ T cells following IL-2 stimulation. (D) quantification of pSTAT5 over time (cell type, time and interaction all p<0.0001). E) MYC protein accumulation in CD4+ and CD8+ T cells. F) Quantification of Myc over first two hours of IL-2 stimulation (cell type p<0.0001, time p=0.045, interaction n.s.). Representative histograms from 3 (A) 11 (C), or 4 (E) independent experiments. Plotted as mean +/− SEM of 3 (B) or 4 (F) independent experiments or mean with 95% confidence interval from 11 independent experiments (D). Time courses compared by repeated measures ANOVA with Bonferroni multiple comparison test (significant p<xxx: ** p<0.01, *** p<0.001 for CD4s vs CD8s at a given time point).
In CD4+ T cells, IL-2 triggers a biphasic STAT5 phosphorylation response, with an initial peak of phosphorylation 15–30 minutes after onset of IL-2 stimulation and a second peak at 2–12 hours of stimulation (5). This prompted us to ask whether CD8+ T cells displayed a similar pattern of STAT5 phosphorylation (pSTAT5). To our surprise, CD8+ and CD4+ T cells exhibited very different signaling dynamics (Fig. 2, C and D). Unlike CD4+ T cells, there was a single, sustained peak of pSTAT5 in CD8+ T cells without the characteristic decrease in phosphorylation seen at 1 hour in CD4+ T cells. The fraction of pSTAT5+ cells was significantly higher in CD8+ T cells at 45 minutes, 1 hour, and 2 hours after onset of IL-2 stimulation, but similar before and after those time points. These patterns persisted at a range of physiologic IL-2 concentrations from 1 unit/mL to 250 units/mL (fig. S1, C and D).
To understand whether differences in the first two hours of signaling could contribute to the CD8+ hyper-proliferative phenotype, we examined the protein product of the immediate early gene Myc, induction of which is essential for cell cycle progression. Studies have shown that both CD4+ and CD8+ T cells require a minimum threshold of Myc protein to divide (11, 22). Within the first two hours of IL-2 stimulation, Myc protein increased almost 4-fold over baseline in CD8+ T cells compared to 1.5-fold in CD4+ T cells (Fig. 2, E and F). Because IL-2–driven Myc induction depends on STAT5 (2), this early Myc increase in CD8+ but not CD4+ T cells likely reflects the sustained pSTAT5 seen in CD8+ T cells.
IL-2–dependent proliferation in T-cell blasts also requires activation of the mechanistic target or rapamycin (mTOR) signaling but not ERK-MAPK signaling (5). As a proxy for mTOR activity, we monitored phosphorylated ribosomal protein S6 (pS6) over time in CD4+ and CD8+ T-cell blasts. Whereas CD4+ T cells required ~2 hours before a majority of cells had activated pS6, almost all CD8+ T cells activated S6 immediately after stimulation. The mean fluorescence intensity of pS6 also increased throughout the time course for CD8+ T cells (fig. S2, A and B). Because maximal, sustained pS6 depends on STAT5 activation (23, 24), this delayed, weaker pS6 activation in CD4+ cells likely reflects the early attenuation of pSTAT5.
Sustained signaling requires protein synthesis in CD4+ but not CD8+ T cells
Could these distinct signaling dynamics explain the difference in proliferative responses to IL-2? To address this question, we first needed to understand the mechanistic basis for these patterns. Washout experiments suggested that both the second wave of pSTAT5 in CD4+ and sustained signaling in CD8+ T cells required the continued presence of IL-2, perhaps to initiate new signaling complexes between IL-2 and unoccupied receptors (Fig. 3, A and B). After adding a small molecule inhibitor of JAK3 to terminate new signal generation, pSTAT5 decayed rapidly in both cell types, indicating that STAT5 is dephosphorylated at similar rates in CD4+ and CD8+ T cells (Fig. 3, C and D).
Figure 3. Requirements for Sustained IL-2 Signaling.

(A) Cells were stimulated for 30 minutes and then IL-2 was washed out and phosphorylated STAT5 (pSTAT5) monitored by phosphoflow cytometry. (B) Quantification of washout. (C, D) Cells were stimulated with IL-2, with a pharmacological inhibitor of JAK3 (JAK3i) added at t=15 minutes, and signal decay was monitored (C) and quantified (D) (E, F) Cells were stimulated simultaneously with IL-2 and brefeldin A as pSTAT5 was monitored over time (E) and quantified (F). Cell type, time and interaction all p<0.0001. (G, H) Cells were stimulated simultaneously with IL-2 and cycloheximide as pSTAT5 was monitored over time (G) and quantified (H). In all experiments, cell type, time, and interaction all p<0.0001. Data are representative of 3 independent experiments (A, C, E, G) or plotted as mean +/− SEM of 3 independent experiments (B, D, F, H). Time courses compared by repeated measures ANOVA with Bonferroni multiple comparison test (significant p<xxx: ** p<0.01, *** p<0.001 for CD4s vs CD8s at a given time point).
The washout and inhibitor studies led us to suspect that the attenuation of pSTAT5 in CD4+ T cells was due to a relative loss of active signaling complexes rather than differential phosphatase activity. At a minimum, STAT5 phosphorylation requires the IL-2Rβ and IL-2Rγ chains and the cytoplasmic kinases JAK1 and JAK3. Upon IL-2 binding, the IL-2 receptor complex is rapidly internalized, and IL-2Rβ and IL-2Rγ are degraded (13, 14). This creates a shortage of receptors to generate new pSTAT5, absent trafficking and possibly synthesis of new receptors. Both cell types required vesicular transport to sustain signaling, because co-treatment with IL-2 and the Arf-guanine nucleotide exchange factor (GEF) inhibitor brefeldin A gave a transient pSTAT5 peak that rapidly decayed after 15 minutes (Fig. 3, E and F). To distinguish between vesicular transport of newly synthesized signaling components and a pre-synthesized intracellular pool, we treated cells with both IL-2 and the protein synthesis inhibitor cycloheximide. Strikingly, pSTAT5 was completely unaffected by cycloheximide in CD8+ T cells. By contrast, pSTAT5 decayed in CD4+ T cells after the initial peak, indicating that protein synthesis was required for the second wave of signaling in CD4+ T cells (Fig. 3, G and H). These results suggested that CD8+ T cells had sufficient quantities of extant signaling components in an intracellular pool to sustain STAT5 phosphorylation, whereas CD4+ T cells needed to re-synthesize a limiting component or components and transport it to the cell surface for the second wave of STAT5 phosphorylation to occur.
IL-2Rβ and IL-2Rγ are twice as abundant in CD8+ T cells as in CD4+ T cells
Given the known short half-life of signaling-competent receptors and the distinct requirement for new protein synthesis, we suspected that differences in initial receptor abundance could underlie the distinct signaling dynamics in CD4+ and CD8+ T cells. Surface staining of IL-2 receptor chains revealed that CD8+ T cells have approximately twice as much IL-2Rβ and IL-2Rγ as CD4+ T cells, with no significant difference in IL-2Rα abundance (Fig. 4, A and B). To probe the intracellular pool specifically, we bound surface receptors with unlabeled antibodies prior to fixation and membrane permeabilization and then stained for each receptor with the same fluorophore-antibody conjugate as used in the surface-only experiments. Using this method, we observed that intracellular IL-2Rα staining was much weaker than surface staining, suggesting a predominantly surface localization, and was not significantly different between CD4+ and CD8+ T cells (Fig. 4, C and D). In contrast to IL-2Rα, there was strong intracellular staining for IL-2Rβ and IL-2Rγ, which indicated substantial intracellular pools, and these pools were significantly larger in CD8+ T cells, by approximately 2-fold and 1.5-fold respectively (Figure 4, C and D). For IL-2Rβ, this difference appeared to be encoded at the mRNA level. Differences in transcript abundance did not correlate with protein for either IL-2Rα or IL-2Rγ, which suggests that there may be important differences in posttranscriptional regulation of these subunits between CD4+ and CD8+ T cells (Fig. 4E).
Figure 4. IL-2R Subunit Abundance.

(A) Representative histograms of IL-2Rα, IL-2Rβ and IL-2Rγ staining on the surface of CD4+ and CD8+ T cells. (B) Quantification of receptor abundance relative to mean fluorescence intensity (MFI) on CD4+ T cells in each experiment. (C) Representative histograms of IL-2Rα, IL-2Rβ and IL-2Rγ intracellular staining in CD4+ and CD8+ T cells. (D) Quantification of intracellular receptor abundance relative to MFI on CD4+ T cells in each experiment. (E) Quantification of il2ra, il2rb, and il2rg transcripts in resting CD4+ and CD8+ T cell blasts by qPCR. Data are representative of 7 (A) and 3 (C) independent experiments. Pooled data from 7 (B) and 3 (D) independent experiments, plotted +/− SEM. Paired T-Test: n.s. not significant; * p<0.05, **p<0.01, ***p<0.001. (E) mRNA from 3 biological replicates plotted with 95% confidence interval.
IL-2Rβ–dependent IL-15 signaling is biphasic in CD4+ but not CD8+ T cells
The increased IL-2Rβ and IL-2Rγ on CD8+ T cells suggested that differences in abundance of either or both receptor chains could explain the distinct signaling dynamics observed in response to IL-2. Within the common γ-chain family of cytokines (γc), two cytokines (IL-2 and IL-15) utilize the IL-2Rβ chain, with other members of this family signaling through a unique chain that pairs with IL-2Rγ (Fig. 5A). To test the respective roles of IL-2Rβ and IL-2Rγ, we compared signaling dynamics in response to IL-7 (which signals through IL-7Rα and IL-2Rγ) and IL-15 (which signals through IL-2Rβ and IL-2Rγ). If IL-2Rγ were limiting, IL-2, IL-7 and IL-15 would all exhibit biphasic signaling, whereas limiting amounts of IL-2Rβ would only impact IL-2 and IL-15. Following stimulation of CD4+ T cells with IL-7, pSTAT5 was strong and sustained, with no loss of signal until 5 hours after IL-2 addition (Fig. 5, B and C). In striking contrast, stimulation with IL-15 produced a biphasic response, with a significant loss of signal at 1 hour and subsequent partial recovery (Fig. 5, B and C). When CD8+ T cells were stimulated with either cytokine, pSTAT5 was strong and sustained (Fig. 5, D and E). Taken together, these results are consistent with a limiting pool of IL-2Rβ in CD4+ T cells underlying the rapid decline of pSTAT5 after stimulation with IL-2 or IL-15.
Figure 5. Comparative dynamics of IL-7 and IL-15.

(A) Schematic of IL-2, IL-7 and IL-15 receptor components. Note IL-15 is typically presented in trans from IL-15Rα on a neighboring cell. (B) Representative time course histograms of phosphorylated STAT5 (pSTAT5) in CD4+ T cells stimulated with IL7 or IL-15. (C) Fraction of pSTAT5+ cells in response to IL-7 and IL-15. Cytokine, time and interaction p<0.0001. (D) Representative time course histograms of pSTAT5 in CD8+ T cells stimulated with IL7 or IL-15. (E) Quantification of pSTAT5 in response to IL-7 or IL-15. Cytokine n.s., time p<0.0001, interaction p=0.0004. Data is representative of (B, D) or pooled from (C, E) 3 independent experiments. Plotted +/− SEM in (C, E). Time courses compared by repeated measures ANOVA with Bonferroni multiple comparison test (significant p<xxx: ** p<0.01, *** p<0.001 for IL-7 vs IL-15 at a given time point).
Reduction of IL-2Rβ abundance converts signaling in CD8+ T cells to a CD4+-like pattern
These results suggested a model in which the dynamics of STAT5 phosphorylation can be tuned by IL-2Rβ abundance. A prediction of this model is that reducing IL-2Rβ in CD8+ T cells would result in a CD4+ T cell–like signaling pattern. Because il2rb+/− T cells have roughly similar IL-2Rβ abundance to wild-type T cells (24), we transduced primary CD8+ T-cell blasts with lentiviruses expressing green fluorescent protein (GFP) and one of a panel of 4 small hairpin RNAs (shRNAs) targeting il2rb. These hairpins gave a range of knockdown efficiencies from 22–77% based on surface staining (Fig. 6A and fig. S3A). Reduction of IL-2Rβ in CD8+ T cells by ~50% with IL2RB shRNA #2 or #3 resulted in a biphasic CD4+-like signaling pattern, with a loss of pSTAT5 at 1 hour, followed by rapid recovery (Fig. 6, B and C and fig. S3B). Scrambled shRNA or a marginal reduction with IL2RB shRNA #4 had a minimal effect on signaling dynamics (Fig. 6, B and C and fig. S3B). Knockdown to below 25% of wild-type abundance with IL2RB shRNA #1 markedly impaired signaling, producing a very weak pSTAT5 response at all time points (Fig. 6C and fig. S3B). Two-fold reduction in IL-2Rβ with shRNA #2 had only a small effect on the maximal pSTAT5 response observed at 15 minutes (fig. S3C), which was comparable to the difference observed between CD4+ and CD8+ T cells (Fig. 2D).
Figure 6. Knockdown of IL2RB in CD8 T Cell blasts.

(A) Effect of expressing 4 different shRNAs targeting IL2RB relative to scrambled shRNA on surface IL-2Rβ abundance in CD8+ T cells, as assessed by FACS. (B) pSTAT5 over time in IL-2–treated CD8+ T cells expressing the scrambled IL2RB shRNA or IL2RB shRNA #2. (C) Fraction of pSTAT5+ cells over time (Repeated Measures ANOVA shRNA p<0.0001, time p<0.0001, interaction p=0.0097. Bonferroni Post test of Scramble shRNA vs. shRNA#1: 0.25 to 3 hour p<0.001, vs shRNA#2: 0.75 and 1 hour p<0.001, vs shRNA#3 0.75 hour p<0.05, 1 hour p<0.001, all other comparisons to scramble n.s.). (D) S-phase entry, assessed by EdU incorporation, after 13 hours of IL-2 stimulation in briefly rested CD8+ T cell blasts expressing the indicated shRNAs (GFP+) or untransduced (GFP-) *** p<0.001 by One-way ANOVA with Bonferroni Multiple comparisons test relative to scramble shRNA. Data are representative of (B) or pooled from 4 (A) or 3 (B, C, D) independent experiments. Plotted as mean +/− SEM.
We next asked whether this reduction in IL-2Rβ abundance contributed to the weaker proliferative response of CD4+ T cells. Sustaining primary CD8+ T cells in culture was challenging in the context of impaired IL-2 signaling. After culture for 3 days in the presence of IL-2, the frequency of cells expressing IL2RB shRNAs #2 and #3 had decreased by 6 and 3-fold, respectively, but there was no substantial change in the frequency of scrambled shRNA–expressing cells (fig. S3D). This result could be explained by defects in either proliferative or survival signaling, so we next assessed S-phase entry in cells that were activated for 3 days, rested overnight, and then stimulated with IL-2 for 13 hours. Under these conditions, a significantly smaller fraction of IL2RB shRNA–expressing cells entered S-phase compared to untransduced cells in the same sample (Fig. 6D). Although additional mechanisms likely contribute, our results indicate that even a modest two-fold difference in IL-2Rβ abundance is sufficient to confer a proliferative advantage CD8+ T cells relative to CD4+ T cells in response to IL-2 stimulation.
Tregs have increased IL-2Rβ abundance and CD8+-like signaling dynamics
To test whether IL-2Rβ abundance was sufficient to tune signaling dynamics, we attempted to overexpress IL-2Rβ in CD4+ T-cell blasts. Unfortunately, this proved technically challenging, so we instead assessed IL-2Rβ abundance in other CD4+ T-cell subsets to see if any had a CD8+-like IL-2Rβ pool. Intracellular IL-2Rβ was approximately two-fold more abundant in freshly isolated splenic Tregs than in effector CD4+ T-cell blasts (Fig. 7, A and B). This intracellular pool was similar to that found in CD8+ T-cell blasts, which our model predicts is sufficient to support a strong and sustained IL-2 pSTAT5 response. Upon IL-2 stimulation, we observed a rapid increase in pSTAT5 that was sustained throughout a 4-hour time course (Fig. 7, C and D). Although only correlative, these results provide further evidence of a crucial role for IL-2Rβ abundance in tuning the dynamics of the IL-2 pSTAT5 response.
Figure 7. Treg IL-2Rβ Abundance and IL-2 signaling dynamics.
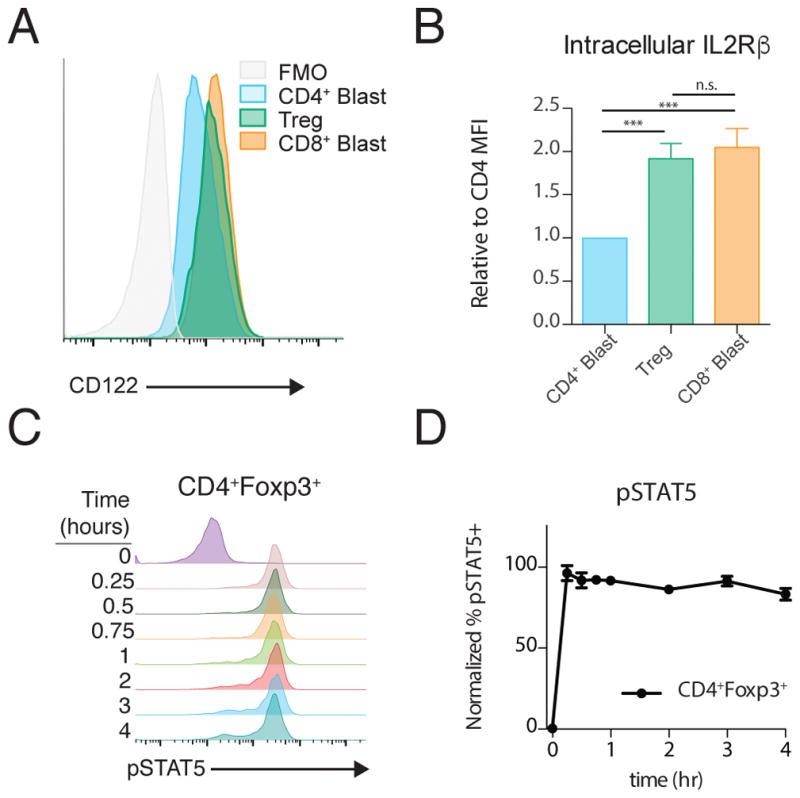
(A) Representative Intracellular staining for IL-2Rβ in CD4+ T cell blasts, CD4+Foxp3+ Tregs and CD8+ T cell blasts. (B) Quantification of IL-2Rβ abundance relative to CD4+ T cell blasts. (C) Representative histograms of STAT5 phosphorylation over time following IL-2 stimulation of CD4+Foxp3+ Tregs in freshly isolated splenocytes, quantified in (D). All graphs plotted as mean +/− SEM. In (B), Tregs are from 4 separate mice and blasts are from 4 separate preparations. *** p<0.001 by one-way ANOVA with Bonferroni multiple comparison test. In (D), data are pooled from 5 mice.
Discussion
Research in multiple systems has recognized the important role that signaling dynamics play in quantitatively altering cell responses (25), as in the case of epidermal growth factor (EGF) and tumor necrosis factor α (TNFα) signaling (26, 27). In these previous studies, the authors altered signaling dynamics by modulating the stimulus. Here, we observe different signaling dynamics in two closely related cell types in response to the same stimulus. A 50% lower abundance of one component of the IL-2 receptor complex, IL-2Rβ, appears to explain the altered dynamics and contribute to the decreased proliferative response of CD4+ T cells to IL-2. In the context of receptor assembly, this is the logical step at which to specifically regulate the pro-proliferative and growth signals of IL-2 and IL-15 without impacting other γc-cytokines. Although there is debate as to whether IL-2 first binds a preformed IL-2Rα/IL-2Rβ heterodimer (28, 29) or first to IL-2Rα and then IL-2Rβ (6, 30) before IL-2Rγ is recruited, in either model a shortage of IL-2Rβ would disfavor the formation of the final quaternary IL-2/IL-2Rα/IL-2Rβ/IL-2Rγ complex required for signaling (29). On CD4+ T-cell blasts, there appears to be sufficient IL-2Rβ to maximally phosphorylate STAT5 at early time points after IL-2 stimulation. However, our data suggest that between 15 minutes and 1 hour, the number of competent signaling complexes falls below the threshold needed to counteract phosphatase activity and maintain STAT5 phosphorylation. Synthesis of new IL-2Rβ molecules enables the second wave of STAT5 phosphorylation in CD4+ T cells. By contrast, the total pool of IL-2Rβ on CD8+ T cells is large enough to maintain surface receptor abundance above this threshold even in the absence of new protein synthesis and thus sustain STAT5 phosphorylation continuously.
Although CD4+ T cells do ultimately have a second, sustained wave of STAT5 phosphorylation, the early loss of pSTAT5 occurs at a crucial time for induction of immediate early genes. In fibroblasts, small differences in the duration of ERK activity (90 vs. 120 minutes) profoundly impact the induction of Myc and the time to cell cycle progression (31). Here, we observe higher Myc abundance in CD8+ T cells after just two hours. This likely has a cascading effect on many genes required for cell cycle entry and contributes to the hyper-proliferative response.
Why are these two cell types programmed to respond to IL-2 with distinct signaling dynamics? We speculate that this may serve as an additional check against the unintentional or mislocalized expansion of CD4+ T cells in response to IL-2. Because IL-2 has a very short effective range in vivo (<20 μm) (32, 33), only a T cell that maintains close proximity with an IL-2–producing source (such as a peptide-APC-bound T cell) would receive sufficient exposure for a first and second wave of STAT5 phosphorylation. This would prevent the expansion of a CD4+ T cell that once saw antigen (perhaps self-antigen) and by chance circulated through an area of active infection. CD4+ T cells respond to infections by recruiting and activating other immune cells, many of which are non-specific and have the potential to cause harmful inflammation (34). This mechanism both necessitates tight control over CD4+ expansion to prevent autoimmunity and minimizes the need for massive expansion because other cells will amplify the response. By contrast, CD8+ T cells primarily respond by directly killing virally infected cells with lytic granules (35). Unlike the non-specific effectors downstream of CD4+ T cells, CD8+ T cell–mediated killing requires recognition of the correct peptide-bound major histocompatibility complex (MHC) on target cells, which mitigates the need for tightly regulated IL-2 responses. In fact, this direct killing mechanism necessitates the opposite: a rapid and large expansion of CD8+ T cells is required because there is little amplification by other effector cells.
In order to focus on the consequences of IL-2 signaling specifically, we studied pre-activated T-cell blasts that were no longer receiving TCR stimulation. Recent work from our lab suggests that IL-2Rβ abundance correlates with responses in the context of simultaneous TCR and IL-2 signals (11). T cells integrate both of these signals at the level of Myc when deciding whether or how much to proliferate (11, 22). However, IL-2 augments TCR-driven Myc induction only in CD8+ T cells (11). This enables CD8+, but not CD4+, T cells to proliferate in response to suboptimal TCR stimulation. Even 24 hours after TCR activation, CD4+ T cells have a very minimal STAT5 phosphorylation response to IL-2 (similar to 75% knockdown of IL-2Rβ in Fig. 5c). These activated CD4+ T cells express very little IL-2Rβ and may represent a physiologic example of the IL-2Rβ tuning described here.
Our data and prior reports show that there is significant heterogeneity in IL-2Rβ abundance, both between and within cell populations, which has an important impact on signaling and cell fate decisions. At the low end, naïve CD4+ T cells have little or no IL-2Rβ, whereas at the high end natural killer (NK) cells have enormous quantities (36). Here, we showed that for Tregs, and CD4+ and CD8+ effectors, mean IL-2Rβ abundance at the population level tunes IL-2 signaling dynamics. Recently, Jansen et al. examined IL-2 signaling dynamics in human NK cells and found, as our model would predict, that these also had a sustained response over a 24 hr period (36). Future work should explore this relationship in other cell types and, when reagents permit, correlate IL-2Rβ abundance to the STAT5 phosphorylation response within specific populations to further understand this heterogeneity.
Cell fate decisions in response to cytokines and other stimuli require sustained signaling. Our results highlight how a focus on immediate signaling can be deceptive when attempting to understand events that occur hours later. Here, a mere 2-fold difference in receptor abundance yields distinct dynamics that contribute to quantitatively different proliferative outcomes.
Material and Methods
Mice
C57BL/6J mice (Jackson) used in this study were housed in the specific pathogen–free facilities at the University of California, San Francisco, and were treated according to protocols approved by the Institutional Animal Care and Use Committee in accordance with US National Institutes of Health guidelines. Both male and female mice, all aged 6–12 weeks, were used as a source of primary T cells.
Reagents
The JAK3-selective inhibitor (JAK3i) was synthesized as previously described (5) and used at a concentration of 500 nM. Recombinant human IL-2 (rhIL-2) was from the NIH AIDS Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, NIH: Maurice Gately (Hoffmann-La Roche). Recombinant murine IL-7 and IL-15 were purchased from Peprotech.
Primary T-Cell Culture
T cells were purified from single-cell suspensions of spleen and lymph nodes from male and female mice aged 6–12 weeks by negative selection with biotinylated antibodies recognizing CD8 or CD4, CD19, B220, CD11b, CD11c, DX5, Ter119, and CD24 (UCSF Monoclonal Antibody Core) and magnetic anti-Biotin beads (MACSi Beads, Miltenyi Biotec). For IL-2 stimulation, purified T cells were pre-activated on 6-well plates coated with antibodies specific for CD3 (2C11) and CD28 (37.51) for 72 hours, removed, and cultured with rhIL-2 (100 u/mL, Roche) for 36 hours, and then cultured without rhIL-2 for the 36 hours prior to all experiments.
IL-2 stimulation of T cells
After resting for 36 hours without IL-2, T cells were washed in fresh media, live/dead stained as appropriate (see Flow Cytometry), and resuspended in warm, fresh RPMI complete media at 1–5 × 106/mL. Cells were then stimulated by adding rhIL-2 (20× stock in RPMI complete, 50 units/mL final concentration) and assayed as detailed below. To washout IL-2 after stimulation, cells were washed 3 times with 1 mL of warmed media and resuspended at the original concentration.
T-Cell Proliferation
Pre-activated T cells were cultured in 24-well plates for 10–30 hours with IL-2 and then pulsed with 10 μM 5-ethynyl-2’-deoxyuridine (EdU) for 1 hour and assayed per the manufacturer’s procedure (Click-IT Plus EdU, Life Technologies). Alternatively, cells were loaded with Cell Trace Violet (Life Technologies), plated into 96-well plates, and stimulated with IL-2 for 72 hours. To maintain viability in the undivided population, control (non-IL-2–stimulated) cells were treated with low dose IL-7 (0.1 ng/mL). Division was then analyzed by quantifying EdU incorporation by flow cytometry.
Flow Cytometry
Cells were live/dead-stained using the Live/Dead Fixable Near IR Dead Cell Stain Kit or Live/Dead Fixable Violet Dead Cell Stain Kit (Life Technologies). For surface stains, cells were stained for 30 minutes on ice with the indicated antibodies. Information on antibody clones, sources, and working dilutions are listed in Table S1. For intracellular stains, samples were fixed at the indicated time after stimulation in 2% paraformaldehyde, surface stained with CD8a-BUV737 and CD4-BUV395, fixed again, then permeabilized with ice cold 90% methanol at −20 °C overnight. Samples were then barcoded using Pacific Orange-NHS ester (0.33 or 5 μg/mL), Pacific Blue-NHS Ester (0.67 or 10 μg/mL), and AlexaFluor (AF) 488-NHS Ester (0.26 or 2 μg/mL) (Life Technologies), as previously described(5). Intracellular antigens were then stained for 30 minutes at 23 °C with antibodies indicated in Table S1. Samples were acquired on a BD LSR Fortessa SORP and analyzed in FlowJo (Tree Star).
Quantitative PCR (qPCR)
RNA was isolated from 1–2 × 106 T-cell blasts per condition using RNAeasy kit (Qiagen) and cDNA was synthesized using qScript (Quanta Biosciences). mRNA was detected by Primetime (IDT) or Taqman (Life Technologies) predesigned qPCR assays. Primer & probe sequences can be found in Table S2. Data are from 3 replicates collected on a QuantStudio 12k (Life Technologies), plotted with 95% confidence intervals as calculated by Quantstudio (Life Technologies).
IL2RB Knockdown
Four shRNAs targeting IL2RB and one scrambled shRNA were purchased (pLKO1.0, The RNAi Consortium / Sigma Aldrich). Sequences of the shRNAs are provided in Table S3. The puromycin selection cassette was replaced with enhanced GFP (eGFP) by restriction enzyme cloning. Lentivirus was produced in HEK293T cells in 10 cm dishes transfected with pCMV-dr8.91 (9 μg), pMD2.G (0.9 μg), and pLKO (9 μg) in the presence of Viral Boost (Alstem). CD8+ T cells were isolated as described above and incubated for 1 day on TCR-coated plates prior to spin transduction (1 hr, 2200 RPM, RT) with viral supernatant in the presence of IL-2 (100 u/mL). Cells were left on TCR plates through day 3 and treated as per culture conditions described above. Assays were conducted as described above, except anti-GFP-AF488 was used to detect GFP in the EDU incorporation assay for S-phase entry.
Statistical Analysis
Data were analyzed in Prism (Graphpad). Three types of statistical analysis were used: 1) Repeated Measures ANOVA with Bonferroni Multiple Comparisons post test was used to compare signaling time courses between cell types, stimulatory cytokines, or shRNA transformations. 2) Paired T-test was used to compare IL-2R subunit levels between CD4 and CD8 T cells (paired samples were stained & collected together on the same day in the same tube). 3) One-way ANOVA with Bonferroni Multiple Comparisons post test was used to compare IL-2Rb levels between different shRNA constructs and to compare S-phase entry between different shRNA constructs.
Supplementary Material
Figure S1. STAT5 phosphorylation in IL-2–stimulated CD4+ and CD8+ T cells
Figure S2. PI3K-mTOR Signaling in IL-2–stimulated CD4+ and CD8+ T cells
Figure S3. IL2RB shRNAs
Table S1. FACS Antibodies
Table S2. qPCR Assays
Table S3. IL2RB shRNAs
Acknowledgments
We thank J. Carelli, B. Au-Yeung and J. Zikherman for helpful discussions and feedback on the manuscript, K. Taylor for statistical advice, and A. Roque for animal husbandry.
Funding: This work was supported by grants from NIH NIAID to GAS (F30AI120517) and to AW (R37AI114575), Rosalind Russell and Ephraim P. Engleman Rheumatology Research Center to AW, and the Howard Hughes Medical Institute to JT & AW.
Footnotes
Author contributions: GAS, JT and AW designed the project. GAS performed experiments and analyzed data. GAS, JT and AW wrote the manuscript.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: Plasmids are available on request.
References and Notes
- 1.Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by γc family cytokines. Nature Reviews Immunology. 2009;9:480–490. doi: 10.1038/nri2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin J-X, Li P, Liu D, Jin HT, He J, Ata Ur Rasheed M, Rochman Y, Wang L, Cui K, Liu C, Kelsall BL, Ahmed R, Leonard WJ. Critical Role of STAT5 transcription factor tetramerization for cytokine responses and normal immune function. Immunity. 2012;36:586–599. doi: 10.1016/j.immuni.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weist BM, Kurd N, Boussier J, Chan SW, Robey EA. Thymic regulatory T cell niche size is dictated by limiting IL-2 from antigen-bearing dendritic cells and feedback competition. Nature Immunol. 2015;16:635–641. doi: 10.1038/ni.3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cantrell DA, Smith KA. The interleukin-2 T-cell system: a new cell growth model. Science. 1984;224:1312–1316. doi: 10.1126/science.6427923. [DOI] [PubMed] [Google Scholar]
- 5.Smith GA, Uchida K, Weiss A, Taunton J. Essential biphasic role for JAK3 catalytic activity in IL-2 receptor signaling. Nat Chem Bio. 2016;12:373–379. doi: 10.1038/nchembio.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X, Rickert M, Garcia KC. Structure of the quaternary complex of interleukin-2 with its alpha, beta, and gammac receptors. Science. 2005;310:1159–1163. doi: 10.1126/science.1117893. [DOI] [PubMed] [Google Scholar]
- 7.Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nature Reviews Immunology. 2012;12:180–190. doi: 10.1038/nri3156. [DOI] [PubMed] [Google Scholar]
- 8.Rao BM, Rao Balaji M, Driver Ian, Driver I, Douglas A, Lauffenburger A, Lauffenburger DA, Wittrup KD, Dane Wittrup K. High-affinity CD25-binding IL-2 mutants potently stimulate persistent T cell growth. Biochemistry. 2005;44:10696–10701. doi: 10.1021/bi050436x. [DOI] [PubMed] [Google Scholar]
- 9.Damjanovich S, Bene L, Matkó J, Alileche A, Goldman CK, Sharrow S, Waldmann TA. Preassembly of interleukin 2 (IL-2) receptor subunits on resting Kit 225 K6 T cells and their modulation by IL-2, IL-7, and IL-15: a fluorescence resonance energy transfer study. Proc Natl Acad Sci USA. 1997;94:13134–13139. doi: 10.1073/pnas.94.24.13134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pillet A-H, Lavergne V, Pasquier V, Gesbert F, Thèze J, Rose T. IL-2 Induces Conformational Changes in Its Preassembled Receptor Core, Which Then Migrates in Lipid Raft and Binds to the Cytoskeleton Meshwork. J Mol Biol. 2010;403:671–692. doi: 10.1016/j.jmb.2010.08.056. [DOI] [PubMed] [Google Scholar]
- 11.Au-Yeung BB, Smith GA, Mueller JL, Heyn CS, Jaszczak RG, Weiss A, Zikherman J. IL-2 Modulates the TCR Signaling Threshold for CD8 but Not CD4 T Cell Proliferation on a Single-Cell Level. J Immunol. 2017;198:2445–2456. doi: 10.4049/jimmunol.1601453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cho JH, Kim HO, Kim KS, Yang DH, Surh CD, Sprent J. Unique features of naive CD8+ T cell activation by IL-2. J Immunol. 2013;191:5559–5573. doi: 10.4049/jimmunol.1302293. [DOI] [PubMed] [Google Scholar]
- 13.Hémar A, Subtil A, Lieb M, Morelon E, Hellio R, Dautry-Varsat A. Endocytosis of interleukin 2 receptors in human T lymphocytes: distinct intracellular localization and fate of the receptor alpha, beta, and gamma chains. J Cell Biol. 1995;129:55–64. doi: 10.1083/jcb.129.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lamaze C, Dujeancourt A, Baba T, Lo CG, Benmerah A, Dautry-Varsat A. Interleukin 2 Receptors and Detergent-Resistant Membrane Domains Define a Clathrin-Independent Endocytic Pathway. Mol Cell. 2001;7:661–671. doi: 10.1016/s1097-2765(01)00212-x. [DOI] [PubMed] [Google Scholar]
- 15.Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, Ahmed R. Prolonged Interleukin-2Ra Expression on Virus-Specific CD8. Immunity. 2010;32:91–103. doi: 10.1016/j.immuni.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 16.Traverse S, Seedorf K, Paterson H, Marshall CJ, Cohen P, Ullrich A. EGF triggers neuronal differentiation of PC12 cells that overexpress the EGF receptor. Current Biology. 1994;4:694–701. doi: 10.1016/s0960-9822(00)00154-8. [DOI] [PubMed] [Google Scholar]
- 17.Viola A, Lanzavecchia A. T cell activation determined by T cell receptor number and tunable thresholds. Science. 1996;273:104–106. doi: 10.1126/science.273.5271.104. [DOI] [PubMed] [Google Scholar]
- 18.Gallegos AM, Xiong H, Leiner IM, Sušac B, Glickman MS, Pamer EG, van Heijst JWJ. Control of T cell antigen reactivity via programmed TCR downregulation. Nature Immunol. 2016;17:379–386. doi: 10.1038/ni.3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gesbert F, Moreau JL, Thèze J. IL-2 responsiveness of CD4 and CD8 lymphocytes: further investigations with human IL-2Rbeta transgenic mice. Int Immunol. 2005;17:1093–1102. doi: 10.1093/intimm/dxh289. [DOI] [PubMed] [Google Scholar]
- 20.Foulds KE, Zenewicz LA, Shedlock DJ, Jiang J, Troy AE, Shen H. Cutting Edge: CD4 and CD8 T Cells Are Intrinsically Different in Their Proliferative Responses. J Immunol. 2002;168:1528–1532. doi: 10.4049/jimmunol.168.4.1528. [DOI] [PubMed] [Google Scholar]
- 21.de Boer RJ, Homann D, Perelson AS. Different dynamics of CD4+ and CD8+ T cell responses during and after acute lymphocytic choriomeningitis virus infection. J Immunol. 2003;171:3928–3935. doi: 10.4049/jimmunol.171.8.3928. [DOI] [PubMed] [Google Scholar]
- 22.Heinzel S, Giang TB, Binh Giang T, Kan A, Marchingo JM, Lye BK, Corcoran LM, Hodgkin PD. A Myc-dependent division timer complements a cell-death timer to regulate T cell and B cell responses. Nature Immunol. 2017;18:96–103. doi: 10.1038/ni.3598. [DOI] [PubMed] [Google Scholar]
- 23.Lockyer HM, Tran E, Nelson BH. STAT5 is essential for Akt/p70S6 kinase activity during IL-2-induced lymphocyte proliferation. J Immunol. 2007;179:5301–5308. doi: 10.4049/jimmunol.179.8.5301. [DOI] [PubMed] [Google Scholar]
- 24.Yu A, Zhu L, Altman NH, Malek TR. A Low Interleukin-2 Receptor Signaling Threshold Supports the Development and Homeostasis of T Regulatory Cells. Immunity. 2009;30:204–217. doi: 10.1016/j.immuni.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Purvis JE, Lahav G. Encoding and Decoding Cellular Information through Signaling Dynamics. Cell. 2013;152:945–956. doi: 10.1016/j.cell.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Albeck JG, Mills GB, Brugge JS. Frequency-Modulated Pulses of ERK Activity Transmit Quantitative Proliferation Signals. Mol Cell. 2013;49:249–261. doi: 10.1016/j.molcel.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kellogg RA, Tian C, Lipniacki T, Quake SR, Tay S, Davis R. Digital signaling decouples activation probability and population heterogeneity. eLife. 2015;4:e08931. doi: 10.7554/eLife.08931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu Z, Johnson KW, Choi Y, Ciardelli TL. Ligand binding analysis of soluble interleukin-2 receptor complexes by surface plasmon resonance. J Biol Chem. 1995;270:16045–16051. doi: 10.1074/jbc.270.27.16045. [DOI] [PubMed] [Google Scholar]
- 29.Liparoto Stefano F, Myszka David G, Wu Zining, Goldstein Byron, Thomas A, Laue M, Ciardelli Thomas L. Analysis of the Role of the Interleukin-2 Receptor γ Chain in Ligand Binding†. Biochemistry. 2002;41:2543–2551. doi: 10.1021/bi011692m. [DOI] [PubMed] [Google Scholar]
- 30.Stauber DJ. Crystal structure of the IL-2 signaling complex: Paradigm for a heterotrimeric cytokine receptor. Proc Natl Acad Sci USA. 2006;103:2788–2793. doi: 10.1073/pnas.0511161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murphy LO, MacKeigan JP, Blenis J. A Network of Immediate Early Gene Products Propagates Subtle Differences in Mitogen-Activated Protein Kinase Signal Amplitude and Duration. Molecular and cellular biology. 2004;24:144–153. doi: 10.1128/MCB.24.1.144-153.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Busse D, de la Rosa M, Hobiger K, Thurley K, Flossdorf M, Scheffold A, Höfer T. Competing feedback loops shape IL-2 signaling between helper and regulatory T lymphocytes in cellular microenvironments. Proc Natl Acad Sci USA. 2010;107:3058–3063. doi: 10.1073/pnas.0812851107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thurley K, Gerecht D, Friedmann E, Höfer T. Three-Dimensional Gradients of Cytokine Signaling between T Cells. PLoS Comput Biol. 2015;11:e1004206. doi: 10.1371/journal.pcbi.1004206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood. 2008;112:1557–1569. doi: 10.1182/blood-2008-05-078154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barry M, Bleackley RC. Cytotoxic T lymphocytes: all roads lead to death. Nature Reviews Immunology. 2002;2:401–409. doi: 10.1038/nri819. [DOI] [PubMed] [Google Scholar]
- 36.Jensen H, Potempa M, Gotthardt D, Lanier LL. Cutting Edge: IL-2–Induced Expression of the Amino Acid Transporters SLC1A5 and CD98 Is a Prerequisite for NKG2D-Mediated Activation of Human NK Cells. J Immunol. 2017;199:ji1700497–1972. doi: 10.4049/jimmunol.1700497. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. STAT5 phosphorylation in IL-2–stimulated CD4+ and CD8+ T cells
Figure S2. PI3K-mTOR Signaling in IL-2–stimulated CD4+ and CD8+ T cells
Figure S3. IL2RB shRNAs
Table S1. FACS Antibodies
Table S2. qPCR Assays
Table S3. IL2RB shRNAs