


Abstract
A hallmark of defense mechanisms based on clustered regularly interspaced short palindromic repeats (CRISPR) and associated sequences (Cas) are the crRNAs that guide these complexes in the destruction of invading DNA or RNA. Three separate CRISPR-Cas systems exist in the cyanobacterium Synechocystis sp. PCC 6803. Based on genetic and transcriptomic evidence, two associated endoribonucleases, Cas6-1 and Cas6-2a, were postulated to be involved in crRNA maturation from CRISPR1 or CRISPR2, respectively. Here, we report a promiscuity of both enzymes to process in vitro not only their cognate transcripts, but also the respective non-cognate precursors, whereas they are specific in vivo. Moreover, while most of the repeats serving as substrates were cleaved in vitro, some were not. RNA structure predictions suggested that the context sequence surrounding a repeat can interfere with its stable folding. Indeed, structure accuracy calculations of the hairpin motifs within the repeat sequences explained the majority of analyzed cleavage reactions, making this a good measure for predicting successful cleavage events. We conclude that the cleavage of CRISPR1 and CRISPR2 repeat instances requires a stable formation of the characteristic hairpin motif, which is similar between the two types of repeats. The influence of surrounding sequences might partially explain variations in crRNA abundances and should be considered when designing artificial CRISPR arrays.
INTRODUCTION
Roughly 30% of bacterial and 70% of archaeal publicly available genomes encode clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (Cas) proteins (1,2). These CRISPR-Cas systems provide an adaptable and inheritable immune system against viruses and other foreign genetic elements (3,4). Most CRISPR-Cas loci consist of an array of alternating identical repeat and varying spacer sequences and a set of genes encoding Cas proteins (1), which have been categorized into two major classes, five major types I–V and into at least 16 subtypes (5). Although there is a complex relationship between repeats and types, a strong correlation between Cas1 and structural motifs and sequence families was found and 4719 repeats were clustered into 18 structural motifs and 24 sequence families (2,6).
The repeat-spacer array gives rise to a long precursor transcript named pre-crRNA (7,8) that is, in Type I and III systems, processed by an endoribonuclease into intermediate crRNAs (70–80 nt) and, in some Type I and III systems, in a second ribonucleolytic step into the mature crRNAs (40–50 nt) (8–11). The known primary endoribonucleases in subtype I-A, I-B, I-E, I-F and Type III systems belong to the Cas6 family of proteins, whereas the enzymatic activity for the second ribonucleolytic step is unknown (8,12). In contrast, Cas5, which possesses a structural role in the interference complex of most other CRISPR subtypes, serves as the dedicated endoribonuclease in subtype I-C systems (13,14).
It is only poorly understood how different Cas6 endoribonucleases, present in organisms with multiple CRISPR systems, differentiate between their targets (15). Cai et al. reported that about 70% of available cyanobacterial genomes possess various types of CRISPR-Cas systems (16). Three separate CRISPR-Cas systems exist in the cyanobacterium Synechocystis sp. PCC 6803, which were named CRISPR1, CRISPR2 and CRISPR3. From these, CRISPR2 and 3 are subtype III-D and III-B systems, based on the presence of a Cas10 protein (17) and the most recent classification system (5). CRISPR1 was classified as a subtype I-D CRISPR-Cas system, characterized by the presence of the type-specific protein Cas3 and the subtype-specific protein Cas10d (18); I-D systems are currently poorly described in the literature. All three CRISPR-Cas-systems (CRISPR1–3) are encoded on the ∼100 kb plasmid pSYSA (17), which in addition encodes at least nine distinct toxin–antitoxin systems, characterizing it as the major defense plasmid of this organism (19,20).
Three of the Synechocystis sp. PCC 6803 cas genes encode enzymes of the Cas6 endoribonuclease family: slr7014, slr7068, sll7075 (17). These were named cas6-1, cas6-2a and cas6-2b according to their location upstream of the CRISPR1 and CRISPR2 arrays, respectively. Deleting cas6-1 abolished the accumulation of CRISPR1 pre-crRNA, processing intermediates and mature crRNAs; whereas in the Δcas6-2a mutant CRISPR2 transcripts >200 nt overaccumulated, but shorter intermediates and mature crRNAs were lacking (17). These phenotypes are consistent with a function of Cas6-1 and Cas6-2a as endoribonucleases. Cas6-1 and Cas6-2a are only 16.5% identical at the amino acid level (Supplementary Figure S1). Although closely related proteins exist in other cyanobacteria, the relation of Cas6-1 and Cas6-2a to biochemically characterized RNA endonucleases is vague and requires genetic and biochemical analysis.
The Synechocystis sp. PCC 6803 CRISPR1 and CRISPR2 hairpins are structurally similar and the last 11 nt of the repeat sequences are identical (Figure 1D). This is relevant because many Cas6 proteins cleave within CRISPR repeats that form hairpin structures: e.g. Cas6e of Escherichia coli (7,18), MmCas6b of Methanococcus maripaludis (21), Cas6f of Pseudomonas aeruginosa (22,23) and several enzymes from Sulfolobus solfataricus (24–26). In other cases, Cas6 proteins also bind an unstructured repeat sequence, e.g. PfCas6 of Pyrococcus furiosus (27).
Figure 1.

Cas6-1- and Cas6-2a-mediated cleavage of synthetic CRISPR repeat fragments. (A) A total of 25 nM of synthetic 5΄ 32P labeled CRISPR1 (C1) or CRISPR2 (C2) oligoribonucleotides were incubated in the presence (+) of enzyme (1 μM Cas6-1 or 2 μl of Cas6-2a elution fraction 3) in reaction buffer A for 1 h. (B) A total of 100 nM of synthetic 5΄ 32P labeled C2 RNA was incubated for 1 h with 1–4 μM Cas6-1. Incubation with Cas6-2a (2 μl of Cas6 2a elution fraction 3) served as a positive control using cleavage buffer B. (C) A total of 100 nM of synthetic 5΄ 32P labeled C1 RNA was incubated for 1 h with increasing concentrations of Cas6-1 in cleavage buffer B. (D) Predicted secondary structures of the Synechocystis sp. PCC 6803 CRISPR1 and CRISPR2 repeat RNAs (C1 and C2). The determined cleavage site (17) of the processing endoribonucleases within the CRISPR1 or CRISPR2 repeats is located 8 nt upstream of the 3΄ repeat end, indicated by an arrow. The RNA secondary structures were predicted with the RNAfold web server (34) and drawn using VARNA (40). (E) Titration of 5΄ labeled C1 RNA cleavage by Cas6-1 through the addition of unlabeled C1 RNA (C1NL). To show that cleavage of 5΄ labeled C1 RNA (C1L) by Cas6-1 is inhibited by addition of unlabeled C1 RNA, 0.025 pmol of C1L and increasing amounts of C1NL (0.0025–0.25 pmol) were incubated with 500 nM Cas6-1 in reaction buffer A for 15 min. All reactions were separated by denaturing 8 M urea 15% polyacrylamide gel electrophoresis (PAGE) and bands were visualized by autoradiography. Three different negative controls were performed, by adding elution buffer 1 (Eb1), elution buffer E (EbE) or an aliquot from a mock purification from Escherichia coli cells containing the respective vector without an inserted gene (M). A byproduct of C1 oligonucleotide synthesis is labeled by the asterisk (*) in panels A, C and E. MA: Low Molecular Weight DNA Marker (Affymetrix); MDNA: radiolabeled oligonucleotides of the respective sizes serving as size markers.
Analysis of Synechocystis sp. PCC 6803 RNA-seq data led to the identification of a putative cleavage site within the repeat sequences of CRISPR1 and 2 (17). The cleavage at this site generates in both cases intermediate crRNAs with a length of 68–83 nt consisting of a single spacer sequence as well as an 8-nt-repeat handle at the 5΄ end and a 29 nt repeat fragment at the 3΄ end. However, in vivo data from northern hybridizations and RNA-seq showed the mature crRNAs to be shorter. They are 39 and 45 nt in case of CRISPR1 and 36 or 37 nt for CRISPR2 (17). Thus, in a second, so far uncharacterized step the crRNA intermediates are processed further into the mature crRNAs. Such a further processing by an unknown trimming nuclease that removes 3΄ portions of the crRNA is also known from several Type III and at least one subtype I-A system (9,28–30).
Here, we demonstrate biochemically that Cas6-1, encoded within a cassette of subtype I-D cas genes in Synechocystis sp. PCC 6803, is the endoribonuclease that generates the 8-nt-repeat handle of CRISPR1 mature crRNAs and that Cas6-2a, encoded within a different cassette of cas gene (belonging to subtype III-D), is the endoribonuclease that processes the crRNAs of CRISPR2. We detected a promiscuity of both enzymes to process not only their cognate CRISPR1 or CRISPR2 transcripts, but to also cleave the transcripts from the other locus. This promiscuity is in striking contrast to the in vivo specificity of these enzymes found in the analysis of deletion mutants (17).
Moreover, cleavage of the non-cognate substrates was less efficient and not all possible cleavage sites were recognized. Bioinformatics analysis of a series of in vitro experiments suggested the successful cleavage to depend on the stable formation of a hairpin motif, which is similar between CRISPR1 and CRISPR2. However, the sequences of adjacent spacers can lead to alternative structures that inhibit stable folding of the hairpin motif and thus are incompatible with the cleavage reaction. The influence of surrounding sequences might partially explain variations in crRNA abundances in vivo and should be considered when designing artificial CRISPR arrays.
MATERIALS AND METHODS
Cloning, expression and purification of cyanobacterial Cas6 endonucleases
The genes slr7014 and slr7068 that encode Cas6-1 and Cas6-2a (17) were amplified by polymerase chain reaction (PCR) using primers containing BamHI and SphI or PstI and SacI restriction sites (Supplementary Table S1) and 10 ng of Synechocystis sp. PCC 6803 genomic DNA. PCR fragments were subcloned in E. coli DH5α after ligation into vector pJET1.2/blunt (CloneJET PCR Cloning Kit, Thermo Fisher Scientific). The slr7014-containing BamHI/SphI restriction fragment was isolated and ligated into the corresponding sites of vector pQE70 (QIAGEN) and transformed into E. coli M15[pREP4], whereas the slr7068–containing PstI/SacI fragment was recloned into vector pASK-IBA7plus (IBA-Solutions for Life Sciences) and transformed into E. coli Rosetta(DE3)pLysS. In this way, the reading frame of Cas6-1 was prolonged by six additional histidine residues at the C-terminus (His6-tag), whereas Cas6-2a is featured with an N-terminal Strep-Tactin® affinity tag (Strep-tag® II). The cloned fragments were verified by DNA sequencing (GATC Biotech).
Escherichia coli M15[pREP4]/pQE70::slr7014 was grown in LB medium (31) in a culture volume of 400 ml (100 μg/ml ampicillin, 50 μg/ml kanamycin) at 37°C to an OD600nm of ∼0.8. Protein expression was induced by the addition of 1 mM IPTG at 30°C for 3 h. Cells were pelleted by centrifugation at 6500 g and 4°C for 15 min and frozen at −20°C, or immediately resuspended in 5 ml of lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole; pH 8) in the presence of protease inhibitor (cOmplete Protease Inhibitor Cocktail Tablets, Roche). Cells were disrupted by sonication (Sonifier 250, Branson) and debris was removed by centrifugation at 11000 g and 4°C for 30 min.
Expression of Cas6-2a was induced in E. coli Rosetta(DE3)pLysS/pASK-IBA7plus::slr7068 in a culture volume of 400 ml (100 μg/ml ampicillin, 34 μg/ml chloramphenicol) at an OD550nm of ∼0.6 by the addition of 200 ng/ml anhydrotetracycline. Cultures were grown at 22°C with 180 rpm overnight. Cells were pelleted as for Cas6-1 but then resuspended in 4 ml of buffer W (100 mM Tris–HCL, pH 8, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), pH 8). Cells were disrupted in 7 ml tubes with 250 μl glass beads (Ø 0.5 mm) with a tissue homogenizer (Precellys24 with Cryolys-N2-cooling, 6* 10 s, 6500 rpm with 5 s of break between each interval, Bertin Technologies). Debris was removed by centrifugation at 13000 g and 4°C for 15 min.
For the purification of Cas6-1, Ni2+-NTA-agarose (QIAGEN) and chromatography columns (Poly-Prep®, BIO-RAD) were used. The protein purification was performed under native conditions as recommended by the the manufacturer (The QIAexpressionist, QIAGEN). A bed volume of 300 μl was used. The wash buffer contained 40 mM imidazole. To elute the bound proteins, elution buffers 1 and 2 (50 mM NaH2PO4, 300 mM NaCl, 250 mM or 500 mM imidazole, pH 8) were used consecutively. The elution fractions 1 and 2 of purified Cas6-1 protein were 10-fold concentrated (Supplementary Figure S2A) using Amicon Ultra-0.5 or Ultra-2 Centrifugal Filter Units with Ultracel-10 membrane (Merck Millipore). Thereby the buffer was exchanged to PBS (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4; pH 7.3). The protein concentrations were determined using the Direct Detect® Spectrometer (Merck Millipore).
For the purification of Cas6-2a, Strep-Tactin® Sepharose (IBA-Solutions for Life Sciences) and chromatography columns (Poly-Prep®, BIO-RAD) were used. The purification was performed under native conditions according to the manufacturer's recommendations (IBA-Solutions for Life Sciences) using a bed volume of 800 μl. The column was washed 5 times with 4 ml of buffer W and bound protein was eluted with 6* 400 μl of elution buffer E (100 mM Tris–HCl, pH 8, 150 mM NaCl, 1 mM EDTA, 2.5 mM desthiobiotin). Purified proteins were analyzed via sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) (6% polyacrylamide (PAA) stacking gel, 15% PAA separating gel), visualized by GelCode Blue Safe Protein staining (Thermo Fisher Scientific) for 1 h, aliquoted and stored at −80°C until use. The purified Cas6-2a protein was further verified by western blot detection (Supplementary Figure S2B) using the Strep-Tactin® HRP conjugate according to the manufacturer's instructions (IBA-Solutions for Life Sciences) and detecting the chemiluminescence signal with the FUSION SL™ imaging system (peQlab). Additionally, the vectors and tags pQE30, pQE70 (His6-tag, from QIAGEN) and pET28a(+) (His6-tag, Merck Millipore), pGEX-6P-1 (GST-tag, GE Healthcare Life Sciences), pASK-IBA6 and pASK-IBA43plus (Strep-tag® II, IBA-Solutions for Life Sciences) were tested for the purification of Cas6-2a. As additional negative controls, all purification steps were repeated for both proteins with an empty vector E. coli control strain.
Generation of radiolabeled synthetic RNA oligonucleotides
Synthetic oligoribonucleotides C1 and C2 (SIGMA-ALDRICH®) correspond to the CRISPR1 and CRISPR2 repeat RNAs (Supplementary Table S1). A total of 12.5 pmol of each oligoribonucleotide were used for 5΄ end-labeling with 32P using 50 μCi of [γ32P] ATP (3000 Ci/mmol, Hartmann Analytic) and 25 U of T4 polynucleotide kinase (Thermo Fisher Scientific) in a reaction volume of 50 μl. As size markers, DNA oligonucleotides of the respective sizes (MDNA) or the Low Molecular Weight Marker (MA, Affymetrix), that is also composed of DNA, were analogously 5΄ end-labeled. Unbound nucleotides were removed with RNA Clean & Concentrator-5 (Zymo Research) and labeled RNA was eluted in 50 μl of nuclease free water.
RNase cleavage assays with synthetic RNA oligonucleotides
Cleavage reactions were performed in a volume of 10 μl in cleavage buffer A (20 mM HEPES-KOH, pH 8, 250 mM KCl, 1 mM DTT, 2 mM MgCl2) or cleavage buffer B (20 mM Tris–HCL, pH 7.8, 400 mM KCl), with 25–100 nM 5΄ labeled RNA and 0.01–4 μM Cas6-1 or, when indicated, with 2 μl of elution fraction 3 of the Cas6-2a purification. As negative controls served elution buffer 1 (Eb1, for Cas6-1 experiments), elution buffer E (EbE, Cas6-2a experiments) or analogous purifications with cells harboring the respective plasmid without an inserted gene (empty vector (mock) controls, MC). Reactions were incubated for 15–60 min at 37°C, stopped by the addition of 2× RNA loading dye (95% formamide, 0.025% SDS, 0.025% bromophenol blue, 0.025% xylene cyanol FF, 0.5 mM EDTA, pH 8) and stored on ice. Before loading onto denaturing 8 M urea 15% PAA gels, the reactions were incubated at 95°C for 5 min and cooled down on ice. Gels were exposed to a phosphor imaging screen (BIO-RAD) and radiolabeled RNA was visualized by phosphor imaging (Molecular Imager PharosFX™ Plus, BIO-RAD) and analyzed using Quantity One® software (BIO-RAD).
In vitro transcription and purification
In vitro transcription of CRISPR1, 2 and 3 was performed with the MEGAshortscript T7 transcription kit (Ambion®, Thermo Fisher Scientific). Suitable templates were PCR-amplified and thereby tagged with a T7 promoter as part of the primer sequences (Supplementary Table S1). The amplified and purified (NucleoSpin® Gel and PCR Clean-up, MACHEREY-NAGEL) fragments were used for in vitro transcription according to the manufacturer's specifications. All in vitro transcribed RNAs carry two additional guanidine nucleotides at their 5΄ ends originating from the T7 promoter. The in vitro transcripts were used directly (products CRISPR1, CRISPR1*, CRISPR2 and CRISPR3; used for experiments shown in Figure 2) or after gel purification (products CRISPR1 I–IX and CRISPR2 I–IX; used for experiments shown in Figure 3). In the latter case, transcripts were size-fractionated by denaturing 8 M urea 10% PAGE, visualized with ethidium bromide under ultraviolet (UV) light and excised at the appropriate size. Transcripts were eluted for 18–24 h at 37°C by adding 300 μl of transcript elution buffer (20 mM Tris–HCL, pH 7.5, 250 mM sodium acetate, pH 5.2, 1 mM EDTA, pH 8). Afterward, the elution buffer containing the eluted in vitro transcript was transferred to a fresh reaction tube and the transcript was precipitated for 18–72 h at −20°C by adding two volumes of ethanol (99.8%). The RNA was pelleted at 11 000 g and 4°C for 30 min, washed once with 100 μl of ethanol (70%), pelleted again for 5 min and resuspended in 50 μl of nuclease free water.
Figure 2.

Incubation of in vitro transcripts of CRISPR1, 2 and 3 with Cas6-1. (A) A total of 2.3 μM of CRISPR1 transcript is cleaved by 6.2 μM Cas6-1 by incubation for 1 h. (B) A total of 2 μM of CRISPR2 transcript is cleaved by 6.2 μM Cas6-1 by incubation for 1 h. (C) A total of 3.2 μM of CRISPR3 transcript is not cleaved by 6.5 μM Cas6-1 by incubation for 2 h. (D) The cleavage of 6.6 μM of the shorter CRISPR1* in vitro transcript by 5.8 μM Cas6-1 monitored over a time of 2 h 20 min. Since no finally mature crRNA (17) was detected, the in vivo presence of an unknown ribonuclease is suggested. All reactions were performed at 37°C in a reaction volume of 5 μl. RNA was separated by 8 M urea 10% PAGE and bands were visualized by ethidium bromide staining. Diamonds represent repeats and rectangles spacer sequences. Transcripts were not gel purified, explaining the appearance of fragments smaller than the full length transcripts of CRISPR1 and 2 in absence of Cas6-1. Eb1, elution buffer 1 used as negative control. Molecular markers: MN, Low Range ssRNA Ladder (NEB); MF, RiboRuler Low Range RNA Ladder (Thermo Fisher Scientific).
Figure 3.

Cleavage of CRISPR1 or CRISPR2 in vitro transcripts by Cas6-1 or Cas6-2a. (A and B) Processing of CRISPR1 or CRISPR2 transcripts I–IX by Cas6-1. In vitro transcripts were incubated with 750 nM Cas6-1 in reaction buffer B. (C and D) Processing of CRISPR1 or CRISPR2 transcripts I-IX by Cas6-2a. In vitro transcripts of CRISPR1 were incubated in buffer C with 2 μl of Cas6-2a elution fraction 3. For the reactions shown in (D) only 1 μl of Cas6-2a elution fraction 3 was used. All in vitro transcripts were gel purified and used in a final concentration of 300 nM. The reactions were performed in a reaction volume of 10 μl in the presence or absence of enzyme at 37°C for 30 min. Reactions were separated on denaturing 8.3 M urea 10% PAA sequencing gels and bands were visualized with SYBR® Gold Nucleic Acid Gel Stain. The sizes of cleavage products are indicated in nucleotides next to the respective fragments. MN, MF, MA: Molecular Weight Markers as in Figures 1 and 2. MOH: alkaline hydrolysis ladder; X: empty lanes.
RNase cleavage assays with in vitro transcripts
Cleavage assays of Cas6-1 were performed in cleavage buffer B (experiments shown in Figure 3A and B) or by adding Cas6-1 directly to the in vitro transcript (experiments shown in Figure 2) at 37°C for 0.5 h if not specified otherwise. Cleavage assays of Cas6-2a were performed in cleavage buffer C (10 mM HEPES-KOH, pH 8.0, 125 mM KCl, 0.5 mM DTT, 0.94 mM MgCl2) with 1 or 2 μl of Cas6-2a elution fraction 3 at 37°C for 0.5 h. To stop the reactions, 2× RNA loading dye was added. Before loading onto denaturing 8 M urea 10% PAA gels, reactions were incubated at 95°C for 5 min. As size markers served the Low Range ssRNA Ladder from NEB (MN), the RiboRuler Low Range RNA Ladder from Thermo Fisher Scientific (MF) and the Low Molecular Weight Marker from Affymetrix (MA). RNA was visualized after ethidium bromide staining under UV light (254 nm) in a gel documentation system (E-Box-3026, peQlab). For size determination of RNA fragments generated in cleavage assays with Cas6-1 or Cas6-2a separation of fragments was performed with a sequencing gel electrophoresis apparatus (Model S2, Biometra). The denaturing 8.3 M urea 10% PAA gel with a size of 31 × 38 cm was prerun at constant power (65 W) for 1 h and with a surface temperature of 42–46°C. After sample loading the gel was run for additional 4.5 h at 65 W. An alkaline hydrolysis ladder was produced by incubation of 20 pmol of a 358 nt in vitro transcript in a buffer containing 50 mM Tris–HCl, pH 8.5 and 20 mM MgCl2 for 48 h at 30°C.
For staining of sequencing gel-separated RNA, SYBR® Gold Nucleic Acid Gel Stain (Thermo Fisher Scientific) was used in a 1:10000 dilution with 0.5 × Tris-Borate-EDTA (TBE) buffer. The image was taken with the Laser Scanner Typhoon FLA 9500 (GE Healthcare Life Sciences) with the following settings: excitation: 473 nm, emission filter long pass blue ≥ 510 nm, photomultiplier value: 450 or 500.
Predicting stabilities of CRISPR hairpin motifs within their natural context
The functional consensus structure motifs for CRISPR1 and CRISPR2, as shown in Figure 1D, were taken from reference (17), where local sequence context was considered and thus the repeat structure that is most stable across the entire CRISPR array was determined. By this definition, the consensus motif is a local structure that consists of the base pairs defined in the consensus motif. We estimate the quality of formation of this local functional repeat structure in a specific fragment by determining the accuracy of this structure as previously defined (32). The accuracy of a local structure consisting of a set of base pairs 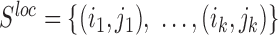 in an RNA-sequence
in an RNA-sequence  is defined as the expected overlap of the local structure
is defined as the expected overlap of the local structure 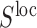 with all possible global structures
with all possible global structures 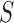 of the sequence
of the sequence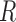 :
:
 |
where  is the Boltzmann probability of the global structure
is the Boltzmann probability of the global structure 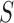 in the ensemble of all structures of
in the ensemble of all structures of 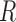 . Since this would require a summation over an exponential number of structures, this cannot be directly calculated this way. However, as shown in reference (32), this quantity is equivalent to:
. Since this would require a summation over an exponential number of structures, this cannot be directly calculated this way. However, as shown in reference (32), this quantity is equivalent to:

which can easily be calculated. Here, 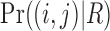 is the base pair probability of the base pair
is the base pair probability of the base pair 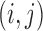 in the sequence
in the sequence 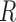 as determined by the McCaskill approach, as e.g. implemented in the Vienna RNA package with RNAfold –p.
as determined by the McCaskill approach, as e.g. implemented in the Vienna RNA package with RNAfold –p.
In the case of longer fragments, the problem of long-range base pairs occurs. It is well known that predictions of these long-range base pairs are especially unreliable and noisy. To minimize this effect, and to also account for possible other effects like co-transcriptional folding or intermediate processing, we followed a local folding approach for determining base pair probabilities (see reference (32) for a discussion of various local folding approaches). The idea is to calculate base pair probabilities as usual as the sum of probabilities of structures that contain this base pair, but to restrict the set of possible structures to local structures by restricting the maximal span of a base pair. This implies, that in all possible structure considered in this calculation, the distance between the left and right end of any base pair is restricted. In our case, we used 80 nt as maximal span of a base pair. Technically, this is achieved by using RNAplfold (33) from Vienna package 1.8.4, where we set the window size (W) equal to the fragment size and the maximum base-pair span (L) equal to 80 nt. In addition, we used the option –noLP to disallow lonely base pairs, which usually improves the prediction quality. Dot plots were calculated for the repeat structure by taking the average of the sub-matrices for each repeat instance, where the base-pair probability matrix is computed for each window separately and then averaged over all windows using RNAfold (34), Vienna package version 1.8.4, with parameters ‘-p -d2 –noLP’. The RNA secondary structures were drawn using VARNA (40).
RESULTS
Cas6-1 mediated cleavage of synthetic oligoribonucleotides
We cloned, expressed and purified recombinant Synechocystis sp. PCC 6803 Cas6-1 and Cas6-2a as soluble proteins (Supplementary Figure S2). Recombinant Cas6-1 and Cas6-2a cleaved their cognate synthetic repeat oligoribonucleotides C1 or C2 completely, whereas both enzymes cleaved their non-cognate repeats (C2 for Cas6-1 and C1 for Cas6-2a) only weakly (Figure 1A–C). Both enzymes cleave their respective targets at a single position, resulting in an 8 nt shorter 5΄ 32P labeled RNA product (29 nt). For both tested repeats, this product is consistent with the cleavage between repeat positions G29 and A30 (Figure 1D) that was determined by RNA-seq analysis (17). We verified 5΄ 32P labeled C1 RNA (C1L) as a substrate of Cas6-1 by titrating the cleavage reaction through addition of increasing amounts of unlabeled substrate C1NL. The addition of a 5- to 10-fold excess of C1NL over C1L caused a decrease of C1L cleavage by Cas6-1 since the protein likely reached a limit of saturation with substrate (Figure 1E).
Promiscuity in the cleavage of CRISPR precursor transcripts by Cas6-1 and Cas6-2a
Since a repeat sequence does not exist on its own but is part of a pre-crRNA transcript, the endoribonuclease activity of Cas6-1 on longer precursors was studied. We incubated precursors containing multiple repeat-spacer units of CRISPR1–3 with the purified protein in vitro and analyzed cleavage products by gel electrophoresis (Figure 2). Transcripts of CRISPR1 (364 nt and a shorter version CRISPR1* of 168 nt) were cleaved to products of the expected sizes (Figure 2A and D; Supplementary Table S2). Surprisingly, in this assay the in vitro transcript of CRISPR2 was cleaved by Cas6-1 with the similar efficiency as the CRISPR1 transcript (Figure 2A and B). In contrast, the CRISPR3 transcript was not cleaved by Cas6-1 (Figure 2C), consistent with the results of genetic analyses (17).
In the following, we characterized the ectopic Cas6-1-mediated processing of CRISPR2 transcripts by systematic substrate variation. In addition, we tested if Cas6-2a could possibly mediate processing of CRISPR1 transcripts as well. Each RNA fragment represented a subsequence of the original CRISPR1 or CRISPR2 array with a different number of repeats and spacers. The CRISPR1 and 2 fragments I–IX were incubated in vitro in the presence or absence of Cas6-1 or Cas6-2a and resulting cleavage fragments were analyzed by denaturing gel electrophoresis (Figure 3). Detected fragment sizes were consistent with the expected lengths when assuming a cleavage 8 nt upstream of the 3΄ end of each repeat instance in the CRISPR1 or 2 fragments (Supplementary Table S2). Both enzymes delivered very similar patterns for the respective substrates, suggesting that the identical sites were recognized and cleaved. However, we noticed for both enzymes that for CRISPR1 all but for CRISPR2 not all theoretically possible fragments (Supplementary Table S2) were observed, consistent with the idea that they could generate some but not all of the theoretically possible products. The presence of potential contaminating RNase activities in the preparations is considered very low because there was no RNA processing or degradation in parallel incubations with empty-vector mock preparations.
Adjacent spacer sequences influence the formation of the substrate structure
Computational analysis of CRISPR structure suggested that adjacent spacer sequences can influence the formation of the repeat structure motif (17). To test whether surrounding sequence context influences Cas6-1 and Cas6-2a cleavage of CRISPR1 and 2 transcripts, we calculated the accuracies (see ‘Materials and Methods’ section) of the local functional repeat motifs for all the products obtained experimentally (Figure 3), each representing a subsequence of the original CRISPR1 or CRISPR2 array with a different number of repeats and spacers (Figure 4). As can be clearly seen, the accuracy of the functional local repeat structure is significantly lower for the non-cleaved compared to the cleaved fragments. To illustrate this further, we chose the CRISPR2 repeats R6 and R7 within fragment VIII as an example. We observed that repeat R7 was cleaved in this fragment, whereas repeat R6 was not cleaved as part of the same fragment VIII (Figure 5A), indicated by the lack of the 123, 76 and 67 nt fragments for CRISPR2-VIII in Figure 3B and D. Predictions of the secondary structure revealed that only the local functional repeat structure of R7 is formed in fragment VIII, whereas the associated positions are blocked by the alternative secondary structure in case of the non-cleaved repeat R6 (Figure 5B). The latter case is especially interesting: while all repeat instances are of identical sequence, the adjacent spacer sequences differ. Thus, this finding illustrated the possible relevance of local basepairing interactions between a repeat and its adjacent spacers. Therefore, we measured the predicted stability of the hairpin motif from Figure 1D for each repeat instance, using the base pair accuracy: a value close to 1 or 0 corresponds to a high or low predicted structure stability, respectively. We observed a very clear separation of base pair accuracies with respect to the presence or absence of cleavage events (Figures 3 and 6). In summary, the base pair accuracy could explain 43 out of 50 experimental cleavage outcomes for CRISPR2 (Supplementary Tables S3 and 4). These results justify using the base pair accuracy to predict cleavage events that depend on the stability of local structure motifs.
Figure 4.

The structure stability of the CRISPR2 hairpin, measured as the base pair accuracy (y-axis), is compared between repeat instances that were cleaved (left) and not cleaved (right) by Cas6-1 and Cas6-2a in the in vitro experiments in Figure 3B and D. High base pair accuracies correspond to successful cleavage events, whereas low base pair accuracies explain repeats that were not cleaved. For both enzymes only 3 out of 25 experimental observations were not explained by the base pair accuracy (Supplementary Table S3).
Figure 5.

Systematic analysis of CRISPR2 cleavage by Cas6-1. (A) Schematic overview of full length CRISPR2 transcripts and positions of cleavage by Cas6-1 as determined by the experiment shown in Figure 3B. All data are also summarized in Supplementary Table S3. (B) Prediction of a global MFE structure to determine the most probable structure for the complete CRISPR2 fragment VIII. We have indicated the positions covered by the local functional repeat structure in turquoise and the remaining repeat sequence in red (R6) or blue (R7). Spacers are colored in yellow (S5), green (S6) or orange (S7). The local functional repeat structure is formed in the cleaved repeat R7, whereas the associated position is blocked by other stems in the non-cleaved repeat R6 of fragment VIII.
Figure 6.

Average dot plot of (A and B) CRISPR1 or (C and D) CRISPR2 repeats that are cleaved or not cleaved in the artificial fragments. Each dot represents the base pairing potential of the nucleotides that can be read from the respective rows and columns. In the bottom-left triangle, we show the base pairs involved in the functional motif (highlighted in red). The top-right triangle represents the average base pair probability of each repeat instance in the respective fragments. Adjacent to the repeats are the average base pair probabilities with respect to the position in the adjacent spacer. (A and C) Repeat instances that were cleaved. (B and D) Repeat instances that were not cleaved. We observe that in (B and D) there are many more base pairs in the surrounding context than in (A and C) and that the base pairs of the functional motif are more probable on average in (A and C) than in (B and D). Note that we represent the spacers by their mode length.
DISCUSSION
Mature crRNAs are integrated into large ribonucleoprotein complexes with their cognate Cas proteins and guide these complexes to invading foreign RNA (9) or DNA sequences (7,9,10,35). Therefore, the accurate processing of crRNA precursors is an essential step in the CRISPR-Cas antiviral defense mechanism. However, the variation in mechanisms and involved factors is amazing. RNases play also a key role in the control of mRNA stability and gene expression mediated by bacterial sRNAs (36) and host RNases are able to perform crucial functions in the maturation of CRISPR transcripts, too. For example, in Type II systems a trans-activating RNA (tracrRNA) together with the endogenous RNase III is the key enzyme for the maturation of crRNAs (37), while a CRISPR element in Listeria monocytogenes is processed by the endogenous polynucleotide phosphorylase (38). However, also the opposite situation exists, in which a native CRISPR-Cas system regulates the expression of an endogenous transcript encoding a bacterial lipoprotein requiring Cas9, together with tracrRNA and the scaRNA sRNA (39). These finding illustrate that it is worthwhile to study CRISPR-Cas systems of different subtypes and different organisms.
Here, we provide the first biochemical analysis of pre-crRNA processing by Cas6 proteins in cyanobacteria and in a subtype I-D CRISPR-Cas system. The enzyme, Cas6-1, is able to specifically process synthetic repeat RNA of its corresponding CRISPR1 repeat-spacer array in vitro, but also CRISPR2 RNA. However, when the cas6-2a gene, which is located upstream of the CRISPR2 array, is knocked out, CRISPR2 RNA accumulates to lengths mainly >200 nt (17). An RNA-seq analysis of a cas6-2a mutant confirmed that no repeat-specific processing of the CRISPR2 array occurs (Supplementary Figure S3). The implication of these results is that Cas6-1 does not process CRISPR2 transcripts in the absence of Cas6-2a in vivo, despite its observed in vitro cleavage activity. Strikingly, we observed a similar promiscuity in the ability of Cas6-2a to correctly process CRISPR1-derived transcripts in vitro, whereas it did not substitute the missing Cas6-1 activity in deletion mutants of cas6-1 in vivo (17). Interestingly, when analyzing the cleavage of only a single artificial repeat sequence in vitro, Cas6-1 could cleave CRISPR1 (oligonucleotide C1 in Figure 1), as did Cas6-2a with the CRISPR2 repeat substrate (oligonucleotide C2 in Figure 1). Conversely, Cas6-1 and Cas6-2a cleaved the single non-cognate repeats only inefficiently (Figure 1A). These substrate specificities are consistent with the specificity for the two enzymes for either CRISPR1 or CRISPR2 in vivo (17) and, in view of the identical secondary structures, must be caused by the differences in the respective repeat sequences. Therefore, it is a possibility that these enzymes possess a higher affinity to their cognate CRISPR transcripts in vivo, which thus would outcompete the non-cognate substrates. However, that does not explain the results of the genetic analyses when one of the respective endonuclease genes was deleted (17). Another explanation is that the cleavage of the respective precursor transcripts requires the specific binding of a further Cas protein that is assembled into a complex only with the correct endonuclease. The latter explanation fits well with the observed accumulation of longer CRISPR2 transcripts in absence of Cas6-2a (Supplementary Figure S3) that could be stabilized by the specific binding of a second Cas protein or a complex of proteins. Furthermore, the CRISPR1 and 2 in vitro transcript cleavage assays also confirmed that Cas6-1 and Cas6-2a are not sufficient to generate the CRISPR1 mature crRNA species detected in vivo (17): only fragments corresponding to intermediate crRNAs with a length of 72–76 nt were observed. Therefore, these intermediate crRNAs are expected to be processed in a second step into mature crRNAs by a so far unknown ribonuclease.
We noticed for both enzymes when incubating them with CRISPR repeat-spacer fragments of varying lengths that cleavage occurred in most repeat instances, but not in all (Figure 3). To shed light on the reasons why a repeat is cleaved or not cleaved depending on the position within a transcript, local secondary structure predictions were performed on the whole transcript VIII of CRISPR2 taking the influence of adjacent sequences into account. In Figure 5B we exemplify this for the repeat instances R6 and R7 and the cleavage behavior with Cas6-1: For the 5΄ part of fragment VIII we see long helical regions that form between repeat R6 and the preceding spacer sequence that obstruct the formation of the characteristic hairpin structure motif and cover the cleavage site within the repeat R6, making it inaccessible for the enzyme. Indeed, repeat instance R6 was not cleaved. In contrast, the functional hairpin motif is clearly formed the 3΄ part of fragment VIII and accessible for the enzyme, consistent with the cleavage of R7.
These findings can be generalized for all repeat instances. In Figure 4, the distribution for the accuracy of the local functional repeat motif from Figure 1D for cleaved and non-cleaved fragments, for CRISPR2, is shown. As can be clearly seen, the accuracy of the function local repeat structure is significantly lower for non-cleaved fragments. The reason for this low accuracy is a competition between the local functional repeat structure and competing stable stems, as shown for repeat R6 of fragment VIII in Figure 5B. To visualize this effect for all cleaved and non-cleaved artificial fragments, we averaged the dot plots (plus a context of 35 nt) of repeats that are cleaved or not cleaved in the artificial fragments of CRISPR1 and CRISPR2. In comparison, we observed that among uncleaved fragments (Figure 6B and D) there are many more base pairs in the surrounding context than among cleaved fragments (Figure 6A and C) and that the base pairing of the functional motif has a much higher average probability for the cleaved fragments (Figure 6B and D). We conclude that Cas6-1 and Cas6-2a are both enzymes that require the formation of the hairpin motif in repeats for substrate recognition.
We could successfully explain cleavage events by measuring the predicted structure stability of the hairpin motif in each repeat instance: high predicted stabilities led to a cleavage and low stabilities did not. Further computational investigation into these results showed that the sequence context (i.e. spacers) surrounding a repeat instance could form stable structures with the repeat sequence that sequester the formation of the functional hairpin motif. Despite each repeat instance always having the same adjacent spacers, some repeat instances were both cleaved and not cleaved, depending on the fragment length. This implies that long-range effects on the repeat structure exist that go beyond the directly adjacent spacer sequences.
In summary, we describe the dependency of Cas6-mediated cleavage on the RNA secondary structure by analyzing the cleavage patterns of nine CRISPR1 and nine CRISPR2 in vitro transcripts, varying in length and sequence, which revealed that a specific repeat is not necessarily always cleaved by Cas6-1 or Cas6-2a. A successful cleavage was furthermore influenced by the context of adjacent (and even more distantly located) sequences and was thereby dependent on secondary structure formation in the direct neighborhood of the repeat. The influence of surrounding sequences might lead to variations in crRNA abundances and should be taken into account when designing artificial CRISPR arrays.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Thomas Wallner for his support in the preparation of alkaline hydrolysis ladders and sequencing gels.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
German Research Foundation (DFG) program FOR1680 ‘Unravelling the Prokaryotic Immune System’ [HE 2544/8-2, BA 2168/5-2 to W.R.H., R.B.]. Funding for open access charge: Deutsche Forschungsgemeinschaft grants [HE 2544/8-2 and BA 2168/5-2].
Conflict of interest statement. None declared.
REFERENCES
- 1. Jansen R., van Embden J.D.A., Gaastra W., Schouls L.M.. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 2002; 43:1565–1575. [DOI] [PubMed] [Google Scholar]
- 2. Lange S.J., Alkhnbashi O.S., Rose D., Will S., Backofen R.. CRISPRmap: an automated classification of repeat conservation in prokaryotic adaptive immune systems. Nucleic Acids Res. 2013; 41:8034–8044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bhaya D., Davison M., Barrangou R.. CRISPR-Cas systems in Bacteria and Archaea: versatile small RNAs for adaptive defense and regulation. Annu. Rev. Genet. 2011; 45:273–297. [DOI] [PubMed] [Google Scholar]
- 4. Grissa I., Vergnaud G., Pourcel C.. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics. 2007; 8:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Makarova K.S., Wolf Y.I., Alkhnbashi O.S., Costa F., Shah S.A., Saunders S.J., Barrangou R., Brouns S.J.J., Charpentier E., Haft D.H. et al. An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 2015; 13:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Alkhnbashi O.S., Costa F., Shah S.A., Garrett R.A., Saunders S.J., Backofen R.. CRISPRstrand: predicting repeat orientations to determine the crRNA-encoding strand at CRISPR loci. Bioinformarmatics. 2014; 30:i489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brouns S.J.J., Jore M.M., Lundgren M., Westra E.R., Slijkhuis R.J.H., Snijders A.P.L., Dickman M.J., Makarova K.S., Koonin E.V., van der Oost J.. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008; 321:960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hale C., Kleppe K., Terns R.M., Terns M.P.. Prokaryotic silencing (psi)RNAs in Pyrococcus furiosus. RNA. 2008; 14:2572–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hale C.R., Zhao P., Olson S., Duff M.O., Graveley B.R., Wells L., Terns R.M., Terns M.P.. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009; 139:945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Karginov F.V., Hannon G.J.. The CRISPR system: small RNA-guided defense in Bacteria and Archaea. Mol. Cell. 2010; 37:7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Przybilski R., Richter C., Gristwood T., Clulow J.S., Vercoe R.B., Fineran P.C.. Csy4 is responsible for CRISPR RNA processing in Pectobacterium atrosepticum. RNA Biol. 2011; 8:517–528. [DOI] [PubMed] [Google Scholar]
- 12. Carte J., Wang R., Li H., Terns R.M., Terns M.P.. Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev. 2008; 22:3489–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nam K.H., Haitjema C., Liu X., Ding F., Wang H., DeLisa M.P., Ke A.. Cas5d protein processes pre-crRNA and assembles into a cascade-like interference complex in subtype I-C/Dvulg CRISPR-Cas system. Structure. 2012; 20:1574–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Punetha A., Sivathanu R., Anand B.. Active site plasticity enables metal-dependent tuning of Cas5d nuclease activity in CRISPR-Cas type I-C system. Nucleic Acids Res. 2014; 42:3846–3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hochstrasser M.L., Doudna J.A.. Cutting it close: CRISPR-associated endoribonuclease structure and function. Trends Biochem. Sci. 2015; 40:58–66. [DOI] [PubMed] [Google Scholar]
- 16. Cai F., Axen S.D., Kerfeld C.A.. Evidence for the widespread distribution of CRISPR-Cas system in the phylum Cyanobacteria. RNA Biol. 2013; 10:687–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Scholz I., Lange S.J., Hein S., Hess W.R., Backofen R.. CRISPR-Cas systems in the cyanobacterium Synechocystis sp. PCC6803 exhibit distinct processing pathways involving at least two Cas6 and a Cmr2 protein. PLoS One. 2013; 8:e56470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Makarova K.S., Haft D.H., Barrangou R., Brouns S.J.J., Charpentier E., Horvath P., Moineau S., Mojica F.J.M., Wolf Y.I., Yakunin A.F. et al. Evolution and classification of the CRISPR–Cas systems. Nat. Rev. Microbiol. 2011; 9:467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kopfmann S., Hess W.R.. Toxin antitoxin systems on the large defense plasmid pSYSA of Synechocystis sp. PCC6803. J. Biol. Chem. 2013; 288:7399–7409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kopfmann S., Roesch S.K., Hess W.R.. Type II toxin-antitoxin systems in the unicellular cyanobacterium Synechocystis sp. PCC 6803. Toxins. 2016; 8:E228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Richter H., Zoephel J., Schermuly J., Maticzka D., Backofen R., Randau L.. Characterization of CRISPR RNA processing in Clostridium thermocellum and Methanococcus maripaludis. Nucleic Acids Res. 2012; 40:9887–9896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Haurwitz R.E., Jinek M., Wiedenheft B., Zhou K., Doudna J.A.. Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science. 2010; 329:1355–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Haurwitz R.E., Sternberg S.H., Doudna J.A.. Csy4 relies on an unusual catalytic dyad to position and cleave CRISPR RNA. EMBO J. 2012; 31:2824–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lintner N.G., Kerou M., Brumfield S.K., Graham S., Liu H., Naismith J.H., Sdano M., Peng N., She Q., Copie V. et al. Structural and functional characterization of an archaeal clustered regularly interspaced short palindromic repeat (CRISPR)-associated complex for antiviral defense (CASCADE). J. Biol. Chem. 2011; 286:21643–21656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shao Y., Li H.. Recognition and cleavage of a nonstructured CRISPR RNA by its processing endoribonuclease Cas6. Structure. 2013; 21:385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sokolowski R.D., Graham S., White M.F.. Cas6 specificity and CRISPR RNA loading in a complex CRISPR-Cas system. Nucleic Acids Res. 2014; 42:6532–6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang R., Preamplume G., Terns M.P., Terns R.M., Li H.. Interaction of the Cas6 riboendonuclease with CRISPR RNAs: recognition and cleavage. Structure. 2011; 19:257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Plagens A., Tjaden B., Hagemann A., Randau L., Hensel R.. Characterization of the CRISPR/Cas subtype I-A system of the hyperthermophilic crenarchaeon Thermoproteus tenax. J. Bacteriol. 2012; 194:2491–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Plagens A., Tripp V., Daume M., Sharma K., Klingl A., Hrle A., Conti E., Urlaub H., Randau L.. In vitro assembly and activity of an archaeal CRISPR-Cas type I-A Cascade interference complex. Nucleic Acids Res. 2014; 42:5125–5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang J., Rouillon C., Kerou M., Reeks J., Brugger K., Graham S., Reimann J., Cannone G., Liu H., Albers S.-V. et al. Structure and mechanism of the CMR complex for CRISPR-mediated antiviral immunity. Mol. Cell. 2012; 45:303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bertani G. Studies on lysogenesis. J. Bacteriol. 1951; 62:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lange S.J., Maticzka D., Mohl M., Gagnon J.N., Brown C.M., Backofen R.. Global or local? Predicting secondary structure and accessibility in mRNAs. Nucleic Acids Res. 2012; 40:5215–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bernhart S.H., Hofacker I.L., Stadler P.F.. Local RNA base pairing probabilities in large sequences. Bioinformatics. 2006; 22:614–615. [DOI] [PubMed] [Google Scholar]
- 34. Hofacker I.L. Vienna RNA secondary structure server. Nucleic Acids Res. 2003; 31:3429–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Marraffini L.A., Sontheimer E.J.. CRISPR interference: RNA-directed adaptive immunity in Bacteria and Archaea. Nat. Rev. Genet. 2010; 11:181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Saramago M., Bárria C., Dos Santos R.F., Silva I.J., Pobre V., Domingues S., Andrade J.M., Viegas S.C., Arraiano C.M.. The role of RNases in the regulation of small RNAs. Curr. Opin. Microbiol. 2014; 18:105–115. [DOI] [PubMed] [Google Scholar]
- 37. Deltcheva E., Chylinski K., Sharma C.M., Gonzales K., Chao Y., Pirzada Z.A., Eckert M.R., Vogel J., Charpentier E.. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011; 471:602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sesto N., Touchon M., Andrade J.M., Kondo J., Rocha E.P.C., Arraiano C.M., Archambaud C., Westhof É., Romby P., Cossart P.. A PNPase dependent CRISPR System in Listeria. PLoS Genet. 2014; 10:e1004065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sampson T.R., Saroj S.D., Llewellyn A.C., Tzeng Y.-L., Weiss D.S.. A CRISPR/Cas system mediates bacterial innate immune evasion and virulence. Nature. 2013; 497:254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Darty K., Denise A., Ponty Y.. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics. 2009; 25:1974–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.