

SUMMARY
CD4+ T cells are tightly regulated by microbiota in the intestine, but whether intestinal T cells interface with host-derived metabolites is less clear. Here, we show that CD4+ T effector (Teff) cells upregulated the xenobiotic transporter, Mdr1, in the ileum to maintain homeostasis in the presence of bile acids. Whereas wild-type Teff cells upregulated Mdr1 in the ileum, those lacking Mdr1 displayed mucosal dysfunction and induced Crohn’s disease-like ileitis following transfer into Rag1−/− hosts. Mdr1 mitigated oxidative stress and enforced homeostasis in Teff cells exposed to conjugated bile acids (CBAs), a class of liver-derived emulsifying agents that actively circulate through the ileal mucosa. Blocking ileal CBA reabsorption in transferred Rag1−/− mice restored Mdr1-deficient Teff cell homeostasis and attenuated ileitis. Further, a subset of ileal Crohn’s disease patients displayed MDR1 loss-of-function. Together, these results suggest that coordinated interaction between mucosal Teff cells and CBAs in the ileum regulate intestinal immune homeostasis.
In Brief (eTOC blurb)
The role of host-derived intestinal metabolites in mucosal immune regulation is poorly understood. Here, Cao et al. show that effector CD4+ T cells upregulate expression of the xenobiotic transporter, Mdr1, in the ileum to safeguard immune homeostasis, revealing an important immunologic consequence of bile acid reabsorption in the ileum.
INTRODUCTION
Adaptive immunity requires naïve CD4+ T helper (Th) cell differentiation into cytokine-secreting effector (Teff) subsets (Nakayamada et al., 2012). Teff cells, which include interferon-gamma (IFNγ)-producing Th1 cells and interleukin-17 (IL-17)-secreting Th17 cells, down-regulate lymphoid-homing receptors and up-regulate tissue-homing chemokine receptors and integrins to egress lymphoid organs and infiltrate non-lymphoid tissues. However, the presence of Teff cells in non-lymphoid tissues is potentially deleterious, and elevated Teff cell cytokine expression in tissues is both a hallmark and important therapeutic target in many inflammatory disorders, including inflammatory bowel diseases (IBDs) (Kaser et al., 2010).
Intestinal microbiota play key roles in suppressing pro-inflammatory Teff cell function in the gut. For example, commensal microbes prevent trafficking of mucosal phagocytes to mesenteric lymph nodes to limit T cell priming (Diehl et al., 2013), produce short-chain fatty acids or polysaccharide A to stimulate peripheral Forkhead box P3 (Foxp3)+ T regulatory (pTreg) cell development or IL-10 expression, respectively (Arpaia et al., 2013; Mazmanian et al., 2008), and convert intestinal pTregs into CD4+CD8αα + intraepithelial lymphocytes (IELs) (Sujino et al., 2016). Still other enteric species, such as segmented filamentous bacteria, induce pro-inflammatory Th17 cell function in the gut (Atarashi et al., 2015; Sano et al., 2015). Thus, major translational initiatives have been launched to characterize the human microbiome in human health and disease, and test microbiota-directed therapies in IBDs.
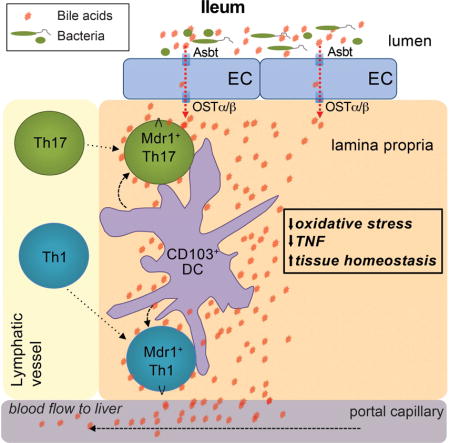
Far less is known about host-derived intestinal metabolites in mucosal immune regulation. Like the microbiota, bile acids (BAs) constitute a unique and fundamental aspect of gastrointestinal physiology (Hofmann and Hagey, 2014). BAs are detergent-like metabolites that are synthesized in the liver, stored in the gall bladder, and secreted into the proximal small intestine (i.e., duodenum) upon food intake. BAs emulsify dietary lipids in the small intestinal lumen, and are reabsorbed in the distal small intestine (i.e., ileum) by epithelial cells expressing the apical sodium-dependent bile acid transporter, Asbt (Dawson et al., 2003). Reabsorbed BAs accumulate in the lamina propria and enter portal circulation for return to the liver (Hofmann and Hagey, 2014). Still, immunologic consequences of BA reabsorption in the ileum remain unknown.
MDR1 (ABCB1) is a membrane-associated, ATP-dependent efflux pump recognized for transporting chemotherapeutic drugs out of tumor cells (Gottesman et al., 2002). We previously reported that MDR1 is preferentially expressed in intestinal vs. circulating human Teff cells (Ramesh et al., 2014). Intriguingly, mice lacking Abcb1a – the mouse ortholog of human ABCB1 – are prone to spontaneous colitis (Panwala et al., 1998), and ABCB1 polymorphisms have been associated with human IBDs (Annese et al., 2006). However, MDR1 remains widely considered a dedicated “drug handler” in mammals (Borst and Schinkel, 2013).
Here we determined that Mdr1 was also expressed in mouse Teff cells at steady-state, where it was increased in the small intestine lamina propria, highest in the ileum, and induced by CD103+ dendritic cells (DCs). Consistent with these findings, Teff cells lacking Mdr1 displayed mucosal dysfunction in the ileum and transferred Crohn’s disease-like ileitis in Rag1−/− hosts. Mdr1 acted during BA exposure to limit oxidative stress and enforce Teff cell homeostasis, and blocking ileal BA reabsorption in transferred Rag1−/− mice restored Mdr1-deficient Teff cell homeostasis and attenuated ileitis. Further, a subset of ileal Crohn’s disease patients displayed MDR1 loss-of-function. Together, these data suggest that circulating Teff cells upregulate Mdr1 in the ileum to compensate for BA reabsorption and safeguard intestinal homeostasis.
RESULTS
Effector CD4+ T Cells Upregulate Mdr1 Expression in the Ileum
Considering that physiologic MDR1 functions will involve interaction with unknown metabolites, we investigated the function of Mdr1 in T cells using in vivo mouse models rather than in vitro human cell culture. Using efflux of the fluorescent Mdr1 substrate, rhodamine 123 (Rh123) (Ludescher et al., 1992), we first confirmed that Mdr1 was expressed in a portion of endogenous mouse Teff cells at steady state (Figure 1A). Teff cell Rh123 efflux was due to Mdr1, as: (i) Rh123 efflux was abolished by ex vivo treatment with the Mdr1 antagonist, elacridar (Hyafil et al., 1993); (ii) Rh123 efflux was absent in Teff cells from Mdr1-deficient mice (FVB.Abcb1a−/−Abcb1b−/−; abbreviated hereafter as FVB.Abcb1a/1b−/−) (Schinkel et al., 1997); and (iii) wild-type Teff cells isolated by fluorescence activated cell sorting (FACS) based on Rh123 efflux displayed enrichment for Abcb1a expression, but not other multidrug transporter messenger (m)RNAs (e.g., Abcc1, Abcc3, Abcg2) (Figure 1A, S1A-B). As in humans (Ramesh et al., 2014), Rh123 efflux was absent in mouse naïve T cells, and higher in Teff cells vs. Tregs (Figure 1A, S1A). Within Teff cells, Rh123 efflux was evident in both Th17 and non-Th17 (mostly IFNγ-expressing Th1) cells, as determined in studies using Th17 fate-mapping reporter (Il17aCreR26LSL-hCD2) mice (Figure S1C-F), where Cre recombinase-induced human CD2 expression stably marks Teff cells that have expressed Il17a during development.
Figure 1. Mdr1 is Expressed in CD4+ Effector T cells and Upregulated in the Ileum.
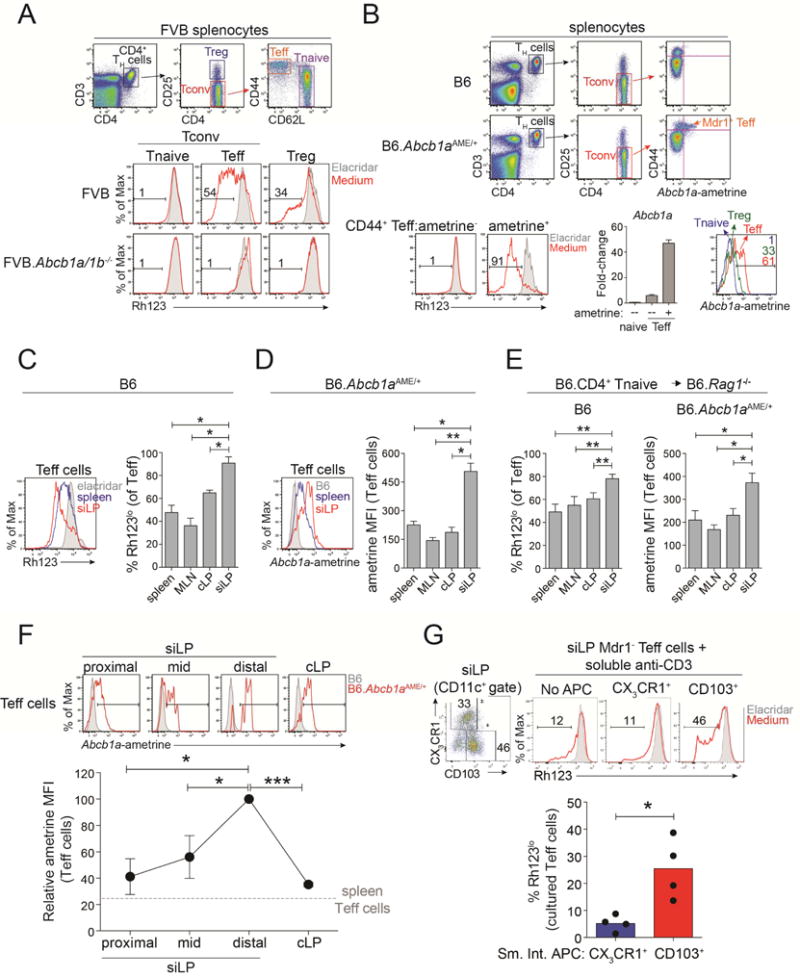
(A) Top, spleen CD4+ TH cell subsets in FVB wild-type mice. Tnaive, naïve T cells; Treg, T regulatory cells; Teff, effector T cells. Bottom, Rh123 efflux in spleen TH cell subsets from FVB wild-type or Mdr1-deficient (Abcb1a/1b−/−) mice in the presence (shaded) or absence (red) of elacridar. Representative of 10 experiments.
(B) Top, spleen TH cell subsets in C57Bl/6 (B6) wild-type or Mdr1-reporter (Abcb1aAME/+) mice. Ametrine expression in CD44hi Teff cells is shown at right. Bottom left, Rh123 efflux in ametrine+/− Teff cells from Abcb1aAME/+ reporter spleens in the presence (shaded) or absence (red) of elacridar. Bottom middle, mean relative Abcb1a expression (+ SEM; n =3), by qPCR, in ex vivo-sorted Tnaive or ametrine +/− Teff cells from Abcb1aAME/+ reporter spleens. Bottom right, ametrine expression in Tnaive, Treg, or Teff cells from Abcb1aAME/+ reporter spleens. Representative of 5 experiments.
(C) Left, Rh123 efflux in B6 wild-type spleen (blue) or small intestine lamina propria (siLP; red) Teff cells. Elacridar-treated spleen Teff cells (shaded) show background. Representative of 4 experiments. Right, mean percentages of Rh123lo Teff cells (± SEM; n = 4) from tissues of wild-type B6 mice. MLN, mesenteric lymph nodes; cLP, colon lamina propria.
(D) Left, ametrine expression in spleen (blue) or siLP (red) Teff cells from of Abcb1aAME/+ reporter mice. Wild-type (B6) spleen Teff cells (shaded) show background. Representative of 4 experiments. Right, ametrine mean fluorescence intensity (MFI) in Abcb1aAME/+ reporter Teff cells (mean ± SEM; n = 4) from different tissues.
(E) Mean (± SEM; n = 4) percentages of Rh123lo wild-type Teff cells (left) or ametrine MFIs in Abcb1aAME/+ reporter Teff cells (right) from tissues of transferred B6.Rag1−/− mice after 6-weeks.
(F) Top, ametrine expression in B6 wild-type (shaded) or Abcb1aAME/+ (red) Teff cells from siLP sections or cLP. Representative of 3 experiments. Bottom, ametrine MFIs (mean + SEM; n = 3) in Abcb1aAME/+ Teff cells from intestinal sections as above.
(G) Top left, CX3CR1 and CD103 expression in siLP CD11c+ antigen-presenting cells (APC) from B6 wild-type mice. Top right, Rh123 efflux in ex vivo-sorted Mdr1− siLP Teff cells from B6 wild-type mice 4 days after anti-CD3-stimulation in the absence (no APC) or presence of sorted APC subsets. Representative of 4 experiments. Bottom, percentages of Rh123lo Teff cells (n = 4) after co-culture with siLP APC subsets as above.
* P < .05, ** P < .01, *** P < .001, student’s t test. See also Figure S1–3.
However useful, Rh123 is an indirect measure of Mdr1 expression that requires ex vivo manipulation, and no commercial antibodies permit direct detection of mouse Mdr1 by flow cytometry. Therefore, we generated a fluorescent Mdr1-reporter mouse (Abcb1aAME/+) using CRISPR/Cas9 gene editing in wild-type C57Bl/6 (B6) zygotes (Yang et al., 2013). We replaced the Abcb1a stop codon with a bi-cistronic reporter cassette containing a P2A peptide sequence and the fluorescent transgene, ametrine (Figure S2A–B). Although an in-frame 9-nucleotide (3-amino acid) deletion was found near the 3′ end of the targeted Abcb1a allele (Figure S2C–D), Rh123 efflux remained intact in Abcb1aAME/+ T cells (Figure S2E). Ametrine mean fluorescence intensity (MFI) within Abcb1aAME/+ T cells was highest in CD25−CD44hi Teff cells, and quantitatively reflected both Rh123 efflux activity and Abcb1a gene expression (Figure 1B, S2F–G, data not shown).
Whether assessed by Rh123 efflux in wild-type Teff cells or ametrine expression in Abcb1aAME/+ Teff cells, Mdr1 expression was increased in Teff cells from small intestine lamina propria (siLP) compared to those from either colon lamina propria (cLP) or other tissues (Figure 1C–D, data not shown). Increased Mdr1 expression in siLP Teff cells was evident by Rh123 efflux on several wild-type backgrounds (e.g., C57BL/6, FVB, BALB/c) (Figure 1C, data not shown), and in transferred Rag1−/− mice receiving wild-type or Abcb1aAME/+ naïve T cells (Figure 1E). Within siLP, Mdr1 expression increased in Teff cells in a gradient fashion from proximal to distal, with highest expression occurring in the ileum (Figure 1F).
Increased Mdr1 expression in siLP Teff cells could be due to selective survival or local upregulation. In support of the latter, Mdr1 expression in siLP Teff cells was transient, and induced by intestinal dendritic cells (DCs). First, Teff cells generated in vitro from anti-CD3-and anti-CD28-stimulated naïve precursors did not express Mdr1 (Figure S1A, S3A). Second, FACS-sorted Mdr1+ Teff cells lost Mdr1 expression upon ex vivo culture (Figure S3B). Third, FACS-sorted Mdr1− Teff cells acquired Mdr1 expression upon re-transfer into naïve Rag1−/− mice, which was again highest in siLP (Figure S3B). Fourth, Mdr1 expression in ex vivo-isolated Mdr1− Teff cells was induced by co-culture with intestinal CD11c+ DCs, but not with intestinal CD11b+CD11c− macrophages or CD11c+ DCs from spleen (Figure S3C). Whereas intestinal CD11c+ DCs that induced Teff cell Mdr1 expression in vitro were heterogeneous for CD103 and chemokine, CX3C motif, receptor 1 (CX3CR1) expression (Figure 1G), only purified CD103+ DCs induced Mdr1 expression in co-cultured Teff cells (Figure 1G). Finally, Mdr1 expression in siLP Teff cells was independent of microbiota, as high-level Rh123 efflux was observed in siLP Teff cells from germ free (GF) mice (Figure S3D). Accordingly, intestinal DCs from both specific pathogen-free (SPF) and GF mice induced Mdr1 expression in co-cultured Teff cells (Figure S3E). Thus, circulating Teff cells upregulate Mdr1 expression upon migration into the ileal mucosa, and do so irrespective of microbiota.
Mdr1 Regulates Effector T Cell Function in the Ileum
Mdr1-deficient T cells induced more aggressive weight loss than wild-type counterparts upon transfer into FVB.Rag1−/− mice (Figure 2A). Inflammation in this model is generally restricted to the colon (Powrie et al., 1994), and both wild-type and Mdr1-deficient T cells induced severe colitis (Figure 2B–C). However, Mdr1-deficient T cells also induced ileitis, whereas wild-type cells did not (Figure 2B–C).
Figure 2. Mdr1-Deficient T Cells Transfer Ileitis in Rag1−/− Hosts.
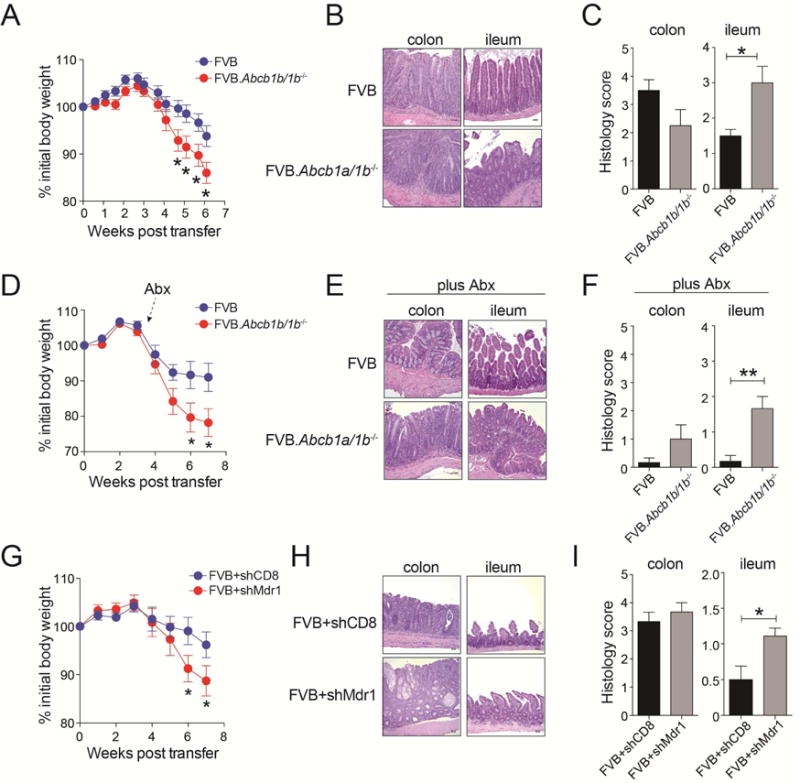
(A) Mean weight loss (+ SEM; n = 28 over 5 experiments) in co-housed FVB.Rag1−/− mice injected with FVB wild-type or Mdr1-deficient (FVB.Abcb1a/1b−/−) naïve T cells.
(B) H&E-stained intestinal sections (colon, ileum) from co-housed FVB.Rag1−/− mice transferred as in (A). Representative of 6–8 mice over 3 experiments.
(C) Mean histology scores (+ SEM) in colon (n = 8; left) or ileum (n = 6; right) of transferred and co-housed FVB.Rag1−/− mice as in (A–B). Incorporates data from 3 experiments.
(D) Mean weight loss (+ SEM; n = 15 over 3 experiments) in FVB.Rag1−/− mice injected with FVB or FVB.Abcb1a/1b−/− naïve T cells and treated with antibiotics (Abx).
(E) H&E-stained intestinal sections (colon, ileum) from co-housed FVB.Rag1−/− mice transferred and treated with Abx as in (D). Representative of 6–8 mice over 3 experiments.
(F) Mean histology scores (+ SEM) in colon (n = 8; left) or ileum n = 6; right) of Abx-treated FVB.Rag1−/− mice as in (D–E). Incorporates data from 3 experiments.
(G) Mean weight loss (+ SEM; n = 21 over 3 experiments) in co-housed FVB.Rag1−/− mice injected with FVB wild-type T cells transduced in vitro to express shRNAs against Cd8a (shCD8) or Abcb1a and Abcb1b (shMdr1).
(H) H&E-stained intestinal sections (colon, ileum) from transferred and co-housed FVB.Rag1−/− mice as in (G). Representative of 8–10 mice over 3 experiments.
(I) Mean histology scores (± SEM) in colon (n = 8–10); left) or ileum (n = 8–10; right) of co-housed FVB.Rag1−/− mice as in (G–H). Incorporates data from 3 experiments.
(A, D, G) Weights relative to day 0.
(B, E, H) 20x magnification, scale bars = 50 μM.
* P < .05, ** P < .01, student’s t test. See also Figure S4–5A.
Mucosal inflammation in transferred Rag1−/− mice requires antigens from the enteric flora (Jones-Hall and Grisham, 2014). However, non-microbial factors contributed to the propagation of Mdr1-deficient T cell-driven ileitis. First, Rag1−/− mice used in these studies were co-housed, indicating that Mdr1-deficient T cell-induced ileitis was not transmissible through coprophagia. Second, ileitis in this model was not associated with dysbiosis, as judged by 16S ribosomal DNA sequencing (Figure S4A-C). Third, Rag1−/− mice receiving Mdr1-deficient T cells displayed increased weight loss and ileitis compared with wild-type T cell recipients, even after broad-spectrum antibiotic therapy (Figure 2D–F, S4D). In addition, ileitis in this model was not indirectly due to aberrant T cell development or function in FVB.Abcb1a1b−/− mice, as short hairpin (sh)RNA-mediated depletion of Abcb1a and Abcb1b (shMdr1) in wild-type T cells (Figure S5A) also increased T cell-driven weight loss and ileitis in Rag1−/− mice, compared with a control shRNA against Cd8a (shCD8) (Figure 2G–I).
Consistent with small bowel pathology and irrespective of antibiotic therapy, Mdr1-deficient Teff cells isolated from siLP of transferred Rag1−/− mice displayed increased tumor necrosis factor (TNF) and IFNγ expression relative to wild-type counterparts (Figure 3A–B, S5B–D). RAR-related orphan receptor gamma-t (RORγt)+ siLP Teff cells lacking Mdr1 also showed elevated IL-17A expression (Figure S5C–D). By contrast, expression of these cytokines was similar in wild-type and Mdr1-deficient Teff cells outside of the small intestine, including in the colon (Figure 3A–B, S5C–D). Like Mdr1 expression (Figure 1F), increased TNF and IFNγ expression in Mdr1-deficient vs. wild-type siLP Teff cells was most pronounced in the ileum (Figure 3C, data not shown). In addition, shRNA-mediated depletion of Mdr1 in wild-type Teff cells recapitulated the pro-inflammatory phenotype of Abcb1a/1b−/− Teff cells in siLP (Figure 3D, S5A). Still, many other parameters of T cell differentiation and function were not affected by Mdr1-deficiency in vivo, including: (i) development of RORγt+ Teff cells and Foxp3+ pTregs; (ii) expression of gut-homing receptors (CCR6, CXCR3, CCR9, α4β7); and (iii) expression of IL-10 and IL-22 (Figure S5E, data not shown).
Figure 3. Mdr1 Regulates Effector T Cell Function in the Ileum.
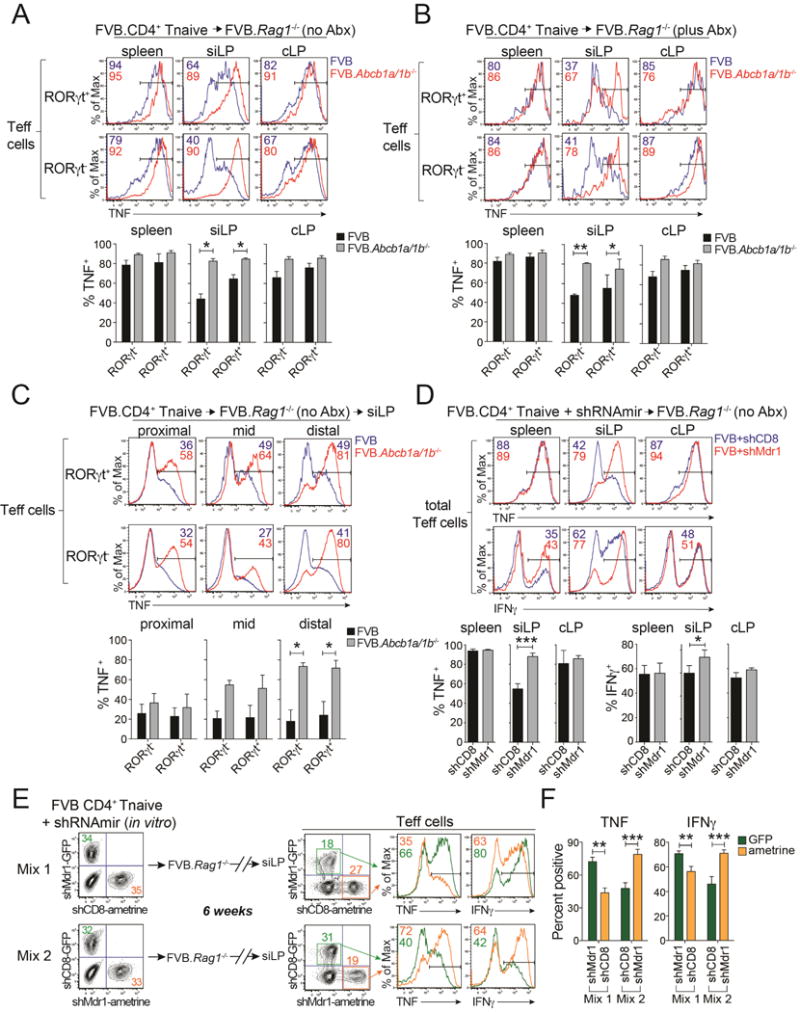
(A) Top, TNF expression in wild-type (FVB; blue) or Mdr1-deficient (FVB.Abcb1a/1b−/−; red) RORγt+/− effector T cells (Teff cells) from tissues of co-housed, transferred, and untreated (no antibiotics [no Abx]) FVB.Rag1−/− mice. siLP, small intestine lamina propria; cLP, colon lamina propria. Representative of 6 mice over 2 experiments. Bottom, mean percentages of TNF+ Teff cells (+ SEM; n = 6) as above.
(B) Top, TNF expression in FVB wild-type (blue) or FVB.Abcb1a/1b−/− (red) RORγt +/− Teff cells from tissues of co-housed and transferred FVB.Rag1−/− mice treated with antibiotics (plus Abx). Representative of 6 mice over 2 experiments. Bottom, mean percentages of TNF+ Teff cells (+ SEM; n = 6) as above.
(C) Top, TNF expression in wild-type (FVB; blue) or FVB.Abcb1a/1b−/− (red) RORγt +/− Teff cells from siLP sections of transferred, co-housed and untreated (no Abx) FVB.Rag1−/− mice. Representative of 3 mice over 2 experiments. Bottom, mean percentages of TNF+ Teff cells (± SEM; n = 3) as above.
(D) Top, TNF or IFNγ expression in FVB wild-type Teff cells expressing shRNAs against Cd8a (shCD8) or Abcb1a and Abcb1b (shMdr1) from tissues of transferred and co-housed FVB.Rag1−/− mice. Representative of 4 mice over 2 experiments. Bottom, mean percentages of TNF+ Teff cells (+ SEM; n = 4) as above.
(E) Untransduced, shCD8- or shMdr1-expressing FVB wild-type T cells distinguished by GFP or ametrine retroviral reporters were FACS-sorted and mixed in vitro, and transferred into FVB.Rag1−/− mice. Cells were re-isolated from siLP for analysis 6-weeks post-transfer. Representative of 6 mice over 2 experiments.
(F) Mean percentages (± SEM; n = 6) of TNF+ (left) or IFNγ+ (right) shCD8- or shMdr1-expressing siLP Teff cells from transferred and co-housed FVB.Rag1−/− as in (E).
(A–F) Intracellular cytokine expression was analyzed 6-to 8-weeks post transfer after ex vivo-stimulation. Color-matched text indicate percentages of cytokine+ Teff cells.
* P < .05, ** P < .01, *** P < .001, student’s t test. See also Figure S5.
To assess cell-intrinsic regulation of siLP Teff cell function by Mdr1, we developed a congenic transfer system in which FVB.Rag1−/− mice received mixtures of untransduced, shCD8-expressing, and shMdr1-expressing wild-type T cells discriminated by GFP vs. ametrine retroviral reporters (Figure 3E); we used shRNAs for these studies because congenic alleles are not available in FVB mice. Indeed, Mdr1 depletion intrinsically increased siLP Teff cell expression of TNF and IFNγ (Figure 3E–F), suggesting that Mdr1 upregulation by mucosal Teff cells in the ileum limits pro-inflammatory cytokine expression and collateral tissue damage.
Conjugated Bile Acids Modulate Mdr1 Function
In vitro incubation of wild-type splenic Teff cells with sterile, soluble luminal content (LC) from small intestine (siLC), but not colon (cLC), inhibited Rh123 efflux (Figure 4A), suggesting that components within siLC modulate Mdr1 function. Given both the high-level Mdr1 expression in mucosal Teff cells from GF mice (Figure S3D) and the persistent ileitis in antibiotic-treated Rag1−/− mice receiving Mdr1-deficient T cells, we hypothesized that Mdr1 interfaces primarily with host-derived metabolites. As noted above, bile acids (BAs), are restricted to the small intestine due to active reabsorption in the ileum (Hofmann and Hagey, 2014). BAs are also cytotoxic to cells at high concentrations, as in cholestatic liver diseases where BA accumulation induces hepatocellular necrosis (Poupon et al., 2000). Indeed, siLC from wild-type mice, but not cLC, was cytotoxic to cultured T cells (data not shown). Two results confirmed that BAs are the principal Mdr1-modulating metabolites within siLC. First, BA concentrations were equivalent in siLC from GF and SPF mice (Figure S6A), and both inhibited Rh123 efflux (Figure S6B). Second, depleting BAs from either GF or SPF siLC with the BA sequestering resin, cholestyramine (Arnold et al., 2014), abolished siLC-mediated inhibition of Rh123 efflux (Figure 4A, S6B). In line with these results, siLC BA concentrations in transferred Rag1−/− mice were not affected by antibiotic therapy (Figure S6C), and siLC from both control and antibiotic-treated Rag1−/− mice inhibited Rh123 efflux (Figure S6D–E).
Figure 4. Mdr1 Regulates Effector T Cell Function in the Presence of Bile Acids.
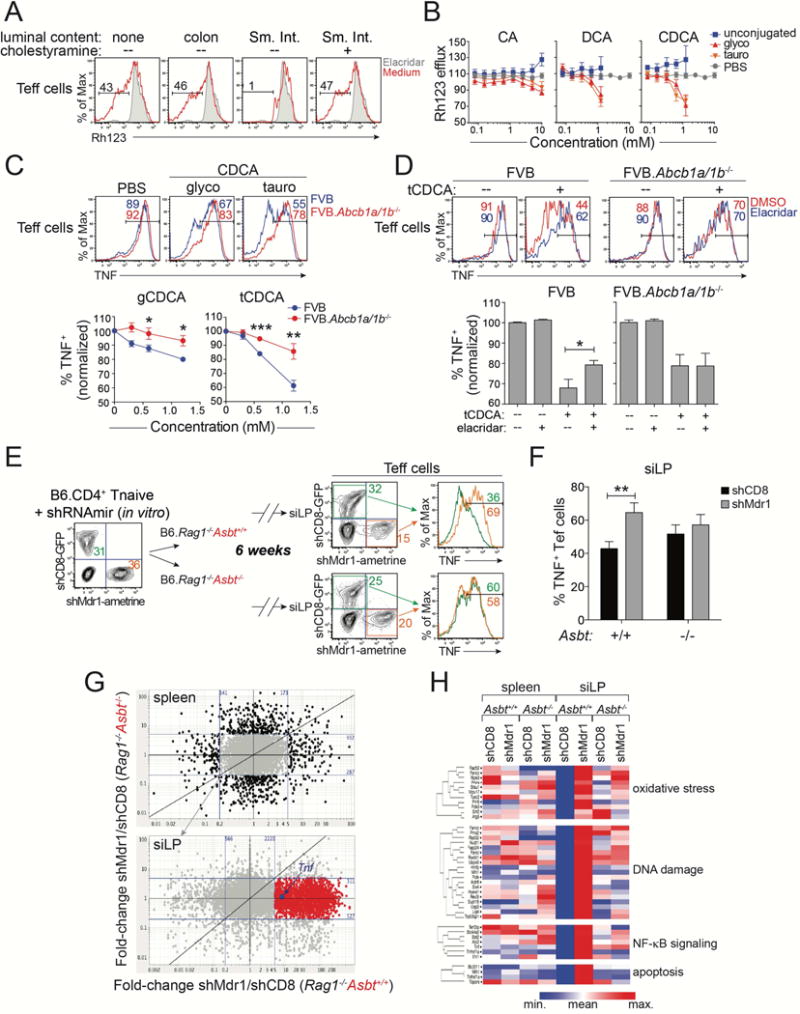
(A) Rh123 efflux in splenic FVB wild-type CD4+ effector T cells (Teff cells) after in vitro treatment without (none) or with lumen content from colon or small intestine (Sm. Int.) (red). Sm. Int. content was pre-treated +/− cholestyramine to deplete bile acids. Elacridar-treated cells (shaded) show background. Representative of 5 experiments.
(B) Dose-responses (+ SEM; n = 3) of synthetic bile acids on splenic FVB wild-type Teff cell Rh123 efflux. Unconjugated (blue), glycine-(glyco; red), or taurine-(tauro; orange) conjugated bile acids: cholic acid (CA), deoxycholic acid (DCA), chenodeoxycholic acid (CDCA). Vehicle (PBS)-treated Teff cells are shown (grey); data are normalized to cells in media alone. Incorporates data from 3 experiments
(C) Top, TNF expression in in vitro-stimulated splenic wild-type (FVB; blue) or Mdr1-deficient (FVB.Abcb1a/1b−/−; red) Teff cells after pre-treatment with vehicle (PBS), or glycine (g)- or taurine (t)-conjugated CDCA. Representative of 4 experiments. Bottom, dose responses of CDCA species on TNF expression (+ SEM; n = 4) in wild-type or FVB.Abcb1a/1b−/− Teff cells as above. Data are normalized to vehicle (PBS)-treated cells.
(D) Top, TNF expression in in vitro-stimulated splenic FVB wild-type (left) or FVB.Abcb1a/1b−/− (right) Teff cells treated +/− tCDCA in the presence (blue) or absence (red) of elacridar (0.1 μM). Representative of 8 experiments. Bottom, Percentages of TNF+ Teff cells (± SEM; n = 8) as above. Data are normalized to vehicle (PBS)-treated cells.
(E) Untransduced, shCD8- or shMdr1-expressing B6 wild-type T cells were mixed and transferred into co-housed Asbt-sufficient or Asbt-deficient B6.Rag1−/− mice. Cells were re-isolated from siLP and analyzed 6-weeks post-transfer. Representative of 6 mice over 2 experiments.
(F) Mean percentages (+ SEM; n = 6) of TNF+ shCD8- or shMdr1-expressing siLP Teff cells from transferred and co-housed Asbt+/+ or Asbt−/− B6.Rag1−/− mice in (E).
(G) Gene expression, by RNA-seq, in ex vivo-isolated shCD8- or shMdr1-expressing Teff cells from spleen (top) or siLP (bottom) of congenically-transferred and co-housed Asbt+/+ or Asbt−/− B6.Rag1−/− mice as in (E-F). Fold-change/fold-change plots are shown (blue lines = 5-fold change). Genes changing < 5-fold in spleen (n = 7,341; grey) are shown in siLP. 1659 genes (red), including Tnf (blue), are > 5-fold higher in shMdr1- vs. shCD8-expressing siLP Teff cells from Asbt+/+, but not Asbt−/−, Rag1−/− mice.
(H) Pathway analysis of genes highlighted red in (G).
(G–H) Gene expression values are means from 5 mice per group in one experiment.
* P < .05, ** P < .01, *** P < .001, student’s t test. See also Figure S6-7.
Five major BA species are present in mammals: cholic acid (CA), chenodeoxycholic acid (CDCA), and β-muricholic acid (βMCA) are primary BAs synthesized by hepatocytes in the liver, whereas deoxycholic acid (DCA) and lithocholic acid (LCA) are secondary BAs that derive from enzymatic actions of microbiota on primary BAs (Hofmann and Hagey, 2014). Each BA species can exist in a free/unconjugated form, or as conjugated to the amino acids taurine or glycine (Hofmann and Hagey, 2014). However, the majority of intestinal BAs in vivo are membrane-impermeant conjugated BAs, maintained by enterohepatic circulation through the ileum (Hofmann and Hagey, 2014). Each BA species was cytotoxic to wild-type T cells in vitro at or above their critical micelle concentration (~2.5–10 mM), and each inhibited Mdr1-dependent Rh123 efflux in a dose-dependent manner at lower concentrations (0.5–2 mM) (Figure 4B, S6F). However, only glycine- or taurine-conjugated BAs (CBAs) inhibited Rh123 efflux (Figure 4B, S6F). Further, both mouse siLC and commercially-synthesized CBAs also inhibited Rh123 efflux mediated by human MDR1 (Figure S6G). Thus, the specific interplay between Mdr1 and CBAs predicts our observations above that phenotypes of Mdr1-deficient Teff cells in vivo are amplified at site of CBA reabsorption, the ileum.
Mdr1 Enforces T Cell Homeostasis in the Presence of Conjugated Bile Acids
To assess direct impacts of Mdr1 and CBAs on Teff cell function, we cultured splenocytes from wild-type or Mdr1-null mice in the presence or absence of synthetic CBAs. After a 4 hr incubation period, we stimulated these cells and analyzed TNF expression in Teff cells by flow cytometry. In line with our in vivo results (Figure 3), both wild-type and Mdr1-deficient Teff cells expressed high levels of TNF in the absence of CBAs (e.g., glycine (g)-, or taurine (t)-conjugated CDCAs), whereas Mdr1-deficient Teff cells displayed elevated TNF expression vs. wild-type counterparts upon CBA exposure (Figure 4C). Mdr1 transport activity per se was important for limiting TNF expression in the presence of CBAs, as: (i) inhibiting Mdr1 with elacridar increased TNF expression in tCDCA-treated wild-type Teff cells, but had no impact on TNF expression in the absence of either tCDCA or Mdr1 (Figure 4D), and (ii) ectopic expression of human MDR1 (hMDR1), but not a transport-deficient mutant (ΔhMDR1; Y401A-Y1044A) (Kim et al., 2006), reduced TNF expression in tCDCA-treated EL4 cells, a mouse thymoma cell line that lacks endogenous Mdr1 expression (Figure S7A-E, data not shown). EL4 cells were used because ectopic MDR1 expression was not tolerated in primary T cells (data not shown).
Exposure of siLP Teff cells to CBAs in vivo should require Asbt-dependent reabsorption by ileocytes (Dawson et al., 2003). Therefore, we tested if Asbt-dependent CBA reabsorption promotes mucosal dysfunction of Mdr1-deficient siLP Teff cells in vivo. We crossed B6.Asbt-deficient (Slc10a2−/−) mice with B6.Rag1−/− mice, and then injected these mice with congenic mixtures of untransduced, shCD8-expressing, or shMdr1-expressing B6 T cells (Figure 4E). As in FVB Teff cells (Figure 3D–F), shRNA-mediated depletion of Mdr1 increased TNF expression in B6 siLP Teff cells from Asbt-sufficient Rag1−/− mice (Figure 4E–F). By contrast, both control and Mdr1-depleted Teff cells displayed equivalent TNF expression in siLP from Asbt-deficient Rag1−/− mice, where CBA reabsorption was ablated (Figure 4E–F).
To gain insight into the mechanism by which CBAs and Mdr1 regulate siLP Teff cell function, we performed RNA-sequencing (seq) on ex vivo-isolated control (shCD8-expressing) or Mdr1-depleted Teff cells from spleen or siLP of congenically-transferred Asbt-sufficient or Asbt-deficient Rag1−/− mice. Compared to bystander control cells, Mdr1-depleted Teff cells over-expressed more than 2,000 transcripts in CBA-replete siLP, including Tnf (Figure 4G, bottom). These transcripts were: (i) regulated selectively in siLP vs. spleen; (ii) reduced, as a rule, in Mdr1-deficient siLP Teff cells by Asbt ablation; and (iii) enriched for genes whose encoded proteins are involved in the oxidative stress response, DNA damage and nucleotide excision repair, apoptosis and p53 signaling, and NF-κB signaling (Figure 4G-H). Given that oxidative stress involves lipid peroxidation and the generation of highly reactive lipid aldehydes (e.g., malondialdehyde, 4-hydroxy-2-nonenal, etc.) (Ayala et al., 2014), which damage cells by forming adducts with proteins and genomic DNA and lead to activation of DNA repair, p53 and NF-κB pathways (Ayala et al., 2014; Kabe et al., 2005), these data suggest that Mdr1 protects Teff cells from CBA-driven oxidative stress in the ileum.
Consistent with a mechanism involving oxidative stress, Mdr1 was also important for Teff cell survival in the presence of CBAs. First, Mdr1 depletion by shRNAs led to systemic defects in Teff cell persistence in transferred Rag1−/− mice (Figure 5A–B, data not shown), which is consistent with the extensive circulation of Teff cells in and out of the gut (Morton et al., 2014). Second, the same concentration of tCDCA (1.2 mM) that drove TNF over-expression in cultured Mdr1-deficient vs. wild-type Teff cells after 8 hr (Figure 4C–D), killed 80–90% of wild-type T cells after 24 hr (Figure 5C); those that survived were uniformly Mdr1+ Teff cells, whether judged by Rh123 efflux in wild-type cells (Figure 5C), or ametrine expression in Abcb1aAME/+ reporter cells (Figure 5D). Third, neither elacridar-treated wild-type T cells nor Mdr1-null T cells survived overnight culture with tCDCA (Figure 5E–F). Finally, ectopic expression of hMDR1, but not the transport-deficient mutant, promoted durable survival of EL4 cells in the face of chronic tCDCA treatment (Figure S7F-G). Thus, the initial pro-inflammatory response of Mdr1-deficient Teff cells to CBA-driven oxidative stress is cytotoxic over time.
Figure 5. Mdr1 Promotes T Cell Survival During Conjugated Bile Acid Exposure.
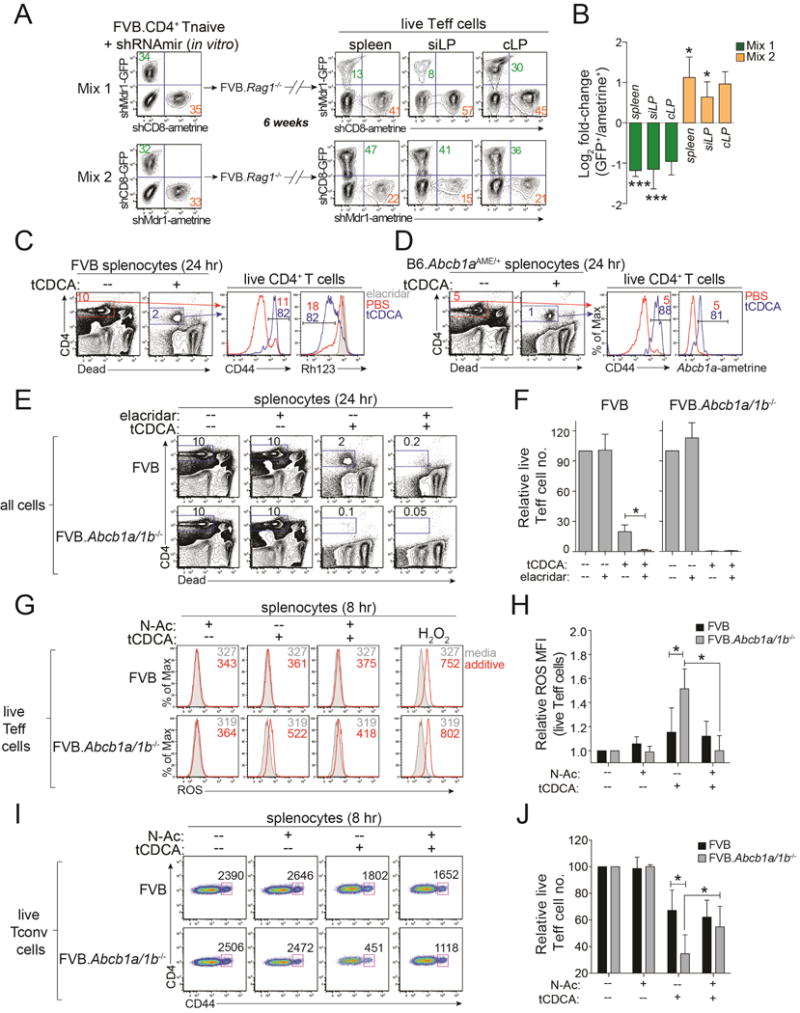
(A) Untransduced, shCD8- or shMdr1-expressing FVB wild-type T cells were mixed and transferred into FVB.Rag1−/− mice (as in Figure 3E). Frequencies of live shCD8- or shMdr1-expressing CD4+ effector T cells (Teff cells) from tissues were determined by ex vivo FACS analysis after 6 weeks. MLN, mesenteric lymph nodes; siLP, small intestine lamina propria; cLP, colon lamina propria. Representative of 5 mice over 2 experiments.
(B) Mean Log2 fold-change (± SEM; n = 5) in frequencies of live shCD8- or shMdr1-expressing Teff cells from tissues of transferred FVB.Rag1−/− mice as in (A) compared to pre-transfer input.
(C) CD44 expression and Rh123 efflux in splenic FVB wild-type CD4+ T cells after 24 hr culture in media +/− taurine-conjugated chenodeoxycholic acid (tCDCA; 1.2 mM); viable CD4+ T cells are gated as at left. Numbers indicate percentages of CD44+ or Rh123lo cells. Representative of 3 experiments.
(D) CD44 and ametrine expression in splenic Abcb1aAME/+ reporter CD4+ T cells after 24 hr in culture +/− tCDCA as in (C). Numbers indicate percentages of CD44+ or ametrine+ cells Representative of 3 experiments.
(E) Survival of wild-type (FVB) or Mdr1-deficient (FVB.Abcb1a/1b−/−) splenic CD4+ T cells after 24 hr in culture +/− tCDCA (1.2 mM) +/− elacridar (0.1 μM). Representative of 4 experiments.
(F) Mean relative numbers (+ SEM; n = 4) as in (E). Data are normalized to cells in media alone.
(G) FACS analysis of reactive oxygen species (ROS) in wild-type (FVB) or FVB.Abcb1a/1b−/− splenic Teff cells after 8 hr in media alone (shaded), or media containing N-acetyl cysteine (N-Ac; 125 μM), tCDCA (1.2 mM), or tCDCA+N-Ac (red). H2O2 treated cells (30 min) are a positive control; numbers indicate ROS MFI. Representative of 4 experiments.
(H) Mean relative ROS MFI (± SEM; n = 4) as in (G). Data are normalized to cells cultured in media alone.
(I) Numbers of live Teff cells after 8 hr in culture +/− tCDCA +/− N-Ac as in (G). Live CD4+CD25− (Tconv) cells are shown; numbers indicate cell numbers quantified over 1 min. Representative of 4 experiments.
(J) Mean relative numbers (+ SEM; n = 4) as in (I). Data are normalized to media alone.
* P < .05, student’s t test. See also Figure S6-7.
Increased concentrations of reactive oxygen species (ROS) were also evident by flow cytometry in tCDCA-treated Mdr1-deficient Teff cells (Figure 5G–H). By contrast, ROS concentrations were similarly low in untreated wild-type and Mdr1-deficient Teff cells, and strongly induced in both genotypes by hydrogen peroxide (H2O2) (Figure 5G). Accordingly, the small molecule anti-oxidant, N-acetyl-L-cysteine (N-Ac), decreased ROS concentrations in tCDCA-treated Mdr1-deficient Teff cells and restored their survival to wild-type levels (Figure 5I–J). Teff cells lacking Mdr1 also displayed increased ROS concentrations and decreased survival in the presence of 13-hydroperoxy-octadecadienoic acid (13-HpODE), an oxidized lipid derived from lineoleic acid, but not in the presence of mitochondrial destabilizers (e.g., H2O2 and menadione) (Figure S7H–K) (Schneider et al., 2001; Thorpe et al., 2004; Gerasimenko et al., 2002). Thus, these results suggest that CBAs induce, and Mdr1 mitigates, lipid peroxidation.
Bile Acid Sequestration Restores Mdr1-deficient Effector T Cell Homeostasis
Bile acid sequestrants, which bind CBAs in the intestinal lumen and prevent their reabsorption into the ileal lamina propria (Arnold et al., 2014), could be pharmacologically useful to restore mucosal homeostasis in Mdr1-deficient Teff cells. To test this, we administered cholestyramine (CME) to FVB.Rag1−/− mice receiving wild-type or Mdr1-deficient T cells. CME-fed Rag1−/− mice displayed increased fecal BA excretion (Figure 6A), which was similar to Asbt-deficient mice and indicative of reduced ileal CBA reabsorption (Dawson et al., 2003). Further, CME treatment reduced both weight loss and ileitis in Rag1−/− mice receiving Mdr1-deficient T cells, without influencing either the normal small bowel histology in Rag1−/− mice receiving wild-type T cells, or the severe colitis present in both groups of Rag1−/− recipients (Figure 6B–D).
Figure 6. Bile Acid Sequestration Restores Mucosal Homeostasis of Mdr1-Deficient T Cells.
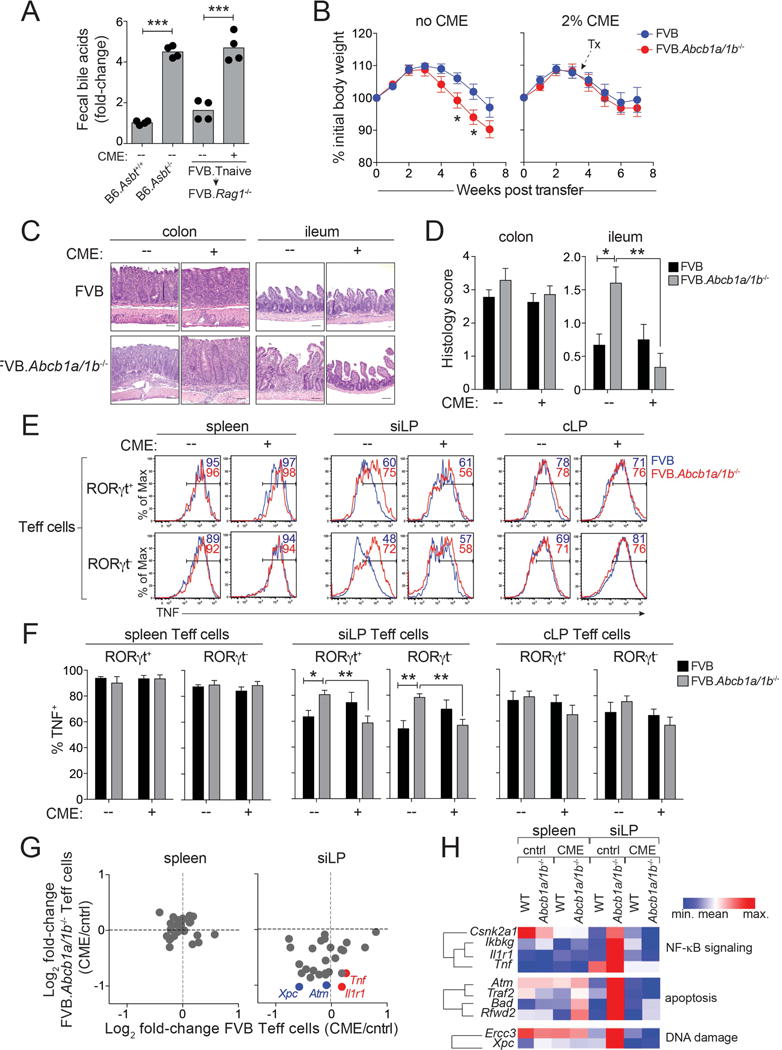
(A) Fecal bile acid concentrations in B6 wild-type or Asbt-deficient (B6.Slc10a2−/−) mice, or in transferred FVB.Rag1−/− mice (FVB.Tnaive ➔ FVB.Rag1−/−) treated +/− cholestyramine (CME) for 8 days. 4 mice per group are shown from one experiment.
(B) Mean weight loss (± SEM) in untreated (no CME; left; n = 18-19) or CME-treated (plus CME; right; n = 20–21) co-housed FVB.Rag1−/− mice injected with FVB wild-type (blue) or FVB.Abcb1a/1b−/− (red) naïve T cells. Weights relative to day 0. Incorporates data from 5 experiments.
(C) H&E-stained intestinal sections (colons, ilea) from transferred and co-housed FVB.Rag1−/− mice injected with FVB or FVB.Abcb1a/1b−/− Tnaive cells and treated +/− CME as in (B). Scale bars = 50 μM; 20x magnification. Representative of 7–8 mice over 3 experiments.
(D) Mean histology scores (+ SEM; n = 7–8) in colon (left) or ileum (right) of transferred FVB.Rag1−/− mice treated +/− CME as in (B). Incorporates data from 3 experiments.
(E) TNF expression in ex vivo-stimulated wild-type (FVB; blue) or FVB.Abcb1a/1b−/− (red) RORγt+/− effector T cells (Teff cells) from spleen, small intestine lamina propria (siLP) or colon lamina propria (cLP) of transferred FVB.Rag1−/− mice treated +/− CME as in (B). Representative of 5 mice over 2 experiments.
(F) Mean percentages of TNF+ Teff cells (± SEM; n = 5) as in (E).
(G) Gene expression, by nanostring, in ex vivo-isolated wild-type (FVB) or Mdr1-deficient (FVB.Abcb1a/1b−/−) Teff cells from spleen (left) or siLP (right) of transferred FVB.Rag1−/− mice treated +/− CME as in (B). 34 genes from Figure 4G–H were selected for analysis; examples of NF-κB-(red) or DNA damage-(blue) associated genes are annotated.
(H) Pathway analysis of genes analyzed in (G).
(G–H) Gene expression values are means from 4 mice per group over 2 experiments.
* P < .05, ** P < .01, *** P < .001 student’s t test.
Consistent with these therapeutic effects and similar to Asbt ablation (Figure 4E–F), CME treatment reduced both TNF production and oxidative stress-related gene expression in Mdr1-deficient, but not wild-type, siLP Teff cells (Figure 6E–H). By contrast, CME had no influence on cytokine production or gene expression in either wild-type or Mdr1-deficient Teff cells outside of the small intestine (Figure 6E–H, data not shown). Although bile acid sequestration alters liver metabolism (Bhat et al., 2003), both the selective regulation of Mdr1-deficient siLP Teff cell function and the similar pharmacodynamic effects of CME treatment and Asbt ablation suggest that CME acts locally in this model to prevent the pathogenic interaction between Mdr1-deficient Teff cells and CBAs in the ileum.
MDR1 Loss-of-Function in a Subset of Human Crohn’s Disease Patients
These results in mice suggest that MDR1 function may be impaired in some patients with ileal Crohn’s disease (CD). Whereas loss-of-function mutations in the human MDR1 gene, ABCB1, are rare and not generally associated with human IBDs (Jostins et al., 2012), we reasoned that identifying even a handful of MDR1-deficient ileal CD patients might inform both mechanistic understanding and personalized therapy. Therefore, we profiled MDR1-dependent Rh123 efflux in peripheral blood T cells from 183 healthy adults and IBD patients (Table S1). We determined the combined frequency of Rh123lo cells within 4-major MDR1-expressing CD4+ and CD8+ T cell subsets (Figure 7A–B), and we ensured assay reliability over time by analyzing Rh123 efflux in a control stock of healthy donor PBMC in parallel with each batch of samples (Figure 7C).
Figure 7. MDR1 Loss-of-Function in a Subset of Ileal Crohn’s Disease Patients.
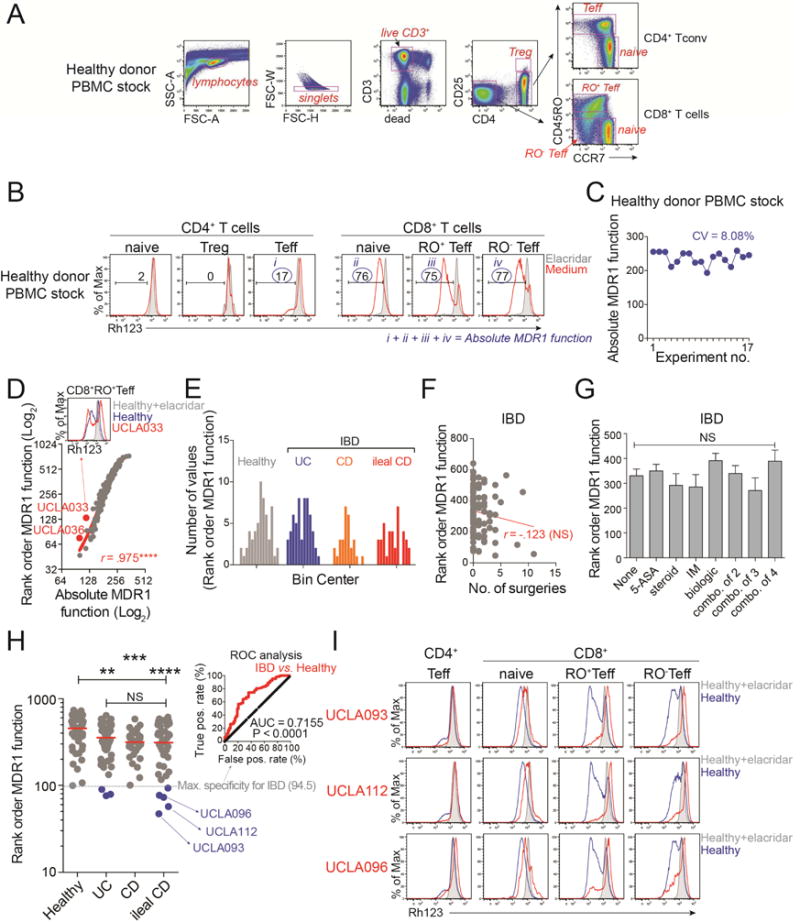
(A) FACS-based identification of CD4+ and CD4− (CD8+) human T cell subsets in human PBMC samples. Teff, effector T cells; Treg, T regulatory cells. Representative of 17 experiments.
(B) Rh123 efflux in T cell subsets (gated as in (A)) in the presence (shaded) or absence (red) of elacridar. The cumulative frequency of Rh123lo (MDR1+) cells within the four subsets indicated by blue circles reflects absolute MDR1 function. Representative of 17 experiments.
(C) Absolute MDR1 function in a control stock of healthy donor PBMC, determined over 17 independent experiments as in (A, B). The coefficient of variation (CV) is shown.
(D) Absolute (x-axis) vs. rank-order (y-axis) MDR1 function in 183 healthy controls or IBD patients. Outliers (red) represent patients in which MDR1 function is reduced in some, but not all, T cell subsets (example above).
(E) Distribution of rank-order MDR1 function within all healthy adults (n = 58), ulcerative colitis (UC) patients (n = 58), Crohn’s disease (CD) patients (n = 29), and ileal CD patients (n = 38). All data passed the D’Agostino-Pearson omnibus normality test.
(F) Correlation between rank-order MDR1 function and number (no.) of bowel surgeries in all IBD patients (n = 125). Red text indicates Pearson correlation (r). NS, not significant.
(G) MDR1 function in all IBD patients (n = 125) segregated based on medication history; NS, not significant, one-way ANOVA. 5-ASA, 5-aminosalicylic acid; IM, immuno-modulator (e.g., azathioprine, methotrexate); combo., combinations of 2, 3, or 4 medicines.
(H) Rank-order MDR1 function in healthy adults or IBD patient groups. Horizontal lines indicate median; dashed grey line indicates maximum (Max.) specificity (100%) for IBD patients (rank order MDR1 function = 94.5), by receiver operating characteristic (ROC) analyses (shown at top right). 8 IBD patients (3 UC, 5 ileal CD; highlighted blue) are below this threshold.
(I) Rh123 efflux in T cell subsets from three MDR1lo ileal CD patients (red peaks) highlighted blue in (H). Rh123 efflux in corresponding healthy control samples analyzed the same day is shown in the presence (shaded) or absence (blue) of elacridar. Data are from one experiment per patient.
** P < .01, *** P < .001, **** P < .0001, one-way ANOVA. See also Table S1.
While robust, this measure of “absolute” MDR1 function placed unequal weight on the 4 T cell subsets, with CD4+ Teff cells accounting for ≤ 10% of total MDR1 activity in most individuals (Figure 7B). Therefore, to more accurately reflect germline-encoded MDR1 function we established a rank-order scoring system that assigns a normalized value between 1 (lowest) and 183 (highest) for MDR1 function within each T cell subset; the sum of these values, or “rank-order” MDR1 function, lends equal weight (25%) to MDR1 function within each subset. Whereas absolute and rank-order MDR1 function strongly correlated within individuals (r = 0.975, P < .0001), rank-order scoring reduced the “false discovery” of patients where MDR1 function was compromised in some, but not all, T cell subsets (Figure 7D). We confirmed that rank-order MDR1 function was normally distributed within all control and patient groups (Figure 7E), and not generically influenced by bowel surgeries or medications (Figure 7F–G).
We found that MDR1 function was reduced, on aggregate, in all IBD patient groups relative to healthy controls, but was not different between discrete forms of IBD (Figure 7H). This result may reflect a general loss of mucosal homeostasis in all IBDs that impacts MDR1 regulation in the gut. At the same time, we identified 8 IBD patients (5 ileal CD, 3 UC) that displayed overt MDR1 loss-of-function below a maximum specificity threshold for IBD patients defined by receiver operating characteristic (ROC) analysis (Figure 7H). The area under curve (AUC) in ROC analysis was 0.72 with a confidence interval of 0.63–0.80, which was significantly different from 0.5 (P < .0001) indicating that rank-order MDR1 function could descriminate IBD patients from controls (Figure 7H). Therefore, we used a 100% specificity threshold for IBD patients (sensitivity = 6.4%; rank-order MDR1 function ≤ 94.5) to identify MDR1 “low-outliers” (Figure 7H). MDR1 low-outliers showed impaired Rh123 efflux in all 4 MDR1-expressing T cell subsets and were enriched in ileal CD (13%; 5/38), compared with either UC (5%; 3/58) or non-ileal CD (0%; 0/29) (Figure 7H–I, data not shown). Thus, these data in humans are consistent with our findings in mice that Mdr1 preferntially regulates immune function in the ileum.
DISCUSSION
Here we show that pro-inflammatory CD4+ Teff cells “adapt” upon migration into the ileum. Ileal adaptation involves upregulation of Mdr1, and permits homeostasis in the presence of locally-reabsorbed CBAs. These findings in Teff cells are consistent with the specialized functions acquired by Foxp3+ Tregs in several non-lymphoid tissues, including visceral adipose tissue, lung, skeletal muscle, and colon (Arpaia et al., 2015; Burzyn et al., 2013; Cipolletta et al., 2012; Schiering et al., 2014). Thus, adaptation in non-lymphoid tissues may be a general feature of circulating lymphocytes.
Our study reveals that BAs play a key role in shaping Teff cell function in the ileum. Due to their primary roles in digestion, CBAs are synthesized in the liver and reabsorbed in the ileum in manners that are influenced by, but not strictly dependent on, microbiota (Hofmann and Hagey, 2014). Accordingly, Mdr1 was highly expressed in intestinal Teff cells from germ free mice and required for mucosal Teff cell homeostasis in transferred Rag1−/− mice treated with antibiotics. Still, BAs are known to shape the enteric flora due to bactericidal and bacteriostatic activities (Lorenzo-Zuniga et al., 2003), and microbiota directly alter luminal BAs through enzymatic activities (Albenberg et al., 2012). Therefore, our results reveal a third-dimension in the interplay between BAs, the microbiota, and the mucosal immune system.
The size and composition of the mucosa-associated CBA pool in the ileum remains ill-defined. Current estimates consider CBAs in the ileal lamina propria at high micromolar concentrations, between those documented in the ileal lumen (1–10 mM) and portal vein (~ 50 μM) (Hofmann and Hagey, 2014). In line with these estimates, synthetic CBAs (e.g., tCDCA) modulated Mdr1 function and Teff cell homeostasis between 250-1,200 μM, and endogenous BAs from the intestinal lumen interacted with Mdr1 at even lower concentrations (25–100 μM). In vitro tCDCA treatment recapitulated several aspects of Mdr1-deficient Teff cell mucosal dysfunction in vivo, including oxidative stress, TNF over-expression, and decreased persistence. Nevertheless, understanding the full scope of mucosal Teff-CBA interactions in vivo will require new insights into the endogenous CBA pool, the duration of Teff cell persistence in the ileum, and how these parameters change during disease, dysbiosis, and dietary alteration.
Our results indicate that Mdr1 mitigates CBA-driven oxidative stress in Teff cells. This notion is supported by previous reports showing that CBAs kill transformed epithelial cells through oxidative stress (Ignacio Barrasa et al., 2011), and that Mdr1 protects against oxidative stress in intestinal epithelial cells (Ho et al., 2017). The only endogenous Mdr1 transport substrates described to date are short-chain phospholipids (van Helvoort et al., 1996), and these are generated during lipid peroxidation (Ayala et al., 2014). Thus, these data support a model in which Mdr1 limits CBA-driven lipid peroxidation by removing oxidized lipids from the T cell plasma membrane. This model predicts a fundamental role for Mdr1 in cellular protection from lipid peroxidation, which is consistent with both the broad expression of MDR1 in mammalian cells and the evolutionary conservation of MDR1 orthologs in prokaryotes. Indeed, Mdr1 also suppressed oxidative stress in Teff cells exposed to 13-HpODE – an oxidized derivative of linoleic acid – but had little impact on stress induced by other reactive oxygen species. A less likely possibility is that Teff cells internalize, and Mdr1 directly effluxes, CBAs.
Whatever the case, mucosal homeostasis coordinated by MDR1 and CBAs appears to be critical for immune homeostasis in the ileum, and may be compromised in a subset of ileal Crohn’s disease (CD). We identified 5 MDR1lo patients with aggressive ileal CD that included 2 males and 3 females, spanned three ethnic backgrounds, and ranged in age between 27–60. Whether MDR1 loss-of-function in these patients is due to genetic perturbation is not yet clear, though the breadth and magnitude of MDR1-deficiency in these patients is consistent with underlying genetic mutations. Given our data in mice, it will be important to determine whether bile acid sequestration is an effective therapy for MDR1lo CD patients. Whereas BA sequestrants have yet to be formally tested in CD, a recent study found that cholestyramine induced mucosal healing in a small cohort of IBD patients with primary sclerosing cholangitis (Pavlidis et al., 2015). Our data suggest a mechanistic basis for this clinical result, and encourage expanded efforts to test BA sequestrants in human CD.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Alexa Fluor 700 anti-mouse CD45, Clone 30-F11 | BioLegend | Cat# 103128 |
| BV650 anti-mouse CD3, Clone 17A2 | BioLegend | Cat# 100229 |
| BV711 anti-mouse CD4, Clone RM4-5 | BioLegend | Cat# 100557 |
| BV605 anti-mouse CD25, Clone PC61 | BioLegend | Cat# 102036 |
| Percp-Cy5.5 anti-mouse CD44, Clone IM7 | BioLegend | Cat# 103031 |
| BV605 anti-mouse CD62L, Clone MEL-14 | BioLegend | Cat# 104438 |
| APC anti-mouse IFNγ, Clone XMG1.2 | BioLegend | Cat# 505810 |
| APC anti-mouse F4/80, Clone BM8 | BioLegend | Cat# 123116 |
| APC anti-mouse CD4, Clone RM4-5 | BioLegend | Cat# 100515 |
| FITC anti-mouse IL-17A, Clone TC11-18H10.1 | BioLegend | Cat# 506907 |
| Percp-Cy5.5 anti-mouse IL-17a, Clone TC11-18H10.1 | BioLegend | Cat# 506919 |
| PE-Cy7 anti-mouse TNFα, Clone MP6-XT22 | BioLegend | Cat# 506323 |
| PE anti-mouse IL-4, Clone 11B11 | BioLegend | Cat# 504103 |
| PE anti-mouse CD45, Clone 30-F11 | BioLegend | Cat# 103105 |
| PE anti-mouse CD63, Clone NVG-2 | BioLegend | Cat# 143903 |
| PE anti-mouse CX3CR1, Clone SA011F11 | BioLegend | Cat# 149005 |
| Pacific Blue anti-mouse CD11b, Clone M1-70 | BioLegend | Cat# 101223 |
| Mouse Fc Block (purified anti-CD16/32), Clone 24G2 | BD Biosciences | Cat# 553141 |
| PE-CF594 anti-mouse CD25, Clone PC61 | BD Biosciences | Cat# 562694 |
| PE-CF594 anti-mouse RORγt, Clone Q31-378 | BD Biosciences | Cat# 562684 |
| PE anti-mouse Siglec-F, Clone E50-2440 | BD Biosciences | Cat# 552126 |
| PE anti-mouse Ly-6G, Clone RB6-8C5 | BD Biosciences | Cat# 108407 |
| FITC anti-mouse Ly-6G, Clone RB6-8C5 | BD Biosciences | Cat# 108405 |
| FITC anti-mouse CD103, Clone M290 | BD Biosciences | Cat# 557494 |
| APC anti-mouse CD11c, Clone N418 | BD Biosciences | Cat# 117309 |
| APC anti-mouse CD45, Clone 30-F11 | BD Biosciences | Cat# 559864 |
| eFluor 450 anti-mouse FOXP3, Clone FJK-16S | Thermo Fisher Scientific (eBioscience) | Cat# 48577382 |
| FITC anti-mouse IAb, Clone M5-114.15.2 | Thermo Fisher Scientific (eBioscience) | Cat# 11532185 |
| FITC anti-mouse CD80, Clone 16-10A1 | Thermo Fisher Scientific (eBioscience) | Cat# 11080182 |
| BV605 anti-human CD3, Clone OKT3 | BioLegend | Cat# 317321 |
| BV421 anti-human CD4, Clone OKT4 | BioLegend | Cat# 317433 |
| PE-Cy7 anti-human CD45RO, Clone UCHL1 | BioLegend | Cat# 304229 |
| Percp-Cy5.5 anti-human CCR7, Clone G034E3 | BioLegend | Cat# 353405 |
| PE-CF594 anti-human CD25, Clone M-A251 | BD Biosciences | Cat# 562525 |
| Soluble anti-mouse CD3ε, Clone 145-2C11 α | Bio X Cell | Cat# BE00011 |
| Soluble anti-mouse CD28, Clone 37.51 | Bio X Cell | Cat# BE00151 |
| Goat anti-syrian hamster IgG, Clone RB2153191 | Thermo Fisher Scientific (eBioscience) | Cat# 31115 |
| Purified anti-mouse IL-2, clone JES6-1A12 | R&D Systems, Inc. | MAB702 |
| Purified anti-mouse IL-4, clone 30340 | R&D Systems, Inc. | MAB404 |
| Purified anti-mouse IFNγ, clone 37895 | R&D Systems, Inc. | MAB485 |
| Biotin anti-mouse CD25 (IL-2Rα), clone PC61.5 | Thermo Fisher Scientific (eBioscience) | Cat# 13-0251-82 |
| Biological Samples | ||
| Human Blood Samples (Healthy volunteers) | OneBlood | https://www.oneblood.org/ |
| Human Blood Samples (IBD Patients) | The University of Miami | N/A |
| Human Blood Samples (Healthy volunteers, IBD patients) | The University of California Los Angeles | N/A |
| Chemicals, Peptides and Recombinant Proteins | ||
| Rhodamine 123 | Sigma-Aldrich | Cat# R8004 |
| Elacridar | Sigma-Aldrich | Cat# SML0486 |
| Fixable viability dye eFluor® 450 | eBioscience | Cat# 65086314 |
| Fixable viability dye eFluor® 506 | eBioscience | Cat# 65086614 |
| Fixable viability dye eFluor® 660 | eBioscience | Cat# 65086414 |
| 7-ADD viability staining solution | BioLegend | Cat# 420403 |
| Phorbol 12-myristate 13-acetate (PMA) | Sigma-Aldrich | Cat# P8139 |
| Ionomycin | Sigma-Aldrich | Cat# I9657 |
| Brefeldin A | Sigma-Aldrich | Cat# B5936 |
| DMEM | Thermo Fisher Scientific | Cat# 11995040 |
| Fetal Bovine Serum | BioFluid Technologies | Cat# BT201500D |
| Paraformaldehyde | Sigma-Aldrich | Cat# P6148 |
| Saponin | Sigma-Aldrich | Cat# S7900 |
| L-Glutamine (200mM) | Thermo Fisher Scientific | Cat# 25030081 |
| 2-Mercaptoethanol | Sigma-Aldrich | Cat# M3148 |
| MEM Vitamin Solution | Thermo Fisher Scientific | Cat# MT25020CI |
| MEM Non-Essential Amino Acids Solution (100X) | Thermo Fisher Scientific | Cat# 11140050 |
| Sodium Pyruvate (100mM) | Thermo Fisher Scientific | Cat# 11360070 |
| L-Arginine | Sigma-Aldrich | Cat# A5006100G |
| L-Asparagine | Sigma-Aldrich | Cat# A4159100G |
| Folic Acid | Sigma-Aldrich | Cat# F875825G |
| HEPES Buffer (1M) | Thermo Fisher Scientific | Cat# BP299100 |
| PmeI | New England BioLabs | Cat# R0560S |
| NotI | New England BioLabs | Cat# R3189S |
| Formalin solution, neutral buffered, 10% | Sigma-Aldrich | Cat# HT501128 |
| Ampicillin | Fisher Scientific | Cat# 1712545GM |
| Vancomycin | Sigma-Aldrich | Cat# 861987-1G |
| Metronidazole | VWR Intl | Cat# IC15571025 |
| Neomycin | Fisher Scientific | Cat# BP26695 |
| Streptomycin | Sigma-Aldrich | Cat# I9657 |
| Cholestyramine | Sigma-Aldrich | Cat# C4650 |
| tert-Butanol | Sigma-Aldrich | Cat# 24127 |
| DMEM without phenol red | Thermo Fisher Scientific | Cat# 21063029 |
| DTT (Dithiothreitol) | Sigma-Aldrich | Cat# DTTRO |
| EDTA | Amresco | Cat# E177 |
| Liberase TL | Sigma-Aldrich | Cat# 5401020001 |
| RNase-free DNaseI | Sigma-Aldrich | Cat# 4716728001 |
| Percoll | Sigma-Aldrich | Cat# P1644 |
| Paraformaldehyde | Sigma-Aldrich | Cat# 158127 |
| Cholic Acid | Sigma-Aldrich | Cat# C1129 |
| Glyco-Cholic Acid | Sigma-Aldrich | Cat# G2878 |
| Tauro-Cholic Acid | Sigma-Aldrich | Cat# T4009 |
| Deoxycholic Acid | Sigma-Aldrich | Cat# D2510 |
| Glycol-Deoxycholic Acid | Sigma-Aldrich | Cat# G7132 |
| Tauro-Deoxycholic Acid | Sigma-Aldrich | Cat# T0557 |
| Chenodeoxycholic Acid | Sigma-Aldrich | Cat# C9377 |
| Glyco-Chenodeoxycholic Acid | Sigma-Aldrich | Cat# G0759 |
| Tauro-Chenodeoxycholic Acid | Sigma-Aldrich | Cat# T6260 |
| Hydrogen Peroxide | Thermo Fisher Scientific | Cat# H325 |
| Menadione | MP Biomedicals | Cat# 0210225925 |
| 13-hydroperoxy-octadecadienoic acid (13-HpODE) | Cayman Chemical | Cat# 33964759 |
| N-acetyl-L-cysteine (N-Ac) | Sigma-Aldrich | Cat# A9165-5G |
| Buffer RLT | Qiagen | Cat# 79216 |
| Abcb1a-Mouse-Taqman Gene Expression Assays | Thermo Fisher Scientific | Assay ID Mm00440761_m1 |
| GAPDH-Mouse-Taqman Gene Expression Assays | Thermo Fisher Scientific | Assay ID Mm999999915_g1 |
| Recombinant human IL-2 | NIH AIDS Reagent Program | Cat# 11697 |
| Recombinant mouse IL-12 | R&D Systems, Inc. | Cat# 419-ML |
| Recombinant mouse IL-4 | R&D Systems, Inc. | Cat# 404-ML |
| Recombinant human TGF β1 | R&D Systems, Inc. | Cat# 240-B |
| Recombinant mouse IL-6 | R&D Systems, Inc. | Cat# 406-ML |
| Recombinant human IL-1β | R&D Systems, Inc. | Cat# 201-LB |
| Recombinant human IL-23 | R&D Systems, Inc. | Cat# 1290-IL |
| Critical Commercial Assays | ||
| Mouse T cell-activator CD3/CD28 magnetic beads | Thermo Fisher Scientific | Cat# 11456D |
| EasySep Mouse CD4+ T Cell Isolation Kit | StemCell Technologies | Cat# 19852 |
| High-Capacity RNA-to-cDNA kit | Thermo Fisher Scientific | Cat# 4387406 |
| Power SYBR Green PCR Master Mix | Life Technologies | Cat# 4368706 |
| Taqman Universal PCR Master Mix | Thermo Fisher Scientific | Cat# 4304437 |
| QuickChange Lightning Site-Directed Mutagenesis Kit | Agilent Technologies | Cat# 210518 |
| Total Bile Acids Assay Kit | Diazyme Laboratories | Cat# DZ092A-K |
| QIAamp DNA Stool Mini Kit | Qiagen | Cat#51504 |
| Ion 16S Metagenomics kit | Thermo Fisher Scientific | Cat# A26216 |
| eBioscience™ Foxp3 / Transcription Factor Staining Buffer Set | Thermo Fisher Scientific | Cat# 00-5523-00 |
| Cellular Reactive Oxygen Species (ROS) Detection Assay Kit (Deep Red Fluorescence) | Abcam | Cat# AB186029 |
| RNeasy Mini Kit | Qiagen | Cat#74104 |
| SMART-Seq v4 Ultra Low Input Kit | Clontech Inc. | Cat# 634888 |
| NEB Next Ultra II DNA Library Prep Kit | New England Biolabs | Cat# E7645S |
| Deposited Data | ||
| N/A | ||
| Experimental Models: Cell Lines | ||
| Platinum-E (PLAT-E) | Dr. Matthew Pipkin The Scripps Research Institute | N/A |
| NIH-3T3 | Dr. Matthew Pipkin The Scripps Research Institute | N/A |
| EL4 | Dr. Jun-Li Luo The Scripps Research Institute | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | Jackson Laboratory | Stock# 000664 |
| Mouse: B6.Rag1 −/− | Jackson Laboratory | Stock# 002216 |
| Mouse: B6.Il17aCre | Jackson Laboratory | Stock# 016879 |
| Mouse: B6.ROSA26LSL-hCD2 | Dr. Klaus Rajewsky Max Delbruck Center for Molecular Medicine | N/A |
| Mouse: B6.Il17a+/Cre ROSA26+/hCD2 | This paper | N/A |
| Mouse: FVB/N | Taconic | Model# FVB |
| Mouse: FVB.Abcb1a/1b −/− | Taconic | Model# 1487 |
| Mouse: FVB.Rag1−/− | Dr. Allan Bieber Mayo Clinic | N/A |
| Mouse: BALB/c | Osaka University | CLEA Japan, Inc. |
| Mouse: B6.Abcb1aAME/+ | New York University | Generated at the NYU Rodent Genetic Engineering Core |
| Mouse: B6.Slc10a2−/− | Dr. Paul Dawson Emory University | N/A |
| Mouse: B6.Rag1−/− Slc10a2−/− | This paper | N/A |
| Oligonucleotides | ||
| Abcb1a ametrine 5′ CRISPR gRNA: 5′-GCUUCUCAAUGGUCAGUGUGCUGUUUUA GAGCUAGAAAUAGCAAGUUAAAAUAAGG CUAGUCCGUUAUCAACUUGAAAAAGUGG CACCGAGUCGGUGCUUUU-3′ | PNA Bio Inc. | N/A |
| ametrine Abcb1a 3′ CRISPR gRNA: 5′-GAAAAUACUUAACAUCUUACAUGUUUUA GAGCUAGAAAUAGCAAGUUAAAAUAAGG CUAGUCCGUUAUCAACUUGAAAAAGUGG CACCGAGUCGGUGCUUUU-3′ | PNA Bio Inc. | N/A |
| Universal 16S rDNA Forward: 5′-ACTCCTACGGGAGGCAGCAGT-3′ | Integrated DNA Technologies | Ayres et al., 2012 |
| Universal 16S rDNA Reverse: 5′-ATTACCGCGGCTGCTGGC-3′ | Integrated DNA Technologies | Ayres et al., 2012 |
| Abcb1a ametrine 5′ Genotype Primer: 5′-AGTTTAACGTGTCTGCAGCTGG-3′ | Integrated DNA Technologies | N/A |
| Abcb1a ametrine 3′ Genotype Primer: 5′-AGCCTGCAGGATCTGTCTG-3′ | Integrated DNA Technologies | N/A |
| Recombinant DNA | ||
| Plasmid: LMPd-ametrine | Chen et al., 2014 | Dr. Matthew Pipkin |
| Plasmid: LMPd-GFP | This paper | N/A |
| Plasmid: pSTBlue-1 | Novagen | Cat# 70199 |
| shRNAmir: Cd8a | TransOMIC Technologies | Cat# TLMSU1400-12525 |
| shRNAmir: Abcb1a | TransOMIC Technologies | Cat# TLMSU1400-18671 |
| shRNAmir: Abcb1b | TransOMIC Technologies | Cat# TLMSU1400-18669 |
| pHaMDRwt | Pastan et al., 1988 | Addgene Plasmid #10957 |
| LMPd.hMDR1-ametrine (containing wild-type human MDR1) (NCBI reference sequence NM_001348945.1) | This paper | N/A |
| LMPd. hMDR1-ametrine (containing transport-deficient human MDR1; Y401A, Y1044A) α | This paper | Kim et al., 2006 |
| Software and Algorithms | ||
| Graphpad Prism 7 | GraphPad Software | http://www.graphpad.com |
| FlowJo (Version 9.9.4) | TreeStar, Inc. | http://www.flowjo.com |
| GenePattern (Version 3.9.10) | Broad Institute | https://genepattern.broadinstitute.org |
| nSolver Analysis Software (Version 1.1) | Nanostring | https://www.nanostring.com |
| Other | ||
| NanoString nCounter Sundrud_03 chip | This paper | https://www.nanostring.com |
| NanoString nCounter Sundrud_04 chip | This paper | https://www.nanostring.com |
Contact for Reagent and Resource Sharing
Further information and requests for reagents should be directed to and will be fulfilled by the Lead Contact, Mark Sundrud (msundrud@scripps.edu).
Experimental Model and Subject Details
Mice
Wild-type C57Bl/6 (B6; stock no. 000664), B6.Rag1−/− (stock no. 002216) and B6.Il17aCre (stock no. 016879) mice were purchased from The Jackson Laboratory. Wild-type FVB/N (model no. FVB) and FVB.Abcb1a−/−Abcb1b−/− (abbreviated throughout as FVB.Abcb1a/1b−/−; model no. 1487) mice were purchased from Taconic. BALB/c mice were purchased from CLEA (Japan) and maintained under specific pathogen-free (SPF) or germ-free (GF) conditions at Osaka University. ROSA26LSL-hCD2 transgenic mice were provided by Dr. Klaus Rajewsky (Max Delbruck Center for Molecular Medicine, Berlin, Germany). B6.Slc10a2−/− mice were obtained from Dr. Paul Dawson (Emory University). FVB.Rag1−/− mice were a gift of Dr. Allan Bieber (Mayo Clinic, Rochester, MN). Mdr1-reporter (Abcb1aametrine) mice were generated at the Rodent Genetic Engineering Core (RGEC) of New York University School of Medicine (NYUSM). Il17aCre mice were crossed with ROSA26LSL-hCD2 transgenic animals to produce Il17a+/CreROSA26+/hCD2 Th17 fate-mapping reporter mice. Asbt-deficient (B6.Slc10a2−/−) mice were crossed with B6.Rag1−/− mice to generate B6.Rag1−/− Slc10a2−/− animals. In vitro experiments used T cells isolated from 8-to 14-week old male and female mice. T cell transfer colitis experiments used 6-to 10-week old female donor mice, and 8- to 12-week old syngeneic female recipients. This study was approved by Institutional Animal Care and Use Committees at The Scripps Research Institute (TSRI) Florida, Osaka University, and NYU Medical Center.
Human blood samples
Human blood samples were collected and analyzed in accordance with protocols approved by Institutional Review Boards at TSRI, OneBlood (Orlando, Florida), The University of Miami, and The University of California Los Angeles (UCLA). Blood was obtained after informed written consent, and consenting volunteers willingly shared clinical history and demographic data prior to phlebotomy. Institutional Review Boards at OneBlood, The University of Miami, and UCLA approved all procedures and forms used in obtaining informed consent, and all documentation for consenting volunteers is stored at OneBlood, The University of Miami or UCLA. Cryopreserved PBMC samples were stored in de-identified vials, and shipped to TSRI Florida for analyses.
Cell lines
EL4 cells are a mouse thymic lymphoma cell line, and were obtained from Dr. Jun-Li Luo (TSRI Florida). Human platinum-E (PLAT-E) cells are derived from the HEK-293 human embryonic kidney fibroblasts and engineered for improved retroviral packaging efficiency. NIH-3T3 cells are a mouse embryonic fibroblast cell line. PLAT-E and NIH-3T3 cells used for this study were provided by Dr. Matthew Pipkin (TSRI Florida). All cell lines were tested to be mycoplasma free, and cultured in DMEM plus 10% FBS, 2 mM L-glutamine, 50 uM 2-mercaptoethanol, 1% HEPES, and 100 U/mL Pen-Strep. We are not aware of any reports that disclose the gender of origin for EL4, PLAT-E, or NIH-3T3 cells.
Primary cell cultures
Single-cell suspensions of splenocytes magnetically purified CD4+ T cells from wild-type or T cell-transferred Rag1−/− mice, were cultured in DMEM plus 10% fetal bovine serum (BioFluid Technologies), 2mM L-glutamine, 50uM 2-mercaptoethanol, 1% MEM vitamin solution, 1% MEM non-essential amino acids solution, 1% Sodium Pyruvate, 1% Arg/Asp/Folic acid, 1% HEPES and 100u/ml Pen-Strep.
Method Details
Generation of Mdr1-Reporter (Abcb1aametrine) Mice
Mdr1-reporter (Abcb1aametrine) mice were generated using the Cas9-cleavase system in wild-type C57Bl/6 zygotes. Briefly, two guide RNAs (gRNAs) were identified using a publically available CRISPR design tool (http://crispr.mit.edu), which are predicted to introduce single-stranded nicks on opposite DNA strands 22 bp upstream and 10 bp downstream of the Abcb1a stop codon; synthetic gRNAs were purchased (PNA Bio) and Cas9-dependent cleavage at the Abcb1a locus was confirmed in vitro prior to zygote microinjection as previously described (Kim et al., 2014). The sequences of the gRNAs used in this targeting are listed in the Key Resources Table above. To facilitate reporter transgene insertion, we constructed a homology-directed DNA repair (HDR) template. 500 bp arms of homology (AOH; 5′ and 3′) were purchased (IDT); the 5′ AOH contains sequences corresponding to all of Abcb1a exon 28 (204 bp) – minus the stop codon – plus the last 204 bp of intron 27–28; the 3′ AOH contains the first 500 bp of the 3′ untranslated region (UTR) downstream of the stop codon. Synonymous point mutations were introduced into both 5′ and 3′ AOH at the protospacer-adjacent motifs (PAMs) to remove Cas9-cleavase activity towards the HDR template. AOHs were then assembled into an ampicillin-resistant cloning vector (pSTBlue-1, Novagen) together with bicistronic reporter elements in the following order: 5′ AOH (no Abcb1a stop codon) – glycine-serine-glycine (GSG) linker (in-frame, 5′-GGATCCCGA-3′) –P2A peptide (in-frame) – ametrine open reading frame (in-frame, with stop codon) − 3′ AOH. Correct assembly was confirmed by sequencing, and the linear HDR template was excised from pSTBlue-1 via restriction digest (PmeI/NotI), and gel purified for use in zygote injections. Approximately 150 C57Bl/6 zygotes were generated by in vitro fertilization (IVF), and micro-injected with: gRNAs (10 μg each), linearized HDR template (10 μg), and Cas9-cleavase mRNA (10 μg). 76 zygotes were gown to the 400-cell blastocyst stage and implanted into 8-week old female surrogate mothers; live pups were analyzed for transgene insertion using genotyping primers that anneal outside of the targeted region (see Key Resources Table above). 19 live pups were recovered from the targeting; one female founder pup showed correct insertion of the full transgene by genotyping.
T Cell Transfer Colitis
For T cell transfer experiments using wild-type FVB or FVB.Abcb1a/1b−/− T cells, 0.5 × 106 FACS-sorted naïve CD4+ T cells were injected intraperitoneally (i.p.) into syngeneic FVB.Rag1−/− recipients. For transfers of shCD8- or shMdr1-expressing T cells, magnetically isolated wild-type (FVB or B6) CD4+CD25− T cells were activated in vitro using plate-bound anti-CD3/anti-CD28 antibodies, transduced 24 hr later with shRNAmir-expressing retroviruses, and expanded until day 5 in media supplemented with recombinant human IL-2. In some experiments, 0.5 × 106 unsorted T cells, containing both transduced and untransduced T cells, were transferred into syngeneic Rag1−/− mice. For experiments assessing weight loss and/or intestinal inflammation by histology, day 5-expanded and transduced T cells were FACS sorted as live (viability dye−) ametrine+ cells prior to transfer. For congenic transfer experiments, T cells transduced in vitro with GFP- or ametrine-labelled shCD8- or shMdr1-expressing retroviruses as above, were FACS-sorted on day 5, mixed at 1:1:1 ratios (untransduced:GFP+:ametrine+), checked by FACS to confirm equal ratios, and then transferred (0.3−0.6 × 106 mixed cells/mouse) into syngeneic Rag1−/− mice. All Rag1−/− recipients were weighed immediately prior to T cell transfer to determine baseline weight, and then weighed twice weekly after T cell transfers for the duration of the experiment. Transferred Rag1−/− mice were euthanized upon losing 20% of pre-transfer baseline weight. Rag1−/− mice receiving different donor T cell genotypes (i.e., wild-type vs. Mdr1-deficient T cells; shCD8- vs. shMdr1-expressing T cells) were co-housed throughout to normalize microflora exposure.
Histology
Colon (proximal, distal) or small intestine (proximal, mid, distal/ileum) sections (~ 1 cm) were cut from euthanized Rag1−/− mice 6-to 8-weeks post-T cell transfer. In some experiments, 10 cm segments of distal small intestine and whole colon were dissected from mice and fixed intact. All tissues were fixed in 10% neutral buffered formalin, embedded into paraffin blocks, cut for slides, and stained with hematoxylin and eosin (H&E). H&E-stained sections were analyzed and scored blindly; colons as in (Wirtz et al., 2007), and small intestines/ilea as in (Kosiewicz et al., 2001).
Treating transferred Rag1−/− mice with antibiotics
Rag1−/− mice receiving different donor T cell genotypes (i.e., wild-type vs. Mdr1-deficient T cells; shCD8- vs. shMdr1-expressing T cells) were separated upon initial onset of weight loss (typically 3- to 4-weeks post-T cell transfer) into cages that received water alone, or water containing antibiotics: ampicillin (1 M), vancomycin (0.5 M), metronidazole (1 M), neomycin (1 M), streptomycin (2 M). Bottles containing water with or without antibiotics were replaced twice weekly during assessment of weights.
Treating transferred Rag1−/− mice with cholestyramine
Cholestyramine (CME) was administered as a dietary supplement to transferred Rag1−/− mice. Briefly, 1 kg of standard chow pellets (2920X Teklad irradiated global soy protein free extruded rodent diet, Envigo) were pulverized to a fine powder using a Professional Series 200 food processor (VitaMix), weighed, and supplemented with CME (2% w:w). An equal amount of ground powder food without CME was used as the control diet. One week prior to initiating treatment (typically 2-weeks post-T cell transfer), solid food was removed and replaced with control powder food (no CME) provided in covered feeding containers (Ancare Corporation). Upon onset of weight loss, transferred Rag1−/− mice were separated into cages receiving powder food with or without CME. Each cage contained mixtures of Rag1−/− mice receiving wild-type or Mdr1-null T cells. Covered feeding containers were exchanged, cleaned, and autoclaved weekly.
Bile acid measurements
Bile acid concentrations in feces (for CME treatment experiments) or small intestine luminal content (siLC) were quantified using the Total Bile Acids Assay Kit (Diazyme Laboratories) per manufacturer’s instructions. For bile acid measurements in CME treatment experiments, bile acids were extracted with tert-Butanol as previously described (Bhat et al., 2003).
Microbiome analyses
Genomic DNA from fecal stool of transferred Rag1−/− mice was purified by QIAamp DNA Stool Mini Kit (Qiagen) according to manufacturer’s instructions. Total 16S rDNA was quantified by real-time PCR performed on a StepOnePlus real time PCR instrument (Life Technologies/Applied Biosystems), using Power SYBR green mastermix and universal 16S rDNA primers listed above in the Key Resources Table. Bacterial species were quantified by 16S ribosomal DNA sequencing using the Ion 16S Metagenomics kit (Life Technologies). Genomic DNA from cecal stool was amplified utilizing two primer sets that selectively amplify the hypervariable regions against 16S rRNA gene V2-4-8 and V3-6, 7-9, respectively. Amplicons for each sample were pooled and adaptor ligated with barcoded adaptors specific for Ion Torrent platforms. Samples were then pooled at equimolar ratios and sequenced using 400 bp chemistry on Ion Torrent Personal Genome Machine using 316 and 318 chips. Data was analyzed with Ion Reporter Software Ion 16S Metagenomics kit analysis module (Life Technologies).
Mononuclear cell isolation
Single-cell suspensions were prepared from spleen, peripheral lymph nodes, or mesenteric lymph nodes (MLN) by flushing through 70 μm nylon filters (BD Biosciences). For isolating cells from intestinal tissues, whole small intestines (duodenum to terminal ileum), small intestinal segments (proximal 1/3, middle 1/3, distal 1/3), or whole colons (cecum to anus) were removed, flushed with PBS to remove the fecal contents, and opened longitudinally; peyers patches were excised from small intestines. Tissues were cut into small (~ 3 cm) segments, and incubated for 30 min at room temperature in DMEM media without phenol red (Thermo Fisher Scientific) plus 0.15% DTT (Sigma-Aldrich) to remove mucus. After washing with media, intestines were incubated for 30 min at room temperature in media containing 1 mM EDTA (Amresco) to remove the epithelium. After washing again with media, lamina propria was digested in media containing 0.25 mg/mL liberase TL and 10 U/mL RNase-free DNaseI (both from Roche) in a bacterial incubator (Environ Shaker; Labline) for 15–25 min at 37°C. Single cell suspensions were passed through 70 μm nylon filters and mononuclear cells were isolated by 70/30% percoll gradient centrifugation (Sigma-Aldrich). Mononuclear cells were washed twice in complete T cell medium, counted, and resuspended for FACS analysis or sorting.
CD4+ T cell isolation
CD4+CD25− T cells were magnetically purified from spleen mononuclear cells using an EasySep T cell negative isolation kit (Stem Cell Technologies, Inc.), Manufacturer’s instructions were followed, except for the addition of a separate biotin anti-mouse CD25 antibody (0.5 μg/mL; eBioscience) along with the supplied antibody cocktail. For Rag1−/− transfer experiments, magnetically-enriched CD4+CD25− T cells from spleens of donor mice were FACS-sorted to obtain pure naïve T cells (CD3+CD4+CD25−CD62LhiCD44lo).
Isolation of antigen-presenting cells
Mononuclear cells from spleen or small intestinal lamina propria (siLP) were recovered as previously described (Atarashi et al., 2008), incubated with blocking antibodies (anti-CD16/32) for 5 min at 4°C, stained with antibodies against CD45, CD11b, and CD11c, and sorted based on differential CD11b/CD11c expression within the live (viability dye−) CD45+ gate. Spleen dendritic cells (DCs) were sorted as CD45+CD11c+CD11b−. For some experiments, Rh123hi (Mdr1−) or Rh123lo (Mdr1+) Teff cells (CD45+CD3+CD4+CD25−CD62LhiCD44lo) from MLN or siLP were FACS-sorted, following ex vivo Rh123 labeling and efflux; background Rh123 efflux in cells treated with elacridar (0.1 μM) was used to set sorting gates.
Cell surface FACS analysis
Cell surface FACS staining was performed in PBS by incubating cells with fluorochrome-conjugated antibodies and fixable viability dyes (see Key Resources Table above) for 20 min at room temperature. At completion, cells were washed once with PBS, resuspended in either PBS (for Rh123-labelled samples), or PBS plus 1% PFA, and acquired on LSRII or FACSCantoII cytometers (BD Biosciences). The Cellular Reactive Oxygen Species (ROS) Detection Assay Kit (Deep Red Fluorescence; Abcam) was used to assess ROS production. The ROS probe was added together with viability dyes and surface antibody cocktails in PBS, and incubated for 30 min at room temp. Stained cells were washed with PBS, fixed in PBS plus 4% PFA, and run immediately on the flow cytometer. For Rh123 labelling and efflux, whole mononuclear cells or purified T cells were labelled with complete medium containing 1 μg/mL Rh123 for 30 min on ice. Labelled cells were washed with cold media, resuspended in room temp media with or without elacridar (0.1 μM), and then placed in a 37°C tissue culture incubator for 30–60 min to permit Mdr1-dependent efflux. After efflux, cells were washed and stained with viability dyes and surface antibodies in ice-cold PBS for 20 min. After staining, cells were washed and resuspended in cold PBS, and acquired immediately on a cytometer. For Rh123 labelling and efflux experiments in the presence of mouse intestinal contents or synthetic bile acids, cells were kept in PBS (not media) throughout the protocol.
Intracellular FACS analysis
For intracellular stains including transcription factors (e.g., RORγt, Foxp3), cells were washed in PBS after surface staining, and then fixed and permeabilized using a Foxp3 intracellular staining kit (eBioscience) in accordance with manufacturer’s instructions. For intracellular stains not including transcription factors (i.e., cytokines only), surface-stained cells were washed in PBS and fixed in PBS plus 4% PFA (15–20 min, room temp). After washing again in PBS, fixed cells were permeabilized in PBS plus 0.5% saponin and 1% FBS (10 min, room temp). Fluorochrome-conjugated anti-cytokine antibodies (see Key Resources Table above) were added directly to fixed cells and incubated at room temp for another 20 min. At completion, cells were washed once with PBS, resuspended in PBS plus 1% PFA, and acquired as above. All intracellular staining was performed after stimulating cells (4 hr) in media containing phorbol 12-myristate 13-acetate (PMA; 10 nM), ionomycin (1 μM), and brefeldin A (10 μg/mL) (all from Sigma-Aldrich).
FACS sorting
For cell sorting, stained cells were passed through 70 μm nylon filters, resuspended in DMEM media plus 2% serum, and sorted on a FACS AriaII machine (BD Biosciences). Sorted cells were collected in serum-coated tubes containing complete T cell media plus 50% serum. Gates used to sort Mdr1+/− T cells based on Rh123 efflux were set based on background Rh123 efflux in elacridar-treated cells.
FACS analysis
FACS data was analyzed using FlowJo software (TreeStar, Inc.). Throughout, Mdr1 transport activity was quantified by dividing the mean fluorescence intensity (MFI) of Rh123 in elacridar-treated cells by that of cells incubated in media alone, or media containing mouse intestinal content or synthetic bile acids.
Naïve CD4+ T cell differentiation
Magnetically-purified CD4+CD25− T cells from spleens of wild-type C57Bl/6 (B6) mice were activated in sterile, tissue-culture-treated 96-well flat-bottom plates (Falcon). Wells were coated for 1–2 hr at 37°C with goat-anti-hamster whole IgG (0.3 mg/mL). After washing with PBS, T cells resuspended in media were added to coated wells along with hamster-anti-mouse CD3ε (0.3 μg/mL) and hamster-anti-mouse CD28 (0.5 μg/mL). For non-polarizing conditions (ThN), T cell activation was performed in media alone. For polarization into Th1, Th2, iTreg, or non-pathogenic or pathogenic Th17 cells, the following sets of neutralizing antibodies and recombinant cytokines (see Key Resources Table above) were added at the time of anti-CD3/anti-CD28 stimulation:
Th1 – 20 ng/mL mouse IL-12 plus 0.5 μg/mL anti-mouse IL-4; Th2 – 20 ng/mL mouse IL-4 plus 0.5 μg/mL anti-mouse IFNγ; iTreg – 1 ng/mL human TGFβ1 plus 0.5 μg/mL anti-mouse IL-4 and 0.5 μg/mL anti-mouse IFNγ; non-pathogenic Th17 – 1 ng/mL human TGFβ1 plus 10 ng/mL mouse IL-6, 0.5 μg/mL anti-mouse IL-4, 0.5 μg/mL anti-mouse IFNγ and 1 μg/mL anti-mouse IL-2; pathogenic Th17 – 1ng/mL human IL-1β plus 10 ng/mL mouse IL-6, 20ng/mL human IL-23, 0.5 μg/mL anti-mouse IL-4, 0.5 μg/mL anti-mouse IFNγ and 1 μg/mL anti-mouse IL-2. Activated T cells were removed from antibody-coated wells after 48 hr (2 days), and expanded in uncoated wells until day 4 or 5. All cytokines and neutralizing antibodies were re-added at day 2 together with 10 IU/mL recombinant human IL-2 (for ThN, Th1, Th2, and iTreg conditions). No exogenous IL-2 was added to non-pathogenic or pathogenic Th17 cultures.
Induction of Mdr1 expression in CD4+ T cells by antigen-presenting cells
FACS-sorted Rh123hi (Mdr1−) CD4+ effector/memory (Teff) cells from small intestinal lamina propria (siLP) were co-cultured in U-bottom 96-well plates with FACS-sorted antigen-presenting cell (APC) subsets from siLP or spleen at a 1:2 ratio and stimulated with soluble anti-CD3 (1 μg/ml). Mdr1 expression was analyzed on day 4 post-stimulation by Rh123 labeling and efflux, followed by staining with 7-AAD and anti-CD4.
Retroviral plasmids and transductions
All recombinant DNAs used in this study are listed in the Key Resources Table above. shRNAmirs against mouse CD8 (Cd8a) and Mdr1 (Abcb1a, Abcb1b) were purchased from TransOMIC Technologies. shRNAmirs were PCR amplified and cloned into GFP- or ametrine-expressing murine retroviral vectors (LMPd) containing the enhanced miR-30 cassette (Fellman et al., 2013). HA-tagged human ABCB1 (hMDR1) was purchased from Addgene. Full-length ABCB1 was PCR-amplified without the HA-tag, and cloned into an ametrine-expressing LMPd vector without the miR-30 expression cassette. An empty LMPd.ametrine vector was generated by cutting out ABCB1 from the modified vector, blunting the restriction sites, and re-ligating. Transport-defective mutant MDR1 (ΔMDR1, Y401A/Y1044A; Kim et al., 2006) was generated using a QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies). All retroviral constructs were confirmed by sequencing prior to use in cell culture experiments. Retroviral particles were produced in PLAT-E cells; harvested supernatants were filtered through a 45 μm filter (BD Biosciences), concentrated by centrifugation (6,000 × g; 18 hr; 4°C) and used to transduce NIH-3T3 fibroblasts or 24 hr-stimulated CD4+CD25− T cells.
Isolation of intestinal luminal contents
Feces from colons or small intestines were collected in 15 mL conical tubes, weighed, and diluted in media or PBS (2 parts media or PBS:1 part feces). Contents were mixed thoroughly by high-speed vortexing for 2 min, and feces was pelleted by centrifugation (1,000 × g; 10 min; room temperature). Supernatants were transferred to clean eppindorf tubes and cleared of debris by another round of high-speed centrifugation (13,000 × g; 20 min; room temperature). Supernatants were re-collected, sterile-filtered through 0.2 μm syringe filters (BD Biosciences), aliquoted, and stored at −80°C. Lumen content from small intestine (siLC) or colon (cLC) was added to Rh123-labelled splenocytes or purified CD4+CD25− T cells at dilutions ranging from 1–16% (2-fold dilutions) of total culture volume (v:v). Unless otherwise stated, results show T cells treated with 4% or 8% siLC or cLC. In some experiments, bile acids were depleted in siLC by pre-treatment with cholestyramine (CME). Here, siLC was treated for 1 hr with PBS, or PBS plus 50 μg/mL CME. CME-bound material was pelleted by centrifugation (1 min; 13,000 × g; room temperature), and supernatants were collected and used to treat Rh123-labelled T cells.
Bile acids
Synthetic bile acids: cholic acid (CA), glyco-cholic acid (gCA), tauro-cholic acid (tCA), deoxycholic acid (DCA), glyco-deoxycholic acid (gDCA), tauro-deoxycholic acid (tDCA), chenodeoxycholic acid (CDCA), glyco-chenodeoxycholic acid (gCDCA), or tauro-chenodeoxycholic acid (tCDCA) (all from Sigma-Aldrich) were reconstituted in PBS, aliquoted, and stored at 4°C.
Modulation of T cell cytokine expression by bile acids
Single cell suspensions of whole splenocytes from FVB wild-type or Mdr1-null (Abcb1a/1b−/−) mice, or EL4 cells, were pre-treated with or without glycine- or taurine-conjugated CDCA in complete T cell media for 4 hr prior to PMA/ionomycin stimulation (4 hr; see Intracellular FACS analysis above) In some experiments, tCDCA (1.2 mM) was added alone, or together with elacridar (0.1 μM) for 4 hr prior to stimulation and staining.
Bile acid-induced cytotoxicity
Single cell suspensions of whole splenocytes from FVB wild-type or Mdr1-deficient mice, B6 wild-type or Mdr1-reporter mice, or EL4 cells, were treated with or without tCDCA (1.2 mM) in the presence or absence of elacridar (0.1 μM) for 18–24 hr. In other experiments, splenocytes were treated for 8 hr with or without 1.2 mM tCDCA, 0.5 mM hydrogen peroxide (H2O2), 15 μM menadione, or 50 μM 13-hydroperoxy-octadecadienoic acid (13-HpODE) +/− 125 μM N-acetyl-L-cysteine (N-Ac).
RNA sequencing
Two next-generation RNA-sequencing (RNA-seq) experiments were performed. For RNA-seq on FACS-sorted Rh123hi (Mdr1−) or Rh123lo (Mdr1+) effector/memory T cells (Teff cells: viability dye−CD45+CD3+CD4+CD25−CD44hi) from mesenteric lymph nodes of FVB.Rag1−/− mice injected 6 weeks prior with wild-type (FVB) naïve T cells, total RNA was isolated using RNeasy columns (Qiagen), with on-column DNase treatment. 500 ng of total RNA was used to prepare libraries with TrueSeq total RNA-Seq (Illumina), according to manufacturer’s instructions. For RNA-seq on FACS-sorted wild-type (B6) Teff cells expressing control (shCD8) or Mdr1 (shMdr1; shAbcb1a+shAbcb1b) shRNAmirs from spleen or small intestine lamina propria (siLP) of B6.Rag1−/− or B6.Rag1−/− Slc10a2−/− mice, approximately 10,000 sorted cells were processed directly to generate cDNA using the Clontech SMART-Seq v4 Ultra Low Input Kit (Clontech, Inc.). The generated cDNA was size selected using beads to enrich for fragments > 400 bp. The enriched cDNA was converted to Illumina-compatible libraries using the NEBNext Ultra II DNA kit (New England Biolabs, Inc.) using 1ng input. All libraries used for sequencing were quantified using Qubit, and validated on Bioanalyzer DNA high sensitivity chips (Agilent) and qPCR (Kappa Biosystems). Barcoded libraries were pooled at equimolar ratios and sequenced using single-end 75 bp reads on a NextSeq 500 instrument (Illumina). Raw sequencing reads (fastq files) were mapped to the mm9 genome using TopHat (Trapnell et al., 2009). The number of reads falling into each gene defined by RefSeq gene annotations were quantified using HTSeq-count. DESeq software (Anders and Huber, 2010) was used to detect differentially expressed genes between samples. Normalized gene expression was analyzed using the GenePattern software suite (http://genepattern.broadinstitute.org), and visualized using MultiPlot or Hierarchical Clutering Viewer modules; only annotated genes displaying normalized expression (RPKM) values ≥ 1.5 are shown for each experiment. Pathway analysis of differentially expressed gene sets was performed using the DAVID Functional Annotation Tool (https://david.ncifcrf.gov/summary.jsp).
Nanostring
Cell pellets (10–30k cells/condition) were lysed in 5 μL RLT buffer containing fresh 2-mercaptoethanol (Qiagen). Frozen lysates were sent to Nanostring Technologies, Inc. (Seattle, WA) or the University of Miami Genomics Core Facility (Miami, FL) for analysis on nCounter platforms using custom codesets. Raw data were normalized and analyzed using nSolver software (Version 1.1; Nanostring Technologies, Inc.). RNA counts were normalized using 3 housekeeping genes (Rps19, Gapdh, Actinb), with a cutoff for expression based on the highest negative control on the chip. Normalized RNA counts were exported to Microsoft Excel and presented using GraphPad Prism Software.
qPCR
RNA was isolated from cultured or ex vivo-isolated cells using RNeasy columns with on-column DNase treatment (Qiagen); this was used to synthesize cDNA via a high capacity cDNA reverse transcription kit (Life Technologies/Applied Biosystems). Taqman qPCR was performed on a StepOnePlus real time PCR instrument (Life Technologies/Applied Biosystems) using commercial taqman primer/probe sets for Abcb1a (Mm00607939_s1) and Gapdh (mm99999915_g1) from Life Technologies.
Quantification and Statistical Analyses
Statistical analyses were performed using Prism (GraphPad). P values were determined by paired or unpaired student’s t tests, one-way ANOVA, and ROC analyses as appropriate. Statistical tests are indicated throughout the Figure legends. Differences were considered significant when P < 0.05 (* P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001), and levels of significance are specified throughout the Figure legends. Data are shown as mean values ± S.E.M throughout. For histological analyses, certified veterinary pathologists were ‘blinded’ to the allocation of animals and experimental groups.
Supplementary Material
Highlights.
CD4+ effector T cells upregulate Mdr1 expression in the ileum
Mdr1 protects effector T cells in the ileum from bile acid-driven oxidative stress
Bile acid sequestration restores Mdr1-deficient T cell homeostasis in the ileum
A subset of ileal Crohn’s disease patients display MDR1 loss-of-function
Acknowledgments
We thank the Core Facilities at The Scripps Research Institute (TSRI) Florida for technical support, and Dr. Paul Dawson for critical discussions. This work was funded by TSRI Florida via the State of Florida (to M.S.S.), the National Institute of Allergy and Infectious Diseases (R21AI119728, to M.S.S.), the National Institute of Diabetes and Digestive and Kidney Diseases (5R01DK099076-07, to M.T.A.; P01DK071176, to R.G.L.), the Crohn’s and Colitis Foundation of America (#422515, to M.S.S.; #3786, to M.T.A.; #26971, to R.G.L.), the Broad Medical Foundation (#IBD-0389R, to M.T.A.), and the Core Research for Evolutional Science and Technology, Japan Science and Technology Agency; the Ministry of Education, Culture, Sports, Science and Technology; and the Ministry of Health, Labour and Welfare (to K.T). H.K. was supported by a Lotte Research Promotion Grant, and the Nagao Memorial Fund. M.T.A and D.W.H have received research grants and consultancy fees from: AbbVie, Amgen, Asana Medical, Inc., Ferring Pharmaceuticals, Focus Medical Communications, Genentech, Genova Diagnostics, GI Health Foundation, Gilead, GSK Holding Americas, Inc., Hospira, Inc., Janssen, Mucosal Health Board, Pfizer, Prometheus Laboratories, Prova Education, Inc., Sanofi Aventis, Shire, Takeda, UCB, and WebMD Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental information includes seven figures and one table and can be found with this article online at
AUTHOR CONTRIBUTIONS
M.S.S designed the study, and wrote the manuscript. W.C., H.K., M.L.C., A.D., A.S., S.Y.K., E.S.R., and K.M. performed experiments. A.P.B., C.B.J., G.C., G.J.M., S.Y.K., S.B.K., T.I., and K.T. analyzed data. A.O., P.N.L., and D.W.H. contributed resources. R.G.L., A.R.P., F.C., M.T.A., D.W.H., S.B.K., and K.T. provided critical discussion. H.K., M.L.C., G.C., G.J.M., M.T.A., D.W.H., S.B.K., and K.T. revised the manuscript.
References
- Albenberg LG, Lewis JD, Wu GD. Food and the gut microbiota in inflammatory bowel diseases: a critical connection. Curr Opin Gastroenterol. 2012;28:314–320. doi: 10.1097/MOG.0b013e328354586f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome biology. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annese V, Valvano MR, Palmieri O, Latiano A, Bossa F, Andriulli A. Multidrug resistance 1 gene in inflammatory bowel disease: a meta-analysis. World journal of gastroenterology : WJG. 2006;12:3636–3644. doi: 10.3748/wjg.v12.i23.3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold MA, Swanson BJ, Crowder CD, Frankel WL, Lam-Himlin D, Singhi AD, Stanich PP, Arnold CA. Colesevelam and Colestipol: Novel Medication Resins in the Gastrointestinal Tract. The American journal of surgical pathology. 2014 doi: 10.1097/PAS.0000000000000260. [DOI] [PubMed] [Google Scholar]
- Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, Rudensky AY. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504:451–455. doi: 10.1038/nature12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, Treuting PM, Rudensky AY. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell. 2015;162:1078–1089. doi: 10.1016/j.cell.2015.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, Narushima S, Suda W, Imaoka A, Setoyama H, Nagamori T, et al. Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell. 2015;163:367–380. doi: 10.1016/j.cell.2015.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala A, Munoz MF, Arguelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev. 2014;2014:360438. doi: 10.1155/2014/360438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat BG, Rapp SR, Beaudry JA, Napawan N, Butteiger DN, Hall KA, Null CL, Luo Y, Keller BT. Inhibition of ileal bile acid transport and reduced atherosclerosis in apoE−/− mice by SC-435. Journal of lipid research. 2003;44:1614–1621. doi: 10.1194/jlr.M200469-JLR200. [DOI] [PubMed] [Google Scholar]
- Borst P, Schinkel AH. P-glycoprotein ABCB1: a major player in drug handling by mammals. The Journal of clinical investigation. 2013;123:4131–4133. doi: 10.1172/JCI70430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, Sefik E, Tan TG, Wagers AJ, Benoist C, Mathis D. A special population of regulatory T cells potentiates muscle repair. Cell. 2013;155:1282–1295. doi: 10.1016/j.cell.2013.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Belanger S, Frederick MA, Li B, Johnston RJ, Xiao N, Liu YC, Sharma S, Peters B, Rao A, et al. In Vivo RNA Interference Screens Identify Regulators of Antiviral CD4(+) and CD8(+) T Cell Differentiation. Immunity. 2014;41:325–338. doi: 10.1016/j.immuni.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, Benoist C, Mathis D. PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. 2012;486:549–553. doi: 10.1038/nature11132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson PA, Haywood J, Craddock AL, Wilson M, Tietjen M, Kluckman K, Maeda N, Parks JS. Targeted deletion of the ileal bile acid transporter eliminates enterohepatic cycling of bile acids in mice. The Journal of biological chemistry. 2003;278:33920–33927. doi: 10.1074/jbc.M306370200. [DOI] [PubMed] [Google Scholar]
- Diehl GE, Longman RS, Zhang JX, Breart B, Galan C, Cuesta A, Schwab SR, Littman DR. Microbiota restricts trafficking of bacteria to mesenteric lymph nodes by CX(3)CR1(hi) cells. Nature. 2013;494:116–120. doi: 10.1038/nature11809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimenko JV, Gerasimenko OV, Palejwala A, Tepikin AV, Petersen OH, Watson AJ. Menadione-induced apoptosis: roles of cytosolic Ca(2+) elevations and the mitochondrial permeability transition pore. J Cell Sci. 2002;115:485–497. doi: 10.1242/jcs.115.3.485. [DOI] [PubMed] [Google Scholar]
- Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nature reviews. Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- Ho GT, Aird RE, Liu B, Boyapati RK, Kennedy NA, Dorward DA, Noble CL, Shimizu T, Carter RN, Chew ET, et al. MDR1 deficiency impairs mitochondrial homeostasis and promotes intestinal inflammation. Mucosal immunology. 2017 Apr;12 doi: 10.1038/mi.2017.31. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann AF, Hagey LR. Key discoveries in bile acid chemistry and biology and their clinical applications: history of the last eight decades. Journal of lipid research. 2014;55:1553–1595. doi: 10.1194/jlr.R049437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyafil F, Vergely C, Du Vignaud P, Grand-Perret T. In vitro and in vivo reversal of multidrug resistance by GF120918, an acridonecarboxamide derivative. Cancer research. 1993;53:4595–4602. [PubMed] [Google Scholar]
- Ignacio Barrasa J, Olmo N, Perez-Ramos P, Santiago-Gomez A, Lecona E, Turnay J, Antonia Lizarbe M. Deoxycholic and chenodeoxycholic bile acids induce apoptosis via oxidative stress in human colon adenocarcinoma cells. Apoptosis. 2011;16:1054–1067. doi: 10.1007/s10495-011-0633-x. [DOI] [PubMed] [Google Scholar]
- Jones-Hall YL, Grisham MB. Immunopathological characterization of selected mouse models of inflammatory bowel disease: Comparison to human disease. Pathophysiology. 2014;21:267–288. doi: 10.1016/j.pathophys.2014.05.002. [DOI] [PubMed] [Google Scholar]
- Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabe Y, Ando K, Hirao S, Yoshida M, Handa H. Redox regulation of NF-kappaB activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxid Redox Signal. 2005;7:395–403. doi: 10.1089/ars.2005.7.395. [DOI] [PubMed] [Google Scholar]
- Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annual review of immunology. 2010;28:573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim IW, Peng XH, Sauna ZE, FitzGerald PC, Xia D, Muller M, Nandigama K, Ambudkar SV. The conserved tyrosine residues 401 and 1044 in ATP sites of human P-glycoprotein are critical for ATP binding and hydrolysis: evidence for a conserved subdomain, the A-loop in the ATP-binding cassette. Biochemistry. 2006;45:7605–7616. doi: 10.1021/bi060308o. [DOI] [PubMed] [Google Scholar]
- Kosiewicz MM, Nast CC, Krishnan A, Rivera-Nieves J, Moskaluk CA, Matsumoto S, Kozaiwa K, Cominelli F. Th1-type responses mediate spontaneous ileitis in a novel murine model of Crohn’s disease. The Journal of clinical investigation. 2001;107:695–702. doi: 10.1172/JCI10956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo-Zuniga V, Bartoli R, Planas R, Hofmann AF, Vinado B, Hagey LR, Hernandez JM, Mane J, Alvarez MA, Ausina V, Gassull MA. Oral bile acids reduce bacterial overgrowth, bacterial translocation, and endotoxemia in cirrhotic rats. Hepatology. 2003;37:551–557. doi: 10.1053/jhep.2003.50116. [DOI] [PubMed] [Google Scholar]
- Ludescher C, Thaler J, Drach D, Drach J, Spitaler M, Gattringer C, Huber H, Hofmann J. Detection of activity of P-glycoprotein in human tumour samples using rhodamine 123. British journal of haematology. 1992;82:161–168. doi: 10.1111/j.1365-2141.1992.tb04608.x. [DOI] [PubMed] [Google Scholar]
- Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008;453:620–625. doi: 10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- Morton AM, Sefik E, Upadhyay R, Weissleder R, Benoist C, Mathis D. Endoscopic photoconversion reveals unexpectedly broad leukocyte trafficking to and from the gut. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:6696–6701. doi: 10.1073/pnas.1405634111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayamada S, Takahashi H, Kanno Y, O’Shea JJ. Helper T cell diversity and plasticity. Current opinion in immunology. 2012;24:297–302. doi: 10.1016/j.coi.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panwala CM, Jones JC, Viney JL. A novel model of inflammatory bowel disease: mice deficient for the multiple drug resistance gene, mdr1a, spontaneously develop colitis. Journal of immunology. 1998;161:5733–5744. [PubMed] [Google Scholar]
- Pavlidis P, Heneghan M, Hayee B. Cholestyramine treats primary sclerosing cholangitis-associated inflammatory bowel disease. Journal of Crohn’s & colitis. 2015;9:210. doi: 10.1093/ecco-jcc/jju020. [DOI] [PubMed] [Google Scholar]
- Poupon R, Chazouilleres O, Poupon RE. Chronic cholestatic diseases. Journal of hepatology. 2000;32:129–140. doi: 10.1016/s0168-8278(00)80421-3. [DOI] [PubMed] [Google Scholar]
- Powrie F, Correa-Oliveira R, Mauze S, Coffman RL. Regulatory interactions between CD45RBhigh and CD45RBlow CD4+ T cells are important for the balance between protective and pathogenic cell-mediated immunity. The Journal of experimental medicine. 1994;179:589–600. doi: 10.1084/jem.179.2.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh R, Kozhaya L, McKevitt K, Djuretic IM, Carlson TJ, Quintero MA, McCauley JL, Abreu MT, Unutmaz D, Sundrud MS. Pro-inflammatory human Th17 cells selectively express P-glycoprotein and are refractory to glucocorticoids. The Journal of experimental medicine. 2014 doi: 10.1084/jem.20130301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano T, Huang W, Hall JA, Yang Y, Chen A, Gavzy SJ, Lee JY, Ziel JW, Miraldi ER, Domingos AI, et al. An IL-23R/IL-22 Circuit Regulates Epithelial Serum Amyloid A to Promote Local Effector Th17 Responses. Cell. 2015;163:381–393. doi: 10.1016/j.cell.2015.08.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiering C, Krausgruber T, Chomka A, Frohlich A, Adelmann K, Wohlfert EA, Pott J, Griseri T, Bollrath J, Hegazy AN, et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature. 2014;513:564–568. doi: 10.1038/nature13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinkel AH, Mayer U, Wagenaar E, Mol CA, van Deemter L, Smit JJ, van der Valk MA, Voordouw AC, Spits H, van Tellingen O, et al. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:4028–4033. doi: 10.1073/pnas.94.8.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C, Tallman KA, Porter NA, Brash AR. Two distinct pathways of formation of 4-hydroxynonenal. Mechanisms of nonenzymatic transformation of the 9-and 13-hydroperoxides of linoleic acid to 4-hydroxyalkenals. The Journal of biological chemistry. 2001;276:20831–20838. doi: 10.1074/jbc.M101821200. [DOI] [PubMed] [Google Scholar]
- Sujino T, London M, Hoytema van Konijnenburg DP, Rendon T, Buch T, Silva HM, Lafaille JJ, Reis BS, Mucida D. Tissue adaptation of regulatory and intraepithelial CD4(+) T cells controls gut inflammation. Science. 2016;352:1581–1586. doi: 10.1126/science.aaf3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorpe GW, Fong CS, Alic N, Higgins VJ, Dawes IW. Cells have distinct mechanisms to maintain protection against different reactive oxygen species: oxidative-stress-response genes. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:6564–6569. doi: 10.1073/pnas.0305888101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Helvoort A, Smith AJ, Sprong H, Fritzsche I, Schinkel AH, Borst P, van Meer G. MDR1 P-glycoprotein is a lipid translocase of broad specificity, while MDR3 P-glycoprotein specifically translocates phosphatidylcholine. Cell. 1996;87:507–517. doi: 10.1016/s0092-8674(00)81370-7. [DOI] [PubMed] [Google Scholar]
- Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nature protocols. 2007;2:541–546. doi: 10.1038/nprot.2007.41. [DOI] [PubMed] [Google Scholar]
- Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013;154:1370–1379. doi: 10.1016/j.cell.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.