

Abstract
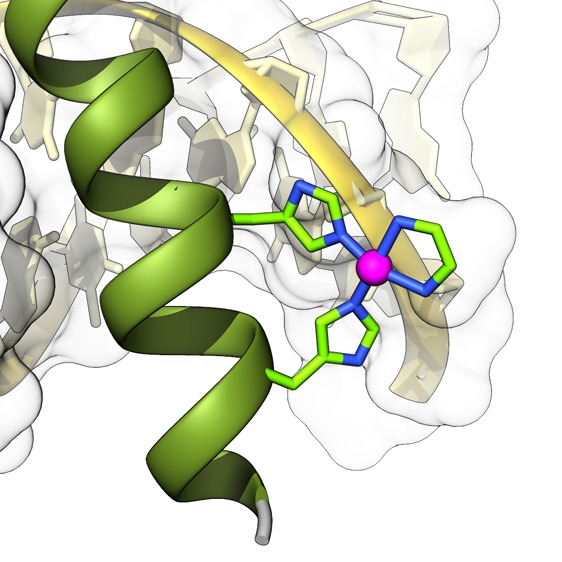
A fragment of the DNA basic region (br) of the GCN4 bZIP transcription factor has been modified to include two His residues at designed i and i+4 positions of its N-terminus. The resulting monomeric peptide (brHis2) does not bind to its consensus target DNA site (5′-GTCAT-3′). However, addition of Pd(en)Cl2 (en, ethylenediamine) promotes a high-affinity interaction with exquisite selectivity for this sequence. The peptide–DNA complex is disassembled by addition of a slight excess of a palladium chelator, and the interaction can be reversibly switched multiple times by playing with controlled amounts of either the metal complex or the chelator. Importantly, while the peptide brHis2 fails to translocate across cell membranes on its own, addition of the palladium reagent induces an efficient cell internalization of this peptide. In short, we report (1) a designed, short peptide that displays highly selective, major groove DNA binding, (2) a reversible, metal-dependent DNA interaction, and (3) a metal-promoted cell internalization of this basic peptide.
Introduction
Protein expression depends on the concerted action of transcription factors (TFs), which are specialized proteins that specifically bind to regulatory DNA sequences, thereby modulating the transcription of downstream genes.1 Over the past few years, there has been a great interest in the development of miniaturized models of TFs capable of reproducing their DNA-binding properties, as they might open interesting biomedical opportunities.2 These peptides interact with the DNA mainly through the major groove and, thus, might provide a compelling alternative to classic DNA binding agents that interact by intercalation or by insertion into the minor groove.3 However, the design and preparation of this type of minimalistic peptides has proven extremely challenging.2,4
The most successful examples have been inspired by the DNA binding domain of bZIP proteins such as GCN4, an archetypal member of this family of TFs that specifically binds to palindromic ATF/CREB (5′-ATGAC-GTCAT-3′) or AP1 (5′-ATGA(c)TCAT-3′) sites as a leucine zipper-mediated dimer of uninterrupted α-helices.5 The DNA interaction occurs through the N-terminal basic region (br), which folds into an α-helix upon insertion in the major groove of the target DNA site.6
Following the seminal work by Kim and co-workers in 1990,7 a number of groups have demonstrated that the complete leucine zipper of the GCN4 DNA binding domain could be replaced by artificial dimerizing elements without significantly compromising the DNA recognition properties of the system.8 In contrast, monomeric GCN4 peptides display very low DNA binding affinity,9 probably because the high entropic cost associated with the folding of the peptide chain into the α-helical conformation, required for DNA binding, is not compensated by the enthalpic gain of a monomeric interaction.10,11 A couple of reports have shown that introducing lactam bridges or covalent staples can promote DNA binding, although the affinity and selectivity of these modified systems are rather modest.12 Better results have been obtained by grafting key DNA binding residues from the bZIP basic regions into stable peptide scaffolds that support the α-helical conformation.13 In an alternative approach, our group has demonstrated that tethering the peptides to prosthetic minor groove binders that establish accessory stabilizing interactions can also lead to stable DNA complexes.14 Furthermore, we have shown that such tethering can also be achieved through a nickel-mediated coordination with bisbenzamidine–bipyridine conjugates.15 Tethering metal-based intercalators to peptides has also allowed cooperative binding to DNA.16 Despite these important advances, an efficient and selective major groove DNA interaction by simple, minimalist peptides, featuring natural amino acids, has not been demonstrated.
Herein we show that a designed bis-histidine grafted peptide featuring 23 amino acids of the basic region of GCN4 binds its consensus DNA site (5′-GTCAT-3′) with high affinity and specificity when treated with a slight excess of [Pd(en)Cl2. The palladium complex works as a stapling agent that favors the required α-helical folding and, ultimately, the insertion of the peptide into the major groove of the target DNA (Figure 1). Noticeably, the interaction can be reversibly switched by controlling the relative amounts of the palladium complex and a metal sequestering agent. Overall, we report the shortest, high-affinity, sequence-selective DNA binding natural peptide described so far, the first on–off, cyclically switchable peptide DNA binder, and the use of a palladium complex to control a selective DNA interaction.
Figure 1.
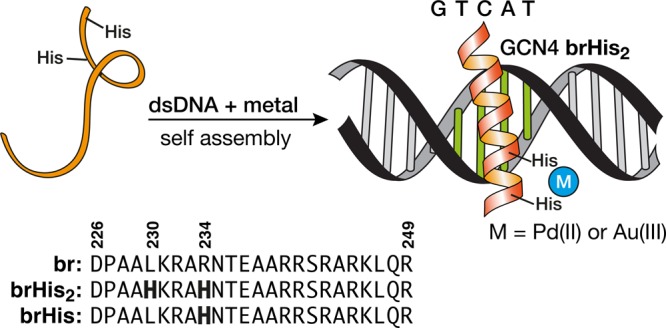
Top: Schematic structure of the supramolecular assembly into specific DNA sites of a modified GCN4 basic region featuring two His residues (His230 and His234, brHis2) in the presence of a metal clip. Bottom: Sequences of the natural GCN4 basic region and the mutated peptides brHis2 and brHis. Mutated residues are in bold.
Importantly, we have also found that the palladium clip triggers the cellular uptake of otherwise nonpenetrating peptides. To the best of our knowledge, this represents the first demonstration of a metal-promoted cell internalization process.
Results and Discussion
Design and Synthesis
As reference for our design we used a fragment of the GCN4 basic region comprising residues Asp226 to Arg249.5,17 Inspection of the X-ray structure of the GCN4/DNA complex revealed that residues Leu230 and Arg234 are oriented toward the solvent on the outer face of the α-helix, and their substitution by His residues should not affect the DNA contacting surface of the peptide.3a,18 Thus, we synthesized the peptide brHis2, featuring two His residues, as well as the natural basic region peptide (br) and the control peptide brHis, in which only one residue is replaced by His (Arg234 → His).
DNA Binding of the Peptide brHis2
The DNA binding properties were studied by electrophoretic mobility assays (EMSA) in polyacrylamide gel under nondenaturing conditions, using SybrGold as the DNA stain.19 As expected, incubation of the peptide brHis2 with the double-stranded (ds) oligonucleotide GTCAT containing the consensus binding site (5′-GTCAT-3′) does not induce the formation of retarded bands (Figure 2a, lane 2), even when using a large excess of the peptide. This is in consonance with the poor DNA affinity of such monomeric basic regions. However, addition of 20 equiv of Pd(en)Cl2 (en, ethylenediamine) to this mixture gives rise to a neat, new (more retarded) band that is consistent with the formation of the desired peptide–DNA complex GTCAT/[(brHis2)Pd(en)]2+ (Figure 2a, lane 3).
Figure 2.

DNA binding properties of brHis2. (a) Incubation with the target ds-oligonucleotide GTCAT and switching the DNA binding by addition of DEDTC. (b) Incubation with mutated dsDNA MUT. Concentration of the components when present: 75 nM dsDNA, 2 μM brHis2, 20 μM Pd(en)Cl2. (a) Lane 1: dsDNA; lane 2: dsDNA and brHis2; lane 3: previous mixture and Pd(en)Cl2; lane 4: mixture in lane 3 after addition of 50 equiv of DEDTC; lane 5: addition of 60 equiv of Pd(en)Cl2 to the mixture in lane 4; lane 6: mixture in lane 5 after addition of 150 equiv of DEDTC; lane 7: addition of 200 equiv of Pd(en)Cl2 to the mixture in lane 6. All the equiv are expressed relative to brHis2. Samples were resolved on a 10% nondenaturing polyacrylamide gel and 0.5× TBE buffer over 40 min at 25 °C and stained with SyBrGold (5 μL in 50 mL of 1× TBE) for 10 min, followed by fluorescence visualization. Oligonucleotide sequences (only one strand shown, brHis2 binding site in bold): GTCAT: 5′-CGC GTCAT AATTGAGAG CGC-3′; MUT: 5′-CGC GTGAT AATTGAGAG CGC-3′.
Importantly, the complex can be dismantled by the addition of a small excess of an external Pd chelator (diethyldithiocarbamate, DEDTC, Figure 2a, lane 4), a result that supports the role of the palladium reagent in promoting the DNA binding. Furthermore, the DNA complex can be reversibly assembled or dismantled by controlling the relative amounts of the Pd complex and the DEDTC (Figure 2a, lanes 4–7). To our knowledge, reversible cyclic DNA binding of such minimalist synthetic peptides lacks any precedent.20,21
The metal-promoted interaction is highly selective; therefore, mutation in a single position of the target site was enough to completely abolish the DNA binding (Figure 2b and Figure S11). It is also important to note that neither the natural GCN4 basic region (br) nor the peptide containing a single His mutation (brHis) gives rise to new bands in the presence of palladium (Figure S11), which confirms the requirement of the two coordinating histidines.
The above data are consistent with the proposed DNA binding by a peptide monomer. Binding by a dimeric species is unlikely, as it would require a specific arrangement of two basic regions that are difficult to envision. Nevertheless, to fully discard the presence of such dimeric species binding to the DNA target, we carried out control experiments with a disulfide dimer of the basic region peptide [br2(SS)], similar to that used by Kim et al.17a As shown in Figure 3, the interaction of this dipeptide with a DNA featuring either its consensus dimeric target site (AP-1) or the monomeric half site (GTCAT) leads to slower migrating bands than those observed for the DNA complex with [(brHis2)Pd(en)]2+. The retarded band observed in lane 8 with AP1, in the presence of brHis2 and the Pd reagent, is consistent with the insertion of two monomeric peptides in their respective half-sites. ESI/MS analysis of the peptide-Pd(en)2 complexes (Figure S19) and UV/vis experiments with Pd(bipy)Cl2 in the presence of the target DNA (Figure S17) also support the formation of DNA complexes with monomeric peptides (see the Supporting Information).
Figure 3.

Comparative EMSA experiment with monomeric brHis2 and br2(SS). Lanes 1–4: 75 nM GTCAT; lanes 2, 3: 2 μM brHis2; lane 3: 20 μM Pd(en)Cl2; lane 4: 1.2 μM br2(SS); lanes 5–8: 75 nM AP-1; lane 6: 1.2 μM br2(SS); lanes 7, 8: 2 μM brHis2; lane 8: 20 μM Pd(en)Cl2. The dsDNA AP-1 features two binding sites in neighbor sequences. We have used this ds-oligonucleotide: 5′-TGGAG ATGA cg TCAT CTCGT-3′ (only one strand shown). Each peptide of the disulfide br2(SS) features the sequence of peptide br (Figure 1), with the C-terminal Arg (249) substituted by Gly-Gly-Cys.
DNA Binding Is Only Observed in the Presence of Specific Metal Complexes
At this point we checked whether other metal ions could also promote DNA binding, or if the induction of the DNA interaction was exclusive for the palladium reagent. As shown in Figure 4a, mixing the target dsDNA GTCAT with brHis2 in the presence of several Ni2+, Zn2+, Cu2+, Co2+, or Fe2+ salts failed to give rise to new bands in the EMSA (Figure 4a, lanes 4–8). It is important to highlight the failure of NiCl2, as this metal complex had been successfully used for promoting bivalent major–minor groove interactions in cooperation with bisbenzamidine–bipyridine conjugates.15 However, it is not useful in the case of the more challenging monovalent peptide–DNA interactions.
Figure 4.

DNA properties of brHis2 with different metal salts. Lane 1: 75 nM GTCAT; all other lanes: 75 nM GTCAT, 2 μM brHis2, 20 μM metal complexes (same conditions as in Figure 2a and b). Note: The gold dichloride complexes present a positive charge.
Not surprisingly, trans-[Pd(PPh3)2Cl2] also failed to induce DNA binding, while Pd(bipy)Cl2 was roughly as effective as Pd(en)Cl2 (Figure S12). We also proved that an analogous complex featuring nitrate instead of chloride ligands Pd(bipy)(NO3)2 is also a very efficient binding trigger (Figure S12). Therefore, it appears that square planar cis-Pd(II) complexes present the ideal coordination properties to promote an effective folding, and interaction to the target DNA.22 Notably, similar PtCl2 complexes were ineffective, most probably because of the low reactivity and kinetic stability of the precursor salts (Figure 4b). However, electrophilic Au(III) complexes, particularly [Au(bipy)Cl2]+, which exhibit a square planar geometry similar to that of Pd(II), can also promote the DNA binding (Figure 4c). This result is quite appealing as, to the best of our knowledge, gold complexes had never been used in related bio-supramolecular strategies.
Spectroscopic Characterization of the Palladium-Promoted Interaction between brHis2 and the DNA
In order to obtain more information on the DNA binding process, we performed fluorescence anisotropy titrations by adding brHis2 to a solution containing 20 μM cis-[Pd(en)Cl2] and a tetramethylrhodamine (TMR)-labeled ds-oligonucleotide containing the target sequence. The addition of brHis2 led to a progressive increase in the fluorescence anisotropy of the TMR label. The data could be fitted to a simplified 1:1 binding model by formally considering the complex [(brHis2)(Pd(en))]2+ as a single DNA binding unit, with an apparent KD ≈ 24 ± 17 nM (Figure 5, left).
Figure 5.

Left: Representative fluorescence anisotropy titration of a 25 nM solution of the TMR-labeled target ds-oligonucleotide TMR-GTCAT (TMR-5′-CGCGTCATAATTGAGAGCGC-3′, only one strand shown) in the presence of 20 μM Pd(en)Cl2 and increasing concentrations of brHis2. The best fit to a 1:1 binding model is also shown. Measurements were made in 20 mM Tris-HCl buffer pH 7.5 and 100 mM NaCl. Right: CD of a 5 μM solution of brHis2 (dotted line), after the subsequent addition of 1 equiv of the target dsDNA (dashed line), of the same solution after the addition of cis-Pd(en)Cl2 (thick solid line), and after addition of DEDTC demonstrating the reversibility (narrow solid line). All experiments were carried out at 25 °C, in 10 mM phosphate buffer pH 7.5 and 100 mM of NaCl. The contribution of DNA to the CD spectrum has been subtracted for clarity. Mean residue molar ellipticity (MRE) was calculated with respect to the 24-mer brHis2.
As expected for a poorly structured peptide, the circular dichroism (CD) spectrum of brHis2 presents a relatively weak negative signal at 222 nm, even in the presence of the consensus DNA GTCAT (Figure 5 right). Addition of Pd(en)Cl2 to the mixture promoted a significant increase in the negative ellipticity at 222 nm, which is consistent with the folding of the peptide into an α-helix (Figure 5 right, thick solid line).23 In accordance with the results obtained by EMSA, addition of DEDTC to the mixture resulted in a drastic decrease in the helicity of the peptide (Figure 5 right, narrow solid line), which correlates with the disruption of the DNA complex. Not surprisingly, NiCl2 or Pt(en)Cl2 complexes did not promote the peptide α-helical folding in the presence of the target DNA (Figure S14). Interestingly, adding Pd(en)Cl2 to brHis2, in the absence of the target DNA, gives rise only to a relatively small increase in helicity (Figure S15). This result raised the question of whether the palladium is coordinating the peptide in the absence of the DNA template. UV–vis spectra and HPLC-MS analysis of the mixture of the peptide and the palladium complex were fully consistent with the formation of the expected metal-clipped peptide (Figures S17 and S19). Furthermore, fluorescence anisotropy experiments carried out by adding Pd(en)Cl2 to a solution of a TMR-labeled brHis2 resulted in a progressive decrease in the anisotropy of the TMR fluorophore at 559 nm, which agrees with the peptide acquiring a more compact conformation (Figure S13).
The above data indicate that the Pd(II) complex does coordinate the histidine residues, but the peptide only acquires a high proportion of helicity after addition of the target DNA. The palladium clip is not inducing a permanent helical conformation as traditional helix staples,10,24 but rather it enhances the local helical propensity, which thus facilitates the subsequent folding in the presence of the DNA.25,10,24 Therefore, the system maintains the folding-upon-binding behavior of the parent GCN4 transcription factor, which might be the reason that the [(brHis2)Pd(en)]2+ complex shows such an excellent DNA selectivity.26
Internalization Studies: brHis2 Only Translocates in the Presence of Pd(II)
A key factor when considering potential biological applications of this type of peptides has to do with their ability to cross cell membranes. It is known that basic peptides derived from transcription factors, such as the Antennapedia, can be efficiently internalized.27 Moreover, previous studies with covalently stapled peptides suggest that the conformational restriction associated with the stapling favors the cell uptake,12,28 and therefore we were curious to know if the palladium clipping might also influence the cell penetration ability of brHis2.
Incubation of mammalian Vero or HeLa cells with TMR-brHis2 and 2-fold washing with phosphate-buffered saline (PBS) revealed that despite the basic character of the peptide, it shows a rather poor internalization.29 Gratifyingly, and also quite surprisingly, addition of 1 equiv of Pd(en)Cl2 led to the appearance of bright intracellular emission, which was mainly localized in endosomes (Figures 6, S21, and S22).30 Importantly, neither the TMR-labeled basic region peptide (TMR-br) nor the single mutated basic region (TMR-brHis) showed appreciable cell translocations upon incubation using similar conditions (Figure S23).
Figure 6.
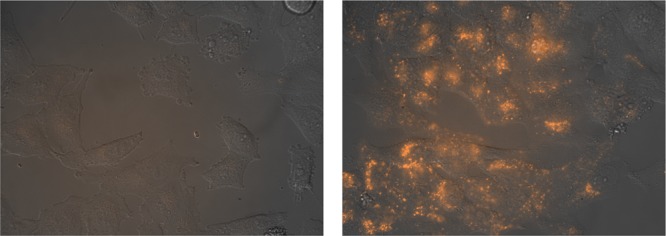
Fluorescence micrographies of HeLa cells. Bright-field images are superimposed to the red emission channel after incubation with 5 μM TMR-brHis2 for 30 min at 37 °C (left) and the same experiment in the presence of 5 μM Pd(en)Cl2 (right). The complex was preincubated (1:1) in water for 10 min before the addition. The cells were washed twice with PBS before being observed in a fluorescence microscope. All incubations were made in Dulbecco’s modified Eagle medium completed with 10% fetal bovine serum. TMR: tetramethylrhodamine.
While obtaining exact information on the molecular mechanism of the cell internalization process will require a detailed study, we have carried out preliminary experiments with modified peptides that shed light on the structural requirements of the peptide for an effective metal-switched uptake. Thus, we found that the positively charged TMR fluorophore is not required for the metal-induced internalization, so that when this fluorophore was replaced by the anionic fluorescein (Figure S35, FITC-brHis2), the resulting peptide replicates the palladium-induced internalization displayed by the parent TMR-brHis2. Similarly, the metal switch is perfectly operative in specifically modified peptides, such as the double mutant peptides Leu247 → Ala, Gln248 → Ala or Thr236 → Ala, Ala238 → Thr, as well as the single mutant Ala238 → Thr. Importantly, the cell entrance of longer peptides that include amino acid insertions, such as one containing a Leu-Ser fragment between Ala229 and His230, can be also regulated by the Pd(II) reagent. Not surprisingly, the cell uptake is sensitive to the presence of arginine residues, which is in consonance with the well-known role of these amino acids in cell-penetrating peptides.31 Thus, the double mutant Arg245 → Ser; Arg249 → Ala peptide failed to translocate across the plasmatic membrane, even in the presence of the palladium clip (Figures 7, S33, and S34).
Figure 7.
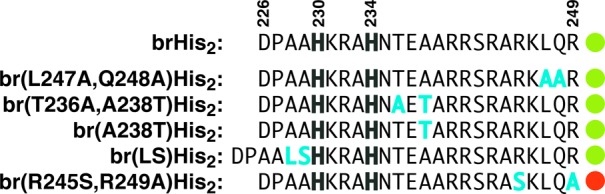
Sequences of modified peptides tested in the metal-switched cell uptake process, highlighting the mutated residues (blue) and whether the peptide is internalized (green dot) or does not show an observable translocation under the same conditions (red dot).
Although unraveling the exact structural requirements for the cell uptake switch requires further work, these early results indicate that the internalization requires the metal clip and the presence of several arginine residues in the sequence, but is highly tolerant to other changes. The underlying internalization mechanism is also unknown, but it is likely that the clipping effect of the palladium promotes a change in the amphiphilicity of the system, which favors the membrane crossing. Our peptides will require further engineering to promote endosomal escape; however the above results represent the first examples of a metal-triggered cell internalization of designed basic peptides and should foster further research in this area.32
Conclusion
In summary, we have introduced a new approach for achieving a highly selective recognition of dsDNA with a minimal (23 amino acids long), synthetic peptide that is entirely made of natural residues. Key for the success of the approach is the introduction of histidines in suitable i, i+4 positions in the sequence and the addition of square-planar, Pd(II) or Au(III) reagents. In contrast with other covalently stapled α-helical peptides, our palladium–peptide complex is mostly unstructured in solution and acquires a predominantly helical conformation only in the presence of the target DNA sequence. The kinetic lability of the metal coordination facilitates the disassembly of the supramolecular structure upon addition of external agents that sequester the palladium.
Importantly, the DNA binding and disassembly processes can be repeated several times, therefore providing for a fully reversible switchable system. Also importantly, we show that while the apo-peptide presents a very poor cellular uptake, the addition of the palladium reagent triggers an efficient internalization. This is the first demonstration of triggering the cell penetration of a short, basic peptide using a metal complex; this tactic promises to find further applications in dynamic cellular delivery.
Acknowledgments
This work has received financial support from the MINECO (SAF2013-41943-R, SAF2016-76689-R, and CTQ2015-70698-R), the Xunta de Galicia (2015-CP082, ED431C 2017/19, and Centro Singular de Investigación de Galicia Accreditation 2016–2019, ED431G/09), the European Union (European Regional Development Fund, ERDF), and the European Research Council (Advanced Grant No. 340055). Support of COST CM1306 and the orfeo-cinqa network are also acknowledged. J.R. thanks the Xunta de Galicia for a Ph.D. fellowship. We also wish to acknowledge the generous support by the Fundación AECC (IDEAS197VAZQ grant). Finally, we also thank R. Menaya-Vargas for her technical assistance with cell experiments, and Dr. J. Mosquera for the initial synthesis of brHis2
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b07422.
Peptide synthesis and characterization; detailed protocols for EMSA experiments, fluorescence anisotropy, circular dichroism parameters, and control experiments; cell internalization and cytotoxicity studies (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Latchman D. S.Eukaryotic Transcription Factors, 5th ed.; Elsevier: London, 2011; Chapter 1, p 1. [Google Scholar]; b Garvie C. W.; Wolberger C. Mol. Cell 2001, 8, 937–946. 10.1016/S1097-2765(01)00392-6. [DOI] [PubMed] [Google Scholar]; c Todeschini A.-L.; Georges A.; Veitia R. A. Trends Genet. 2014, 30, 211–219. 10.1016/j.tig.2014.04.002. [DOI] [PubMed] [Google Scholar]
- a Vázquez M. E.; Caamaño A. M.; Mascareñas J. L. Chem. Soc. Rev. 2003, 32, 338–349. 10.1039/B206274G. [DOI] [PubMed] [Google Scholar]; b Pazos E.; Mosquera J.; Vázquez M. E.; Mascareñas J. L. ChemBioChem 2011, 12, 1958–1973. 10.1002/cbic.201100247. [DOI] [PubMed] [Google Scholar]; c Strekowski L.; Wilson B. Mutat. Res., Fundam. Mol. Mech. Mutagen. 2007, 623, 3–13. 10.1016/j.mrfmmm.2007.03.008. [DOI] [PubMed] [Google Scholar]; d Arora P. S.; Ansari A. Z.; Best T. P.; Ptashne M.; Dervan P. B. J. Am. Chem. Soc. 2002, 124, 13067–13071. 10.1021/ja0208355. [DOI] [PubMed] [Google Scholar]; e Fechter E. J.; Olenyuk B.; Dervan P. B. Angew. Chem., Int. Ed. 2004, 43, 3591–3594. 10.1002/anie.200454231. [DOI] [PubMed] [Google Scholar]; f Rodríguez J.; Mosquera J.; Vázquez O.; Vázquez M. E.; Mascareñas J. L. Chem. Commun. 2014, 50, 2258–2260. 10.1039/c3cc47599a. [DOI] [PubMed] [Google Scholar]
- a Neidle S. Nat. Prod. Rep. 2001, 18, 291–309. 10.1039/a705982e. [DOI] [PubMed] [Google Scholar]; b Strekowski L.; Wilson B. Mutat. Res., Fundam. Mol. Mech. Mutagen. 2007, 623, 3–13. 10.1016/j.mrfmmm.2007.03.008. [DOI] [PubMed] [Google Scholar]; c Vázquez O.; Sánchez M.; Martínez-Costas J.; Vázquez M. E.; Mascareñas J. L. Org. Lett. 2010, 12, 216–219. and references therein 10.1021/ol902501j. [DOI] [PubMed] [Google Scholar]
- Boga S.; Bouzada D.; García Peña D.; Vázquez López M.; Vázquez M. E. Eur. J. Org. Chem. 2017, 10.1002/ejoc.201700988. [DOI] [Google Scholar]
- a Ellenberger T. E.; Brandl C. J.; Struhl K.; Harrison S. C. Cell 1992, 71, 1223–1237. 10.1016/S0092-8674(05)80070-4. [DOI] [PubMed] [Google Scholar]; b König P.; Richmond T. J. J. Mol. Biol. 1993, 233, 139–154. 10.1006/jmbi.1993.1490. [DOI] [PubMed] [Google Scholar]; c Keller W.; König P.; Richmond T. J. J. Mol. Biol. 1995, 254, 657–667. 10.1006/jmbi.1995.0645. [DOI] [PubMed] [Google Scholar]
- Weiss M. A.; Ellenberger T.; Wobbe C. R.; Lee J. P.; Harrison S. C.; Struhl K. Nature 1990, 347, 575–578. 10.1038/347575a0. [DOI] [PubMed] [Google Scholar]
- Talanian R. V.; McKnight C. J.; Kim P. S. Science 1990, 249, 769–771. 10.1126/science.2389142. [DOI] [PubMed] [Google Scholar]
- a Cuenoud B.; Schepartz A. Science 1993, 259, 510–513. 10.1126/science.8424173. [DOI] [PubMed] [Google Scholar]; b Morii T.; Saimei Y.; Okagami M.; Makino K.; Sugiura Y. J. Am. Chem. Soc. 1997, 119, 3649–3655. 10.1021/ja963280k. [DOI] [Google Scholar]; c Aizawa Y.; Sugiura Y.; Ueno M.; Mori Y.; Imoto K.; Makino K.; Morii T. Biochemistry 1999, 38, 4008–4017. 10.1021/bi9828829. [DOI] [PubMed] [Google Scholar]; d Carrette L. L. G.; Morii T.; Madder A. Eur. J. Org. Chem. 2014, 2014, 2883–2891. 10.1002/ejoc.201301854. [DOI] [Google Scholar]; e García Y. R.; Vladimir Pabon-Martinez Y.; Edvard Smith C. I.; Madder A. Chem. Commun. 2017, 53, 6653–6656. 10.1039/C7CC01705G. [DOI] [PubMed] [Google Scholar]; f Caamaño A. M.; Vázquez M. E.; Martínez-Costas J.; Castedo L.; Mascareñas J. L. Angew. Chem. 2000, 112, 3234–3237. . [DOI] [PubMed] [Google Scholar]; g Bullen G. A.; Tucker J. H. R.; Peacock A. F. A. Chem. Commun. 2015, 51, 8130–8133. 10.1039/C5CC01618E. [DOI] [PubMed] [Google Scholar]; h Mosquera J.; Sánchez M. I.; Vázquez M. E.; Mascareñas J. L. Chem. Commun. 2014, 50, 10975–10978. 10.1039/C4CC04512B. [DOI] [PubMed] [Google Scholar]
- Wang X.; Cao W.; Cao A.; Lai L. Biophys. J. 2003, 84, 1867–1875. 10.1016/S0006-3495(03)74994-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M. I. N.; Wu B.; Zhao H.; Taylor J. W. J. Pept. Sci. 2002, 136, 125–136. 10.1002/psc.377. [DOI] [PubMed] [Google Scholar]
- Park C.; Campbell J. L.; Goddard W. A. J. Am. Chem. Soc. 1996, 118, 4235–4239. 10.1021/ja950653t. [DOI] [Google Scholar]
- Lyer A.; Van Lysebetten D.; Ruiz García Y.; Louage B.; De Geest B. G.; Madder A. Org. Biomol. Chem. 2015, 13, 3856–3862. 10.1039/C4OB02659D. [DOI] [PubMed] [Google Scholar]
- a Montclare J. K.; Schepartz A. J. Am. Chem. Soc. 2003, 125, 3416–3417. 10.1021/ja028628s. [DOI] [PubMed] [Google Scholar]; b Zondlo N. J.; Schepartz A. J. Am. Chem. Soc. 1999, 121, 6938–6939. 10.1021/ja990968z. [DOI] [Google Scholar]
- a Vázquez M. E.; Caamaño A. M.; Martínez-Costas J.; Castedo L.; Mascareñas J. L. Angew. Chem., Int. Ed. 2001, 40, 4723–4725. . [DOI] [PubMed] [Google Scholar]; b Blanco J. B.; Vázquez M. E.; Castedo L.; Mascareñas J. L. ChemBioChem 2005, 6, 2173–2176. 10.1002/cbic.200500294. [DOI] [PubMed] [Google Scholar]; c Vázquez O.; Vázquez M. E.; Blanco J. B.; Castedo L.; Mascareñas J. L. Angew. Chem., Int. Ed. 2007, 46, 6886–6890. 10.1002/anie.200702345. [DOI] [PubMed] [Google Scholar]; d Sánchez M. I.; Vázquez O.; Martínez-Costas J.; Vázquez M. E.; Mascareñas J. L. Chem. Sci. 2012, 3, 2383. 10.1039/c2sc00027j. [DOI] [Google Scholar]; e Blanco J. B.; Vázquez M. E.; Martinez-Costas J.; Castedo L.; Mascareñas J. L. Chem. Biol. 2003, 10, 713–722. 10.1016/S1074-5521(03)00172-8. [DOI] [PubMed] [Google Scholar]; f Rodríguez J.; Mosquera J.; Couceiro J. R.; Vázquez M. E.; Mascareñas J. L. Chem. Sci. 2015, 6, 4767–4771. 10.1039/C5SC01415H. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Rodríguez J.; Mosquera J.; García-Fandiño R.; Vázquez M. E.; Mascareñas J. L. Chem. Sci. 2016, 7, 3298–3303. 10.1039/C6SC00045B. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Chakraborty M.; Roy S. Chem. Commun. 2017, 53, 376–379. 10.1039/C6CC08086C. [DOI] [PubMed] [Google Scholar]
- a Sánchez M. I.; Mosquera J.; Vázquez M. E.; Mascareñas J. L. Angew. Chem., Int. Ed. 2014, 53, 9917–9921. 10.1002/anie.201405726. [DOI] [PubMed] [Google Scholar]; b Rodríguez J.; Mosquera J.; Vázquez M. E.; Mascareñas J. L. Chem. - Eur. J. 2016, 22, 13474–13477. 10.1002/chem.201602783. [DOI] [PubMed] [Google Scholar]
- a Sardesai N. Y.; Zimmermann K.; Barton J. K. J. Am. Chem. Soc. 1994, 116, 7502–7508. 10.1021/ja00096a005. [DOI] [Google Scholar]; b Copeland K. D.; Fitzsimons M. P.; Houser R. P.; Barton J. K. Biochemistry 2002, 41, 343–356. 10.1021/bi011793k. [DOI] [PubMed] [Google Scholar]
- a Talanian R. V.; McKnight C. J.; Rutkowski R.; Kim P. S. Biochemistry 1992, 31, 6871–6875. 10.1021/bi00145a002. [DOI] [PubMed] [Google Scholar]
- a König P.; Richmond T. J. J. Mol. Biol. 1993, 233, 139–154. 10.1006/jmbi.1993.1490. [DOI] [PubMed] [Google Scholar]
- a Coin I.; Beyermann M.; Bienert M. Nat. Protoc. 2007, 2, 3247–3256. 10.1038/nprot.2007.454. [DOI] [PubMed] [Google Scholar]; b Pazos E.; Portela C.; Penas C.; Vázquez M. E.; Mascareñas J. L. Org. Biomol. Chem. 2015, 13, 5385–5390. 10.1039/C5OB00318K. [DOI] [PubMed] [Google Scholar]
- A reversible binding of peptide dimers was reported by our group:Mosquera J.; Jiménez-Balsa A.; Dodero V. I.; Vázquez M. E.; Mascareñas J. L. Nat. Commun. 2013, 4, 1874. 10.1038/ncomms2825. [DOI] [PubMed] [Google Scholar]
- There is an earlier report of a Co(II)-responsive GCN4 dimer featuring iminodiacetic acid residues:Azuma Y.; Imanishi M.; Yoshimura T.; Kawabata T.; Futaki S. Angew. Chem., Int. Ed. 2009, 48, 6853–6856. 10.1002/anie.200902888. [DOI] [PubMed] [Google Scholar]
- For examples of metal-promoted helical peptide foldings, see:; a Radford R. J.; Tezcan F. A. J. Am. Chem. Soc. 2009, 131, 9136–9137. 10.1021/ja9000695. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kalek M.; Madsen A. S.; Wengel J. J. Am. Chem. Soc. 2007, 129, 9392–9400. 10.1021/ja071076z. [DOI] [PubMed] [Google Scholar]; c Ghadiri M. R.; Choi C. J. Am. Chem. Soc. 1990, 112, 1630–1632. 10.1021/ja00160a054. [DOI] [Google Scholar]; d Ghadiri M. R.; Fernholz A. K. J. Am. Chem. Soc. 1990, 112, 9633–9635. 10.1021/ja00182a030. [DOI] [Google Scholar]; e Shepherd N. E.; Hoang H. N.; Abbenante G.; Fairlie D. P. J. Am. Chem. Soc. 2005, 127, 2974–2983. 10.1021/ja0456003. [DOI] [PubMed] [Google Scholar]; f Kelso M. J.; Hoang H. N.; Oliver W.; Sokolenko N.; March D. R.; Appleton T. G.; Fairlie D. P. Angew. Chem., Int. Ed. 2003, 42, 421–424. 10.1002/anie.200390128. [DOI] [PubMed] [Google Scholar]; g Smith S. J.; Radford R. J.; Subramanian R. H.; Barnett B. R.; Figueroa J. S.; Tezcan F. A. Chem. Sci. 2016, 7, 5453–5461. 10.1039/C6SC00826G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelton J. T.; McLean L. R. Anal. Biochem. 2000, 277, 167–176. 10.1006/abio.1999.4320. [DOI] [PubMed] [Google Scholar]
- a Bernal F.; Tyler A. F.; Korsmeyer S. J.; Walensky L. D.; Verdine G. L. J. Am. Chem. Soc. 2007, 129, 2456–2457. 10.1021/ja0693587. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kim Y.-W.; Grossmann T. N.; Verdine G. L. Nat. Protoc. 2011, 6, 761–771. 10.1038/nprot.2011.324. [DOI] [PubMed] [Google Scholar]
- Drobnak I.; Gradišar H.; Ljubetič A.; Merljak E.; Jerala R. J. Am. Chem. Soc. 2017, 139, 8229–8236. 10.1021/jacs.7b01690. [DOI] [PubMed] [Google Scholar]
- a Tompa P. FEBS Lett. 2005, 579, 3346–3354. 10.1016/j.febslet.2005.03.072. [DOI] [PubMed] [Google Scholar]; b Uversky V. N. J. Biol. Chem. 2016, 291, 6681–6688. 10.1074/jbc.R115.685859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Derossi D.; Joliot A. H.; Chassaing G.; Prochiantz A. J. Biol. Chem. 1994, 269, 10444–10450. [PubMed] [Google Scholar]; b Derossi D.; Calvet S.; Trembleau A.; Brunissen A.; Chassaing G.; Prochiantz A. J. Biol. Chem. 1996, 271, 18188–18193. 10.1074/jbc.271.30.18188. [DOI] [PubMed] [Google Scholar]
- a Zhao H.; Jiang Y.; Tian Y.; Yang D.; Qin X.; Li Org. Biomol. Chem. 2017, 15, 459–464. 10.1039/C6OB02501C. [DOI] [PubMed] [Google Scholar]; b Nagel Y. A.; Raschle P. S.; Wennemers H. Angew. Chem., Int. Ed. 2017, 56, 122–126. 10.1002/anie.201607649. [DOI] [PubMed] [Google Scholar]; c Peraro L.; Zou Z.; Makwana K. M.; Cummings A. E.; Ball H. L.; Yu H.; Lin Y.-S.; Levine B.; Kritzer J. A. J. Am. Chem. Soc. 2017, 139, 7792–7802. 10.1021/jacs.7b01698. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Lättig-Tünnemann G.; Prinz M.; Hoffmann D.; Behlke J.; Palm-Apergi C.; Morano I.; Herce H. D.; Cardoso M. C. Nat. Commun. 2011, 2, 453. 10.1038/ncomms1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Milletti F. Drug Discovery Today 2012, 17, 85. 10.1016/j.drudis.2012.03.002. [DOI] [PubMed] [Google Scholar]; b Gupta B.; Levchenko T. S.; Torchilin V. P. Adv. Drug Delivery Rev. 2005, 57, 637–651. 10.1016/j.addr.2004.10.007. [DOI] [PubMed] [Google Scholar]; c Goun E. A.; Pillow T. H.; Jones L. R.; Rothbard J. B.; Wender P. A. ChemBioChem 2006, 7, 1497–1515. 10.1002/cbic.200600171. [DOI] [PubMed] [Google Scholar]; d Mosquera J.; Sánchez M. I.; Valero J.; de Mendoza J.; Vázquez M. E.; Mascareñas J. L. Chem. Commun. 2015, 51, 4811–4814. 10.1039/C4CC09525A. [DOI] [PubMed] [Google Scholar]; e Vázquez O.; Blanco-Canosa J. B.; Vázquez M. E.; Martínez Costas J.; Castedo L.; Mascareñas J. L. ChemBioChem 2008, 9, 2822–2829. 10.1002/cbic.200800345. [DOI] [PubMed] [Google Scholar]
- For strategies to promote endosomal escapes, see:; a Erazo-Oliveras A.; Muthukrishnan N.; Baker R.; Wang T. Y.; Pellois J. P. Pharmaceuticals 2012, 5, 1177–1209. 10.3390/ph5111177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbard J. B.; Kreider E.; VanDeusen C. L.; Wright L.; Wylie B. L.; Wender P. A. J. Med. Chem. 2002, 45, 3612–3618. 10.1021/jm0105676. [DOI] [PubMed] [Google Scholar]
- For other stimuli-responsive cell internalization strategies, see:; a Jiang T.; Olson E. S.; Nguyen Q. T.; Roy M.; Jennings P. A.; Tsien R. Y. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 17867–17872. 10.1073/pnas.0408191101. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rodríguez J.; Mosquera J.; Couceiro J. R.; Nitschke J. R.; Vázquez M. E.; Mascareñas J. L. J. Am. Chem. Soc. 2017, 139, 55–58. 10.1021/jacs.6b11103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.