


Abstract
In recent years, new methods for computational RNA design have been developed and applied to various problems in synthetic biology and nanotechnology. Lately, there is considerable interest in incorporating essential biological information when solving the inverse RNA folding problem. Correspondingly, RNAfbinv aims at including biologically meaningful constraints and is the only program to-date that performs a fragment-based design of RNA sequences. In doing so it allows the design of sequences that do not necessarily exactly fold into the target, as long as the overall coarse-grained tree graph shape is preserved. Augmented by the weighted sampling algorithm of incaRNAtion, our web server called incaRNAfbinv implements the method devised in RNAfbinv and offers an interactive environment for the inverse folding of RNA using a fragment-based design approach. It takes as input: a target RNA secondary structure; optional sequence and motif constraints; optional target minimum free energy, neutrality and GC content. In addition to the design of synthetic regulatory sequences, it can be used as a pre-processing step for the detection of novel natural occurring RNAs. The two complementary methodologies RNAfbinv and incaRNAtion are merged together and fully implemented in our web server incaRNAfbinv, available at http://www.cs.bgu.ac.il/incaRNAfbinv.
INTRODUCTION
The design of RNAs with favorable traits is a promising endeavor that can be viewed as part of growing efforts in synthetic biology (1), as well as other applications. For example, it can be used to enhance the search for particular RNAs such as ribozymes and riboswitches in sequenced genomes (2), as well as other non-coding RNAs that may act as regulators of disease (3) or participate in catalysis (4). For riboswitches (5,6), aside of the classical problem of computationally designing transcription regulators and validating them experimentally (7,8) to complement pure experimental designs (9–11), the inverse RNA folding problem that was initially formulated and addressed in (12) can be used as a pre-processing step before BLAST for riboswitch identification (13). This recent use was also worked out for IRES-like structural subdomain identification in (14). It has potential to advance the field described in (15) for conserved RNAs in general.
Thus, computational RNA design is of increasing biological importance. Since the first program for solving the inverse RNA folding problem (or RNA design) called RNAinverse was put forth in (12), several other programs were developed. The approach to solve it by stochastic optimization relies on the solution of the direct problem using software available in RNA folding prediction web servers, e.g. the RNAfold server (16) or mfold (17), by performing energy minimization with thermodynamic parameters (18). Initially, a seed sequence is chosen, after which a local search strategy is used to mutate the seed and apply repeatedly the direct problem of RNA folding prediction by energy minimization. Then, in the vicinity of the seed sequence, a designed sequence is found with desired folding properties according to the objective function in the optimization problem formulation.
In recent years, several programs for RNA design have been developed with the goal of offering added features with respect to the original RNAinverse, most of which are general in purpose for solving similar problems (19–27) and a few are more specialized for nanostructure design and for fixed-backbone 3D design, respectively (28,29). Recently, an extension to the problem was gradually developed (30–32) that allows designing sequences that fold into a prescribed shape, leaving some flexibility in the secondary structure of RNA motifs that do not necessarily possess a known functional role. This extension, when offering a fragment selection to the user, is called ‘fragment-based’ design because it is based on a user-selected secondary structure motif (the fragment) that possesses a functional role and is therefore inserted as a ‘fragment-based’ constraint to the design problem. The shape of the RNA can be represented as a tree-graph (33) that groups together a family of RNA secondary structures, all belonging to the same coarse grained graphical representation.
The aforementioned extension led to a unique inverse RNA folding program called RNAfbinv (32) that to the best of our knowledge is more general in scope than any existing program in its shape-based approach. In that regard a shape-based approach is more general than a structure-based approach by allowing more designed sequences as solutions to the design problem, although other generalizations like pseudoknot inclusion that can be found in several other programs have not yet been implemented in our program. In parallel, controlling nucleotide distribution in RNA design problems was addressed in our presented web server in a unique way by a weighted sampling approach (34). The approach is of general importance for the future of inverse RNA folding because instead of a random start performing a local search, the initial sequence for performing the iterative procedure of solving the inverse problem is carefully picked by using global considerations in a guided manner in search space. The program for the weighted sampling approach called incaRNAation has so far only been exemplified in (34) for RNA design. The incaRNAfbinv web server described herein is a merge between RNAfbinv (32) and incaRNAtion (34). It offers sequence design solutions that to the best of our knowledge are not available in neither the most recently published programs for RNA design, namely antaRNA (26) and RNAiFold (27), nor any other such program that was devised since the seminal program called RNAinverse from the Vienna RNA package (12) was put forth. It should be noted that incaRNAfbinv relies on other programs aside of RNAinverse that are available in the Vienna RNA package such as RNAfold that solves a direct problem at each iteration and RNAdistance.
Moreover, pseudoknots have not yet been implemented in our designed program, as well as some experimental constraints, such as avoiding transcription slippage in the case of consecutive G nucleotides. In the future, it will be desired to add these features and others of experimental type to our program, as more experimental results with designed sequences obtained from our program are accumulated. In the following sections, the incaRNAfbinv web server and its method are described.
WEB SERVER
The incaRNAfbinv web server is available at http://www.cs.bgu.ac.il/incaRNAfbinv. It runs on a Unix Lenovo system x3650 M5 server with Dual Intel(R) Xeon(R) CPU E5-2620 v3 2.4GHz processors containing six logical cores and 15MB L3 cache each.
The backend is written in Java EE and run on Tomcat 8. It dispatches design tasks responsible for running incaRNAtion (34) and RNAfbinv (32). Every design task runs on up to four cores depending on load. The server runs up to ten simultaneous design tasks while the rest wait in a queue.
The frontend is designed using the Bootstrap css framework. Web pages are generated using JSP and JSTL. They utilize JavaScript, Jquery, JSON and ajax.
Input
The input screen of the incaRNAfbinv web server is shown in Figure 1. Initially, the user provides a query combining an input RNA secondary structure in dot-bracket notation, along with optional constraints and parameters. Sequence constraints are expressed using the IUPAC sequence notation. The motif selection constraint, one of the unique features that sets incaRNAfbinv apart from other inverse RNA folding programs, is presented on the right with a question mark until the user inserts the input RNA secondary structure. Once the input structure is available, the question mark is replaced by the secondary structure drawing, and in the motif selection box the user can specify which motif should be preserved exactly. For illustration, in the guanine-binding riboswitch aptamer example, taken from RFAM (35) and used for sequence design in (13), the multi-loop M13 may be selected from the drop-down menu since the ligand is known to bind within the multi-loop motif. incaRNAtion is the default option for the seed generation method, and leads to the execution of the weighted sampling algorithm (34). Alternative options include a random initial guess or a user defined sequence, as described in the original RNAfbinv (32). In combination with the incaRNAtion option, the user is allowed to specify a targeted GC content (default = 50%). Further options can be accessed by checking the Show Advanced Options box, where the target minimum free energy (kcal/mol) and target mutational robustness (neutrality between 0 and 1) can be specified. Optional parameters include the number of simulated annealing iterations (default value is set to 1000) and the number of output sequences (default value is set to 20), along with an email address and query name which can be specified to locate the job later on. The user validates the task by clicking the Submit job button when the form is complete.
Figure 1.

Input screen of the incaRNAfbinv web server, configured for the design of a guanine-binding riboswitch aptamer (5), used as a pre-processing step in a novel riboswitch detection procedure (13). In addition to an input structure and sequence constraints, optional parameters include: motif selection for the fragment-based design, target minimum free energy, target mutational robustness, generation method for the seed (incaRNAtion is the default), GC content, number of simulated annealing iterations and number of output sequences.
Results are sent by email if specified, otherwise the results are available upon completion in an interactive job mode. Aside of the Desgin Form page, a Search Result page is available in the top menu, should the user wish to re-analyze a previously-computed result, using its corresponding query name or identification. A general Help page is also available, as well as contextual tooltips that provide brief explanations for each field.
Output
The results can be accessed through the web link provided to the user, and are guaranteed to be accessible for at least a week following their generation. In addition to keeping the web link for later use, the user has an option to download the results in excel format for further analysis.
After the example parameters in the input screen of Figure 1 are inserted and the form is submitted, the main results screen appearing in Figure 2 is obtained. The query structure and associated sequence constraint appears at the top of the page. Below it are filtering options of the results displayed and further below is a table with a list of results. The table contains all the designed sequences that were generated. Each row provides a designed sequence result and its folded predicted structure in dot-bracket notation (12), its Shapiro tree-graph representation (33), minimum free energy in kcal/mol (calculated using RNAeval from the Vienna RNA package, according to the Turner energy model, 2004 (18)), mutational robustness, base pair distance from input structure, Shapiro distance from input structure, and an option to view a VARNA (36) drawing of the MFE predicted structure. The user can click on Fold Image in each row, and a popup window shows the predicted secondary structure by energy minimization of the designed sequence.
Figure 2.

The results screen of the incaRNAfbinv web server, where the designed sequences are found in a table with options to sort and filter by selected parameters. Each row provides a designed sequence result and its folded predicted structure in dot-bracket notation (12), its Shapiro tree-graph representation (33), minimum free energy in kcal/mol, mutational robustness, base pair distance from input structure, Shapiro distance from input structure and an option to view the secondary structure drawing of its folded predicted structure using VARNA (36).
For the user to have an estimate of run times, given inputs of different lengths, Figure 3 constraints run times for four different structures. The number of sequences designed was 20 by default. Tests were made using the default parameters and are presented in Log-10 s. The fourth structure was taken for timing purposes, although it should be noted that our method is using energy minimization predictions and therefore it is expected to become less accurate for lengths over 150 nt and output results for the fourth structure are not likely to have any biological meaning. There can be structures that may have results of biological meaning over 150 nt and our web server supports inputs of up to 512 nt.
Figure 3.

Runtimes for four example structures: (i) miRNA-146 precursor (65 bases). (ii) Guanine-binding riboswitch aptamer (69 bases). (iii) Cobalamin riboswitch (127 bases). (iv) S14 Ribosomal RNA—Domain 2 (361 bases, for timing purposes). Each of the following structures was tested using five different GC% contents. The number of sequences designed was 20 by default. Tests were made using the default parameters and are presented in Log-10 s.
MATERIALS AND METHODS
The incaRNAfbinv web server consists of two complementary methodologies that are merged together: RNAfbinv and incaRNAtion. In the following we describe these two methodologies.
RNAfbinv
The inverse RNA folding problem for designing sequences that fold into a given RNA secondary structure was introduced in (12). The approach to solve it by stochastic optimization relies on the solution of the direct problem (16–18). Initially, a seed sequence is chosen, after which a local search strategy was used in the original RNAinverse (12) to mutate the seed and repeatedly perform RNA folding prediction by energy minimization. As was mentioned in the Introduction, in the past several years we have been developing an extension to the approach that allows designing sequences that fold into a prescribed shape (30–32), leaving some flexibility in the secondary structure of RNA motifs that do not necessarily possess a known functional role. The shape of the RNA is represented as a tree-graph (33) in our implementation. The RNAfbinv program that implements this type of sequence design, as described in (32), relies on programs from the Vienna RNA package such as RNAfold, RNAinverse, RNAdistance (12).
Most of the constraints are inserted to the objective function in an additive manner with proper weights. This raises compatibility issues with rigid constraints like sequence constraints, which could also be inserted to the objective function in future work although at present they are left as rigid constraints for simplicity.
For any user-provided target structure S⋆, it attempts to find sequences whose MFE folding S minimizes the following objective function:
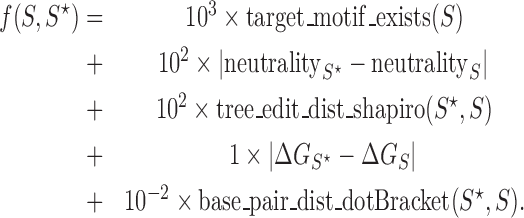 |
The weights are fixed and the rationale for their values is explained below, as well as a description for each one of the terms. To start with, the first term for the target motif existence is a binary term and is the most important constraint in general that should be fulfilled exactly without any compromise. Therefore a much larger weight of 103 relative to all others in the objective function is chosen for this term (32). In our problem of riboswitch identification, we may use it in case we encounter a specific motif such as the multi-branched loop of the guanine-binding aptamer that we would like to preserve. The neutrality for measuring mutational robustness is a number between 0 and 1. Therefore a weight of 102 is assigned to it.
The neutrality of an RNA sequence of length L is calculated by the formula <(L − d)/L >, where d is the base-pair distance between the secondary structure of the original sequence and the secondary structure of the mutant, averaged over all 3L one-mutant neighbors. The base-pair distance is evaluated by the RNAdistance program from the Vienna RNA package. The minimum free energy ΔG is for measuring thermodynamic stability in kcal/mol, therefore a unity weight is assigned. All distances between secondary structures are calculated using RNAdistance in the Vienna RNA package (12) (supporting both the coarse-grain tree graphs called the Shapiro representation (33), and the dot bracket representation of the secondary structure). For the tree edit distance between Shapiro representations, a relatively large weight of 100 is chosen for shape preservation, while for the base pair distance in the last term, a very small weight of 10−2 is assigned. This last term is the one used in the original RNAinverse (12) for preserving the exact secondary structure and its purpose is to protect the solutions from being over-dominated by shape. As explained in (31,32), shape preservation that is controlled by the term with the weight of 100 (minimizing distances between shapes) is an important aspect of our method, allowed with a flexible RNA inverse folding solver. RNAfbinv uses simulated annealing with a 4-nt look ahead local search function.
IncaRNAtion
incaRNAtion (34) addresses RNA design in a complementary way. Rather than preventing the formation of alternative secondary structures (negative design principle), it stochastically produces sequences having high affinity toward the target structure S⋆, as measured by its free-energy (positive design principle). To that purpose, a pseudo-Boltzmann distribution is postulated on the set of sequences compatible with S⋆, where the probability of emitting an RNA ω for a given pseudo-temperature T is proportional to 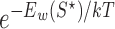 , where Eω(S⋆) is the free-energy of S⋆ upon an RNA sequence ω and k is the suitably-dimensioned Boltzmann constant. A linear-time dynamic programming algorithm is then used to generate sequences at random exactly from the pseudo-Boltzmann distribution, resulting in candidate designs whose affinity toward S⋆ ranges from extreme to reasonable, depending on the value of T. Further terms can be incorporated in the free-energy function, and combined with a provably-efficient rejection step, to control the
, where Eω(S⋆) is the free-energy of S⋆ upon an RNA sequence ω and k is the suitably-dimensioned Boltzmann constant. A linear-time dynamic programming algorithm is then used to generate sequences at random exactly from the pseudo-Boltzmann distribution, resulting in candidate designs whose affinity toward S⋆ ranges from extreme to reasonable, depending on the value of T. Further terms can be incorporated in the free-energy function, and combined with a provably-efficient rejection step, to control the 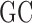 -content of produced sequences.
-content of produced sequences.
Preliminary analyses (34) revealed that incaRNAtion produces sequences that are more diverse than those obtained using competing algorithms. Furthermore, it was shown that sequences designed by incaRNAtion could be used as seeds for algorithms implementing negative design principles, increasing the diversity of their final output, while generally retaining the general properties (high-affinity, prescribed 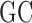 -content...) enforced by incaRNAtion in its initial generation. Figure 4 illustrates the general robustness of this good behavior in combination with RNAfbinv, by showing that sequences generated by incaRNAtion not only have lower free-energy than those produced by the default initialization of RNAfbinv, but that this higher stability persists across its iterative improvements.
-content...) enforced by incaRNAtion in its initial generation. Figure 4 illustrates the general robustness of this good behavior in combination with RNAfbinv, by showing that sequences generated by incaRNAtion not only have lower free-energy than those produced by the default initialization of RNAfbinv, but that this higher stability persists across its iterative improvements.
Figure 4.

Comparison of the free-energies of candidate solutions along the execution of RNAfbinv, for targeted GC contents from 0.1 to 0.9 and using incaRNAtion (solid lines) and the default random initialization of RNAfbinv (dashed lines), for the design of a guanine-binding riboswitch aptamer. Values averaged over 1000 runs.
CONCLUSION
When solving the inverse RNA folding problem, it is important to be able to address biological constraints in the forms of structural constraints, as well as physical observables and sequence constraints. New programs that were recently developed such as antaRNA (26) and RNAiFold (27) attempt to address these constraints but they are yet limited in their scope and cannot handle fragment-based constraints like the ones handled in RNAfbinv (32) or GC-content like in the structured and efficient way it is handled in incaRNAtion (34). These types of constraints can substantially improve targeted design of RNA sequences in the cases when such a biological-driven constraint is known in advance. The uniqueness of the fragment-based design approach together with the weighted sampling approach that traverses the search space in a guided manner merits a user-friendly web server that can accommodate practitioners of various backgrounds. We present a new web server called incaRNAfbinv that fulfills this need. It is based on the methodologies implemented in the programs (none of which is available as a web server) called RNAfbinv (32) and incaRNAtion (34). It offers a significant extension to programs performing RNA design that do not consider the aforementioned advanced constraints and are limited to strictly obeying the RNA secondary structure of the input as in the original and well-accustomed formulation of RNAinverse (12), even if a small deviation from it can produce a designed sequence that can much better accommodate the biological constraint imposed based on prior knowledge.
It should be noted that the allowed flexibility of the fragment-based design approach may also introduce spurious solutions that could be more noticeable in specific cases. Some of these issues could be remedied in the future, especially when more practical experience is gained on biologically-driven problems. As a consequence, the user should not get disappointed in special cases when the imposed constraints do not seem to lead to the desired outcome from the biological standpoint. For example, the fragment-based constraint and the sequence constraints are not fully compatible, and this could introduce designed sequences in which the sequence constraints that were meant to appear inside a certain selected motif appear outside it in adjacent motifs. Such compatibility issues could be alleviated in future versions of our approach by enforcing links between the different types of constraints that are beyond the scope of the present work. At present, non-desired results as a consequence of these issues could be neglected or filtered out in a suitable post-processing step. Extensions for pseudoknot consideration and additional biologically-driven constraints, including varied-length designed sequences, are also left as prospects for future work.
The incaRNAfbinv web server was developed with the goal of making the unique methods of fragment-based design with RNAfbinv and targeted weighted sampling with incaRNAtion available for the entire biological community. The web server is user-friendly and accessible to practitioners, both in terms of ease of use and simplification of the output. We believe that it will serve experimental groups for improving their capability to perform RNA sequence design.
Acknowledgments
We thank Arik Goldfeld and Vitaly Shapira from the computer science laboratory at Ben-Gurion University for their help with our web server.
FUNDING
ISF within the ISF-UGC joint research program framework [9/14]. Funding for open access charge: ISF within the ISF-UGC joint research program framework [9/14].
Conflict of interest statement. None declared.
REFERENCES
- 1.Isaacs F.J., Dwyer D.J., Collins J.J. RNA synthetic biology. Nat. Biotechnol. 2006;24:545–554. doi: 10.1038/nbt1208. [DOI] [PubMed] [Google Scholar]
- 2.Hammann C., Westhof E. Searching genomes for ribozymes and riboswitches. Genome Biol. 2007;8:210. doi: 10.1186/gb-2007-8-4-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taft R.J., Pang K.C., Mercer T.R., Dinger M., Mattick J.S. Non-coding RNAs: regulators of disease. J. Pathol. 2010;220:126–139. doi: 10.1002/path.2638. [DOI] [PubMed] [Google Scholar]
- 4.Strobel S.A., Cochrane J.C. RNA catalysis: ribozymes, ribosomes, and riboswitches. Curr. Opin. Chem. Biol. 2007;11:636–643. doi: 10.1016/j.cbpa.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Breaker R.R. Prospects for riboswitch discovery and analysis. Mol. Cell. 2011;43:867–879. doi: 10.1016/j.molcel.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Serganov A., Nudler E. A decade of riboswitches. Cell. 2013;152:17–24. doi: 10.1016/j.cell.2012.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Findeiß S., Wachsmuth M., Mörl M., Stadler P.F. Design of transcription regulating riboswitches. Methods Enzymol. 2015;550:1–22. doi: 10.1016/bs.mie.2014.10.029. [DOI] [PubMed] [Google Scholar]
- 8.Wachsmuth M., Domin G., Lorenz R., Serfling R., Findeiß S., Stadler P.F., Mörl M. Design criteria for synthetic riboswitches acting on transcription. RNA Biol. 2015;12:221–231. doi: 10.1080/15476286.2015.1017235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soukup G.A., Breaker R.R. Nucleic acid molecular switches. Trends Biotechnol. 1999;17:469–476. doi: 10.1016/s0167-7799(99)01383-9. [DOI] [PubMed] [Google Scholar]
- 10.Chang A.L., Wolf J.J., Smolke C.D. Synthetic RNA switches as a tool for temporal and spatial control over gene expression. Curr. Opin. Biotechnol. 2012;23:679–688. doi: 10.1016/j.copbio.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berens C., Suess B. Riboswitch engineering—making the all-important second and third steps. Curr. Opin. Biotechnol. 2015;31:10–15. doi: 10.1016/j.copbio.2014.07.014. [DOI] [PubMed] [Google Scholar]
- 12.Hofacker I.L., Fontana W., Stadler P.F., Bonhoeffer L.S., Tacker M., Schuster P. Fast folding and comparison of RNA secondary structures. Monatsh. Chem. 1994;125:167–188. [Google Scholar]
- 13.Drory Retwitzer M., Kifer I., Sengupta S., Yakhini Z., Barash D. An efficient minimum free energy structure-based search method for riboswitch identification based on inverse RNA folding. PLoS One. 2015;10:e0134262. doi: 10.1371/journal.pone.0134262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dotu I., Lozano G., Clote P., Martinez-Salas E. Using RNA inverse folding to identify IRES-like structural subdomains. RNA Biol. 2013;10:1842–1852. doi: 10.4161/rna.26994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruzzo W.L., Gorodkin J. De novo discovery of structured ncRNA motifs in genomic sequences. Methods Mol. Biol. 2014;1097:303–318. doi: 10.1007/978-1-62703-709-9_15. [DOI] [PubMed] [Google Scholar]
- 16.Hofacker I.L. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–3431. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mathews D.H., Disney M.D., Childs J.L., Schroeder S.J., Zuker M., Turner D.H. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. U.S.A. 2004;101:7287–7292. doi: 10.1073/pnas.0401799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Busch A., Backofen R. INFO-RNA–a fast approach to inverse RNA folding. Bioinformatics. 2006;22:1823–1831. doi: 10.1093/bioinformatics/btl194. [DOI] [PubMed] [Google Scholar]
- 20.Aguirre-Hernández R., Hoos H.H., Condon A. Computational RNA secondary structure design: empirical complexity and improved methods. BMC Bioinformatics. 2007;8:34. doi: 10.1186/1471-2105-8-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zadeh J.N., Wolfe B.R., Pierce N.A. Nucleic acid sequence design via efficient ensemble defect optimization. J. Comput. Chem. 2011;32:439–452. doi: 10.1002/jcc.21633. [DOI] [PubMed] [Google Scholar]
- 22.Lyngsø R.B., Anderson J.W., Sizikova E., Badugu A., Hyland T., Hein J. Frnakenstein: multiple target inverse RNA folding. BMC Bioinformatics. 2012;13:260. doi: 10.1186/1471-2105-13-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cohen B., Skiena S. Natural selection and algorithmic design of mRNA. J. Comput. Biol. 2003;10:419–432. doi: 10.1089/10665270360688101. [DOI] [PubMed] [Google Scholar]
- 24.Taneda A. Multi-objective genetic algorithm for pseudoknotted RNA sequence design. Front. Gene. 2012;3:36. doi: 10.3389/fgene.2012.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Esmaili-Taheri A., Ganjtabesh M. ERD: a fast and reliable tool for RNA design including constraints. BMC Bioinformatics. 2015;16:20. doi: 10.1186/s12859-014-0444-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kleinkauf R., Mann M., Backofen R. antaRNA: ant colony-based RNA sequence design. Bioinformatics. 2015;31:3114–3121. doi: 10.1093/bioinformatics/btv319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia-Martin J.A., Dotu I., Clote P. RNAiFold 2.0: a web server and software to design custom and Rfam-based RNA molecules. Nucleic Acids Res. 2015;43:W513–W521. doi: 10.1093/nar/gkv460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bindewald E., Afonin K., Jaeger L., Shapiro B.A. Multistrand RNA secondary structure prediction and nanostructure design including pseudoknots. ACS Nano. 2011;5:9542–9551. doi: 10.1021/nn202666w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yesselman J.D., Das R. RNA-Redesign: a web server for fixed-backbone 3D design of RNA. Nucleic Acids Res. 2015;43:W498–W501. doi: 10.1093/nar/gkv465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dromi N., Avihoo A., Barash D. Reconstruction of natural RNA sequences from RNA shape, thermodynamic stability, mutational robustness, and linguistic complexity by evolutionary computation. J. Biomol. Struct. Dyn. 2008;26:147–162. doi: 10.1080/07391102.2008.10507231. [DOI] [PubMed] [Google Scholar]
- 31.Avihoo A., Churkin A., Barash D. RNAexinv: an extended inverse RNA folding from shape and physical attributes to sequences. BMC Bioinformatics. 2011;12:319. doi: 10.1186/1471-2105-12-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weinbrand L., Avihoo A., Barash D. RNAfbinv: an interactive Java application for fragment-based design of RNA sequences. Bioinformatics. 2013;29:2938–2940. doi: 10.1093/bioinformatics/btt494. [DOI] [PubMed] [Google Scholar]
- 33.Shapiro B.A. An algorithm for comparing multiple RNA secondary structures. Comput. Appl. Biosci. 1988;4:387–393. doi: 10.1093/bioinformatics/4.3.387. [DOI] [PubMed] [Google Scholar]
- 34.Reinharz V., Ponty Y., Waldispühl J. A weighted sampling algorithm for the design of RNA sequences with targeted secondary structure and nucleotide distribution. Bioinformatics. 2013;29:i308–i315. doi: 10.1093/bioinformatics/btt217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nawrocki E.P., Burge S.W., Bateman A., Daub J., Eberhardt R.Y., Eddy S.R., Floden E.W., Gardner P.P., Jones T.A., Tate J., Finn R.D. Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 2015;43:D130–D137. doi: 10.1093/nar/gku1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Darty K., Denise A., Ponty Y. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics. 2009;25:1974–1975. doi: 10.1093/bioinformatics/btp250. [DOI] [PMC free article] [PubMed] [Google Scholar]