

Abstract
The Hippo pathway controls organ size and tissue homeostasis through a kinase cascade leading from the Ste20-like kinase Hpo (MST1/2 in mammals) to the transcriptional coactivator Yki (YAP/TAZ in mammals). While previous studies have uncovered positive and negative regulators of Hpo/MST, how they are integrated to maintain signaling homeostasis remains poorly understood. Here we identify a self-restricting mechanism whereby autophosphorylation of an unstructured linker in Hpo/MST creates docking sites for the STRIPAK PP2A phosphatase complex to inactivate Hpo/MST. Mutation of the phospho-dependent docking sites in Hpo/MST or deletion of Slmap, the STRIPAK subunit recognizing these docking sites, results in constitutive activation of Hpo/MST in both Drosophila and mammalian cells. In contrast, autophosphorylation of the Hpo/MST linker at distinct sites are known to recruit Mats/MOB1 to facilitate Hippo signaling. Thus, multisite autophosphorylation of Hpo/MST linker provides an evolutionarily conserved built-in molecular platform to maintain signaling homeostasis by coupling antagonistic signaling activities.
Keywords: Hippo signaling, Hpo, MST1/2, STRIPAK, PP2A, SLMAP, autophosphorylation
eTOC
The Hippo pathway was named after the Ste20-like kinase Hpo/MST but how its activity is regulated remains unclear. Zheng et al identify a self-restricting mechanism whereby autophosphorylation of an unstructured linker in Hpo/MST creates docking sites for the STRIPAK PP2A phosphatase complex to inactivate Hpo/MST in both Drosophila and mammals.
Introduction
The Hippo signaling pathway was first elucidated as a mechanism that restricts the growth of imaginal discs in Drosophila and has since been found to control tissue growth and homeostasis in diverse animals including mammals (Harvey and Hariharan, 2012; Pan, 2010; Yu et al., 2015). Central to this pathway is a core kinase cascade in which the Ste-20 family kinase Hippo (Hpo, MST1/2 in mammals), in conjunction with the scaffold protein Salvador (Sav, SAV1 in mammals), phosphorylates and activates the NDR family kinase Warts (Wts, LATS1/2 in mammals) and its cofactor Mob as tumor suppressor (Mats, MOB1A/B in mammals). Activated Wts/LATS-Mats/MOB1 complex then phosphorylates and inactivates transcriptional coactivator Yorkie (Yki, YAP/TAZ in mammals), leading to phosphorylation-dependent nuclear exclusion and inactivation of Yki/YAP/TAZ. Under conditions of low Hippo signaling, Yki/YAP/TAZ enters the nucleus and partners with the TEF/TEAD family DNA-binding transcriptional factor Scalloped (Sd, TEAD1/2/3/4 in mammals) to drive the expression of growth promoting genes. Under conditions of high Hippo signaling and absence of nuclear Yki/YAP/TAZ, Sd/TEAD functions as a default repressor to repress the transcription of Hippo target genes (Koontz et al., 2013). An emerging paradigm from studies in multiple model systems indicates that the core kinase cascade of the Hippo pathway functions as a signaling module that integrates diverse cell-intrinsic and -extrinsic signals, such as cell adhesion and polarity, mechanical forces, soluble factors and various stress signals (Genevet and Tapon, 2011; Halder et al., 2012; Yu et al., 2015).
Although the Hippo pathway was named after the protein kinase Hpo/MST, our understanding of the molecular mechanisms regulating its kinase activity remains incomplete. Activation of Hpo and its mammalian homologues MST1/2 requires phosphorylation of a key site in the activation loop of the kinase domain (T195 for Hpo, T183/T180 for MST1/2), which can be mediated by trans-autophosphorylation (Creasy et al., 1996; Ni et al., 2013; Praskova et al., 2004) or by an upstream kinase Tao-1 (TAOK1/2/3 in mammals) (Boggiano et al., 2011; Poon et al., 2011). When overexpressed in cells, MST1/2 forms homodimers with elevated kinase activity, suggesting that homodimerization per se is sufficient to trigger autoactivation by trans-autophosphorylation (Creasy et al., 1996; Ni et al., 2013; Praskova et al., 2004). Autoactivation of Hpo/MST requires dimerization elements in both the N-terminal kinase domain and the C-terminal SARAH (Salvador-Rassf-Hpo) domain, as mutations abolishing either the N- or the C-terminal dimerization domain strongly compromise autophosphorylation (Deng et al., 2013; Jin et al., 2012; Ni et al., 2013). SAV1 promotes MST dimerization and activation by forming heterotetramers containing two MST and two SAV1 subunits (Ni et al., 2013). The ability of Hpo/MST to homodimerize and autoactivate poses a potential risk, as activated Hpo/MST is a potent inducer of apoptosis, through Hippo signaling, as well as Hippo-independent events, such as phosphorylation of histone H2B (Cheung et al., 2003) or FOXO transcription factors (Lehtinen et al., 2006). Thus, the kinase activity of Hpo/MST must be carefully regulated to prevent spurious activation in development and physiology.
One way to restrict the kinase activity of Hpo/MST is by engaging negative regulators. Indeed, several negative regulatory mechanisms have been reported that restrict the kinase activity of Hpo/MST. The cell polarity regulator Par-1 in Drosophila and its mammalian homologs microtubule affinity-regulating kinases 1/2/3 (MARK1/2/3) have been reported to physically interact with and antagonizes Hpo/MST activity through unknown mechanisms (Huang et al., 2013; Kwan et al., 2016). Another negative regulator of Hpo/MST is the STRIPAK PP2A phosphatase complex, which has been shown to associate with Hpo/MST in Drosophila and mammalian cells (Couzens et al., 2013; Ribeiro et al., 2010) and restrict Drosophila Hpo kinase activity by dephosphorylating its activation loop motif (Ribeiro et al., 2010). Despite these advances, how positive and negative inputs into Hpo/MST are integrated to maintain signaling homeostasis is poorly understood.
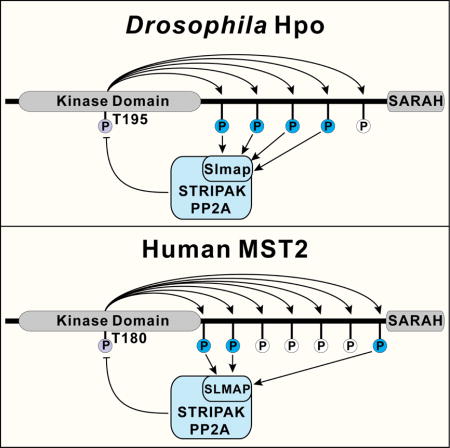
We have previously shown that upon phosphorylation at its activation loop in the kinase domain, Hpo/MST undergoes multisite autophosphorylation in an unstructured linker region between the kinase domain and the SARAH domain (Ni et al., 2015). Some of these sites serve as phospho-dependent docking sites that function together with a hydrophobic motif (HFM) (Figure 1A) to recruit Mats/MOB1 thus promoting Wts/LATS phosphorylation (Ni et al., 2015). While investigating the functional contribution of the remaining phosphorylation sites in the linker region, we found that, contrary to the Mats/MOB1 docking sites, these autophosphorylation sites play an opposite role in Hippo signaling. Further investigation revealed that these sites function as phosphorylation-dependent docking motifs for the STRIPAK PP2A phosphatase complex to inactivate Hpo/MST. Thus, multisite autophosphorylation of the Hpo/MST linker provides a built-in molecular platform to maintain signaling homeostasis by coupling antagonistic signaling outputs. We suggest that strategies aimed at disrupting this self-restricting mechanism may provide a potential approach to activate Hippo signaling for cancer treatment.
Figure 1. The linker region of Hpo contains two types of autophosphorylation sites.

(A) GST pull-down assay of binding between Mats and Hpo variants with mutated linker autophosphorylation sites. Left: a schematic of Hpo protein showing the five autophosphorylation sites and sequence of the linker region. Mats-binding site is labeled by red, non-Mats-binding sites by blue, and the hydrophobic motif recognized by Mats (HFM) by orange. Right: cell lysates were prepared from S2R+ cells transfected with the indicated Hpo variants and then incubated with GST-Mats in GST pull-down assay. Hpo was pulled down by GST-Mats (lane 2), but not GST alone (lane 1). Also note the comparable binding between GST-Mats and wildtype Hpo (lane 2) and Hpo4A/431T (lane 8), and reduced binding between GST-Mats and the remaining Hpo mutants (lanes 3–7).
(B) A schematic illustration of the FLP-out expression system. A FRT y+ FRT cassette containing a transcription STOP signal is inserted between Tubulin α1 promoter and hpo cDNA to prevent hpo expression. A tissue-specific source of FLP is used to excise the FRT y+ FRT cassette through recombination between the two FRT sequences, leading to hpo expression.
(C–F) Images of adult wings expressing the indicated Hpo mutants in the engrailed (en)-expression posterior compartment by the FLP-out expression system. Note that the size of the posterior compartment (highlighted by green color) was greatly decreased by Hpo4A/431T (F). Scale bars are 0.2mm.
(G) Quantification of the size of the posterior compartment relative to the anterior compartment for experiments described in (C–F) (mean ± SEM, n = 15). Wildtype Hpo slightly and Hpo4A/431T greatly decreased the size of the en-expression domain, while Hpo431A expression had no effect.
(H–K) Third instar wing imaginal discs corresponding to experiments described in (C–F) and stained with anti-Diap1 antibodies. Note the reduction of Diap1 protein levels by Hpo4A/431T, but not wildtype or Hpo431A in the GFP-positive en-expression domain (arrows). Scale bars represent 50μm.
Results
The linker region of Hpo contains two types of autophosphorylation sites with opposing effects on Hippo signaling
Previously, we have identified a cluster of autophosphorylation sites in the linker of Hpo/MST, with each site characterized by a Thr residue followed by a hydrophobic Met, Leu, or Val residue (Ni et al., 2015). Hpo contains five such sites (Figure 1A). We have shown previously that aside from a Mats-binding hydrophobic motif (HFM), the T431 site also contributes to Mats binding in a GST pull-down assay (Ni et al., 2015). To further dissect the function of the linker phosphorylation sites, we generated a Hpo mutant replacing Thr by Ala in all five sites (Hpo5A), as well as Hpo mutants containing Ala mutations in all but one of the five phosphorylation sites (Hpo4A/356T, Hpo4A/374T, Hpo4A/387T, Hpo4A/421T and Hpo4A/431T). Each Hpo mutant was expressed in Drosophila S2R+ cells and tested for Mats-binding using bacterially purified GST-Mats fusion protein in a GST pull-down assay. As shown in Figure 1A, comparable amount of wildtype Hpo and Hpo4A/431T was pulled down by GST-Mats, whereas binding was reduced to a similar extent between GST-Mats and all mutants that disabled T431, including Hpo5A, Hpo4A/356T, Hpo4A/374T, Hpo4A/387T and Hpo4A/421T. The residual Mats-binding in these Hpo mutant is likely mediated by the hydrophobic motif and the kinase domain of Hpo (Ni et al., 2015). These results confirmed that T431 contributes to Mats binding (Ni et al., 2015) and further suggested that it is the only linker site mediating this interaction (Figure 1A). Thus, the linker phosphorylation sites can be categorized into two groups, the Mats-binding site T431 and the remaining four non-Mats-binding sites of unknown function.
To investigate the potential function of the two groups of linker phosphorylation sites, we compared the in vivo activity of two Hpo mutants, Hpo431A that lacks the Mats-binding site T431 and Hpo4A/431T that lacks the four non-Mats-binding sites, with that of wildtype Hpo. We reasoned that the commonly used UAS/GAL4 system might not be adequate for comparing their activities because massive overexpression of wildtype Hpo resulting from the UAS/GAL4 system is sufficient to activate Hippo signaling in Drosophila, potentially masking any intrinsic differences between the different Hpo variants. With this consideration, we turned to a conditional expression system driven by the ubiquitous and mild Tubulin α1 promoter. For this purpose, we engineered transgenes in which an FRT y+ FRT cassette (Basler and Struhl, 1994) was placed between the Tub promoter and cDNA of FLAG-tagged Hpo or its variants, thus blocking transcription downstream of the Tub promoter (Figure 1B). Each transgene was inserted into identical landing site by PhiC31-mediated chromosomal integration (Bischof et al., 2007). In the resulting transgenic flies, the FRT y+ FRT cassette can be excised (FLP-out) by a tissue-specific Flippase (FLP) recombinase source, thus allowing the expression of a Hpo variant downstream of the Tub promoter in a tissue-specific manner (Figure 1B).
Using this FLP-out expression system, we first compared the activity of wildtype Hpo, Hpo431A and Hpo4A/431T in the eye using an eye-specific FLP source ey-FLP. While expression of wildtype Hpo or Hpo431A by the Tub promoter caused no discernable phenotypes in adult flies, we failed to recover any adult flies expressing Hpo4A/431T, suggesting that Hpo4A/431T may confer gain-of-function activity (Figure S1A–D). To obtain viable adults for comparison, we turned to engrailed (en)-Gal4 to drive the expression of FLP in the posterior compartment of wing imaginal discs. Expression of wildtype Hpo using this system slightly decreased the size of posterior compartment of adult wings, while expression of the Mats-binding deficient mutant Hpo431A had no effect, suggesting that Hpo431A is less active than wildtype Hpo (Figure 1C–E and Figure 1G). Thus, consistent with our previous results (Ni et al., 2015), the Mats-binding site T431 in the Hpo linker promotes Hippo signaling. In contrast to Hpo431A, expression of Hpo4A/431T markedly reduced the size of the posterior compartment, indicating gain-of-function activity of Hpo4A/431T (Figure 1F and G). Consistent with the adult wing phenotypes, expression of Hpo4A/431T, but not wildtype Hpo or Hpo431A, significantly decreased the expression of the Hippo target diap1 in the posterior compartment in developing wing imaginal discs (Figure 1H–K). To further confirm the gain-of-function activity of Hpo4A/431T, we examined the expression level of Hippo pathway reporter diap1-lacZ. Indeed, only Hpo4A/431T was able to markedly downregulate the expression level of diap1-lacZ in wing discs (Figure S1E–H). Of note, western blotting analysis of wing imaginal discs revealed comparable expression level of the FLAG-tagged Hpo variants, excluding the possibility that different in vivo activities were simply due to different protein levels resulting from the transgenes (Figure S1I). Taken together, these results suggest that the Hpo linker contains two types of autophosphorylation sites with opposing effects on Hippo signaling, with the Mats-binding site T431 promoting while the remaining four non-Mats-binding sites inhibiting Hippo signaling.
The non-Mats-binding sites in the Hpo linker bind to Slmap in a phospho-dependent manner
To understand the biochemical mechanism underlying the gain-of-function activity of the Hpo4A/431T mutant, we expressed it in Drosophila S2R+ cells. Interestingly, compared to wildtype Hpo, Hpo4A/431T showed elevated phosphorylation at its activation loop autophosphorylation site T195 (Figure 2A), suggesting that Hpo4A/431T may possess higher intrinsic kinase activity than wildtype Hpo.
Figure 2. The non-Mats-binding sites in the Hpo linker bind to Slmap in a phospho-dependent manner.
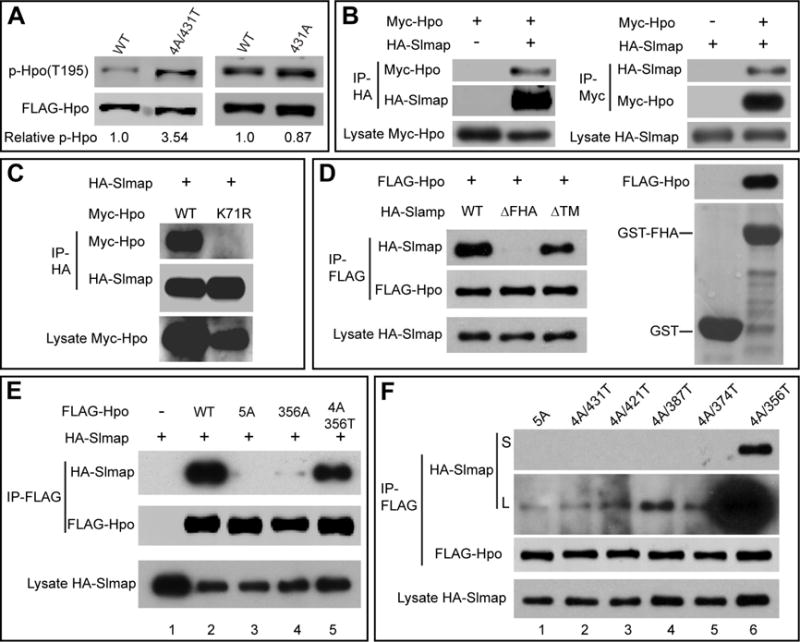
(A) The indicated FLAG-Hpo variants were expressed in S2R+ cells, partially purified by anti-FLAG IP and blotted for T195 phosphorylation. The p-Hpo level over total Hpo level was quantified relative to the wildtype Hpo and indicated below the blot. Note the increased p-Hpo level for Hpo4A/431T compared to wildtype Hpo.
(B) Reciprocal co-IP between Myc-Hpo and HA-Slmap expressed in S2R+ cells.
(C) S2R+ cells expressing the indicted constructs were subjected to co-IP. Note the absence of interaction between Slmap and HpoK71R.
(D) The FHA domain of Slmap is required and sufficient for Hpo binding. Left: co-IP assay showing interaction between Hpo and wildtype Slmap or SlmapΔTM, but not SlmapΔFHA. Right: GST pull-down assay showing binding between GST-FHA and FLAG-Hpo expressed in S2R+ cells. No binding was detected between GST alone and FLAG-Hpo.
(E) Identification of 356T as a strong Slmap binding site in the Hpo linker. S2R+ cells expressing the indicated constructs were analyzed by anti-FLAG immunoprecipitation. Note the absence of binding between Hpo5A and Slmap (lane 3), very weak binding between Hpo356A to Slmap (lane 4), and mostly restored binding between Hpo4A/356T and Slmap (lane 5).
(F) Characterization of additional Slmap binding sites in the Hpo linker. S2R+ cells expressing the indicated constructs were analyzed by anti-FLAG immunoprecipitation. Both short (S) and Long (L) exposures of the same gel were shown to reveal weak binding. Besides the strong binder Hpo4A/356T (lane 6), Hpo4A/421T (lane 3), 4A/387T (lane 4) and 4A/374T (lane 5) also showed slightly higher binding than Hpo5A (lane 1) and Hpo4A/431T (lane 2).
The increased T195 phosphorylation of the Hpo4A/431T mutant suggests that the non-Mats-binding sites may normally recruit a negative regulator(s) to restrict the kinase activity of Hpo. Furthermore, such negative regulator(s) may interact with Hpo through these sites in a phospho-Thr-dependent manner. Three types of negative regulators of Hpo have been identified to date, including Rassf (Polesello et al., 2006), the STRIPAK PP2A phosphatase complex (Ribeiro et al., 2010) and the kinase Par-1 (Huang et al., 2013). A thorough survey of the domain structure of these negative regulators revealed Slmap as a prominent candidate containing a phospho-Thr binding domain. Slmap is a subunit of the STRIPAK PP2A phosphatase complex (Goudreault et al., 2009), an evolutionarily conserved negative regulator of Hpo/MST. Slmap contains a phospho-Thr binding domain known as Forkhead-associated (FHA) domain (Durocher and Jackson, 2002). Indeed, Slmap has been identified as a Hpo-binding protein from two independent unbiased screens in Drosophila, including a genome-wide affinity purification coupled with mass spectrometry analysis (Guruharsha et al., 2011) and a high-throughput yeast two-hybrid (Y2H) screen (Formstecher et al., 2005). We verified Hpo-Slmap interaction by reciprocal coimmunoprecipitation (co-IP) between epitope-tagged Hpo and Slamp in S2R+ cells (Figure 2B). Importantly, no interaction was detected between Slmap and the kinase dead HpoK71R mutant protein (Wu et al., 2003) (Figure 2C), consistent with the hypothesis that Slmap binds to Hpo only after Hpo has undergone autophosphorylation.
Interestingly, the previous high-throughput Y2H screen isolated three overlapping clones of Slmap using Hpo as a bait, and all three clones include the FHA domain of Slamp (Figure S2A) (Formstecher et al., 2005). In agreement with the Y2H results, epitope-tagged Hpo co-precipitated with N-terminal half of Slmap, which contains the FHA domain (SlmapN, aa 1–343), but not with the C-terminal half of Slmap (SlmapC, aa 344–897) (Figure S2B). To further test the importance of the FHA domain, we introduced point mutations in the FHA domain by replacing three conserved residues (R260 N261 H262) with Alanine, resulting in a mutant called SlmapΔFHA. Since Slmap is a tail-anchored protein containing a single predicted transmembrane (TM) domain at the extreme C-terminus (Figure S2A) (Byers et al., 2009), we also generated a mutant deleting the TM segment, SlmapΔTM, as a control. Supporting the importance of the FHA domain, SlmapΔFHA, but not wildtype Slmap or SlmapΔTM, abolished Slmap-Hpo interaction (Figure 2D, left). Direct binding between Hpo and Slmap’s FHA domain was further confirmed by GST pull-down assay between bacterially purified GST-FHA fusion protein and epitope-tagged Hpo expressed in S2R+ cells (Figure 2D, right).
Next, we tested whether Hpo-Slmap interaction is mediated by the non-Mats-binding autophosphorylation sites in the Hpo linker. Mutating all five phosphorylation sites (Hpo5A) abolished Hpo-Slmap interaction in co-IP assays, suggesting that at least some of the linker sites are required for Slmap binding (Figure 2E). Mutating the most C-terminal site T356 (Hpo356A) greatly diminished Hpo-Slmap interaction (Figure 2E), and conversely, a Hpo mutant with all sites mutated except for T356 (Hpo4A/356T), still interacted strongly with Slmap (Figure 2E). To determine whether the residual interaction between Hpo356A and Slmap is due to the remaining sites in the Hpo linker, we compared Hpo5A with Hpo mutants retaining only one of the five linker sites. As shown in Figure 2F, while T356 was most critical, Hpo4A/421T, Hpo4A/387T and Hpo4A/374T also interacted with Slmap at a level above Hpo5A or Hpo4A/431T (Figure 2F, short and longer exposures). Thus, all four non-Mats-binding sites in the Hpo linker contribute to Slmap binding, with T356 being the most important. Taken together, these results support our hypothesis that Slmap, through its phospho-Thr-binding FHA domain, binds to the non-Mats-binding autophosphorylation sites in the Hpo linker, in a phosphorylation-dependent manner.
Slmap mediates the association between Hpo and the STRIPAK PP2A phosphatase complex
The STRIPAK PP2A phosphatase complex is an evolutionarily conserved negative regulator of Hpo/MST in both Drosophila and mammals (Couzens et al., 2013; Ribeiro et al., 2010). Mammalian cells contain two mutually exclusive STRIPAK PP2A complexes, defined by the presence of CTTNBP2-like adaptors or SLMAP in conjunction with SIKE or its paralogue FGFR1OP2 (Goudreault et al., 2009). However, the initial proteomic study, which isolated the STRIPAK PP2A phosphatase complex as a negative regulator of Hpo in Drosophila, did not identify Slmap as a Hpo-associated protein (Ribeiro et al., 2010). To confirm that Slmap is indeed a component of the STRIPAK complex in Drosophila, we depleted Slmap in Drosophila wing imaginal discs by RNAi. Slmap RNAi in the en-expression domain resulted in downregulation of Hippo targets Diap1, diap1-LacZ and ex-LacZ (Figure 3A–F) as well as a reduction in the size of the posterior compartment of adult wings (Figure S3A–C). These phenotypes closely resemble RNAi of Cka or Fgop2, two previously characterized STRIPAK components (Ribeiro et al., 2010), or expression of the Slmap-binding deficient Hpo4A/431T mutant (Figure 1). Furthermore, Slmap RNAi suppressed the elevated Diap1 expression in exe1 clones but not in hpo42–47 clones (Figure S3D–G), consistent with Slmap acting through Hpo to regulate the Hippo pathway.
Figure 3. Slmap mediates the association between Hpo and the STRIPAK PP2A phosphatase complex in Drosophila.
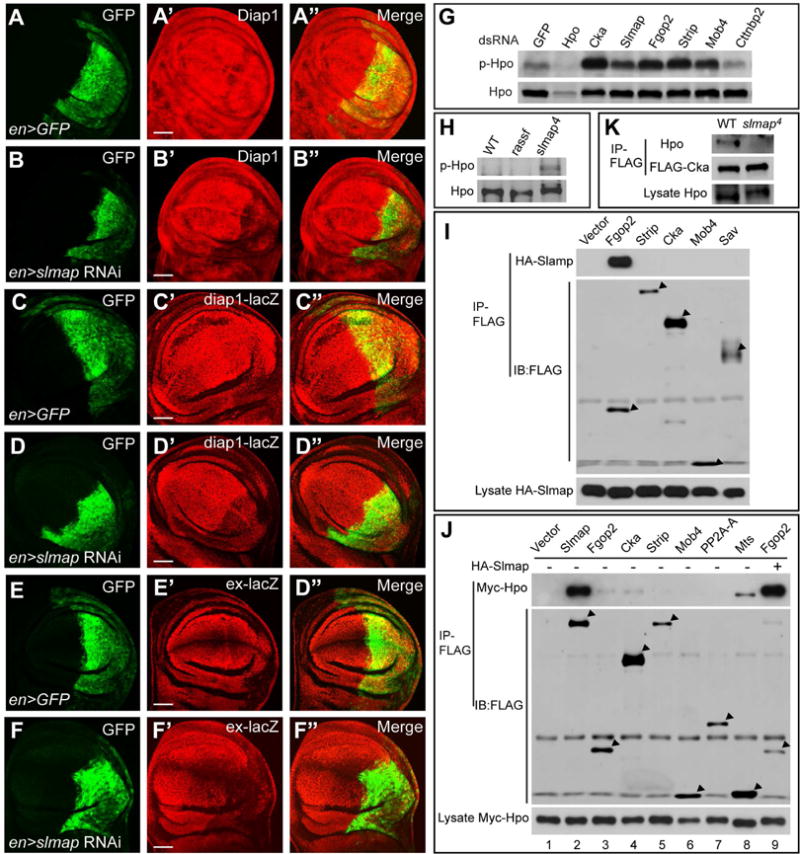
(A–F) Wing imaginal discs expressing UAS-slmap RNAi by the en-Gal4 UAS-GFP driver were examined for the expression of Diap1 (A–B), diap1-LacZ (C–D) and ex-LacZ (E–F). Note the downregulation of these reporters in the en-expression domain (GFP-positive). Scale bars represent 50μm.
(G) The indicated subunits of the STRIPAK complex were depleted by RNAi in S2R+ cells and the cell lysates were examined for T195 phosphorylation of endogenous Hpo. Note increased phospho-T195 level upon RNAi of Cka, Slmap, Fgop2, Strip, or Mob4, but not Cttnbp2.
(H) Eye-antennal imaginal discs were dissected from third instar larvae of wildtype, rassf36/rassf44.2 trans-heterozygotes or slmap4 homozygotes and lysed in 2×SDS loading buffer and examined for phospho-Hpo (T195) level. Note increased p-Hpo level in slmap but not rassf mutant tissues. rassf36 and rassf44.2 are null alleles (Polesello et al., 2006).
(I) S2R+ cells co-expressing HA-Slmap and the indicated FLAG-tagged proteins were analyzed by anti-FLAG immunoprecipitation. Note interactions between Slmap and Fgop2.
(J) Myc-Hpo was co-expressed with the indicated FLAG-tagged subunits of the STRIPAK complex in S2R+ cells, with (lane 9) or without HA-Slmap (lanes 1–8). The interactions between Hpo and these subunits was assayed by anti-FLAG IP. Note strong interactions between Hpo and Slmap, and weak interactions between Hpo and Fgop2, Cka or Mts. Also note the markedly enhanced interaction between Hpo and Fgop2 in the presence of HA-Slmap (compare lane 9 to lane 3).
(K) Tissue lysates from wildtype or slamp4 homozygous larvae, both carrying the same tub-FLAG-cka transgene, were subjected to anti-FLAG immunoprecipitation. Note the presence of co-purified endogenous Hpo from wildtype not slmap mutant animals.
To test whether Slmap is essential for the STRIPAK complex to downregulate the kinase activity of Hpo, we individually depleted several STRIPAK subunits including Slmap in S2R+ cells by RNAi and examined T195 phosphorylation of endogenous Hpo. Depletion of Slmap, Cka, Fgop2, Strip or Mob4 all led to increased T195 phosphorylation, while knockdown of Cttnbp2 showed no effects (Figure 3G). To further confirm these results, we applied CRISPR/Cas9-mediated genome editing to generate the slmap4 allele, which contains a 19 bp deletion predicted to cause a premature stop codon close to the N-terminus of the protein (Figure S3H). slmap4 homozygous flies are pupal lethal, and the lethality was rescued 100% by a slmap transgene driven by the Tubulin α1 promoter. In agreement with the results from S2R+ cells, eye-antennal imaginal discs isolated from 3rd instar larvae of slmap4 homozygotes showed elevated Hpo T195 phosphorylation compared to wildtype control (Figure 3H). In contrast, eye-antennal discs from the null mutants of Rassf, another protein that has been proposed to mediate Hpo-STRIPAK interaction in Drosophila (Ribeiro et al., 2010), did not show obvious elevation of Hpo T195 phosphorylation (Figure 3H). Taken together, these results support a role for Slmap and the STRIPAK complex in suppressing the kinase activity of Hpo.
Next, we investigated how Slmap interacts with the other subunits of the STRIPAK complex by co-IP assays and found that Slmap interacted strongly with Fgop2, but not Cka, Strip or Mob4 (Figure 3I). Consistent with this observation, Slmap was identified as a Fgop2-binding protein in a Y2H-based protein-interaction map of the Drosophila proteome (Giot et al., 2003). Next, we performed co-IP assays between Hpo and each subunit of the STRIPAK complex. In line with a previous study (Ribeiro et al., 2010), we detected weak interactions between Hpo and Fgop2, Cka, or Mts (Figure 3J). Although not identified as a Hpo-binding protein in the same study, Slmap showed much stronger interaction with Hpo under the same experimental condition (Figure 3J). Significantly, co-expression of Slmap markedly enhanced the interaction between Hpo and Fgop2 (Figure 3J, compare lane 9 with lane 3), suggesting that Slmap may function as a bridge between Hpo and Fgop2, and by inference, the rest of the STRIPAK complex. We tested this model by taking advantage of a FLAG-tagged Cka transgene driven by the Tubulin α1 promoter that fully rescued the lethality of Cka mutant flies (Liu et al., 2016). As shown in Figure 3K, FLAG-Cka readily co-immunoprecipitated with endogenous Hpo from protein lysates of wildtype third instar larvae, but not homozygous slmap4 larvae (Figure 3K), suggesting that Slmap is required for the other subunits of the STRIPAK complex to associate with Hpo.
Phospho-dependent recruitment of SLMAP by MST2 in mammalian cells
Having established a mechanism by which autophosphorylation of the Hpo linker recruits Slmap and the STRIPAK complex to suppress Hpo activity, we explored whether such mechanism is conserved in mammals. We noted that co-immunoprecipitation between MST1/2 and SLMAP has been reported in mammalian cells (Couzens et al., 2013). However, neither the physiological outcome of this interaction nor the underlying mechanism of how Slmap brings to STRIPAK to Hpo/MST was elucidated. Like its Drosophila counterpart, the mammalian SLMAP binds to MST2 but not MST2D164A, a kinase dead mutant of MST2 (Chan et al., 2005) (Figure 4A). To test whether loss of SLMAP also leads to activation of MST1/2, we mutated the SLMAP gene using CRISPR-mediated genome editing in the haploid cell line HAP1 (Figure S4A). Indeed, loss of SLMAP led to increased phosphorylation of MST1/2 at its activation loop site (T183/180), accompanied by increased phosphorylation of MOB1 (T35) and YAP (S127 and S397) (Figure 4B). The activation of Hippo signaling upon loss of SLMAP is MST1/2-dependent, since a MST1/2-specific inhibitor XMU-MP-1 (Fan et al., 2016) completely reversed the elevated phosphorylation of MST1/2, MOB1 and YAP in the SLMAP mutant cells (Figure 4B). Consistent with these results, YAP was more concentrated in the cytoplasm in the SLMAP mutant HAP1 cells compared to the wildtype control (Figure 4C–D).
Figure 4. Phospho-dependent recruitment of SLMAP by MST2 in mammalian cells.
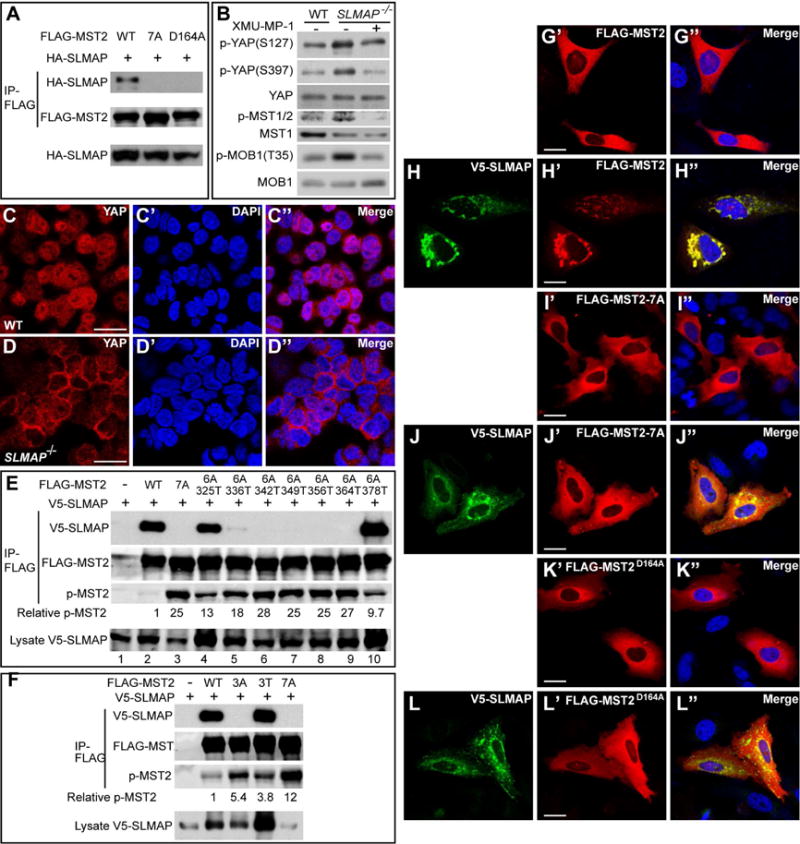
(A) HEK293 cells transfected with the indicated constructs were analyzed by anti-FLAG immunoprecipitation. Note the interaction between SLMAP and wildtype MST2, but not MST2-7A or MST2D164A.
(B) Control and SLMAP mutant HAP1 cells were probed for phosphorylation of MST1/2 (T183/180), YAP (S127 and S397) and MOB1 (T35). Note increased phosphorylation of these proteins in the SLMAP mutant cells, which was completely suppressed by XMU-MP-1.
(C–D) Subcellular localization of YAP in HAP1 cells. Note that YAP was evenly distributed between cytosol and nucleus in the wildtype HAP1 cells, but mostly concentrated in the cytosol in the SLMAP mutant HAP1 cells. Scale bars are 20μm.
(E) Identification of 325T, 336T and 378T as SLMAP binding sites in the MST2 linker. HEK293 cells expressing V5-SLMAP and the indicated linker site mutants of MST2 were subjected to immunoprecipitation and analyzed by the indicated antibodies. The phospho-T180 over total MST2 level was also quantified relative to the wildtype MST2 and indicated below the p-MST2 blot. Note the absence of SLMAP-binding for MST2-7A (lane 3), the significant restoration of SLAMP-binding for MST2-6A/325T (lane 4) and MST2-6A/378T (lane 10), and the weak restoration of SLMAP-binding for MST2-6A/336T (lane 5). Also note the reverse correlation between p-MST2 level and the strength of SLMAP-binding.
(F) Further characterization of 325T, 336T and 378T as SLMAP binding sites. Similar to (E) except that different MST2 mutants were analyzed. Note the absence of SLMAP-binding for MST2-3A and MST2-7A, and the comparable SLMAP-binding for wildtype and MST2-3T. Also note that phospho-T180 level of MST2-3A or MST2-3T is higher than that of wildtype MST2 but lower than MST2-7A.
(G–L) Subcellular localization of MST2 and SLMAP. HeLa cells expressing the indicated FLAG-MST2 variants (red) with or without V5-SLMAP (green) were visualized by FLAG and V5 immunostaining. Note the ubiquitous cytosolic distribution of MST, MST2-7A or MST2D164A in the absence of co-expressed SLMAP (G, I and K). Also note the co-localization of MST2, but not MST2-7A or MST2D164A, to punctate structures in the presence of co-expressed SLMAP (compare H with J and L). Scale bars are 25μm.
The human MST2 linker contains seven autophosphorylation sites, four of which are recognized by MOB1 (Ni et al., 2015). Substitution of all seven sites with Ala (MST2-7A) abolished MST2-SLMAP interaction (Figure 4A and 4E), suggesting that human SLMAP also recognizes the autophosphorylation linker sites in MST2. In further agreement with the results in Drosophila, MST2-7A also showed gain-of-function activity, as revealed by a dramatic increase of MST2 autophosphorylation at its activation loop site T180 (Figure 4E). To assess the contribution of each linker site to MST2-SLMAP binding, we engineered a set of MST2 variants mutating all but one of the seven linker phosphorylation sites (MST2-6A/325T, MST2-6A/336T, MST2-6A/342T, MST2-6A/349T, MST2-6A/356T, MST2-6A/364T, and MST2-6A/378T). Among these mutants, MST2-SLMAP binding was significantly restored in MST2-6A/325T and MST2-6A/378T, and slightly restored in MST2-6A/336T (Figure 4E). Accordingly, these three mutants also showed less T180 phosphorylation than MST2-7A or the rest of the MST2-6A mutants (Figure 4E). Taken together, these results suggest that the T325, T336 and T378 site each contributes partially to SLMAP binding. We note that among these three sites, T378 is one of the four linker sites that have been implicated in MOB1 binding (Ni et al., 2015). Thus, unlike Drosophila Hpo in which the Mats-binding and the Slmap-binding sites are completely separable, T378 is a shared site in MST2 that can bind both MOB1 and SLMAP (Figure S4D).
To further explore the function of T325, T336 and T378 in SLMAP binding, we generated two additional mutants, MST2-3A (325/336/378A-342/349/356/364T) that mutates only these three sites, and MST2-3T (325/336/378T-342/349/356/364A) that retains only these three sites. MST2-3A completely lost SLMAP binding while MST2-3T showed comparable SLMAP binding as wildtype MST2 (Figure 4F), demonstrating that the T325, T336 and T378 sites combinatorically mediate MST2-SLMAP interaction. It is worth noting that although both MST2-3A and MST2-7A abolished SLMAP binding, MST2-7A still had higher T180 phosphorylation than MST2-3A (Figure 4F). Conversely, while MST2-3T and wildtype MST2 showed similar SLMAP binding, MST2-3T showed higher T180 phosphorylation than wildtype MST2 (Figure 4F). The simplest explanation is that the linker phosphorylation sites may engage additional proteins besides SLMAP to restrict MST2 kinase activity. Alternatively, SLMAP may bind to the other linker sites besides T325, T336 and T378, but the binding may be too weak to be revealed by co-IP assays.
To further corroborate our model of phospho-dependent recruitment of SLMAP by MST2, we examined the subcellular localization of the two proteins. For comparison, we also included the linker-sites-defective mutant MST2-7A and the kinase-dead mutant MST2D164A in our analysis. When expressed alone in HeLa cells, the tail-anchored protein SLMAP was predominantly localized to punctate structures positive for mitochondria and ER markers (Figure S4B–C), as reported previously (Byers et al., 2009), while MST2, MST2-7A or MST2D164A was distributed ubiquitously in the cytosol (Figures 4G, 4I and 4K). However, when SLMAP and MST2 were co-expressed, MST2 was predominantly co-localized with SLMAP to punctate structures (Figure 4H). In sharp contrast, MST2-7A or MST2D164A was still ubiquitously distributed in the cytosol in the presence of co-expressed SLMAP (Figures 4J and 4L). These results not only support the critical role of MST2 linker phosphorylation in SLMAP recruitment but also implicate mitochondria and ER surface as potential subcellular sites of STRIPAK action.
DISCUSSION
In this study, we have identified an evolutionarily conserved mechanism by which activated Hpo/MST self-restricts its own kinase activity. This mechanism involves autophosphorylation of an unstructured linker in active Hpo/MST, which creates docking sites for Slmap and the STRIPAK PP2A phosphatase complex to inactivate Hpo/MST (Figure S4D). In support of our model, mutation of the phospho-dependent docking sites in Hpo/MST or loss of Slmap results in constitutive activation of Hpo/MST in both Drosophila and mammalian cells. Interestingly, both the linker phosphorylation motifs in Hpo/MST and the Slmap-STRIPAK complex are conserved even in the single cell organism Capsaspora (Suga et al., 2013). The deep conservation of this mechanism suggests that it represents a primordial and significant mode of regulation that emerged very early in the evolutionary of the Hippo signaling pathway.
Our model provides a molecular explanation for several outstanding findings in the literature. First, an earlier structure-function analysis has identified an inhibitory domain in MST1 between aa 331 and aa 394, as deletion of this region resulted in increased MST1 kinase activity (Creasy et al., 1996). Since this region includes six of the seven linker phosphorylation sites, deletion of this domain is predicted to largely abolish SLMAP-mediated recruitment of STRIPAK and thus result in increased activity of MST1, as we have seen in the MST2-7A mutant. Second, it was reported before that mutation of the activation loop phosphorylation site T180 in MST2 reduced its interactions with SLMAP (Couzens et al., 2013), which we have confirmed (Figure 4A). While in principle this finding can be interpreted as the activation loop phosphorylation site of MST1/2 (T180/183) being the binding site for SLMAP, our studies argue against this possibility. For example, the MST2-7A mutant completely abolished SLMAP-binding despite showing a dramatic increase of T180 phosphorylation, demonstrating that phosphorylated T183/180 per se is not directly involved in SLMAP-binding. Instead, mutation of T183/180 likely affects SLMAP-binding indirectly, by abolishing MST1/2 kinase activity, preventing linker phosphorylation and therefore SLMAP-binding.
We note that our model of Slmap-mediated association between Hpo/MST and the STRIPAK complex differs from a previous model positing that Rassf brings the STRIPAK complex to suppress Hpo activity (Ribeiro et al., 2010). Unlike Slmap, which binds to the linker autophosphorylation sites of Hpo/MST, Rassf binds to Hpo/MST through their C-terminal SARAH domains. These two models, however, are not necessarily exclusive of each other. Dimerization of Hpo/MST, either on its own or assisted by Sav/SAV1, promotes its activation by trans-autophosphorylating the activation loop site in the kinase domain (Ni et al., 2013; Praskova et al., 2004). In contrast, RASSF5 forms heterodimer with MST2, a configuration that precludes trans-autophosphorylation (Ni et al., 2013). Indeed, the Hpo-Sav complex has much higher kinase activity than the Hpo-Rassf complex when isolated from the Drosophila S2 cells (Polesello et al., 2006). Thus, it is possible that the Slmap- and the Rassf-mediated mechanisms may apply to different threshold of Hpo/MST activity. Irrespective of the validity of this model, the fact that loss of Rassf leads to a much milder phenotype in Drosophila (Polesello et al., 2006) than loss of Slmap or mutating the Slmap docking sites in Hpo suggests the Slmap-mediated mechanism is likely more critical in the context of organ development, a proposition that is further supported by our observation of elevated Hpo T195 phosphorylation in Slmap but not Rassf mutant fly tissues (Figure 3H).
Multisite phosphorylation of a single substrate by a single kinase is a common phenomenon in signal transduction (Cohen, 2000; Salazar and Hofer, 2009). Depending on the cooperativity among the phosphorylation sites, it can lead to switch-like behavior (Koivomagi et al., 2011; Nash et al., 2001) or graded response (Pufall et al., 2005; Strickfaden et al., 2007), and therefore increasing the complexity of signal transduction. A unique feature of Hpo/MST is that multisite autophosphorylation both facilitates as well as restricts Hippo signaling (Figure S4D). These opposing effects on signal transduction are mediated by largely non-overlapping autophosphorylation sites that recruit distinct effectors, Mats/MOB1 and STRIPAK. This molecular design provides a most efficient way to couple a “signal-on” state to a “signal-off” state, and may serve to maintain a homeostatic level of Hippo signaling or prevent spurious/prolonged pathway activation that can be detrimental to an organism. Interestingly, multisite phosphorylation with opposing effects was also documented in a recent study of the Elk-1 transcriptional factor (Mylona et al., 2016), suggesting that this may be a common design principle in signal transduction.
PP2A represents the most abundant phosphatase in cells and is predominantly found as a heterotrimeric complex composed of a catalytic C subunit, a scaffolding A subunit, and a regulatory B subunit (Rogers et al., 2016; Slupe et al., 2011). While it is generally believed that the diverse B subunit dictates substrate specificity of the PP2A holoenzyme, we demonstrate here that recognition of Hpo/MST by the STRIPAK PP2A complex is mediated not by the B subunit (which is Cka in Drosophila and Striatin1/3/4 in mammals), but rather by a more periphery subunit Slmap. This recognition is mediated by modular interactions between a dedicated phospho-Thr-binding domain (FHA domain) in Slmap and linker phosphorylation sites in Hpo/MST, in much the same way that modular interactions mediate other cell signaling events, such as the recognition of phosphorylated Tyrosine residues by SH2 domains. The phospho-dependent nature of Hpo/MST-Slmap interactions not only provides a robust mechanism to couple phosphorylation and dephosphorylation in dynamic cell signaling, but also highlights the importance of Slmap as a phosphorylation dependent substrate adaptor protein that targets PP2A to its substrates in a regulated manner. Whether Slmap mediates the recognition of other substrates besides Hpo/MST by PP2A awaits future investigation.
Lastly, we suggest that strategies aimed at disrupting the Slmap-mediated self-restricting mechanism on Hpo/MST may provide a potential approach to activate Hippo signaling for cancer treatment. Most human cancers show activation of YAP/TAZ, yet they generally lack mutations in the canonical tumor suppressors upstream of YAP/TAZ (Yu et al., 2015; Zanconato et al., 2016). This suggests that in most tumors with high YAP/TAZ activity, the canonical Hippo signaling is likely intact. Thus, strategies aimed at inhibiting the activity or the recruitment of the STRIPAK complex to Hpo/MST may in principle lead to activation of Hpo/MST, inactivation of YAP/TAZ and suppression of cancer growth. The modular nature of Hpo/MST-Slmap interactions makes this approach especially appealing. Furthermore, due to its profound effects on Hpo/MST kinase activity, the Slmap-STRIPAK complex may represent an important and underappreciated entry point of regulation by physiological signals. In support of this view, bacteria-induced Toll receptor signaling activates the Hippo pathway in Drosophila fat bodies through degradation of the STRIPAK component Cka (Liu et al., 2016). Identification of other signals that converge on the Slmap-STRIPAK complex will provide important insights into the regulation of Hippo signaling in development and physiology.
Methods and Materials
Plasmids and antibodies
Myc-Hpo, FLAG-Sav (Wu et al., 2003); FLAG-Cka, FLAG-Slmap, FLAG-Fgop2, FLAG-Strip, FLAG-Mob4 (Liu et al., 2016); FLAG-MST2, FLAG-MST2D164A (Chan et al., 2005); Flag-Hpo, and GST-Mats (Ni et al., 2015) expression constructs have been described previously. HA-Slmap was made from Flag-Slmap. FLAG-PP2A-A (LD10247) and FLAG-Mts (LD26077) expression constructs were generated from cDNA clones obtained from Drosophila Genomics Resource Center. V5-SLMAP was made from the cDNA clone BC115701.1. Site-directed mutagenesis was used to generate mutants of linker autophosphorylation sites of Hpo/MST2. A flexible linker GGSG was fused to the N-terminus of Slmap FHA domain (C205-Q343) and the DNA sequence was inserted into pGEX6p-1 to produce GST-FHA proteins in BL21(DE3)-RIPL cells (Agilent).
The following antibodies were used in this study: FLAG and HA (Sigma-Aldrich); Myc (Calbiochem); V5 (Invitrogen); Hpo (Wu et al., 2003); Diap1 (Gift from B. Hay); β-galactosidase (Developmental Biology Hybridoma Bank); YAP (Novus; for immunostaining); GFP, YAP (for Western blotting), p-YAP(S127), p-YAP(S381), p-MST1/2(T181/T180; also used to detect p-Hpo), MOB1, and p-MOB1(T35) (Cell Signaling); Calreticulin (Abcam).
Drosophila and mammalian cell culture
Drosophila S2R+ cells were cultured in Schneider’s medium supplemented with 10% FBS and antibiotics at 25 °C (Invitrogen). 293T and HeLa cells were maintained in DMEM medium, while HAP1 cells in IMDM medium, supplemented with 10% FBS and antibiotics at 37 °C (Invitrogen). Transfection, immunoprecipitation, and western blotting were carried out as described previously (Zheng et al., 2015). RNAi in S2R+ cells was carried out using the bathing protocol obtained from DRSC (http://www.flyrnai.org/DRSC-PRR.html).
Drosophila genetics
Slmap RNAi (8199) line was obtained from the Vienna Drosophila Resource Center (VDRC). The 20XUAS-FLPG5.PEST:attP18 line was obtained from Bloomington Drosophila Stock Center. rassf36 and rassf44.2 were gifts from N. Tapon (Polesello et al., 2006). The tub-FLAG-Cka transgenic line has been described before (Liu et al., 2016). To generate a mutation in slmap, a CRISPR target sites (CGAGGCGTCCACGAAGAACC) was selected by CRISPR Optimal Target Finder (http://tools.flycrispr.molbio.wisc.edu/targetFinder/). Mutants were first selected according the lethal phenotype and then further verified by DNA sequencing and rescue test using slmap cDNA. To generate the FLP-out fly lines, FLAG-Hpo was inserted into the pTubattB vector (Liu et al., 2016) using KpnI/XbaI digestion and then the FRT y+ FRT cassette was inserted into the KpnI site. The resulting constructs were integrated into the attP landing site 89Fb. The following genotypes were used in this study:
Comparing the in vivo activity of Hpo, Hpo431A and Hpo4A/431T:
20XUAS-FLPG5/+; en-Gal4 UAS-GFP; tub-FRT-y+-FRT-FLAG-hpo (wing)
20XUAS-FLPG5/+; en-Gal4 UAS-GFP; tub-FRT-y+-FRT-FLAG-hpo/diap1-lacZ (wing for diap1-lacZ)
ey-Flp; tub-FRT-y+-FRT-FLAG-hpo (eye)
GFP+ hpo− clones:
UAS-GFP hs-Flp; tub-Gal80 FRT42D/FRT42D hpo42–47; tub-Gal4
GFP+ hpo− clones overexpressing slmap RNAi:
UAS-GFP hs-Flp; tub-Gal80 FRT42D/FRT42D hpo42–47; tub-Gal4/UAS-slmap RNAi
GFP+ ex− clones:
UAS-GFP hs-Flp; tub-Gal80 FRT40A/FRT40A exe1; tub-Gal4
GFP+ ex− clones overexpressing slmap RNAi:
UAS-GFP hs-Flp; tub-Gal80 FRT40A/FRT40A exe1; tub-Gal4/UAS-slmap RNAi
slmap− flies expressing FLAG-Cka:
slmap4/ slmap4; tub-FLAG-cka
Immunofluorescence staining
HAP1 cells were seeded on the glass chamber slides (Lab Tek) that were pre-coated with 5 g/mL human plasma fibronectin (EMD Millipore) for 5 hrs before fixation. HeLa cells transfected with expression constructs were cultured for 24 hrs before fixation. Cells were fixed in 4% paraformaldehyde for 15 min and then permeabilized with 0.3% Triton X-100. After blocking in 5% goat serum for 1 hr, slides were incubated with primary antibody diluted in 5% goat serum for overnight at 4°C. After three washes in PBS, slides were incubated with secondary antibody for 1 hr, washed for three times in PBS, incubated with DAPI (1:10000 dilution) for 5 min, then washed and mounted. HeLa cells were incubated with 100 nM of Mitotracker (Cell signaling) for 1 hr before fixation to label mitochondria.
CRISPR/Cas9 genome editing in HAP1 cells
CRISPR target site (GTCCCTGAGGCGGGTTCTTC) of SLMAP was selected by the ATUM gRNA Design Tool (https://www.atum.bio/eCommerce/cas9/input) with a score of 100 out of 100, and was further verified by the online CRISPR Design Tool of MIT (http://crispr.mit.edu:8079/?) which gave a score of 84 out of 100. The target sequence was cloned into vector pSpCas9n(BB)-2A-GFP and used to introduce a mutation to SLMAP gene in HAP1 cells according to the described protocol (Ran et al., 2013).
Drug treatment
The MST1/2 inhibitor XMU-MP-1HAP1 (Y-100526) was purchased from Medchemexpress. Hap1 cells were incubated with 10μM XMU-MP-1 in fresh IMDM medium supplemented with 10% FBS for 1hr before harvest.
Statistical analyses
Two-tailed Student’s t test was used for two sample comparisons. Error bars represent SEM and p < 0.05 was considered significant.
Supplementary Material
Highlights.
The Hpo/MST linker contains inhibitory autophosphorylation sites.
The autoinhibitory sites of Hpo/MST bind Slmap in a phospho-dependent manner.
Slmap recruits the STRIPAK PP2A complex to inactivate Hpo/MST.
Acknowledgments
We thank Dr. Nic Tapon (Francis Crick Institute, UK) for rassf mutant flies and Dr. Xuelian Luo (UT Southwestern Medical Center, US) for sharing unpublished data. We also thank Bloomington Drosophila Stock Center, Vienna Drosophila Resource Center (VDRC, www.vdrc.at), and Drosophila Genomics Resource Center (supported by NIH grant OD010949-10) for fly strains or reagents. This study was supported in part by grants from the National Institutes of Health (EY015708). D.P. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Y.Z. and D.P. initiated the project; Y.Z., B.L., L.W., H.L. and D.P. designed experiments; Y.Z., B.L., L.W., H.L., and K.D.P.P. performed experiments; Y.Z., B.L. and D.P. analyzed data and constructed figures; Y.Z. and D.P. wrote the manuscript.
References
- Basler K, Struhl G. Compartment boundaries and the control of Drosophila limb pattern by hedgehog protein. Nature. 1994;368:208–214. doi: 10.1038/368208a0. [DOI] [PubMed] [Google Scholar]
- Bischof J, Maeda RK, Hediger M, Karch F, Basler K. An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:3312–3317. doi: 10.1073/pnas.0611511104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boggiano JC, Vanderzalm PJ, Fehon RG. Tao-1 phosphorylates Hippo/MST kinases to regulate the Hippo-Salvador-Warts tumor suppressor pathway. Developmental cell. 2011;21:888–895. doi: 10.1016/j.devcel.2011.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers JT, Guzzo RM, Salih M, Tuana BS. Hydrophobic profiles of the tail anchors in SLMAP dictate subcellular targeting. BMC cell biology. 2009;10:48. doi: 10.1186/1471-2121-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EH, Nousiainen M, Chalamalasetty RB, Schafer A, Nigg EA, Sillje HH. The Ste20-like kinase Mst2 activates the human large tumor suppressor kinase Lats1. Oncogene. 2005;24:2076–2086. doi: 10.1038/sj.onc.1208445. [DOI] [PubMed] [Google Scholar]
- Cheung WL, Ajiro K, Samejima K, Kloc M, Cheung P, Mizzen CA, Beeser A, Etkin LD, Chernoff J, Earnshaw WC, et al. Apoptotic phosphorylation of histone H2B is mediated by mammalian sterile twenty kinase. Cell. 2003;113:507–517. doi: 10.1016/s0092-8674(03)00355-6. [DOI] [PubMed] [Google Scholar]
- Couzens AL, Knight JD, Kean MJ, Teo G, Weiss A, Dunham WH, Lin ZY, Bagshaw RD, Sicheri F, Pawson T, et al. Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase- phosphatase interactions Science signaling. 2013;6:rs15. doi: 10.1126/scisignal.2004712. [DOI] [PubMed] [Google Scholar]
- Creasy CL, Ambrose DM, Chernoff J. The Ste20-like protein kinase, Mst1, dimerizes and contains an inhibitory domain. The Journal of biological chemistry. 1996;271:21049–21053. doi: 10.1074/jbc.271.35.21049. [DOI] [PubMed] [Google Scholar]
- Deng Y, Matsui Y, Zhang Y, Lai ZC. Hippo activation through homodimerization and membrane association for growth inhibition and organ size control. Developmental biology. 2013;375:152–159. doi: 10.1016/j.ydbio.2012.12.017. [DOI] [PubMed] [Google Scholar]
- Durocher D, Jackson SP. The FHA domain. FEBS letters. 2002;513:58–66. doi: 10.1016/s0014-5793(01)03294-x. [DOI] [PubMed] [Google Scholar]
- Fan F, He Z, Kong LL, Chen Q, Yuan Q, Zhang S, Ye J, Liu H, Sun X, Geng J, et al. Pharmacological targeting of kinases MST1 and MST2 augments tissue repair and regeneration. Science translational medicine. 2016;8:352ra108. doi: 10.1126/scitranslmed.aaf2304. [DOI] [PubMed] [Google Scholar]
- Genevet A, Tapon N. The Hippo pathway and apico-basal cell polarity. The Biochemical journal. 2011;436:213–224. doi: 10.1042/BJ20110217. [DOI] [PubMed] [Google Scholar]
- Giot L, Bader JS, Brouwer C, Chaudhuri A, Kuang B, Li Y, Hao YL, Ooi CE, Godwin B, Vitols E, et al. A protein interaction map of Drosophila melanogaster. Science. 2003;302:1727–1736. doi: 10.1126/science.1090289. [DOI] [PubMed] [Google Scholar]
- Goudreault M, D’Ambrosio LM, Kean MJ, Mullin MJ, Larsen BG, Sanchez A, Chaudhry S, Chen GI, Sicheri F, Nesvizhskii AI, et al. A PP2A phosphatase high density interaction network identifies a novel striatin-interacting phosphatase and kinase complex linked to the cerebral cavernous malformation 3 (CCM3) protein. Molecular & cellular proteomics : MCP. 2009;8:157–171. doi: 10.1074/mcp.M800266-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder G, Dupont S, Piccolo S. Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nature reviews Molecular cell biology. 2012;13:591–600. doi: 10.1038/nrm3416. [DOI] [PubMed] [Google Scholar]
- Harvey KF, Hariharan IK. The hippo pathway. Cold Spring Harbor perspectives in biology. 2012;4:a011288. doi: 10.1101/cshperspect.a011288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HL, Wang S, Yin MX, Dong L, Wang C, Wu W, Lu Y, Feng M, Dai C, Guo X, et al. Par-1 regulates tissue growth by influencing hippo phosphorylation status and hippo-salvador association. PLoS biology. 2013;11:e1001620. doi: 10.1371/journal.pbio.1001620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Dong L, Lu Y, Wu W, Hao Q, Zhou Z, Jiang J, Zhao Y, Zhang L. Dimerization and cytoplasmic localization regulate Hippo kinase signaling activity in organ size control. The Journal of biological chemistry. 2012;287:5784–5796. doi: 10.1074/jbc.M111.310334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivomagi M, Valk E, Venta R, Iofik A, Lepiku M, Balog ER, Rubin SM, Morgan DO, Loog M. Cascades of multisite phosphorylation control Sic1 destruction at the onset of S phase. Nature. 2011;480:128–131. doi: 10.1038/nature10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koontz LM, Liu-Chittenden Y, Yin F, Zheng Y, Yu J, Huang B, Chen Q, Wu S, Pan D. The Hippo effector Yorkie controls normal tissue growth by antagonizing scalloped-mediated default repression. Developmental cell. 2013;25:388–401. doi: 10.1016/j.devcel.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan J, Sczaniecka A, Heidary Arash E, Nguyen L, Chen CC, Ratkovic S, Klezovitch O, Attisano L, McNeill H, Emili A, et al. DLG5 connects cell polarity and Hippo signaling protein networks by linking PAR-1 with MST1/2. Genes & development. 2016;30:2696–2709. doi: 10.1101/gad.284539.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen MK, Yuan Z, Boag PR, Yang Y, Villen J, Becker EB, DiBacco S, de la Iglesia N, Gygi S, Blackwell TK, et al. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell. 2006;125:987–1001. doi: 10.1016/j.cell.2006.03.046. [DOI] [PubMed] [Google Scholar]
- Liu B, Zheng Y, Yin F, Yu J, Silverman N, Pan D. Toll Receptor-Mediated Hippo Signaling Controls Innate Immunity in Drosophila. Cell. 2016;164:406–419. doi: 10.1016/j.cell.2015.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mylona A, Theillet FX, Foster C, Cheng TM, Miralles F, Bates PA, Selenko P, Treisman R. Opposing effects of Elk-1 multisite phosphorylation shape its response to ERK activation. Science. 2016;354:233–237. doi: 10.1126/science.aad1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash P, Tang X, Orlicky S, Chen Q, Gertler FB, Mendenhall MD, Sicheri F, Pawson T, Tyers M. Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature. 2001;414:514–521. doi: 10.1038/35107009. [DOI] [PubMed] [Google Scholar]
- Ni L, Li S, Yu J, Min J, Brautigam CA, Tomchick DR, Pan D, Luo X. Structural basis for autoactivation of human Mst2 kinase and its regulation by RASSF5. Structure. 2013;21:1757–1768. doi: 10.1016/j.str.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni L, Zheng Y, Hara M, Pan D, Luo X. Structural basis for Mob1-dependent activation of the core Mst-Lats kinase cascade in Hippo signaling. Genes & development. 2015;29:1416–1431. doi: 10.1101/gad.264929.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D. The hippo signaling pathway in development and cancer. Developmental cell. 2010;19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polesello C, Huelsmann S, Brown NH, Tapon N. The Drosophila RASSF homolog antagonizes the hippo pathway. Current biology : CB. 2006;16:2459–2465. doi: 10.1016/j.cub.2006.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon CL, Lin JI, Zhang X, Harvey KF. The sterile 20-like kinase Tao-1 controls tissue growth by regulating the Salvador-Warts-Hippo pathway. Developmental cell. 2011;21:896–906. doi: 10.1016/j.devcel.2011.09.012. [DOI] [PubMed] [Google Scholar]
- Praskova M, Khoklatchev A, Ortiz-Vega S, Avruch J. Regulation of the MST1 kinase by autophosphorylation, by the growth inhibitory proteins, RASSF1 and NORE1, and by Ras. The Biochemical journal. 2004;381:453–462. doi: 10.1042/BJ20040025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pufall MA, Lee GM, Nelson ML, Kang HS, Velyvis A, Kay LE, McIntosh LP, Graves BJ. Variable control of Ets-1 DNA binding by multiple phosphates in an unstructured region. Science. 2005;309:142–145. doi: 10.1126/science.1111915. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nature protocols. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro PS, Josue F, Wepf A, Wehr MC, Rinner O, Kelly G, Tapon N, Gstaiger M. Combined functional genomic and proteomic approaches identify a PP2A complex as a negative regulator of Hippo signaling. Molecular cell. 2010;39:521–534. doi: 10.1016/j.molcel.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Rogers S, McCloy R, Watkins DN, Burgess A. Mechanisms regulating phosphatase specificity and the removal of individual phosphorylation sites during mitotic exit. BioEssays : news and reviews in molecular, cellular and developmental biology. 2016;38(Suppl 1):S24–32. doi: 10.1002/bies.201670905. [DOI] [PubMed] [Google Scholar]
- Slupe AM, Merrill RA, Strack S. Determinants for Substrate Specificity of Protein Phosphatase 2A. Enzyme research. 2011;2011:398751. doi: 10.4061/2011/398751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickfaden SC, Winters MJ, Ben-Ari G, Lamson RE, Tyers M, Pryciak PM. A mechanism for cell-cycle regulation of MAP kinase signaling in a yeast differentiation pathway. Cell. 2007;128:519–531. doi: 10.1016/j.cell.2006.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suga H, Chen Z, de Mendoza A, Sebe-Pedros A, Brown MW, Kramer E, Carr M, Kerner P, Vervoort M, Sanchez-Pons N, et al. The Capsaspora genome reveals a complex unicellular prehistory of animals. Nature communications. 2013;4:2325. doi: 10.1038/ncomms3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Huang J, Dong J, Pan D. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell. 2003;114:445–456. doi: 10.1016/s0092-8674(03)00549-x. [DOI] [PubMed] [Google Scholar]
- Yu FX, Zhao B, Guan KL. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell. 2015;163:811–828. doi: 10.1016/j.cell.2015.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the Roots of Cancer. Cancer cell. 2016;29:783–803. doi: 10.1016/j.ccell.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Wang W, Liu B, Deng H, Uster E, Pan D. Identification of Happyhour/MAP4K as Alternative Hpo/Mst-like Kinases in the Hippo Kinase Cascade. Developmental cell. 2015;34:642–655. doi: 10.1016/j.devcel.2015.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.