

Abstract
Recently, 5-hydroxymethyluracil (5hmU) was identified in mammalian genomic DNA as an oxidative product of thymine by the ten-eleven translocation (TET) proteins. While the biological role of this modification remains unclear, identifying its genomic location will assist in elucidating function. Here we present a rapid and robust method to selectively tag and enrich genomic regions containing 5hmU. This method involves the selective glucosylation of 5hmU residues by the base J glucosyltransferase from trypanosomes creating glucosylhydroxymethyluracil (base J). The base J can then be efficiently and selectively pulled down by antibodies against base J or by J-binding protein 1. DNA that is enriched is suitable for analysis by quantitative PCR or sequencing. We utilized this tagging reaction to provide proof of concept for the enrichment of 5hmU containing DNA from a pool that contains modified and unmodified DNA. Furthermore, we demonstrate that the base J pull-down assay identifies 5hmU at specific regions of the trypanosome genome involved in transcriptional repression. The method described here will allow for a greater understanding of the functional role and dynamics of 5hmU in biology.
Introduction
5-Methylcytosine (5mC) is an important epigenetic mark within the mammalian genome, regulating gene expression during many biological processes. It is generally considered a repressive mark as the presence of methylated cytosines at a promoter region is directly connected with transcriptional repression of a gene [1]. The presence of 5mC at particular regions of the genome is a dynamic and reversible process. The dynamic nature of DNA methylation is crucial for regulation of genes during development [2]. The 5mC can be removed through a passive demethylation mechanism by loss through cell replication, or through an active demethylation pathway catalyzed by the ten-eleven translocation (TET) enzymes [3]. The TET enzymes are 5mC hydroxylases, which catalyze the iterative oxidation of 5mC to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC) [4–6]. 5fC and 5caC can then be excised by a thymine DNA glycosylase followed by base excision repair (BER) to replace 5mC with an unmodified cytosine (C) [6, 7]. Deletion of the TET proteins lead to reduced 5hmC levels in mammalian genomes and defects in gene expression patterns. For example, mice deficient in individual TET enzymes display embryonic abnormalities [8–12] and combined deficiency of all three TET enzymes in mouse embryonic stem cells (mESCs) completely depletes 5hmC levels, impairs differentiation, and does not support embryogenesis [13]. In addition, knockdown of TET1 in embryonic stem cells leads to a loss of 5hmC at specific promoters and a corresponding increase in 5mC at transcriptional start sites of genes regulated by TET1 [14]. In conjunction with interacting proteins, the TET oxidation products 5hmC, 5fC, and 5caC may themselves have unique epigenetic regulatory functions, presumably via regulating gene transcription [15].
Studies have recently revealed that the TET enzymes not only oxidize 5mC, but are also capable of oxidizing thymidine (T) to form 5-hydroxymethyluracil (5hmU) [16, 17]. These characterizations of TET enzymes from mice and the amoeboflagellate Naegleria gruberi indicate that TET activity on thymine is evolutionally conserved. Isotope tracing and quantitative mass spectrometry studies indicate the majority of 5hmU within mESCs is produced by the mammalian TET proteins with very little 5hmU generated through 5hmC deamination or radical oxygen species [16]. Thus, the majority of 5hmU in the genome is matched (5hmU:A) and not mismatched (5hmU:G). Furthermore, they found that the levels of 5hmU change throughout mESC differentiation, suggesting that like the other TET-generated DNA modifications, 5hmU may serve as an important epigenetic mark [16]. Presence of 5hmU in DNA has been shown to perturb DNA–protein interactions and transcription factor binding in vitro [18]. A potential role for 5hmU in transcription has recently been proposed where CpA sites provide a mechanism for inheritance of gene silencing [19]. According to this hypothesis, the presence of a methyl group at C5 of both thymine and 5meC methylation at CpA sites would generate a “fully methylated” dinucleotide. Likewise, further modification by TET enzymes would generate fully modified CpA/TpG sites that could provide a useful mechanism of epigenetic inheritance during replication. To further explore the function of 5hmU, it will be important to localize 5hmU derived from TET hydroxylation. Therefore, an efficient and robust method for determining the position of 5hmU within eukaryotic genomes is required.
New techniques were recently developed for determining the genome-wide distribution of 5hmC utilizing T4 phage β-glucosyltransferase (βGT) to glucosylate 5hmC, which can be enriched by a specific protein [e.g., J-binding protein 1 (JBP1)] or can be chemically modified further with biotin labeling allowing efficient pull down using streptavidin [20–24]. The addition of the glucose moiety also allows single-base-resolution (SMRT) sequencing methods for mapping 5hmC [25]. While 5hmC and 5hmU are structurally similar, βGT is unable to modify matched 5hmU:A [26]. Matched 5hmU, however, is present in trypanosomatid genomic DNA and is converted to β-d-glucosyl-hydroxymethyluridine (base J), by the base J-specific glycosyltransferase (JGT) [27, 28] (Fig. 1A). We have demonstrated that recombinant JGT glucosylates 5hmU in a DNA sequence-independent manner [29], prompting us to design a similar strategy for selectively labeling 5hmU and mapping its location within mammalian genomes (Fig. 1B). In the first scheme, JGT is utilized to install a modified N3-glucose into the hydroxyl group of 5hmU followed by incorporation of the biotin linker through click chemistry and capture of 5hmU-containing DNA fragments via streptavidin pull down. Alternatively, the 5hmU-containing DNA fragments can be captured by immunoprecipitation using the base J-specific antibody [30–33] or by using the base J binding protein (JBP1) [20–22, 33, 34]. The enriched fragments can be applied to deep sequencing to map the location of 5hmU. The resulting glucosylated 5hmU (base J) could also be directly mapped using SMRT sequencing [35].
Figure 1.

Selective labeling and enrichment of 5hmU in genomic DNA using JGT. (A) The hydroxyl group of 5hmU in duplex DNA can be glycosylated by JGT to form β-d-glucosylhydroxymethyluridine (glucosyl-5hmU), also called base J. (B) Overview of the selective labeling and enrichment strategy for 5hmU. Scheme 1: JGT is utilized to selectively label 5hmU in genomic DNA with N3-glucose. After addition of the biotin tag through click chemistry, the 5hmU-containing DNA fragments can be enriched by streptavidin-coupled beads allowing detection and sequencing. Scheme 2: JGT is utilized to selectively label 5hmU with glucose allowing subsequent affinity purification using the base JBP1 or antisera against base J. The glucosylated base can also be directly sequenced using SMRT-sequencing technology.
Base J has been shown to regulate transcription initiation and termination at specific sites along the trypanosomatid genome [32, 33, 36–38]. Specific thymines in the genome are somehow targeted and modified to base J in a two-step reaction [39].The thymine base in DNA is first oxidized by one of the thymidine hydroxylases (TH), JBP1 and JBP2, to generate 5hmU [40, 41]. JGT then transfers glucose from UDP-glucose onto 5hmU forming base J [27]. While both JBP1 and JBP2 contain a TH domain at the N-terminus that has led to the designation of these enzymes as belonging to the TET/JBP subfamily of dioxygenases [4, 40–44], JBP1 has a novel C-terminal domain that allows it to bind J-DNA [34, 45–47]. While both JBP1 and JBP2 stimulate de novo thymidine hydroxylation in vivo, the ability of JBP1 to bind J-DNA allows the enzyme to stimulate additional 5hmU synthesis (and J) in the genome [41, 48, 49]. Analysis of J synthesis in vivo and JGT function in vitro has indicated that JGT is a DNA sequence non-specific enzyme and anywhere 5hmU is located in the trypanosome genome, it will be converted to J [29, 41]. Therefore, it is believed that the JBP enzymes regulate the specific localization of base J in the trypanosomatid genome. In order to test this hypothesis and the new 5hmU mapping procedure, we utilized the glucose tagging/J enrichment method to map the location of 5hmU in the Trypanosoma brucei genome. We demonstrate that 5hmU synthesized by the JBP enzymes is localized at specific sites of the genome where base J is known to function.
Materials and methods
Trypanosome cell culture
The bloodstream form T. brucei cell lines were cultured in HMI9 medium supplemented with 10% heat-inactivated fetal bovine serum and 10% serum plus as described previously [48].
Isolation of genomic DNA
Trypanosoma brucei genomic DNA was isolated as described previously [31]. Genomic mESC DNA was extracted using the Qiagen DNeasy Blood and Tissue Kit following the manufacturer’s instructions (Qiagen). All buffers were supplemented with the antioxidants 3,5-di-tert-butyl-4-hydroxytoluene, deferoxamine mesylate salt, and tetrahydrouridine at a concentration of 200 µM to reduce DNA deamination and oxidation [50]. Amount and purity of DNA was determined by using NanoDrop® ND-1000 Spectrophotometer.
Preparation of recombinant JGT
Expression and purification of the N-terminal 10× His-tagged T. brucei-JGT was performed with BL21-CodonPlus (DE3)-RIL competent cells as previously described [27] with minimal changes. JGT was eluted from the Talon resin with imidazole elution buffer (50 mM HEPES, pH 7.5, 300 mM NaCl, 10 mM β-mercaptoethanol, 5% glycerol, 300 mM imidazole). The eluted protein was diluted 1:5 in dilution buffer (50 mM HEPES, pH 7.5, 50 mM NaCl, 10% glycerol), concentrated with 10 000 MWCO Centricon, and visualized by blue silver-stained SDS polyacrylamide gel electrophoresis (PAGE) and anti-His Western blot.
Generation of DNA substrates
Unmodified oligonucleotides used in this study were purchased from Invitrogen. The 15-bp 5hmU and 5hmC-containing oligos used in the gel-shift assay were synthesized by TriLink Biotechnologies (San Diego, CA, USA) and possess the sequence TTAGGGTXAGGGTTA with either a 5hmU or 5hmC at the position of the bold X. The 36-bp 5hmU and 5hmC-containing oligos used in the filter binding assays were synthesized by TriLink Biotechnologies (San Diego, CA) and possess the sequence CTATACCTCCTCAACTTCXGATCACCGTCTCCGGCG with either a 5hmU or 5hmC at the position of the bold X. DNA duplexes were prepared by annealing complementary oligos in annealing buffer [100 mM potassium acetate, 30 mM HEPES (pH 7.5)] by boiling for 10 min and allowing to cool overnight.
The 5mC, 5hmC, and 5hmU modified and unmodified 75-bp DNA substrates used in the anti-base J immunoprecipitations were generated using PCR. Modified deoxynucleotide triphosphates (dNTPs) were purchased from TriLink Biotechnologies and each modified dNTP completely replaced its respective unmodified dNTP in a PCR reaction mix. PCR reactions were performed using Promega GoTaq® DNA Polymerase (Promega in Green GoTaq® Reaction Buffer (Promega) using the following thermocycling conditions: 95°C 30 s (95°C 15 s, 68°C 15 s) for 30 cycles, and 68°C for 5 min. Template DNA sequence possessed the sequence 5′-GCTATCACA GTCCTGCGCTGAG ATACGAGTTG CT G CCTTGGTGCACTTAGAGGTCATGAGAAGGTTTACTGCCCG-3′ with underlined portions representing the primer annealing sites.
In vitro glucosyltransferase reactions
Filter binding assay
JGT glucosylation reaction consisted of 100 µM UDP-[3H]glucose, ∼0.05 µM recombinant JGT, and 100 µM DNA in JGT reaction buffer [50 mM potassium acetate, 20 mM Tris acetate, 10 mM manganese acetate, and 1 mM DTT (pH 7.9)] in a 50 µl reaction volume. Reactions were incubated at 25°C for 1 h. βGT reactions consisted of 100 µM UDP-[3H]glucose, 5 units recombinant βGT (New England Biolabs), and 100 µM DNA in T4 βGT reaction buffer [50 mM potassium acetate, 20 mM Tris acetate, 10 mM magnesium acetate, and 1 mM DTT (pH 7.9)] in a 50 µl reaction volume. Reactions were incubated at 25°C for 1 h and were applied directly to a 2.5 cm DE81 membrane (GE Healthcare) under vacuum. The filters were then washed three times with 2 ml 0.2 M ammonium bicarbonate, three times with water, and three times with 100% ethanol. Membranes were air-dried and placed in scintillation vials containing 5 ml of scintillation fluid. The solution was mixed, and tritium incorporation was measured for 1 min. Reaction values were corrected for non-specific binding of UDP-[3H]glucose to the membrane. Background values were determined using reactions performed in the absence of enzyme but in the presence of UDP-[3H]glucose.
UDP-Glo assay
UDP-Glo Glycosyltransferase Assay (Promega) is an in vitro bioluminescent UDP detection assay measuring glycosyltransferase activity based on how much UDP is generated following cleavage of UDP glucose. UDP-Glo was used to analyze JGT activity as previously described [29].
Gel shift assay
The 15-bp 5hmU or 5hmC containing oligonucleotides were radioactively labeled using T4 polynucleotide kinase (New England Biolabs) and [γ32P]-ATP. The labeled oligonucleotide was then gel-purified and annealed with a non-labeled 27 bp complimentary oligonucleotide. Radiolabeled double-stranded DNA (dsDNA) substrates were then incubated with JGT or T4 β-GT enzyme and either UDP-glucose or UDP-6-N3-Glucose (Active Motif, catalog #55020) in an in vitro glucosyltransferase reaction. JGT glucosylation reaction consisted of 300 µM UDP-glucose, ∼0.05 µM recombinant JGT, and 1 pmol DNA in JGT reaction buffer [50 mM potassium acetate, 20 mM Tris acetate, 10 mM manganese acetate, and 1 mM DTT (pH 7.9)] in a 10 µl reaction volume. Reactions were incubated at 25°C for 0.5 h. βGT reactions consisted of 300 µM UDP-glucose, 7.5 units recombinant βGT (New England Biolabs), and 0.5 pmol DNA in T4 βGT reaction buffer [50 mM potassium acetate, 20 mM Tris acetate, 10 mM magnesium acetate, and 1 mM DTT (pH 7.9)] in a 7.5 µl reaction volume. Reactions were incubated at 37°C for 1 h. The 125 pmol of unlabeled DNA was added to all reactions to compete for any nuclease activity. Biotinylation reactions were then carried out according to the manufacturer’s instructions in the Hydroxymethyl Collector™ Kit (Active Motif, catalog #55013). Samples were then mixed with 2× formamide loading buffer and electrophoretically separated on a 20% polyacrylamide/7 M urea gel in 1× TBE buffer. Radioactively labeled DNA was then visualized by autoradiography. Efficiency of the glucosylation reaction is expressed as percent conversion, which was determined after phospho-imager quantification of the 15 nt DNA substrate and modified product (glucosylated 15 nt DNA product/15 nt DNA substrate).
DNA dot blots
To quantify the levels of 5hmU after in vitro JGT reactions, anti-base J immunoblots were performed as described previously [31]. Briefly, genomic DNA was serially diluted, treated with 1× volume of 0.6 M sodium hydroxide for 15 min, and then treated with 2× volumes cold 2 M ammonium acetate for 5 min. DNA was then blotted onto a nitrocellulose membrane, followed by incubation with anti-base J antisera. Bound antibodies were detected by a secondary goat anti-rabbit antibody conjugated to HRP and visualized by ECL. As a loading control, the membrane was then stripped and stained with a solution of 0.02% methylene blue in 0.3 M sodium acetate, pH 5.2.
J-DNA enrichment
Base J immunoprecipitations were performed as previously described [30–32, 51]. Briefly, 25 µl protein G beads were pre-blocked with 5 µl 10 mg/ml BSA and 5 µl yeast tRNAs for 15 min. A 500 µl IP reaction containing blocked beads, 1–3 µg DNA, 5 µl 10 mg/ml BSA, 5 µl yeast tRNAs, and 10 µl rabbit α-base J serum were set up for each sample. Genomic DNA samples were sonicated to 0.5–3 kb range prior to J-IP. IP reactions, performed in triplicate, were incubated at room temperature for 1.5 h with rotation. DNA was eluted from beads by the addition of 400 µl TE buffer, 4 µl 10 mg/ml proteinase K, and 4 µl 10% SDS and incubation at 37°C for 30 min. A phenol–chloroform extraction was then performed on each sample and DNA was precipitated with the addition of 800 µl 100% ethanol, 12 µl 5 M NaCl, and 4 µl 20 mg/ml glycogen.
Glucosylation and 5hmU pull down
Spike DNA 5hmU enrichment assay
To determine the specificity of 5hmU pull-down method, JGT reactions were performed on a mixture of DNA containing 5 ng modified or unmodified 75 bp DNA substrate in 1 µg sonicated genomic JGT KO DNA. DNA from this reaction was subjected to the J-IP protocol described above. The amount of 5hmU-DNA pull down was determined by quantitative PCR (qPCR) analysis. The %IP was calculated relative to input DNA. Quantification was performed on an iCycler with an iQ5 multicolor real-time PCR detection system (Bio-Rad Laboratories, Hercules, CA, USA). The reaction mixture contained 5 pmol forward and reverse primers, 2× iQ SYBR green super mix (Bio-Rad Laboratories, Hercules, CA, USA), and 2 µl of template DNA. Primers used in qPCR analyses are listed below. qPCR cycling conditions: 95°C 5 min (95°C 15 s, 60°C 30 s, 72°C 30 s) for 40 cycles, and 95°C for 1 min. Each standard curve generated had an R2 value of at least 0.98, slope of −3, y-intercept of 23, and an efficiency of ∼90–100%.
Genome-wide mapping of 5hmU
To determine the location of 5hmU throughout the T. brucei genome, JGT reactions were performed on JGT KO and JBP1/2 KO T. brucei genomic DNA. Reactions (50 µl) containing 3 µg of genomic DNA, 100 µM UDP-glucose, ∼0.05 µM JGT and JGT reaction buffer [50 mM potassium acetate, 20 mM Tris acetate, 10 mM manganese acetate, and 1 mM DTT (pH 7.9)] were incubated at 25°C for 1 h. Reactions were then proteinase K treated, phenol–chloroform extracted, and ethanol precipitated. Three micrograms of the purified glucosylated DNA was then utilized in a J-IP reaction as described above. Due to the low levels of 5hmU in the JGT KO and JBP1/2 KO genomes, five IP reactions were performed for each replicate and pooled at the final step. The amount of 5hmU-DNA pull-down was determined by qPCR as described above. To help apply this method to future genomic DNA samples, we provide a detailed step-by-step method used to map hmU in the JBP1/2 KO.
Step-by-step method: mapping hmU in the trypanosome genome
Preparation of DNA
Centrifuge 2.5 × 108 T. brucei cells and wash cells in 1× PBS. Resuspend cells in 500 µl lysis buffer [1% SDS (w/v), 25 mM EDTA, 0.4 M NaCl, 50 mM Tris–HCl (pH 7.5) and 400 µg/ml proteinase K] and incubate overnight at 37°C. Phenol–chloroform extract 3×, precipitate DNA with 2 volumes 100% ethanol, 0.1 volume 3 M sodium acetate, and 0.02 volume glycogen (20 mg/ml) and wash DNA pellet 2× in 70% ethanol. Resuspend DNA pellet (∼40 ug) in 800 µl water. Sonicate DNA to ∼0.5–3 kb fragments.
Glucosyltransferase reaction
Set up a glucosyltransferase reaction containing 10 µg sonicated genomic DNA, 1 mM UDP-glucose, 0.05 µM JGT, and 1× GT buffer [50 mM potassium acetate, 20 mM Tris acetate, 10 mM manganese acetate, and 1 mM DTT (pH 7.9)] in a total reaction volume of 50 µl. Incubate reaction for 1 h at 25°C. Add 350 µl TE buffer to stop reaction. Proteinase K treat each sample. Phenol–chloroform extract 3×, precipitate DNA with 2 volumes 100% ethanol, 0.1 volume 3 M sodium acetate, and 0.02 volume glycogen (20 mg/ml) and wash DNA pellet 2× in 70% ethanol. Resuspend DNA pellet in 100 µl water.
Immunoprecipitation
Protein G beads (25 µl) need to be pre-blocked with 5 µl 10 mg/ml BSA and 5 µl yeast tRNAs for 15 min. For each IP reaction, add the following to 25 µl pre-blocked protein G beads: 3 µg sonicated genomic DNA from the glucosyltransferase reaction, 5 µl Yeast tRNAs, 5 µl BSA (10 mg/ml), 10 µl base J antisera, 50 µl 10× TBSTE [1× TBSTE: 150 mM NaCl, 100 mM Tris–HCl (pH 8.0), 0.1% Tween-20, and 20 mM EDTA] in a total reaction volume of 500 µl. Rotate samples at room temperature for 2 h. Wash beads 4× in 1× TBSTE. Proteinase K treat each sample. Phenol–chloroform extract 1× and precipitate DNA with 1.5 volumes 100% ethanol, 0.03 volume 5 M NaCl, and 0.02 volume glycogen (20 mg/ml). Wash DNA pellet 2× with 70% ethanol and resuspend in 50 µl water. As discussed above, for each replicate five IP reactions are pooled and the amount of 5hmU-DNA pull down can be determined by qPCR or high-throughput DNA sequencing methods.
DNA primers
| Primers | Sense | Antisense |
|---|---|---|
| Spike | 5′-GCTATCACAGTCCT GCGCTG-3″ | 5′-CGGGCAGTAAAC CTTCTCATGA-3′ |
| ASF1 | 5′-CTTTCGTGTGGGTCG GTAGT-3′ | 5′-CCCCTAACACTTC CTGCGTA-3′ |
| VSG 224 | 5′-CGATGACGTCAATC CAGATG-3′ | 5′-CCGTTGGTGTCG TGTCTTC-3′ |
| cSSR - 1 | 5′-GTATCACCACAGCCC GAACT-3′ | 5′-GGCAACCGAAAA CAAAGAAA-3′ |
| cSSR - 2 | 5′-AATTCGCCTACTGTC CATGCCGAT-3′ | 5′-TGTGCAGAACGCA CATAAGGCAAC-3′ |
| cSSR - 3 | 5′-GGTAAAGCTGGCGAA GTTGAAGGT-3′ | 5′-TTTCTTCCGGACA CTCGCGATCAT-3′ |
| cSSR - 4 | 5′-GGCCTTTATCCGCCG AAATTGGTT-3′ | 5′-CACTTTGTGGTGA ATCAGCGGCAT-3′ |
| cSSR - 5 | 5′-AACAACAGACTAAT GGCGGG-3′ | 5′-TCGATGAATCTG CGCACTAC-3′ |
| dSSR - 1 | 5′-CCCAATTTCACGGA AGAAAA-3′ | 5′-CTTGTGGACACG TGACTGCT-3′ |
| dSSR - 2 | 5′-CGACCCAGCATAAT GTTCCT-3′ | 5′-GGAAAGTGGACC GTTTTGAA-3′ |
| dSSR - 3 | 5′-AAGCGGCGTCATTA TTTGCAGACG-3′ | 5′-ATTGCTTCCACAC CAACCAACGAC-3′ |
| dSSR - 4 | 5′-TTCACGTGAGAGGT GCATTCCAGT-3′ | 5′-ACCATGCCGAATT CAGTTGTACCG-3′ |
| dSSR - 5 | 5′-CACCCAATCCGTCA TTCCACATCA-3′ | 5′-ACAGTCACAGCTC TCCTTCTCACA-3′ |
| dSSR - 6 | 5′-TTCGTGTCAACAGG AGGTGCACTA-3′ | 5′-ACAGATGCCGTAG GTTCATTCGGT-3′ |
Results
JGT is a 5hmU-specific glucosyltransferase
We first wanted to determine if JGT is specific for 5hmU and if there is any activity on other TET oxidation products, specifically 5hmC. As expected, and consistent with previous results [27, 29], we see that JGT is capable of glucose transfer onto dsDNA substrates containing matched 5hmU (5hmU:A) (Fig. 2A). The other natural, but minor, form of 5hmU in mammalian genomic DNA is such that it is paired with G as a result of deamination of 5hmC [52, 53]. We now report that JGT has the ability to also modify substrates containing mismatched 5hmU:G (Fig. 2A). JGT does not, however, show any activity on 5hmC containing dsDNA substrate even when using DNA concentrations 10-fold above the Km of the enzyme for 5hmU-DNA [29] (Fig. 2A). The specificity of JGT for 5hmU versus 5hmC is confirmed by measuring glucosyltransferase activity based on cleavage of UDP-Glc (Fig. 2C). In contrast, as previously demonstrated, βGT is a glucosyltransferase that acts on its primary substrate 5hmC, with some low relative-level activity on 5hmU, but only when mispaired to guanine (5hmU:G) [26, 54] (Fig. 2B). βGT has no detectable activity on matched 5hmU DNA substrate. These data demonstrate that JGT has the unique ability to modify matched 5hmU in DNA, with no activity towards the other TET oxidation product, 5hmC.
Figure 2.

DNA substrate specificity of JGT. (A) Recombinant JGT or (B) T4 βGT and UDP-[3H]glucose incubated with 36 nt-long dsDNA substrate containing one 5hmU or 5hmC residue, as described in the “Materials and methods” section. The modified base within the dsDNA substrate was present in the context of a matched base pair (5hmU:A, 5hmC:G) or mismatched base pair (5hmU:G). CPM were measured and converted into micromolar glucose transferred. All experiments were performed in triplicate, error bars represent standard deviations. (C) UDP-Glo assay of glucosyltransferase activity of JGT for dsDNA substrates used above as described in the “Materials and methods” section. The amount of UDP Cleaved, indicative of the transfer of glucose to DNA, was estimated from a standard curve of UDP. All experiments were performed in triplicate and error bars are representative of standard deviation.
Selective enrichment of 5hmU DNA using JGT and base J immunoprecipitation
Techniques to map 5hmC within genomes have relied on using βGT transfer of an azide-modified glucose onto 5hmC followed by biotin conjugation and streptavidin pull down. As shown in Fig. 3A, βGT is able to utilize UDP-6-N3-Glc to transfer azido-glucose to 5hmC-modified DNA substrate with high efficiency. However, repeated attempts with JGT have indicated the inability of the enzyme to utilize this modified glucose donor (Fig. 3B). While JGT is able to utilize unmodified glucose to convert 5hmU in DNA with ∼95% efficiency, the enzyme appears completely unable to utilize UDP-6-N3-Glc as demonstrated by the lack of glucosylated-DNA formation and subsequent biotinylation product (Fig. 3B). Since βGT and JGT belong to different glycosyltransferase structural fold families, GT-B and GT-A, respectively [27, 55, 56], it is not surprising that the protein domain and specific interactions involved in binding UDP-glucose might be different between the two enzymes. Until we are able to synthesize other azido-linked Glc donors, we will be unable to evaluate other potential substrates for use in chemical labeling of 5hmU-modified DNA.
Figure 3.

JGT is unable to utilize UDP-6-N3-glucose. Denaturing PAGE for monitoring the reaction mixtures of DNA substrates treated with (A) T4 βGT or (B) JGT and UDP-glucose or UDP-6-N3-glucose (azido-UDP-glucose). The 15-nt-long 32P labeled dsDNA substrate containing either a 5hmU or 5hmC were incubated with the indicated GT enzyme and nucleotide sugar as described in the “Materials and methods” section. The addition of a glucose moiety to the DNA substrate results in a visible shift on PAGE, with an even greater shift upon subsequent addition of biotin. No enzyme control indicates DNA substrate incubated without the addition of the corresponding GT enzyme. The band indicated with an asterisk is a 14-nt-long glucosylated degradation product.
While JGT is unable to utilize UDP-6-N3-Glc, the enzyme is able to utilize unmodified glucose donor to convert 5hmU in DNA up to 95% efficiency. The resulting base J-DNA can be selectively enriched using base J antisera [30, 32, 51] or JBP1 [20–22, 33, 34]. Both enrichment methods have been utilized in high-throughput sequencing of base J in trypanosomatid genomes [32, 33]. To examine whether this labeling method can discriminate between 5hmU and other modifications in a biological sample, we spiked in a 75 bp dsDNA substrate with unmodified bases (T) or containing the modified bases 5hmU, 5mC, or 5hmC into trypanosome genomic DNA lacking base J and performed the JGT reaction and base J immunoprecipitation. The enrichment (%IP) of each substrate was analyzed by qPCR (Fig. 4). A significant enrichment of 5hmU was observed. No enrichment was observed for T, 5mC, or 5hmC substrates. The 5hmU enrichment is dependent on the JGT reaction, indicating both the strict dependence of this method on JGT and the specificity of the base J IP. These data demonstrate that incubation of 5hmU DNA with JGT followed by base J immunoprecipitation allows selective and efficient enrichment of 5hmU.
Figure 4.
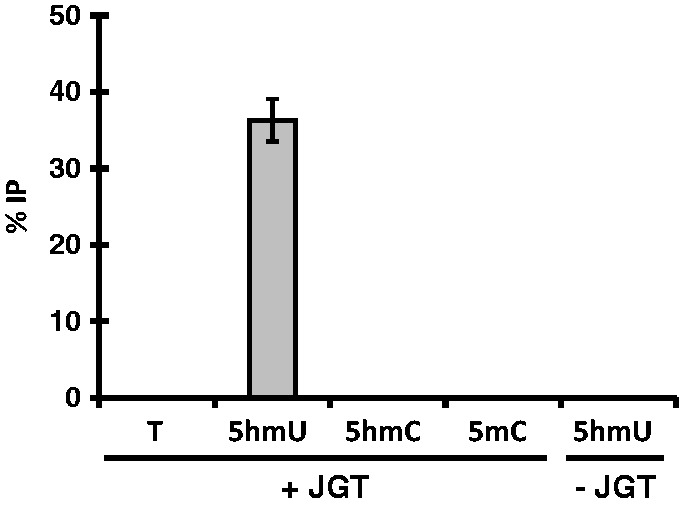
Enrichment test of the 5hmU pull-down assay. The 75-nt-long DNA containing T, 5hmU, 5hmC, or 5mC was added to trypanosome DNA as spike-in controls and the JGT reaction and J-IP was performed as described in the “Materials and methods” section. The 5hmU DNA substrate analyzed without the JGT reaction (5hmU no JGT) is provided as a negative control. The %IP was calculated relative to input DNA. All experiments were performed in triplicate, error bars represent standard deviations.
To begin to assess the feasibility of using this approach on biological samples, we performed JGT conversion of 5hmU in genomic DNA from mESCs. Genomic DNA was sonicated into small fragments (500–3000 bp), treated with JGT in the presence of UDP-glucose to yield base J. Because of the efficiency of this reaction, this method ensures selective modification of most (if not all) 5hmU in genomic DNA. The conversion of 5hmU to base J allows accurate quantification of the amount of 5hmU in a genome using J antisera and HRP. Quantification of 5hmU levels using this approach indicate approximately 2-fold more 5hmU within the R1 mESC line than in the 2i mESC line (Fig. 5), which is consistent with previous quantitative mass spectrometry analyses [16]. Taken together, these results demonstrate our ability to modify and measure 5hmU specifically in synthetic DNA oligonucleotides, PCR amplified DNA, and native genomic DNA.
Figure 5.
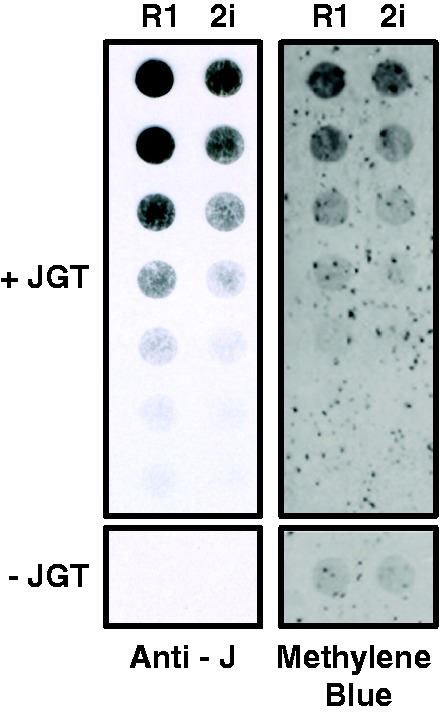
Quantitation of 5hmU in mESC genomic DNA. Genomic DNA isolated from two mESC lines (R1 and 2i) was incubated with JGT and UDP-glucose, spotted onto nitrocellulose in a 2-fold dilution series and levels of glucosyl-5hmU detected by base J antisera (Anti-J). The dependence of the assay on the JGT labeling reaction is indicated below each blot by lack of signal from the highest DNA concentration assayed without the addition of JGT (−JGT). Methylene blue staining of the blot controls for DNA loading. Shown is a representative blot from three independent experiments.
Mapping of 5hmU within the T. brucei genome
Figure 4 demonstrates the ability to specifically enrich for a short 5hmU modified DNA substrate from a pool of DNA, however we would like to use this technique to enrich and map 5hmU-containing DNA within a genome. As a proof of principle, we performed JGT labeling and J enrichment of genomic DNA from trypanosomes, subjecting the enriched fragments to qPCR analysis to allow identification of 5hmU-containing genomic regions. To do this, we took advantage of two different T. brucei cell lines, which are devoid of base J; JBP1/2 KO and JGT KO. According to the J-biosynthesis model, 5hmU will be specifically localized in the genome of the JGT KO similar to base J profile in wild type (WT) trypanosomes, albeit at lower levels, due to JBP oxidation of thymidine [27, 29]. In contrast, the JBP1/2 KO lacks both of the TH enzymes involved in the first step of J-biosynthesis. Thus, the presence of 5hmU in this genome would presumably be due to damage during the genomic DNA isolation procedure and localized non-specifically. WT T. brucei genomic base J pull down provides a positive control for J localization studies.
Base J is localized to particular sites within the trypanosome genome, including silent sub-telomeric localized variant surface glycoprotein (VSG) genes and RNA polymerase II (Pol II) transcription start sites and termination sites [32]. In agreement with previous studies, we observed enrichment of base J in WT T. brucei at a silent VSG gene (VSG 224) versus a genome internally localized gene, ASF1 (Fig. 6A). The no antibody control illustrates the strict dependence of the enrichment on base J antisera (Fig. 6A). In the JGT KO genome we observed a similar specific enrichment of 5hmU that is dependent on the in vitro JGT reaction and no 5hmU detected in the JBP1/2 KO genome (Fig. 6A). Further analyses also reveal that 5hmU is localized within regions involved in Pol II transcription termination (Fig. 6B) and initiation (Fig. 6C) with a profile identical to base J localization in the WT genome. No 5hmU is detected in these regions of the JBP1/2 KO genome. These data indicate that even with low levels of 5hmU in the JGT KO genome, ∼25 5hmU modifications per 106 nucleosides [27], this method allows genome profiling of 5hmU. These observations also provide the first direct evidence that the JBP’s dictate the specific localization of 5hmU (and J) in the T. brucei genome.
Figure 6.

Mapping 5hmU in the T. brucei genome. Genomic DNA from the JGT KO and JBP 1/2 KO T. brucei cell lines was incubated with JGT and UDP-glucose and J-DNA was enriched by anti-J IP. Anti-J IP of WT T. brucei DNA was used as a control to demonstrate the normal distribution of base J. qPCR analysis of J IP for the indicated regions of the genome was performed as described in the “Material and methods” section. The %IP was calculated relative to input DNA. Error bars indicate standard deviation of three independent replicates. (A) Analysis of base J and 5hmU at the silent 224 VSG (VSG) and the ASF1 gene. Specificity of the anti-J IP reaction is indicated by the %IP in WT DNA with and without addition of the J antisera. Specificity of the 5hmU mapping method is indicated by the %IP with and without in vitro JGT incubation in JGT KO and JBP1/2 KO DNA. (B and C) 5hmU profile at a transcription termination site and a transcription initiation site. Above, diagram of a transcription termination site for two convergent polycistronic units of chromosome 10 (region 2500–2530 kb) (B) and a transcription start site for divergent units on chromosome 10 (region 1620–1640 kb) (C). Boxes represent genes on the top and bottom DNA strand, arrows indicate direction of transcription. Location of qPCR primers spanning the known peak of base J in WT cells is indicated. The %IP was calculated relative to input DNA and normalized to the minus base J antisera (WT) or minus JGT control (JGT KO and JBP1/2 KO).
Discussion
In summary, taking advantage of the JGT-mediated glucosylation, we provide a profiling method for 5hmU. This method ensures that only 5hmU-containing genomic regions will be precipitated and identified in subsequent assays. Given that JGT specifically catalyzes the glucosylation of 5hmU and JBP1 and J-antibody specifically recognizes the resulting glucosylated 5hmU base, the DNA pulled down is highly enriched in the 5hmU modification. The enriched DNA is then ready for analysis by real-time qPCR or sequencing by any method, including high-throughput sequencing. This method can be applied, with few exceptions, to any eukaryotic genome. There is no evidence for glucosylated DNA in mammalian genomes and, other than kinetoplastids, only Diplonema and Euglena have been shown to have base J [57, 58]. Background precipitation without JGT also provides a negative control. Furthermore, base J has been detected in synthetic DNA substrates using SMRT technology [35]. Thus, our method has the potential to provide single-base resolution detection of 5hmU in a genome.
This report is, to our knowledge, the first assessment of the location of matched 5hmU in any genome. With the JGT tagging and enrichment procedure presented here, we observed 5hmU present at regions of the T. brucei genome where base J has been shown to regulate Pol II transcription and gene expression [32, 37]. The localization of 5hmU synthesized by the JBP enzymes at known base J sites provides strong support for the specificity of J biosynthesis being regulated by the initial oxidation of specific thymines in the DNA rather than any bias by JGT.
We have demonstrated the ability of the JGT-mediated glucosylation method to detect and quantify levels of 5hmU in eukaryotic genomes, including mESCs. Future experiments will utilize this protocol for epigenetic profiling of 5hmU in mESC genomic DNA. While the majority of 5hmU in the genome is generated by the TET enzymes, a small portion of 5hmU is generated through deamination of cytosine yielding 5hmU:G mispairs [16]. This mispaired 5hmU within the genome is rapidly removed by BER and replaced with a non-modified cytosine base [52, 53]. Therefore, it is thought that 5hmU generated in this way does not contribute significantly to the steady state levels of 5hmU within the genome [16]. While JGT can modify both matched and mismatched 5hmU, mismatched 5hmU can be identified by C-to-T mutation around the identified peaks following deep sequencing. In fact, the C-to-T mutation sites around the peak would both validate the 5hmU peak and mark the exact location of mismatched 5hmU site. These studies highlight the potential use of this method in mapping 5hmU localization and a powerful tool for probing the function of this newly discovered TET oxidation product.
Acknowledgements
We acknowledge Steve Dalton and Ben Boward for culturing the mES cells. We thank the Borst lab for providing base J antisera. We thank Jessica Lopes da Rosa-Spiegler for critical reading of the manuscript.
Conflict of interest statement. None declared.
Funding
This work was supported by the National Institutes of Health (R01AI109108 to R.S. and T32AI060546 to W.B.). Funding for open access charge: National Institutes of Health (R01AI109108 to R.S.).
References
- 1. Jones P. Functions of DNA methylation: islands, start sites, gene bodies, and beyond. Nat Rev Genet 2012;13:484–92. [DOI] [PubMed] [Google Scholar]
- 2. Smith Z, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet 2013;14:204–20. [DOI] [PubMed] [Google Scholar]
- 3. Kohli R, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013;502:472–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009;324:930–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ito S, Shen L, Dia Q, et al. TET proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carbosylcytosine. Science 2011;333:1300–03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. He Y, Li B, Liu P, et al. TET-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011;333:1303–07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maiti A, Drohat A. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implication for active demethylation of CpG sites. J Biol Chem 2011;286:35334–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moran-Crusio K, Reavie L, Shih A, et al. TET2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 2011;20:11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li Z, Cai X, Cai C, et al. Deletion of TET2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 2011;118:4509–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gu T, Guo F, Yang H, et al. The role of TET3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 2011; 477:606–10. [DOI] [PubMed] [Google Scholar]
- 11. Dawlaty M, Ganz K, Powell B, et al. TET1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell Stem Cell 2011;9:166–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dawlaty M, Breiling A, Le T, et al. Combined deficiency of TET1 and TET2 causes epigenetic abnormalities but is compatible with postnatal development. Dev Cell 2013;24:310–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dawlaty M, Breiling A, Le T, et al. Loss of TET enzymes compromises proper differentiation of embryonic stem cells. Dev Cell 2014;29:102–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xu Y, Wu F, Tan L, et al. Genome-wide regulation of 5hmC, 5mC, and gene expression by TET1 hydroxylase in mouse embryonic stem cells. Mol Cell 2011;42:451–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Song C, He C. Potential functional roles of DNA demethylation intermediates. Trends Biochem Sci 2013;38:480–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pfaffeneder T, Spada F, Wagner M, et al. TET oxidizes thymine to 5-hydroxymethyluracil in mouse embryonic stem cell DNA. Nat Chem Biol 2014;10:574–84. [DOI] [PubMed] [Google Scholar]
- 17. Pais JE, Dai N, Tamanaha E, et al. Biochemical characterization of a NaegleriaTET-like oxygenase and its application in single molecule sequencing of 5-methylcytosine. Proc Natl Acad Sci 2015;112:4316–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rogstad D, Liu P, Burdzy A, et al. Endogenous DNA lesions can inhibit the binding of the AP-1 (c-Jun) transcription factor. Biochemistry 2002;41:8093–102. [DOI] [PubMed] [Google Scholar]
- 19. Hashimoto H, Zhang X, Vertino PM, et al. The mechanisms of generation, recognition, and erasure of DNA 5-methylcytosine and thymine oxidations. J Biol Chem 2015;290:20723–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Robertson A, Dahl J, Vågbø C, et al. A novel method for the efficient and selective identification of 5-hydroxymethylcytosine in genomic DNA. Nucleic Acids Res 2011;39:e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Robertson A, Dahl J, Ougland R, et al. Pull-down of 5-hydroxymethylcytosine DNA using JBP1-coated magnetic beads. Nat Protoc 2012;7:340–50. [DOI] [PubMed] [Google Scholar]
- 22. Cui L, Chung T, Tan D, et al. JBP1-seq: A fast and efficient method for genome-wide profiling of 5hmC. Genomics 2014;104:368–75. [DOI] [PubMed] [Google Scholar]
- 23. Song C, Sun Y, Dai Q, et al. Detection of 5-hydroxymethylcytosine in DNA by transferring a keto-glucose by using T4 phage β-glucosyltransferase. ChemBioChem 2011;12:1682–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li Y, Song C, He C, et al. Selective capture of 5-hydroxymethylcytosine from genomic DNA. J Vis Exp 2012;68:4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Song C, Clark T, Lu X, et al. Sensitive and specific single-molecule sequencing of 5-hydroxymethylcytosine. Nat Methods 2012;9:75–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yu M, Song C, He C. Detection of mismatched 5-hydroxymethyluracil in DNA by selective chemical labeling. Methods 2015;72:16–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bullard W, Lopes da Rosa-Spiegler J, Liu S, et al. Identification of the glucosyltransferase that converts hydroxymethyluracil to base J in the trypanosomatid genome. J Biol Chem 2014;289:20273–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sekara A, Merritta C, Baugha L, et al. Tb927.10.6900 encodes the glucosyltransferase involved in synthesis of base J in Trypanosoma brucei. Mol Biochem Parasitol 2014;196:9–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bullard W, Cliffe L, Wang P, et al. Base J glucosyltransferase does not regulate the sequence specificity of J synthesis in trypanosomatid telomeric DNA. Mol Biochem Parasit 2015;204:77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Van Leeuwen F, Kieft R, Cross M, et al. Tandemly repeated DNA is a target for the partial replacement of thymine by B-D-glucosyl-hydoxymethyluracil in Trypanosoma brucei. Mol Biochem Parasitol 2000;109:133–145. [DOI] [PubMed] [Google Scholar]
- 31. Van Leeuwen F, Wijsman ER, Kieft R, et al. Localization of the modified base J in telomeric VSG gene expression sites of Trypanosoma brucei. Genes Dev 1997;11:3232–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cliffe LJ, Siegel TN, Marshall M, et al. Two thymidine hydroxylases differentially regulate the formation of glucosylated DNA at regions flanking polymerase II polycistronic transcription units throughout the genome of Trypanosoma brucei. Nucleic Acids Res 2010;38:3923–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Van Luenen H, Farris C, Jan S, et al. Glucosylated hydroxymethyluracil, DNA base J, prevents transcriptional readthrough in Leishmania. Cell 2013;150:909–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cross M, Kieft R, Sabatini R, et al. The modified base J is the target for a novel DNA-binding protein in kinetoplastid protozoans. EMBO 1999;18:6573–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Genest P, Baugh L, Taipale A, et al. Defining the sequence requirements for the positioning of base J in DNA using SMRT sequencing. Nucleic Acids Res 2015;43:133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ekanayake D, Sabatini R. Epigenetic regulation of polymerase II transcription initiation in Trypanosoma cruzi. Modulation of nucleosome abundance, histone modification, and polymerase occupancy by O-linked thymine DNA glucosylation. Eukaryot. Cell 2011;10:1465–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reynolds D, Cliffe L, Fo ürstner K, et al. Regulation of transcription termination by glucosylated hydroxymethyluracil, base J, in Leishmania major and Trypanosoma brucei. Nucleic Acids Res 2014;42:9717–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Reynolds D, Hofmeister B, Cliffe L, et al. Base J represses genes at the end of polycistronic gene clusters in Leishmania major by promoting RNAP II termination. Mol Microbiol 2016;101:559–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Borst P, Sabatini R., Base J. Discovery, biosynthesis, and possible functions. Annu Rev Microbiol 2008:62:235–51. [DOI] [PubMed] [Google Scholar]
- 40. Yu Z, Genest PA, ter Riet B, et al. The protein that binds to DNA base J in trypanosomatids has features of a thymidine hydroxylase. Nucleic Acids Res 2007;35:2107–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cliffe LJ, Kieft R, Southern T, et al. JBP1 and JBP2 are two distinct thymidine hydroxylases involved in J biosynthesis in genomic DNA of African trypanosomes. Nucleic Acids Res 2009;37:1452–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Iyer LM, Tahiliani M, Rao A, et al. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle 2009;8:1698–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cliffe L, Hirsch G, Wang J, et al. JBP1 and JBP2 Proteins are Fe2+/2-Oxoglutarate-dependent dioxygenases regulating hydroxylation of thymidine residues in trypanosome DNA. J Biol Chem 2012;287:19886–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Reynolds D, Cliffe L, Sabatini R. 2-Oxoglutarate-dependent hydroxylases Involved in DNA base J (β-D-glucopyranosyloxymethyluracil) synthesis. In Christopher J Schofield and Robert P Hausinger, (eds), 2-Oxoglutarate-Dependent Oxygenases, Cambridge, UK: The Royal Society of Chemistry, 309–23. [Google Scholar]
- 45. Toaldo CB, Kieft R, Dirks-Mulder A, et al. A minor fraction of base J in kinetoplastid nuclear DNA is bound by the J-binding protein 1. Mol Biochem Parasitol 2005;143:111–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sabatini R, Meeuwenoord N, van Boom JH, et al. Recognition of base J in duplex DNA by J-binding protein. J Biol Chem 2002;277:958–66. [DOI] [PubMed] [Google Scholar]
- 47. Heidebrecht T, Christodoulou E, Chalmers MJ, et al. The structural basis for recognition of base J containing DNA by a novel DNA binding domain in JBP1. Nucleic Acids Res 2011;39:5715–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. DiPaolo C, Kieft R, Cross M, et al. Regulation of trypanosome DNA glycosylation by a SWI2/SNF2-like protein. Mol Cell 2005;17:441–51. [DOI] [PubMed] [Google Scholar]
- 49. Kieft R, Brand V, Ekanayake D, et al. JBP2, a SWI2/SNF2-like protein, regulates de novo telomeric DNA glycosylation in bloodstream form Trypanosoma brucei. Mol Biochem Parasitol 2007;156:24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Taghizadeh K, McFaline J, Pang B, et al. Quantification of DNA damage products resulting from deamination, oxidation and reaction with products of lipid peroxidation by liquid chromatography isotope dilution tandem mass spectrometry. Nat Protoc 2008;3:1287–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cross M, Kieft R, Sabatini R, et al. J-binding protein increases the level and retention of the unusual base J in trypanosome DNA. Mol Microbiol 2002;46:37–47. [DOI] [PubMed] [Google Scholar]
- 52. Cortellino S, Xu J, Sannai M, et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 2011;146:67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Guo J, Su Y, Zhong C, et al. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 2011;145:423–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ji D, Wang Y. Facile enzymatic synthesis of base J-containing oligodeoxyribonucleotides and an analysis of the impact of base J on DNA replication in cells. PLoS One 2014;9:e103335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vrielink A, Ruger W, Driessen HP, et al. Crystal structure of the DNA modifying enzyme beta-glycosyltransferase in the presence and absence of the substrate uridine diphosphoglucose. EMBO 1994;13:3413–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Iyer L, Zhang D, Burroughs M, et al. Computational identification of novel biochemical systems involved in oxidation, glycosylation and other complex modifications of bases in DNA Nucleic Acids Res 2013;42:7635–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Van Leeuwen F, Taylor MC, Mondragon A, et al. Beta-D-glucosyl-hydroxymethyluracil is a conserved DNA modification in kinetoplastid protozoans and is abundant in their telomeres. Proc Natl Acad Sci 1998;95:2366–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dooijes D, Chaves I, Kieft R, et al. Base J originally found in kinetoplastida is also a minor constituent of nuclear DNA of Euglena gracilis. Nucleic Acids Res 2000;28:3017–21. [DOI] [PMC free article] [PubMed] [Google Scholar]