

Abstract
RNA molecules are attractive therapeutic targets because non-coding RNA molecules have increasingly been found to play key regulatory roles in the cell. Comparing and classifying RNA 3D structures yields unique insights into RNA evolution and function. With the rapid increase in the number of atomic-resolution RNA structures, it is crucial to have effective tools to classify RNA structures and to investigate them for structural similarities at different resolutions. We previously developed the algorithm CLICK to superimpose a pair of protein 3D structures by clique matching and 3D least squares fitting. In this study, we extend and optimize the CLICK algorithm to superimpose pairs of RNA 3D structures and RNA–protein complexes, independent of the associated topologies. Benchmarking Rclick on four different datasets showed that it is either comparable to or better than other structural alignment methods in terms of the extent of structural overlaps. Rclick also recognizes conformational changes between RNA structures and produces complementary alignments to maximize the extent of detectable similarity. Applying Rclick to study Ribonuclease III protein correctly aligned the RNA binding sites of RNAse III with its substrate. Rclick can be further extended to identify ligand-binding pockets in RNA. A web server is developed at http://mspc.bii.a-star.edu.sg/minhn/rclick.html.
INTRODUCTION
Beyond the transfer of genetic information, RNAs play important roles in biological functions such as transcription regulation (1–4), enzymatic reactions (4), and chromosome replication (5). Like proteins, RNAs have to fold into intricate 3D conformations in order to carry out its diverse functions. Because different RNA sequences may share similar functional 3D motifs, aligning and comparing RNA 3D structures is critical to elucidate RNA biology. Also, comparing RNA structures has facilitated making evolutionary and functional connections amongst diverse RNAs. While several methods exist for comparing and classifying protein 3D structures (6–9), few methods for comparing RNA 3D structures are available (10). The protein databank (PDB) (11) now has over 3100 structures of RNAs. Given the speed with which new RNA structures are deposited in the PDB, it is crucial to develop tools to efficiently compare and classify them.
Recently introduced methods for comparing RNA structure fall broadly into two categories: (i) representing the 3D structure as a 1D sequence by using local structure features, and aligning these 1D sequences, and (ii) aligning local sub-structures before extending the local alignment to a global one. In the first category, DIAL (12) and LaJolla (13) use torsion angles to represent nucleotides and align the encoded torsion-angle sequences using a dynamic programming algorithm and an n-gram model. SARA (14,15) represents each nucleotide using a set of unit vectors derived from consecutive nucleotides and aligns two RNA structures based on a unit-vector root-mean-square approach. In the second category, ARTS (16,17) uses backbone phosphate atoms to find a maximum common substructure between two RNA 3D structures. R3D Align (18,19) aligns two RNA structures based on local alignments and then uses a maximum clique algorithm to merge local alignments to form a global alignment. SETTER (20,21) divides an RNA structure into generalized secondary structure units (GSSUs) and aligns two RNA structures based on the 3D similarity of the GSSUs. It has recently been used for superposition of multiple RNA structures (22,23).
Each of these aforementioned approaches have limitations, such as restrictions on the size of aligned RNA structures and alignment inaccuracies resulting from approximations made to reduce run times (19). For example, although R3D Align (18,19) is capable of aligning large RNA structures, its running time is long. SETTER does not produce alignments at the level of single nucleotide resolution (20,21). Previously, we developed the CLICK algorithm (24,25) that can optimally superimpose a pair of protein 3D structures. CLICK uses the Cartesian coordinates of protein structures with the option of using other structural features such as secondary structure, solvent accessible surface area, and residue depth to guide the alignment. However CLICK produces inaccurate alignments for large RNA structures (26).
We introduce an improved version of CLICK for aligning pairs of RNA structures (Rclick), in which small cliques of points from both RNA structures are first matched by a least squares fit. The cliques comprise of three to seven nucleotide residues, each represented by one or more points. The clique-matching step carries out a one-to-one mapping of the equivalent residues in the two structures. A structural superimposition of the equivalent residues yields the final structural alignment. The algorithm does not consider chain connectivity when aligning the cliques of points: Rclick is a topology independent structure superimposition program. Rclick also reports more than one alignment between a pair of RNA structures, if there are detectable conformational changes.
Rclick is benchmarked and compared to other popular methods for RNA structural alignments. In most cases, Rclick alignments are better (in terms of structure overlap) than the other methods. In addition, we demonstrate the broad utility of Rclick to (a) identify conformational changes, (b) compare large RNA structures of ribosomal subunits, (c) superimpose RNA–protein complexes and (d) identify ligand binding pockets. The extensive applicability of Rclick enables novel biological insights that are unattainable with existing RNA alignment methods.
MATERIALS AND METHODS
Alignment measures
Root mean square deviation
The RMSD between the two sets of Cartesian coordinates of representative atoms of RNA structures A and B (after superimposition) is given by
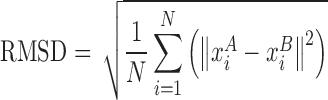 |
(1) |
where 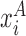 and
and 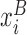 are the coordinates of representative atoms of structurally equivalent/ aligned residues of RNA structures A and B, and N is the number of equivalent residues (14).
are the coordinates of representative atoms of structurally equivalent/ aligned residues of RNA structures A and B, and N is the number of equivalent residues (14).
Structure overlap
Structure overlap (SO; also called equivalent positions) is the percentage of the representative atoms of residues Ai in the structure A that are within 4.0 Å (RMSD_Thr = 4.0Å ) of the representative atoms of equivalent/aligned residues Bj in the superimposed structure B (14).
Algorithm
Rclick detects local structural similarities of RNA structures and RNA–protein complexes, independent of topology. The algorithm consists of the following steps:
Extracting features
A residue in RNA structures and RNA–protein complexes are represented by the Cartesian coordinates of one atom. In this study, we chose C3΄ and Cα as representative atoms for RNA and protein structures, respectively.
Forming cliques
For each of the two RNA structures A and B, we calculate all possible internal pair-wise distances between their representative atoms. A clique is defined as a subset of n residues, where any pair within the clique has its distance within a threshold, dthr (Equation 2). Let Sn be the set of all possible cliques of n residues. If An∈Sn, then all pair-wise distances of representative atoms of residues Ai and Aj of An satisfy
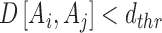 |
(2) |
where D is the Euclidean distance between two representative atoms of Ai and Aj, and Ai, Aj∈An.
Clique matching
We match all possible three-body cliques A3 and B3 (inclusive of all permutations) where A3 and B3∈S3, and identify the list of their equivalent residues. A pair of three-body cliques (A3, B3) is matched if their RMSD after superimposition is smaller than a threshold RMSD_Thr3 (Equation 3). The superimposition of A3 and B3 is performed by 3D least squares fit (27) using their equivalent residues.
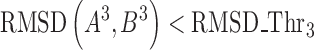 |
(3) |
We extend the matched pair of three-body cliques (A3,B3) to four-body cliques A4 and B4, by including one residue Ai and Bj (Ai∉A3 and Bj∉B3) subject to the criterion of threshold dthr (Equation 2), A4 = A3∪Ai and B4= B3∪Bj. (A4,B4) is matched if their RMSD is smaller than another threshold, RMSD_Thr4 (Equation 4, n = 4).
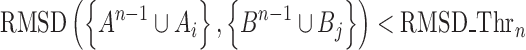 |
(4) |
Next, all matched pairs of four-body cliques (A4,B4) are extended to possible higher order cliques, An and Bn, where An, Bn∈Sn, An = An-1∪Ai and Bn = Bn-1∪Bj, and n>4. Pairs of n-body cliques (An,Bn) are selected if their RMSD is smaller than a threshold RMSD_Thrn (Table 1). In our study, we extend cliques to a maximum of seven residues.
Table 1. Optimal threshold RMSD for different n-body clique size, 3≤n≤7.
| n | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|
| RMSD_Thrn | 0.40 Å | 0.50 Å | 0.60 Å | 0.90 Å | 1.50 Å |
Alignment
Using each matched n-body clique (An,Bn), we identify the other pairs of equivalent/aligned residues (Ai,Bj), Ai∉An and Bj∉Bn, of the two structures A and B after superimposing them by the 3D least squares fit of (An,Bn). For a residue Ai of structure A (Ai∉An), there is possibly more than one residue Bj of structure B (Bj∉Bn) such that the distances of their representative atoms are within 4.0 Å. Rclick selects the residue Bjsuch that the distance of representative atoms of (Ai, Bj) is smallest. Using these equivalent residues (Ai,Bj) and the equivalent residues of the matched n-body clique (An,Bn), a final 3D least squares fit is performed to superimpose the two structures A and B. Since the matching of cliques is independent of the chain connectivity, the superimposition of structures A and B sometimes results in anomalous matches (24). Heuristic rules are applied to correct these anomalous matches (24). After that, a structure overlap of A and B based on the matched n-body clique (An,Bn) (SO of the global alignment from the matched (An,Bn)) is computed.
In our study, the superimposition of two structures A and B based on the matched n-body clique (An, Bn) that yields the best structure overlap (SObest) is selected (Equation 5).
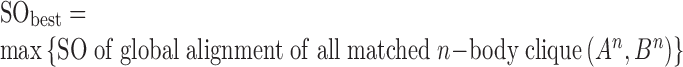 |
(5) |
Detecting conformational changes
When the two RNA structures differ by a conformational change, existing rigid superimposition methods only align the largest similar sub-structures (26). Other methods based on local alignments such as R3D Align (18,19) and SETTER (20,21) have the potential to identify conformational changes. In this study, we show the utility of Rclick to identify conformational changes between pairs of RNA structures.
Consider a pair of RNA structures A and B that have nA and nB residues, respectively. Rclick first identifies the superimposition of A and B that results in the largest SO (see Equation 5). We assume that Ak1and Bk1 are the sub-structures of A and B that have the largest SO with k1 pairs of equivalent/aligned residues (k1≤ nA and k1≤nB), and their RMSD: RMSD(Ak1,Bk1) ≤RMSD_Thr. Rclick shows the first alignment of A and B based on these k1 pairs of aligned residues.
Next, we define {A1} and {B1} to be the list of residues of both RNA structures A and B that are not in the sub-structures Ak1and Bk1, i.e.:
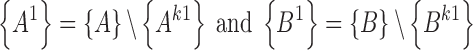 |
where {A} and {B} are the list of residues of structures A and B.
Rclick then identifies the superimposition of structures A1and B1 that results in the largest SO and their RMSD on superimposition is equal and smaller than RMSD_Thr. We assume that Ak2 and Bk2 are the sub-structures from A1 and B1 that have the largest SO with k2pairs of aligned residues and RMSD(Ak2,Bk2) ≤RMSD_Thr. Using these k2 pairs of aligned residues, Rclick shows the second alignment of A and B. This procedure is iterated till the number of unaligned residues is five or lower.
Improved Rclick for superimposing large RNA 3D structures
Given a number of large RNA 3D structures have been determined such as ribosomal subunits of Escherichia coli, Deinococcus radiodurans, Thermus thermophilus, Haloarcula marismortui and Saccharomyces cerevisiae, it is important to develop effective tools for accurately comparing these structures in a reasonable length of time. Current methods restrict the size of aligned RNA structures and/or are inaccuracies for large RNA 3D structures. The original CLICK algorithm compares all possible n-body cliques between two structures to find the optimal global alignment – this approach scales poorly with number of residues. For large RNA structures of thousands of nucleotides, the number of all possible n-body cliques is intractable. Therefore, limited by run time, CLICK is only able to find optimal local alignment for large RNA structures. Rclick reduces overall run time by only considering n-body cliques of two large RNA structures (An and Bn) with identical RNA nucleotides (i.e. in Rclick for large RNA structures, nucleotides of An can only match with the same nucleotides of Bn). The structure overlap of Rclick is better than that of CLICK on the large RNA 3D structure dataset of ribosomal subunits (Figure 1, Table 2a and b).
Figure 1.

The comparison of Rclick against R3D Align, SETTER, and CLICK using structure overlap (SO) scores on the dataset of 3D structures of 5S, 16S, 23S ribosomal subunits (19).
Table 2. a and b: The comparison of SO and RMSD scores of Rclick against R3D Align, SETTER and CLICK on the dataset of 3D structures of 5S, 16S and 23S ribosomal subunits (19).
| Rclick | CLICK | SETTER | R3D Align | |
|---|---|---|---|---|
| Average SO | 78.8% | 38.6% | 61.7% | 66.0% |
| RMSD | 1.92Å | 2.63Å | 1.65Å | 2.04Å |
| Number of alignment with better Rclick SO values | Statistical significance of difference (P-value) | |||
| CLICK | 29 | YES(<10−4) | ||
| SETTER | 29 | YES(<10−4) | ||
| R3D Align | 35 | YES(<10−4) |
Improved Rclick for superimposing RNA–protein complexes
A useful feature of the CLICK algorithm is that it can align different kinds of molecules (24). However, because proteins are often significantly larger than RNAs in RNA–protein complexes, equal weighting of representative atoms when matching n-body cliques in CLICK leads to protein-only alignment: alignment of the RNA–protein interface is overlooked. To solve this problem, Rclick matches the n-body cliques of RNA prior to superimposing protein residues in the global alignment step (i.e. only RNA residues are used in the steps of Forming Cliques and Clique Matching). Using the equivalences of RNA residues, a 3D least squares fit is performed to superimpose the two complexes in the global alignment step. The superimposed protein residues are now identified and used for calculating the SO of two complexes. For example, while Rclick produces accurate alignments of RNA–protein interactions between Ribonuclease III structures from Saccharomyces cerevisiae (28) and Aquifex aeolicus (29) (PDB codes: 1T4L and 2NUE, respectively) (Figure 8A), CLICK only aligns the protein regions of these two complexes, and cannot correctly align the RNA-binding sites (Figure 8C).
Figure 8.
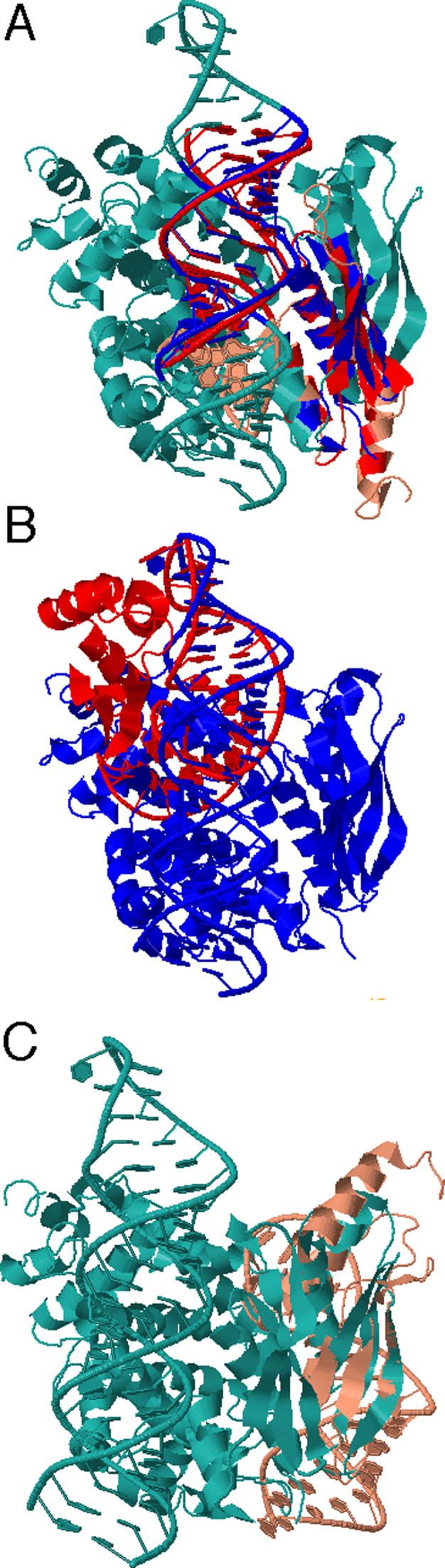
(A) The alignment of Rclick for two Ribonuclease III ofSaccharomyces cerevisiae, 1T4L and Aquifex aeolicus, 2NUE with SO of 56.56% and RMSD of 2.60 Å The superimposed RNA and protein residues of 1T4L and 2NUE are shown in red and blue, respectively. While Rclick produced accurate alignments of RNA–protein interactions between these two structures, SETTER and CLICK could not align correctly RNA-binding sites in (B and C).
Alignment datasets
Ribosomal subunits
Rclick and the other methods were tested on a dataset of 3D structures of 5S, 16S and 23S ribosomal subunits(from E. coli, D. radiodurans, Th. thermophilus, H. marismortui and S. cerevisiae) from R3D Align server (19). These ribosomal subunits have between 117 and 3308 nucleotides. These structures are selected because they are large and highly structured, characterized by several non-Watson–Crick base pairs and long-range interactions (19). Additionally, E. coli, Th. thermophilus, H. marismortui and S. cerevisiae are phylogenetically distant, and therefore have significant differences in sequence and structure (19). In all, this dataset includes 35 pair-wise alignments of ribosomal subunits (http://rna.bgsu.edu/main/r3dalign-help/gallery-of-featured-alignments/). We use the dataset of ribosomal subunits to compare Rclick against R3D Align, SETTER, and CLICK using SO and RMSD scores. SARA and ARTS web servers are incapable of producing alignments of large ribosomal RNA structures (more than 1000 nucleotides).
NR95-HR dataset
This dataset includes 1275 pair-wise RNA alignments that were used to benchmark different methods (14). This dataset (http://structure.biofold.org/sara/pages/datasets/NR95-HR.txt) includes crystal structures with resolution better than 4 Å, between 20 and 320 nucleotides, and non-redundant sequences (95% identity). In our study, this dataset is used to compare Rclick against CLICK, ARTS and SARA using SO and RMSD scores.
Difficult cases of NR95-HR dataset
This third dataset is used to benchmark the different methods when the structural similarity is low, for instance the structures of distant homologues. They include 55 pair-wise alignments from NR95-HR dataset with 30% < SO < 70% and RMSD > 2.5 Å (Table 2). The SO of Rclick is compared to results obtained from R3D Align, SARA, ARTS, CLICK and SETTER for this dataset. This dataset is available at: http://mspc.bii.a-star.edu.sg/minhn/Rclick_55_difficult_pairwises_NR95-HR.txt.
FSCOR dataset
This dataset includes 87 571 pair-wise RNA alignments of FSCOR dataset from the SARA server (15) which is the largest dataset used for comparing RNA structural alignment methods. The FSCOR dataset contains all RNA chains with more than three nucleotides and these are annotated with a unique SCOR functional class. In all, this dataset includes alignments between 419 RNA structures (http://structure.biofold.org/sara/pages/datasets/
FSCOR.txt) each having between 11 and 2774 nucleotide residues. On FSCOR dataset, Rclick results are compared to those of SARA and CLICK. Since SETTER and R3D Align have their web servers, we submitted pair-wise alignments on their web servers using the default parameters for the comparison. Hence, we could not submit the very large number of pair-wise alignments of FSCOR dataset on SETTER and R3D Align web servers.
Implementation of Rclick
Rclick has been implemented in C++. On average Rclick took 1 second to perform a comparison of a pair of RNA structure each of size ∼80 residues on a Ubuntu 10.04 Linux with 3.20 GHz CPU. A web server of Rclick is developed and freely accessible at http://mspc.bii.a-star.edu.sg/minhn/rclick.html. The web server provides options allowing users to choose the representative atom of RNA and to submit both pdb and mmCIF files. Detailed description of using pdb and mmCIF input files is available in the help page of Rclick (http://mspc.bii.a-star.edu.sg/minhn/help_rclick.html). For basepair matching, users can select C1’ atom as a representative atom, or users can define a new atom such that this atom is the middle point of C1’ and N9 atoms for A and G, and middle point of C1’ and N1 atoms for C and U. In addition, users can contact us (http://mspc.bii.a-star.edu.sg/minhn/contacts_rclick.html) to obtain the binary version of Rclick. Since cliques are extended to a maximum of 7 residues, users should submit input structures containing more than 7 residues.
JSMol and Chimera (30) are used to render all atomic, ribbon, and cartoon representation of RNA structures in this study.
Methods compared
Rclick was compared with other RNA structural alignment methods including R3D Align (18,19), SARA (14,15), ARTS (16,17), CLICK (24,25) and SETTER (20,21) on the different RNA benchmark datasets described above. All these web servers and programs were run using default parameters.
Tests for statistical significance
The non-parametric Wilcoxon signed rank test (31) was used to estimate the statistical significance of the comparisons of Rclick with other methods in terms of SO. The software Octave available at http://www.gnu.org/software/octave/index.html) was used for the Wilcoxon tests.
RESULTS
In this section, we begin by optimizing Rclick parameters and then compare Rclick with other methods. Subsequently, we illustrate the utility of Rclick with examples of identifying conformational changes, comparing large RNA structures of ribosomal subunits, and superimposing RNA–protein complexes and RNA–ligand structures.
Optimization of clique size, distance threshold and RMSD
Rclick parameters such as clique size, RMSD cut-off and distance threshold for RNA structure comparisons were optimized using a grid search on 55 difficult pair-wise alignments of NR95-HR dataset. The grid search was performed by varying the number of clique members, n, from three to seven members, and the cut-off distance, dthr, in the range [12 Å, 18 Å] were used for the grid search. At each step, the SO value was computed. The optimal cut-off distance dthr was determined to be 15 Å for n = 7. To identify the appropriate RMSD_Thrn, another grid search was performed with the similar range of n and RMSD_Thrn in the range [0.1 Å, 2.0 Å]. The optimal value of RMSD_Thrn for a particular clique size was chosen as the value above which there was no change in the SO (Table 1).
Comparing Rclick with other methods
Rclick was compared to other RNA structure alignment methods using four datasets: (i) 35 pair-wise alignments of ribosomal subunits (Table 2a and b and Figure 1), (ii) 1275 pair-wise alignments of NR95-HR dataset (Table 3a and b and Figure 2), (iii) 55 pair-wise alignments of the difficult NR95-HR dataset (Table 4a and b and Figure 3) and (iv) 87 571 pair-wise RNA alignments of FSCOR dataset (Table 5a and b). (Detailed description of each dataset is available in the methods section). Since R3D Align (18,19) and SETTER (20,21) make local alignments and do not produce and seek to maximize SO, we computed SO of R3D Align and SETTER for two RNA structures A and B using their output 3D superposition and alignment (the output of SETTER is the list of pairs of aligned residues). C3΄ atom and the cut-off distance of 4Å (RMSD cut-off = 4 Å) are used in this calculation. SARA (14,15) also produces SO using C3΄ atom and RMSD cut-off = 4 Å for its output. SO of ARTS (16,17) is computed using its output 3D superposition and pairs of equivalent/aligned residues.
Table 3. a and b: The comparison of SO and RMSD scores of Rclick against CLICK, ARTS and SARA on the NR95-HR dataset of 1275 pair-wise alignments of RNA structures (14).
| Rclick | SARA | ARTS | CLICK | |
|---|---|---|---|---|
| Average SO | 63.4% | 48.7% | 47.3% | 60.6% |
| RMSD | 1.92 Å | — | — | 1.78 Å |
| Number of alignment with better Rclick SO values | Statistical significance of difference (P-value) | |||
| SARA | 1008 | Yes (<10−4) | ||
| ARTS | 1061 | Yes (<10−4) | ||
| CLICK | 659 | Yes (<10−4) |
Figure 2.

The comparison of Rclick against ARTS and SARA using SO scores on the NR95-HR dataset of 1275 pair-wise alignments of RNA structures (14).
Table 4. a and b: The comparison of SO and RMSD scores of Rclick against R3D Align, SETTER, CLICK, ARTS and SARA on 55 difficult pair-wise alignments with 30%< SO<70% and RMSD>2.5 Å from NR95-HR dataset (14).
| Rclick | SARA | ARTS | CLICK | SETTER | R3D Align | |
|---|---|---|---|---|---|---|
| Average SO | 51.4% | 32.0% | 33.4% | 49.3% | 31.5% | 18.0% |
| RMSD | 2.62Å | — | — | 2.40 Å | 1.75 Å | 3.65 Å |
| Number of alignment with better Rclick SO values | Statistical significance of difference (P-value) | |||||
| SARA | 52 | Yes (<10−4) | ||||
| ARTS | 54 | Yes (<10−4) | ||||
| CLICK | 34 | Yes (<10−4) | ||||
| SETTER | 54 | Yes (<10−4) | ||||
| R3D Align | 53 | Yes (<10−4) |
Figure 3.

The comparison of Rclick against R3D Align, SETTER, CLICK, ARTS and SARA using SO scores on 55 difficult pair-wise alignments with 30% < SO < 70% and RMSD > 2.5 Å from NR95-HR dataset (14).
Table 5. a and b: The comparison SO and RMSD scores of Rclick against CLICK and SARA on the FSCOR dataset of 87 571 RNA pair-wise alignments (15).
| Rclick | SARA | CLICK | |
|---|---|---|---|
| Average SO | 77.3% | 61.4% | 75.3% |
| RMSD | 1.95Å | — | 1.86Å |
| Number of alignment with better Rclick SO values | Statistical significance of difference (P-value) | ||
| SARA | 74 553 | Yes (<10−4) | |
| CLICK | 32 770 | Yes (<10−4) |
The SO values of Rclick alignments are significantly better than those of CLICK, SETTER, and R3D Align for the 35 pair-wise alignments of 5S, 16S, 23S ribosomal subunits. On average the SO scores obtained from Rclick, CLICK, SETTER and R3D Align are 78.8%, 38.6%, 61.7% and 66.0%, respectively (Table 2a). The SO from Rclick alignments is never below 50% in this dataset (Figure 1). Of these 35 pair-wise alignments, Rclick obtained higher SO than R3D Align, SETTER, and CLICK in 35, 29 and 29 cases, respectively (Figure 1 and Table 2b). This dataset shows the ability of Rclick for aligning large RNA structures in reasonable time. For instance, in the case of ribosomal subunits of S. cerevisiae (PDB code 3U5H chains 5 and 8 of 3354 nucleotides (32)) and H. marismortui (PDB code: 1S72 chain 0 of 2922 nucleotides (33)), Rclick took 90 s to perform the alignment with SO of 89.01%. R3D Align (19) took >10 min to perform this alignment. Both alignments from Rclick and R3D Align (19) agree with each other.
In our previous study (24), we have used the criteria of SO and RMSD (30% < SO < 70% and RMSD > 2.5 Å) to benchmark different methods for the case of low structure similarity. In this study, we have used 55 difficult pairwise alignments from NR95-HR dataset with 30% < SO < 70% and RMSD > 2.5 Å to compare Rclick with other methods. Based on SO, Rclick performs better on this dataset than R3D Align, SARA, ARTS, CLICK and SETTER (Figure 3 and Table 4a and b). This suggests that Rclick can align RNA structures even when their structure similarity is low—for instance structures of distant homologues—and therefore Rclick is ideal for identifying non-sequential common substructures.
In our study, Rclick optimally superimposes a pair of RNA structures based on SO. RMSD is then calculated on the optimal superimposition. On these different datasets, the RMSD scores of Rclick are close to those of CLICK and better than those of R3D Align. SETTER obtains the lower RMSD scores than those of Rclick on the datasets of 35 pair-wise alignments of 5S, 16S and 23S ribosomal subunits and 55 difficult pair-wise alignments from NR95-HR (Tables 2a and 4a). The lower RMSD of SETTER could be due to its alignments from the same secondary structure regions.
Identifying conformational changes
A useful feature of Rclick is that it can be used to detect and characterize conformational changes in RNA structures. One example of such flexible alignments is between two RNA aptamer structures, (PDB codes 1OOA chain D (34) and 2JWV chain A (35); Figure 4a and b). The regions, spanning residues 1–8 and 21–29 of 1OOA chain D and 2JWV chain A, respectively, are first aligned with one another (Figure 4a). Following conformational change, Rclick shows a second alignment of the region of residues 10–20 of the RNAs(Figure 4b). Although, other methods such as R3D Align (18,19), SETTER (20,21), SARA (14,15) and ARTS (16,17) have the potential to identify conformational changes, Rclick is the only program that detects this conformational change due to its ability to find two alignments between 1OOA chain D and 2JWV chain A. Other methods and web servers including ARTS, SARA, SETTER and R3D Align produce only one alignment (please refer the link: http://mspc.bii.a-star.edu.sg/minhn/examples_rclick_1ooaD_2jwvA.html to see the alignments of Rclick, ARTS, SARA, SETTER and R3D Align for 1OOA chain D and 2JWV chain A).
Figure 4.

(A) The first alignment of Rclick for two RNA aptamer structures, PDB codes 2JWV chain A (green) and 1OOA chain D (salmon) with SO of 62.07% and RMSD of 1.75 Å. The superimposed residues of 2JWV chain A and 1OOA chain D are shown in blue and red, respectively. The regions, spanning residues 1–8 and 21–29 are aligned with one another. With conformational change, Rclick shows the second alignment of the regions of residues 10–20 of 1OOA chain D and 2JWV chain A with SO of 31.03% and RMSD of 1.91 Å in (B).
Consider the alignment between two structures of ribosomal protein–RNA complex L1 (PDB codes 2VPL chain B (36) and 1U63 chain B (37)). Rclick produces two alignments implying a conformational change. The first alignment is of the regions of residues 2–17 and 29–49 (Figure 5a), and the second is of the regions of 18–28 of 2VPL chain B and 1U63 chain B (Figure 5b).
Figure 5.

(A) The first alignment of Rclick for mRNA for ribosomal protein L1, PDB codes 1U63 chain B (green) and 2VPL chain B (salmon)with SO of 77.08% and RMSD of 1.37 Å. With conformational change, Rclick shows their second alignment with SO of 18.75% and RMSD of 1.96 Å in (B). The superimposed residues of 1U63 chain B and 2VPL chain B are shown in blue and red, respectively.
Comparing large RNA structures of ribosomal subunits
Current approaches are frequently restricted to aligning small RNAs, with significant alignment inaccuracies for large RNAs (19).We tested the ability of Rclick to align large RNA structures using 25S and 5.8S ribosomal subunits of S. cerevisiae (PDB code 3U5H chains 5 and 8 of 3354 nucleotides (32)) and 23S ribosomal subunits of H. marismortui (PDB code 1S72 chain 0 of 2922 nucleotides (33)). S. cerevisiae and H. marismortui have differences in their sequences and structures as they are phylogenetically distant (19). Rclick's topology independent approach (i.e. alignment disregards chain connectivity) enables alignment of disparate chains in S. cerevisae (chains 5 and 8 of 3U5H) to a continuous chain of H. marismortui (chain 0 of 1S72) with a high SO of 89.01% and RMSD of 1.70 Å (Figure 6). This topology independent feature is critical as, in this case, the accurate alignment consists of discontinuous regions (refer the link: http://mspc.bii.a-star.edu.sg/minhn/rclick/141896925472.html for the detailed alignment).
Figure 6.

Shows the first alignment of Rclick for 25S and 5.8S ribosomal subunits of Saccharomyces cerevisiae (PDB code 3U5H chains 5 and 8 of 3354 nucleotides, salmon color) and 23S ribosomal subunits of Haloarcula marismortui (PDB code 1S72 chain 0 of 2922 nucleotides, green color) with SO of 89.01% and RMSD of 1.70Å. The superimposed residues of and 1S72 chain 0 are shown in red and blue. Since Rclick is topology independent structure superimposition program and does not consider chain connectivity, Rclick can align both chains 5 and 8 from 3U5H to chain 0 of 1S72.
Aligning 16S ribosomal subunit from Escherichia coli (PDB code 2AW7 chain A of 1542 nucleotides (38)) and 16S ribosomal subunit from Th. thermophilus (1FJG chain A of 1522 nucleotides (39)) using Rclick showed a first alignment with SO of 78.77% and RMSD of 1.76 Å (Figure 7A). Upon conformational changes, Rclick shows the second alignment of the regions of residues 934–1063 and 1195–1385 of 2AW7 chain A and 1FJG chain A with SO of 11.48% and RMSD of 1.57 Å (Figure 7B, refer the link: http://mspc.bii.a-star.edu.sg/minhn/rclick/141922593176.html for the detailed alignment).
Figure 7.

(A and B) The first and second alignments of 16S ribosomal subunit from E. coli, 2AW7 chain A of 1542 nucleotides (salmon) and 16S ribosomal subunit from Th. thermophilus, 1FJG chain A of 1522 nucleotides (green) with SO of 78.77% and 11.48%, and RMSD of 1.76 and 1.57 Å, respectively. The superimposed residues of 2AW7 chain A and 1FJG chain A are shown in red and blue.
Superimposing RNA–protein complexes
Next, we wanted to test if Rclick can be used to identify structural similarities between two different RNA–protein complexes. We identified several instances of RNA–protein binding sites that are geometrically similar, but belong to proteins with different folds. One particular challenging example is the alignment of Ribonuclease III complexed its RNA substrate from S. cerevisiae (28) (PDB code 1T4L and SCOP entry: d.50.1.1) to Aquifex aeolicus Ribonuclease III (29) (PDB code 2NUE and SCOP entry: a.149.1.1). Sequence alignment resulting from the 3D superimposition of these two structures shows that there are only two RNA residues and three protein residues in the binding sites that are identical, indicating that this is a low complexity binding site. While all of the existing methods failed to aligned the two structures (Figure 8B and C), Rclick successfully aligned them with an SO of 56.56% and RMSD of 2.60 Å (Figure 8A). We observed that the binding regions spanning RNA residues 1–11 and 22–32 of 1T4L chain A are aligned with RNA residues 29–39 and 8–18 of 2NUE chain C, respectively, while the binding regions spanning protein residues 375–390 and 399–444 of 1T4L chain B are aligned with protein residues 159–172 and 184–221 of 2NUE chain A (refer the link: http://mspc.bii.a-star.edu.sg/minhn/rclick/141930893373.html for the detailed sequence alignment). Rclick's ability to perform accurate structural alignment in cases of poor homology demonstrates its utility and potential impact.
Identification of thiamine pyrophosphate (TPP) binding pockets in the PDB
RNA can serve as important cellular sensors (e.g. riboswitches), such as sensing the presence of ligands in the cell, and binding specifically to them (40). Identifying potential ligand pockets in RNA sensors requires accurate 3D alignment of RNA substructures (or motifs). We tested if Rclick can identify TPP binding pockets in a population of diverse RNAs, based on a motif derived from the crystal structure of a known TPP riboswitch.
We first identified a motif of the TPP binding pocket using C2 atoms within 6Å of TPP extracted from the crystal structure (PDB ID: 2CKY (40)). This motif has 16 C2 atoms. The C2 atom was selected in our study as it has the highest number of atoms within 6 Å of TPP. Additionally, C2 allows us to more effectively capture side-chain interactions between the TPP binding pocket with TPP; C2 is also close to the sugar phosphate backbone of the RNA, so should also sufficiently account for backbone-ligand interactions. In contrast, C3’ is located on the backbone of RNA, and is less efficacious in picking up the critical RNA side-chain positions in the binding pocket. We also use the C2 atom to demonstrate the utility of Rclick for using different representative atoms. We used Rclick to search over the PDB (a dataset of 2713 diverse RNA structures) for RNAs with similar binding pocket motif. Fourteen hits were identified with structure overlap (SO) more than 85% and RMSD <2 Å (Table 6). In all of these hits, Rclick perfectly identifies other known TPP binding pockets (Figure 9), indicating that Rclick can accurately detect similar RNA–ligand pockets present in RNA. Moreover, we tested different structural motifs of the TPP binding pocket using C2 atoms within 4, 5, 7 and 8 Å of TPP. The number of true hits identified with SO > 85% and RMSD < 2Å at cut-off distances of 4, 5, 7 and 8 Å are 13, 13, 14 and 14, respectively. We have also used TPP binding pocket motif using C3΄ atoms within cut-off distances of 4, 5, 6, 7 and 8 Å of TPP. The number of true hits identified with SO > 85% and RMSD < 2Å using C3΄ atoms at cut-off distances of 4, 5, 6, 7 and 8 Å are 13, 13, 13, 14 and 14, respectively. As seen, the number of true hits (14) are the same for C2 (at 6, 7 and 8 Å) and C3’ (at 7 and 8 Å).
Table 6. There are 14 hits when the C2 motif of the TPP binding pocket (extracted from 2CKY) was aligned with 2713 RNA structures in the PDB (with SO > 85% and RMSD < 2Å).
| PDB | Structure Overlap(%) | RMSD (Å) | Description |
|---|---|---|---|
| 3D2G | 100.00 | 0.50 | TPP analogues binding to the eukaryotic riboswitch |
| 3D2X | 100.00 | 0.92 | TPP-specific riboswitch bound to oxythiamine pyrophosphate |
| 3D2V | 100.00 | 1.26 | The eukaryotic TPP-specific riboswitch bound to the antibacterial compound pyrithiamine pyrophosphate |
| 3K0J | 100.00 | 1.75 | E. coli ThiM riboswitch in complex with TPP and the U1A crystallization module |
| 2GDI | 93.75 | 0.86 | TPP-specific riboswitch in complex with TPP |
| 2HOM | 93.75 | 0.92 | E. coli thi-box riboswitch bound to thiamine monophosphate |
| 4NYA | 93.75 | 1.73 | E. coli thiM riboswitch in complex with 5-(azidomethyl)-2-methylpyrimidin-4-amine |
| 4NYC | 93.75 | 1.81 | E. coli thiM riboswitch in complex with thieno[2,3-b]pyrazin-7-amine |
| 4NYD | 93.75 | 1.82 | E. coli thiM riboswitch in complex with hypoxanthine |
| 2HOJ | 87.50 | 0.68 | E. coli thi-box riboswitch bound to TPP, manganese ions |
| 2HOK | 87.50 | 0.71 | E. coli thi-box riboswitch bound to TPP, calcium ions |
| 4NYB | 87.50 | 0.75 | E. coli thiM riboswitch in complex with (4-(1,2,3-thiadiazol-4-yl)phenyl)methanamine |
| 2HOL | 87.50 | 1.02 | E. coli thi-box riboswitch bound to TPP, barium ions |
| 2HOO | 87.50 | 1.69 | E. coli thi-box riboswitch bound to benfotiamine |
Figure 9.

Rclick superimposition of the C2 motif is matched correctly with the true TPP binding pocket of 2HOL (41).
We have used this approach to construct a library of TPP binding site geometries defined by the atoms of the binding site residues. Such libraries could be very useful in constructing models of RNA structures that are known to, or speculated to bind specific ligands.
DISCUSSION
We solve the problem of RNA superimposition in Rclick by comparing two sets of points (based on the Cartesian coordinates of representative atoms) in 3D space. The Cartesian coordinates are matched by a many-body least squares fit and a match is only considered relevant if the overall RMSD falls within a defined threshold. Rclick then matches local residue packing in a fashion that is independent of topology/chain connectivity. Additionally, we extended the CLICK algorithm to make Rclick applicable for superimposing large RNA 3D structures and RNA–protein complexes.
We exhaustively compared Rclick with other RNA structural comparison methods including R3D Align, SARA, ARTS and SETTER on four different benchmark datasets. Rclick consistently outperforms these methods in terms of SO. However, this is not an exercise to show the superiority of one method over another, especially because methods like SETTER and R3D Align do not seek to maximize SO. We merely wanted to test Rclick over different datasets to ensure fidelity of alignments. The performance of Rclick over the large RNA structures of ribosomal subunits, the cases of RNA–protein complexes with proteins from different fold families, and the largest RNA benchmark dataset FSCOR of 87 571 pair-wise RNA alignments shows the ability of Rclick to extract structural similarities that are not obvious from the usual sequential or topology-dependent structural comparisons. Moreover, the condition where nucleotides of n-body clique An can only match with the same nucleotides of n-body clique Bn for large RNA structures, may affect sensitivity of the method. However, since we only apply this condition for large RNA structures of thousands of nucleotides, the chance for finding pairs of n-body cliques (An,Bn) where 3≤n≤7 with identical RNA nucleotides is high. On average, 18,200 matched n-body clique (An,Bn; with identical RNA nucleotides (n ≤ 7) of two structures A and B) were found for 25 pair-wise alignments of the large RNA structures of 16S and 23S ribosomal subunits. This number is approximately equal to the number of the matched n-body clique (n ≤ 7) used in aligning two RNA structures with 120 nucleotides. In addition, the number of matched n-body clique is dependent on the distance threshold and RMSD cut-off for different n-body clique size. Rclick provides the option to set these parameters (in the binary version of Rclick), and thus optimize them for different datasets. On the dataset of 35 pair-wise alignments of 5S, 16S, 23S ribosomal subunits, the alignments of large RNA structures agree well between Rclick and R3D Align.
Additionally, we demonstrated that Rclick could yield biological insights by using four different examples. Firstly, we showed that Rclick accurately aligns pairs of RNA structures in cases of conformational changes. These alignments can be used to detect regions in RNA structures around which substructures rearrange. Secondly, we have shown that our method is capable of accurately aligning large RNA structures in reasonable time. In particular, by neglecting chain connectivity during alignment, a more accurate alignment was found between discontinuous chains of ribosomal subunits. Thirdly, we showed that Rclick is suitable for superimposing RNA–protein complexes, even in cases of low protein homology. Rclick produced accurate alignments of RNA–protein complexes between Ribonuclease III structures from Saccharomyces cerevisiae and Aquifex aeolicus (PDB codes: 1T4L and 2NUE), while none of the other methods could. Lastly, we illustrated that Rclick, due to its topology-independent alignment, is ideal for searching ligand-binding pocket motifs in RNA. Using a TPP-binding pocket motif derived from a crystal structure, Rclick successfully identified other known TPP-binding pockets found in the PDB. This example suggests that it is possible to develop methods to characterize ligand binding site geometries and then use Rclick to conduct binding site motif searches in other RNAs.
In conclusion, we demonstrate that Rclick is a highly versatile, efficient and accurate RNA structural alignment algorithm for detecting i) conformational changes between pairs of RNA and ii) similar 3D substructures between RNAs even with low sequence similarity. Rclick has important potential applications in the areas of RNA–protein/ligand structure prediction.
ACKNOWLEDGEMENTS
M.S.M. would like to thank the Wellcome trust-DBT India alliance for a senior fellowship. We offer special thanks to Yong Taipang for his help in setting up, maintaining and improving the Rclick web server. Thanks are also due to various members of the Biomolecular Modeling and Design Division of the Bioinformatics Institute for their feedback.
FUNDING
Agency for Science, Technology and Research (A*STAR) Joint Council Office (JCO) Career Development Award [15302FG145]; Biomedical Research Council (A*STAR), Singapore.
Conflict of interest statement. None declared.
REFERENCES
- 1. Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004; 116:281–297. [DOI] [PubMed] [Google Scholar]
- 2. Dorsett Y., Tuschl T.. siRNAs: applications in functional genomics and potential as therapeutics. Nat. Rev. Drug Discov. 2004; 3:318–329. [DOI] [PubMed] [Google Scholar]
- 3. Doudna J.A. Structural genomics of RNA. Nat. Struct. Biol. 2000; 7(Suppl):954–956. [DOI] [PubMed] [Google Scholar]
- 4. Staple D.W., Butcher S.E.. Pseudoknots: RNA structures with diverse functions. PLoS Biol. 2005; 3:e213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Doudna J.A., Cech T.R.. The chemical repertoire of natural ribozymes. Nature. 2002; 418:222–228. [DOI] [PubMed] [Google Scholar]
- 6. Kolodny R., Koehl P., Levitt M.. Comprehensive evaluation of protein structure alignment methods: scoring by geometric measures. J. Mol. Biol. 2005; 346:1173–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Murzin A.G., Brenner S.E., Hubbard T., Chothia C.. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995; 247:536–540. [DOI] [PubMed] [Google Scholar]
- 8. Cuff A.L., Sillitoe I., Lewis T., Redfern O.C., Garratt R., Thornton J., Orengo C.A.. The CATH classification revisited–architectures reviewed and new ways to characterize structural divergence in superfamilies. Nucleic Acids Res. 2009; 37:D310–D314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stebbings L.A., Mizuguchi K.. HOMSTRAD: recent developments of the Homologous Protein Structure Alignment Database. Nucleic Acids Res. 2004; 32:D203–D207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rother K., Rother M., Boniecki M., Puton T., Bujnicki J.M.. RNA and protein 3D structure modeling: similarities and differences. J. Mol. Model. 2011; 17:2325–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E.. The Protein Data Bank. Nucleic Acids Res. 2000; 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ferrè F., Ponty Y., Lorenz W.A., Clote P.. DIAL: a web server for the pairwise alignment of two RNA three-dimensional structures using nucleotide, dihedral angle and base-pairing similarities. Nucleic Acids Res. 2007; 35:W659–W668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bauer R.A., Rother K., Moor P., Reinert K., Steinke T., Bujnicki J.M., Preissner R.. Fast structural alignment of biomolecules using a hash table, n-Grams and string descriptors. Algorithms. 2009; 2:692–709. [Google Scholar]
- 14. Capriotti E., Marti-Renom M.A.. RNA structure alignment by a unit-vector approach. Bioinformatics. 2008; 24:i112–i118. [DOI] [PubMed] [Google Scholar]
- 15. Capriotti E., Marti-Renom M.A.. SARA: a server for function annotation of RNA structures. Nucleic Acids Res. 2009; 37:W260–W265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dror O., Nussinov R., Wolfson H.. ARTS: alignment of RNA tertiary structures. Bioinformatics. 2005; 21(Suppl. 2):ii47–ii53. [DOI] [PubMed] [Google Scholar]
- 17. Dror O., Nussinov R., Wolfson H.J.. The ARTS web server for aligning RNA tertiary structures. Nucleic Acids Res. 2006; 34:W412–W415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rahrig R.R., Leontis N.B., Zirbel C.L.. R3D Align: global pairwise alignment of RNA 3D structures using local superpositions. Bioinformatics. 2010; 26:2689–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rahrig R.R., Petrov A.I., Leontis N.B., Zirbel C.L.. R3D Align web server for global nucleotide to nucleotide alignments of RNA 3D structures. Nucleic Acids Res. 2013; 41:W15–W21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hoksza D., Svozil D.. Efficient RNA pairwise structure comparison by SETTER method. Bioinformatics. 2012; 28:1858–1864. [DOI] [PubMed] [Google Scholar]
- 21. Čech P., Svozil D., Hoksza D. SETTER: web server for RNA structure comparison. Nucleic Acids Res. 2012; 40:W42–W48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hoksza D., Svozil D.. Multiple 3D RNA Structure Superposition Using Neighbor Joining. IEEE/ACM Trans. Comput. Biol. Bioinformatics. 2014; 12:520–530. [DOI] [PubMed] [Google Scholar]
- 23. Čech P., Hoksza D., Svozil D.. MultiSETTER: web server for multiple RNA structure comparison. BMC Bioinformatics. 2015; 16:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nguyen M.N., Madhusudhan M.S.. Biological insights from topology independent comparison of protein 3D structures. Nucleic Acids Res. 2011; 39:e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nguyen M.N., Tan K.P., Madhusudhan M.S.. CLICK - Topology independent comparison of biomolecular 3D structures. Nucleic Acids Res. 2011; 39:W24–W28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nguyen M.N., Verma C.. Rclick: a web server for comparison of RNA 3D structures. Bioinformatics. 2015; 31:966–968. [DOI] [PubMed] [Google Scholar]
- 27. Kearsley S.K. On the orthogonal transformation used for structural comparisons. Acta Cryst. 1989; A45:208–210. [Google Scholar]
- 28. Wu H., Henras A., Chanfreau G., Feigon J.. Structural basis for recognition of the AGNN tetraloop RNA fold by the double-stranded RNA-binding domain of Rnt1p RNase III. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:8307–8312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gan J., Shaw G., Tropea J.E., Waugh D.S., Court D.L., Ji X.. A stepwise model for double-stranded RNA processing by ribonuclease III. Mol. Microbiol. 2007; 67:143–154. [DOI] [PubMed] [Google Scholar]
- 30. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E.. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 31. Marti-Renom M.A., Madhusudhan M.S., Fiser A., Rost B., Sali A.. Reliability of assessment of protein structure prediction methods. Structure. 2002; 10:435–440. [DOI] [PubMed] [Google Scholar]
- 32. Ben-Shem A., Garreau de Loubresse N., Melnikov S., Jenner L., Yusupova G., Yusupov M.. The structure of the eukaryotic ribosome at 3.0 Å resolution. Science. 2011; 334:1524–1529. [DOI] [PubMed] [Google Scholar]
- 33. Klein D.J., Moore P.B., Steitz T.A.. The roles of ribosomal proteins in the structure assembly, and evolution of the large ribosomal subunit. J. Mol. Biol. 2004; 340:141–177. [DOI] [PubMed] [Google Scholar]
- 34. Huang D.B., Vu D., Cassiday L.A., Zimmerman J.M., Maher L.J. III, Ghosh G.. Crystal structure of NF-kappaB (p50)2 complexed to a high-affinity RNA aptamer. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:9268–9273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Reiter N.J., Maher L.J., Butcher S.E.. DNA mimicry by a high-affinity anti-NF-kappaB RNA aptamer. Nucleic Acids Res. 2008; 36:1227–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tishchenko S., Kljashtorny V., Kostareva O., Nevskaya N., Nikulin A., Gulak P., Piendl W., Garber M., Nikonov S.. Domain II of Thermus thermophilus ribosomal protein L1 hinders recognition of its mRNA. J. Mol. Biol. 2008; 383:301–305. [DOI] [PubMed] [Google Scholar]
- 37. Nevskaya N., Tishchenko S., Gabdoulkhakov A., Nikonova E., Nikonov O., Nikulin A., Platonova O., Garber M., Nikonov S., Piendl W.. Ribosomal protein L1 recognizes the same specific structural motif in its target sites on the autoregulatory mRNA and 23S rRNA. Nucleic Acids Res. 2005; 33:478–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schuwirth B.S., Borovinskaya M.A., Hau C.W., Zhang W., Vila-Sanjurjo A., Holton J.M., Cate J.H.. Structures of the bacterial ribosome at 3.5 A resolution. Science. 2005; 310:827–834. [DOI] [PubMed] [Google Scholar]
- 39. Carter A.P., Clemons W.M. Jr, Brodersen D.E., Morgan-Warren R.J., Wimberly B.T., Ramakrishnan V.. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature. 2000; 407:340–348. [DOI] [PubMed] [Google Scholar]
- 40. Thore S., Leibundgut M., Ban N.. Structure of the eukaryotic thiamine pyrophosphate riboswitch with its regulatory ligand. Science. 2006; 312:1208–1211. [DOI] [PubMed] [Google Scholar]
- 41. Edwards T.E., Ferre-D'Amare A.R.. Crystal structures of the thi-box riboswitch bound to thiamine pyrophosphate analogs reveal adaptive RNA-small molecule recognition. Structure. 2006; 14:1459–1468. [DOI] [PubMed] [Google Scholar]