


Abstract
Module identification is a frequently used approach for mining local structures with more significance in global networks. Recently, a wide variety of bilayer networks are emerging to characterize the more complex biological processes. In the light of special topological properties of bilayer networks and the accompanying challenges, there is yet no effective method aiming at bilayer module identification to probe the modular organizations from the more inspiring bilayer networks. To this end, we proposed the pseudo-3D clustering algorithm, which starts from extracting initial non-hierarchically organized modules and then iteratively deciphers the hierarchical organization of modules according to a bottom-up strategy. Specifically, a modularity function for bilayer modules was proposed to facilitate the algorithm reporting the optimal partition that gives the most accurate characterization of the bilayer network. Simulation studies demonstrated its robustness and outperformance against alternative competing methods. Specific applications to both the soybean and human miRNA-gene bilayer networks demonstrated that the pseudo-3D clustering algorithm successfully identified the overlapping, hierarchically organized and highly cohesive bilayer modules. The analyses on topology, functional and human disease enrichment and the bilayer subnetwork involved in soybean fat biosynthesis provided both the theoretical and biological evidence supporting the effectiveness and robustness of pseudo-3D clustering algorithm.
INTRODUCTION
A biological network exhibits a modular organization. Module identification and analysis is one of the most frequently used approaches to exploring knowledge from complex biological networks. In parallel with the small world, scale-free property and other basic local and/or global characteristics, the modular structure dependent on functional module is of great significance in understanding the organization and dynamics of network functions. Modularization is a ubiquitous phenomenon in various network systems (1). A functional module, composed of many types of interacting molecules, is a discrete local structure whose members have more internal links among themselves than external links with members of other modules (2). Normally, a module's function is separable from those of other modules (3), and many cellular functions are carried out by modules (2). Modular organization has been observed in metabolic (4), transcriptional regulation (5) and protein–protein interaction (PPI) (6) networks. Moreover, the exploration of modular structure has been proposed as a key factor in understanding the complexity of biological systems (7). In the past decade, researchers have proposed a wide variety of module identification or network decomposition methods, which can be broadly classified into six major categories: traditional clustering algorithms (8–19), network topological approaches (20–28), modularity optimization (29–32), seed expansion (33–36), matrix decomposition/factorization (37–41) and comparative network analysis (42–45). A detailed review of the above categories is beyond the scope of this paper and has already been presented by Chen et al. (46).
Nevertheless, there is yet no effective method for bilayer module identification from the increasingly booming bilayer networks, such as gene-disease bilayer network (47,48), gene-phenotype bilayer network (49), drug-target bilayer network (50), miRNA-disease bilayer network (51) and the miRNA-gene bilayer networks that we presented in this paper. A bilayer network often consists of three types of linkages between two kinds of nodes. Specifically, a miRNA-gene bilayer network contains two types of biomolecules (miRNAs and genes) and three types of interacting associations (miRNA–miRNA and gene–gene functional interactions, as well as miRNA–gene regulating relations) (Figure 1A). Existing module identification methods seem to have reported good results for traditional monolayer networks, but in the light of the special topological properties of bilayer networks, there are still many challenges that need to be overcome for bilayer module identification. Consequently, this work aims to present a method (or algorithm) to identify bilayer modules from a bilayer network, with specific applications to the miRNA–gene bilayer networks from both plant and human.
Figure 1.

Bilayer module identification and pseudo-3D clustering. (A) The topological view of a toy miRNA–gene bilayer network; (B) The pseudo-3D view of bilayer module identification using clustering approach.
We constructed one bilayer network each of plant and human, respectively, by combining network topological information based on previously presented gene–gene interactions and miRNA–miRNA interactions, as well as targeting information introduced by miRNAs binding their targets (52–55). Clustering is commonly accepted as a powerful approach to partition the data items into a list of disjoint groups, such that the similarities within each group are maximized and those between different groups are minimized. For such a bilayer network, module identification means simultaneously clustering two kinds of objects and three types of links (Figure 1A). Without loss of generality, the miRNA–gene bilayer network can be mathematically expressed as three adjacency matrices: 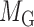 for gene–gene links,
for gene–gene links, 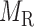 for miRNA–miRNA links and
for miRNA–miRNA links and 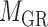 for miRNA–gene regulating relations. From the traditional clustering viewpoint, the bilayer module identification consists of two traditional one-way clustering processes (
for miRNA–gene regulating relations. From the traditional clustering viewpoint, the bilayer module identification consists of two traditional one-way clustering processes (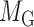 and
and 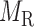 ) and one two-way clustering (biclustering) process (
) and one two-way clustering (biclustering) process (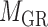 ), which can be organized as the pseudo 3-dimensional (pseudo-3D) view as shown in Figure 1B. Based on this concept, we proposed a method (or algorithm) called pseudo-3D clustering to achieve bilayer module identification, because it is neither a simple combination of traditional one- and two-way clustering methods, nor a real 3D clustering (or triclustering) method used in gene expression data (56).
), which can be organized as the pseudo 3-dimensional (pseudo-3D) view as shown in Figure 1B. Based on this concept, we proposed a method (or algorithm) called pseudo-3D clustering to achieve bilayer module identification, because it is neither a simple combination of traditional one- and two-way clustering methods, nor a real 3D clustering (or triclustering) method used in gene expression data (56).
In addition, the research reported in this paper has its roots in works which study the identification of miRNA–gene (or miRNA–mRNA) regulatory modules. Various methods were proposed to discover miRNA–gene regulatory modules (also named co-modules). However, the miRNA–gene regulatory module identification is different from the miRNA–gene bilayer module detection referred to herein; the former is either based only on the predicted miRNA–gene regulatory relations (57,58), or based only on the (anti-) expression correlations between miRNAs and genes (59–61), or at best incorporates the gene–gene relationships inferred from protein–protein interactions (62). However, the latter aims to respectively clustering the miRNAs and genes in two connecting layers based on three types of relations (Figure 1A). Zhang et al. (62) and Pio et al. (58) have given the detailed reviews of existing methods. Furthermore, specifically for a biological application, the bilayer module identification method has to consider a number of other important issues, of which the most critical is that the resulting bilayer modules should meet the following conditions:
Module overlap, since genes and miRNAs can be involved in multiple biological processes for the pleiotropy. Generally speaking, module overlaps show that nodes or links may belong to two or more modules. Several common clustering algorithms, including CFinder (12), MCL (63), MCODE (15), DetMod (11), ClusterONE (64) and MINE (65) permit overlaps between the modules. The pseudo-3D clustering algorithm will identify the overlaps either on gene layer or miRNA layer.
Hierarchical organization, a fundamental characteristic of many complex networks, implies that small groups of nodes organize in a hierarchical manner into increasingly large groups (66,67). A module at a higher level should contain multiple modules of lower levels. Some algorithms, like hierarchical clustering, can destruct this kind of organization by tuning a cutoff. Hierarchical modules allow studying the intermolecular interactions of different granularity. Bilayer modules should also be hierarchically organized.
High cohesiveness, which means the dense connections within the module, but only sparse connections between different modules. Specific to miRNA–gene bilayer modules, high cohesiveness means that the genes and miRNAs in the same bilayer module should be closely linked in respective layer and show strong regulation from miRNA layer to gene layer.
Taking into account all above considerations, we propose a method, named pseudo-3D clustering algorithm that provides a solution to the issues raised up by the specific module identification task for the mushrooming bilayer biological networks. The pseudo-3D clustering algorithm is demonstrated to be able to identify the overlapping, hierarchically organized and highly cohesive bilayer modules from, but not limited to, the miRNA–gene bilayer networks and further reveal their implications to specific biological process, such as soybean fatty acid synthesis.
MATERIALS AND METHODS
The mathematical representation of bilayer network and necessary definitions
To be algorithm-friendly, some useful definitions are necessary. Figure 2 provides the workflow for construction and mathematical representation of a bilayer network, taking the soybean miRNA–gene bilayer network for instance. In previous studies, we have reconstructed four functional gene networks (FGN) and four miRNA functional networks (miRFN) of soybean (Glycine max), respectively (52,53). Integrating the predicted regulating relations between miRNAs and their target genes, we can easily consolidate a FGN and a miRFN to form a bilayer network such as shown in Figure 1A. For simplicity, we only integrate the most inclusive FGN and miRFN (i.e. FGN–INT and miRFN–INT) without considering all other possible combinations between four FGNs and four miRFNs. The brief statistics of the final bilayer network and the component networks are shown in Supplementary Table S1.
Figure 2.

The construction and mathematical representation of the miRNA–gene bilayer network.
Let 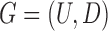 and
and 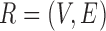 be the FGN and miRFN, where
be the FGN and miRFN, where 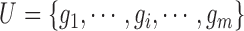 and
and 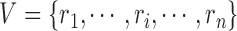 are node sets representing
are node sets representing 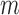 genes and
genes and 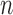 miRNAs, respectively;
miRNAs, respectively; 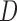 and
and 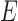 denote edges corresponding to respective gene–gene and miRNA–miRNA interactions. The topology structures can be mathematically captured by their corresponding adjacency matrices
denote edges corresponding to respective gene–gene and miRNA–miRNA interactions. The topology structures can be mathematically captured by their corresponding adjacency matrices 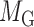 and
and 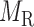 :
:
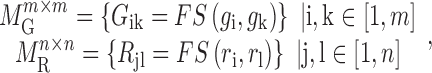 |
(1) |
where 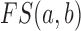 is the functional similarity of the genes (or miRNAs)
is the functional similarity of the genes (or miRNAs) 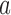 and
and 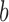 .
.
Let 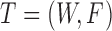 be the miRNA–gene bipartite network, where
be the miRNA–gene bipartite network, where 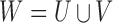 is the union of genes in FGN and miRNAs in miRFN;
is the union of genes in FGN and miRNAs in miRFN; 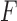 denotes the edge set that with each edge having exact one end vertex in
denotes the edge set that with each edge having exact one end vertex in 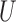 and the other end vertex in
and the other end vertex in 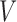 , corresponding to the regulating relations between miRNAs and their target genes. Homoplastically, the topology structure can be mathematically captured by its corresponding adjacency matrix
, corresponding to the regulating relations between miRNAs and their target genes. Homoplastically, the topology structure can be mathematically captured by its corresponding adjacency matrix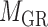 :
:
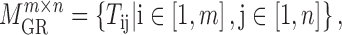 |
(2) |
where 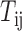 is the regulatory strength value for a given miRNA–gene targeting relation (
is the regulatory strength value for a given miRNA–gene targeting relation ( ), which was defined in our previous work (52).
), which was defined in our previous work (52).
Therefore, we define the bilayer miRNA–gene network 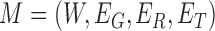 by integrating the three networks
by integrating the three networks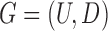 ,
, 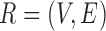 and
and 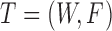 , which share the same set of nodes
, which share the same set of nodes 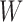 . The bilayer network
. The bilayer network 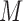 has three types of interactions, where
has three types of interactions, where 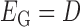 represents the gene-gene interactions within the FGN,
represents the gene-gene interactions within the FGN, 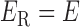 represents the miRNA–miRNA interactions within miRFN and
represents the miRNA–miRNA interactions within miRFN and 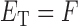 the miRNA-gene targeting information. The topology structure of this bilayer network can be mathematically captured by the block adjacency matrix
the miRNA-gene targeting information. The topology structure of this bilayer network can be mathematically captured by the block adjacency matrix 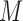 :
:
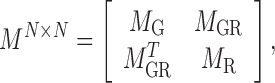 |
(3) |
where 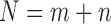 . As an example shown in Supplementary Table S1,
. As an example shown in Supplementary Table S1, 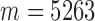 and
and 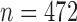 for soybean bilayer network. In the later actual use, the matrix
for soybean bilayer network. In the later actual use, the matrix  is also considered to consist of
is also considered to consist of  row vectors for genes and
row vectors for genes and 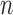 row vectors for miRNAs, namely
row vectors for miRNAs, namely
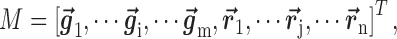 |
(4) |
where each gene (or miRNA) in the bilayer network is represented as a row vector in a (m+n)-dimension space, i.e.
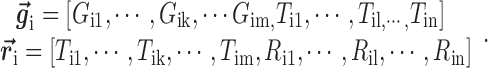 |
(5) |
Let 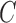 denotes a bilayer module, which is defined in this research as a bilayer sub-network that consists of two objects (genes and miRNAs) and three inter-object relations. Subsequently, a bilayer module containing p (≤ m) genes and q (≤ n) miRNAs can also be represented by a sub-matrix with p + q row vectors in the (m+n)-dimension space. To be noted that, in order to facilitate the description of the psedo-3D algorithm, hereafter a bilayer module
denotes a bilayer module, which is defined in this research as a bilayer sub-network that consists of two objects (genes and miRNAs) and three inter-object relations. Subsequently, a bilayer module containing p (≤ m) genes and q (≤ n) miRNAs can also be represented by a sub-matrix with p + q row vectors in the (m+n)-dimension space. To be noted that, in order to facilitate the description of the psedo-3D algorithm, hereafter a bilayer module 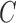 refers specifically to a collection of genes and miRNAs (also known as a bicluster), and does not include the relations between them. Nevertheless, it will not hamper the identification of bilayer modules defined hereinbefore, since each one of these biclusters will be easily reverted to a previously defined module with bilayer topology by decoding the relations from the matrix M. Based on this specific definition of a bilayer module
refers specifically to a collection of genes and miRNAs (also known as a bicluster), and does not include the relations between them. Nevertheless, it will not hamper the identification of bilayer modules defined hereinbefore, since each one of these biclusters will be easily reverted to a previously defined module with bilayer topology by decoding the relations from the matrix M. Based on this specific definition of a bilayer module 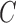 , we use Cg = U ∩ C to denote the gene set of the module, and Cr = V ∩ C to denote the miRNA set.
, we use Cg = U ∩ C to denote the gene set of the module, and Cr = V ∩ C to denote the miRNA set.
The problem description of bilayer module identification in miRNA–gene bilayer network
As aforementioned, our aim in this work is to identify the bilayer network modules with the characteristics of overlap, hierarchy and high cohesiveness. Based on the mathematical representations and definitions, the bilayer module identification problem raised in this research can be described as follows:
Given:
the set of genes
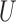 and the set of miRNAs
and the set of miRNAs 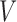 ;
;the block adjacency matrix
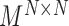 ;
;a cohesiveness function
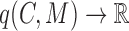 ,
,  is a bilayer module;
is a bilayer module;a cohesiveness threshold
 for
for 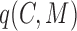 ;
;a modularity function
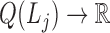 ,
, 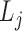 is the j-th level bilayer modules.
is the j-th level bilayer modules.
Find: a list of bilayer modules 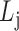 , for each level
, for each level 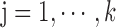 such that:
such that:
for each list
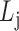 ,
, 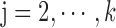 ,
, 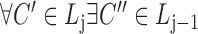 satisfies
satisfies 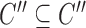 (hierarchy);
(hierarchy);for each list
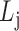 ,
, 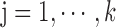 ,
, 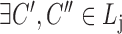 satisfies
satisfies 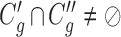 or
or 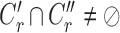 (overlapping);
(overlapping);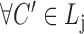 ,
, 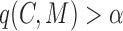 (cohesiveness);
(cohesiveness);the optimal level of bilayer modules
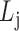 according to
according to 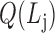 .
.
Pseudo-3D clustering algorithm
The pseudo-3D clustering starts from extracting initial non-hierarchically organized modules and then iteratively decipher the hierarchical organization of modules according to a bottom-up strategy. Based on the above descriptions, we here describe the pseudo-3D clustering algorithm in high-level as Supplementary Figure S1 and following subsections. Supplementary Method M1 provides the full idea and detailed descriptions of the algorithm.
Initial module identification
The pseudo-3D clustering algorithm starts from extraction of a set of initial non-hierarchically organized bilayer modules, namely the lowest-level module partition (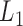 ). Each initial module is a primary bicluster aggregated from bicliques (Figure 3A, left), which are subgraphs extracted from the miRNA-gene bipartite network in two directions, i.e. miRNA-to-gene and gene-to-miRNA. The algorithm starts the identification by extracting a set of initial bicliques and then iteratively aggregates two bicliques into a new one (Figure 3A). The iteration will stop when there are no candidates for aggregation. The resulting initial modules derived from both in miRNA-to-gene and gene-to-miRNA direction are combined to form
). Each initial module is a primary bicluster aggregated from bicliques (Figure 3A, left), which are subgraphs extracted from the miRNA-gene bipartite network in two directions, i.e. miRNA-to-gene and gene-to-miRNA. The algorithm starts the identification by extracting a set of initial bicliques and then iteratively aggregates two bicliques into a new one (Figure 3A). The iteration will stop when there are no candidates for aggregation. The resulting initial modules derived from both in miRNA-to-gene and gene-to-miRNA direction are combined to form 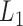 level modules, by simply removing modules that appear more than once and those that are a subset of others. The bidirectional biclique extraction and aggregation process not only cluster the miRNA–gene regulating relations, but also the interactions of miRNA–miRNA and gene–gene, so that it realize the simultaneous clustering of two kinds of objects (genes and miRNA) and their three types of interrelationships, namely the original idea of a pseudo-3D clustering algorithm.
level modules, by simply removing modules that appear more than once and those that are a subset of others. The bidirectional biclique extraction and aggregation process not only cluster the miRNA–gene regulating relations, but also the interactions of miRNA–miRNA and gene–gene, so that it realize the simultaneous clustering of two kinds of objects (genes and miRNA) and their three types of interrelationships, namely the original idea of a pseudo-3D clustering algorithm.
Figure 3.

The schematic view of the biclique aggregation and module merging. (A)The initial bicliques and the process of biclique aggregation (miRNA-to-gene direction); (B) An example of the object distribution of two candidate gene clusters for merging. (C) A toy example of the modularity curve and the corresponding optimal hierarchy level (red arrow).
Iteration of overlap detection and module merging
Overlap detection means to determine whether some objects (miRNAs or genes) belonging to a module appear in another module of the same level. However, not all pairs of overlapping bilayer modules will be merged subsequently. Module merging processes merge the overlapping modules when the spheres in gene (or miRNA) space of two merging candidates are close enough with their distance less than a heuristic threshold (Figure 3B). At each iteration, several pairs of modules can be merged, and an additional level of the hierarchy may or may not be added depending on whether merging is performed. Obviously, a module could be the merging candidate with more than one other module. In order to obtain a result independent of the order in which pairs of modules are considered, merging is actually performed after finding out all merging candidates and guarantees to maximize the cohesiveness of the resulting modules. The iteration will stop when neither overlaps nor merges are performed in the last iteration.
Determination of the optimal module partition
The iteration of overlap detection and merging will produce the partitions of a bilayer network in different levels. We need to further determine the optimal level at which the partition gives a more accurate characterization of the modular organization of the bilayer network. We here, based on the modularity function proposed by Newman (30) specifically for evaluation of a monolayer network partition, give a modularity function for the bilayer network partition. According to the modularity value, the optimal partition is specified as the level that produces the maximum modularity value across all hierarchy levels (Figure 3C).
Time complexity
The time complexity of the pseudo-3D algorithm depends on the time complexity of each single step. The initial module identification occupies a complexity of 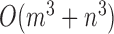 in two directions. One iteration of overlap detection, merging and computing the
in two directions. One iteration of overlap detection, merging and computing the 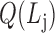 value occupies
value occupies 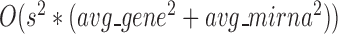 , where
, where 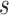 denotes the number of modules. So, for
denotes the number of modules. So, for 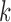 levels of partitions with maximum
levels of partitions with maximum 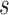 modules at each level, the pseudo-3D algorithm will occupy a total complexity of
modules at each level, the pseudo-3D algorithm will occupy a total complexity of 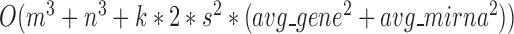 ; it will be reduced to
; it will be reduced to 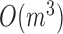 in the most cases that
in the most cases that 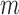 (the number of genes in the bilayer network) is significantly larger than the other parameters. A detailed analysis of time complexity is provided in Supplementary Methods M1.
(the number of genes in the bilayer network) is significantly larger than the other parameters. A detailed analysis of time complexity is provided in Supplementary Methods M1.
RESULTS AND DISCUSSION
Simulation and performance evaluation
To explore the advantages of pseudo-3D clustering algorithm in terms of graph theory before the specific biological applications, we performed the comparison with other network clustering strategies using a stimulated bilayer network (Supplementary Methods, M2). One well-defined bilayer miRNA–gene network with extremely modular structure was simulated (Supplementary Figure S4A, B and Table S1) and three clustering strategies were used for comparison: (i) clustering the gene network and miRNA network separately and match them together via miRNA–gene links (‘Match’); (ii) Combining the three networks as one network and doing clustering (‘Flatten’); (iii) clustering the bilayer network using R-NMTF (68), a method was developed to co-cluster phenotypes and genes, which are also organized as a bilayer network. The performance was evaluated by the extent, to what a clustering method can recover the maintained bilayer modules from a randomly perturbed bilayer network. Based on 1000 perturbations, the pseudo-3D clustering outperforms all three other strategies significantly (P-value < 2.2e-16, Kolmogorov–Smirnov test), followed by R-NMTF (Figure 4A and Supplementary Figure S4C), and the Flatten gets the worst performance. This indicates that, for a bilayer network, the connections between two layers are important to get better module detection and that simultaneously clustering on three types of edges between two types of nodes will reinforce the clustering performance. The superior performance than another bilayer-like module detection method, R-NMTF, indicates the outstanding role of pseudo-3D clustering algorithm specifically for module identification of bilayer networks.
Figure 4.

Pseudo-3D clustering algorithm outperforms other methods based on simulation. (A) The cumulative proportion of recovered modules for each methods based on a simulated bilayer network. (B–F) The performance of each method regarding different node numbers, cluster densities and degree distributions. The (B and C) average degrees and (F) cluster coefficients are of the whole bilayer networks, while the (D and E) node numbers are of the individual bilayer modules.
To investigate how the network density and other relevant topological properties affect the clustering performances and further verify the robustness of pseudo-3D clustering algorithm with respect to the node numbers, cluster densities and degree distributions, we generated 10 simulated miRNA–gene bilayer networks; each bilayer network is composed of 100 well-defined bilayer modules and possess the combination of different node numbers, cluster coefficients and degree distributions (Supplementary Method M2, Supplementary Table S2). Simulation results were provided as boxplots in Supplementary Table S2, shown as the average recovery scores of all four clustering strategies varying coordinately with the nodes number, degree distribution and cluster coefficient, with the same trend that pseudo-3D outperforms other three methods (Figure 4B–F). Specifically, the average degree of the network will facilitate the clustering when the average degree is more than 5 for gene layer and more than 3 for miRNA layer, which indicates that a higher density will produce better performance for all methods (Figure 4B and C). Nevertheless, the absolute node number (either genes or miRNAs) does not contribute consistently to the performance of clustering (Figure 4D and E). Additionally, we found a significantly consistent correlation between the cluster coefficients and recovery scores, indicating that cluster coefficient is the most important determining factor for clustering. This is in line with the expectation that a well modularized network has a larger cluster coefficient and is easier to be partitioned into clusters (modules). Moreover, extremely high cluster coefficient reduces the difference in performance between pseudo-3D and R-NMTF rather than between these two bilayer clustering methods and the other two conventional monolayer clustering methods (Flatten and Match), indicating that considering bilayer structure will improve the clustering performance. Taken together, the simulation studies based on either one benchmark or a series of bilayer networks of the combination of different cluster densities and degree distributions, demonstrated outperformance and robustness of the pseudo-3D clustering method.
The bilayer module identification of the soybean miRNA–gene network
We then applied pseudo-3D clustering to the soybean miRNA–gene network. In the phase of initial module identification, we obtained 472 initial bicliques for ‘miRNA-to-gene’ direction and 5263 for ‘gene-to-miRNA’ direction. After aggregation, all bicliques were consolidated into 300 and 2823 initial modules, respectively. A pruning process was performed to incorporate these two sets of initial modules and generate the first level partition of the miRNA–gene bilayer network, i.e.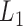 , which contains 1497 modules. The modules at
, which contains 1497 modules. The modules at 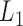 have 83 genes and 6 miRNAs in average, with the average clustering coefficient (0.7363, see Supplementary Table S3) is much higher than the global bilayer network (0.696, see Supplementary Table S1).
have 83 genes and 6 miRNAs in average, with the average clustering coefficient (0.7363, see Supplementary Table S3) is much higher than the global bilayer network (0.696, see Supplementary Table S1).
Taking 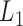 as the input of the iteration for overlap detection and merging, we got another 8 upper levels of module partitions, i.e.
as the input of the iteration for overlap detection and merging, we got another 8 upper levels of module partitions, i.e. 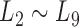 . In other words, the soybean miRNA–gene bilayer network was partitioned into modules that hierarchically organized at 9 different levels. The compositions and main topological properties of the 9 levels of bilayer modules are given in Supplementary Table S3. It shows that, with the increasing of module level from
. In other words, the soybean miRNA–gene bilayer network was partitioned into modules that hierarchically organized at 9 different levels. The compositions and main topological properties of the 9 levels of bilayer modules are given in Supplementary Table S3. It shows that, with the increasing of module level from 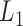 to
to 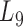 , the average module size increases, the number of modules decreases and the average cohesiveness diminishes, which are consistent with the theoretical characterizations of merging modules into larger ones. However, the average clustering coefficient of the modules at different levels have no significant increasing or decreasing trend (with the variance of 0.000346), indicating that all modules identified by pseudo-3D clustering algorithm are valid. In addition, the average clustering coefficients of modules at all levels (0.7507) are larger than that of the global miRNA–gene bilayer network (0.696, Supplementary Table S1), further illustrating the effectiveness of module partition at each level.
, the average module size increases, the number of modules decreases and the average cohesiveness diminishes, which are consistent with the theoretical characterizations of merging modules into larger ones. However, the average clustering coefficient of the modules at different levels have no significant increasing or decreasing trend (with the variance of 0.000346), indicating that all modules identified by pseudo-3D clustering algorithm are valid. In addition, the average clustering coefficients of modules at all levels (0.7507) are larger than that of the global miRNA–gene bilayer network (0.696, Supplementary Table S1), further illustrating the effectiveness of module partition at each level.
Based on the aforementioned definition, the maximum modularity corresponds to the best module partition. Therefore, 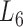 (in bold font, Supplementary Table S3) is the optimal level of module partitions. In the subsequent sections,
(in bold font, Supplementary Table S3) is the optimal level of module partitions. In the subsequent sections, 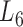 are used to perform deep analyses and discussions on the module topologies, functional enrichments and biological experimental evidences, aiming at getting the insight into the regulatory characteristics between the soybean miRNAs and their target genes at module level and the further evaluation of the effectiveness of the pseudo-3D clustering algorithm.
are used to perform deep analyses and discussions on the module topologies, functional enrichments and biological experimental evidences, aiming at getting the insight into the regulatory characteristics between the soybean miRNAs and their target genes at module level and the further evaluation of the effectiveness of the pseudo-3D clustering algorithm.
The topological analysis of the soybean bilayer modules
At the 6th level (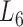 ), the soybean miRNA–gene bilayer network is partitioned into 100 overlapping bilayer modules.
), the soybean miRNA–gene bilayer network is partitioned into 100 overlapping bilayer modules. 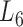 has an average of 333 genes and 72 miRNAs per module, with the average cohesiveness of 0.457 and the average clustering coefficient of 0.7464. Supplementary file 2 (sheet 1) provides the summary statistics of the 100 modules at
has an average of 333 genes and 72 miRNAs per module, with the average cohesiveness of 0.457 and the average clustering coefficient of 0.7464. Supplementary file 2 (sheet 1) provides the summary statistics of the 100 modules at 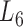 . In average, each miRNA targets 9.3 intra-module genes (i.e.
. In average, each miRNA targets 9.3 intra-module genes (i.e. 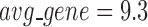 ), and each gene is targeted by 1.5 intra-module miRNAs (i.e.
), and each gene is targeted by 1.5 intra-module miRNAs (i.e. 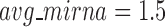 ).
).
Firstly, we analyzed the variation of the clustering coefficients of 100 modules at 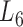 . Shown in Figure 5A and Supplementary File 2 (sheet 1), the most of clustering coefficients vary insignificantly from 0.7 to 0.8 with the average of 0.7464 and the variance of 0.006. To compare, we built a set of 100 randomized modules maintaining as same node numbers and degree distributions for each module as those at L6 by edge perturbations. In contrast, the clustering coefficients of the 100 randomized modules vary significantly from 0.01 to 0.65 with a lower average of 0.2080 and a larger variance of 0.022. There was additional significant difference between the two sets of modules regarding the clustering coefficients (P-value = 1.2e-81 by ANOVA). The above results show that the nodes (genes or miRNAs) of the modules at L6 are connected more closely than those of the randomized ones, which implies the modular characterization of the genes and/or miRNAs in functionality.
. Shown in Figure 5A and Supplementary File 2 (sheet 1), the most of clustering coefficients vary insignificantly from 0.7 to 0.8 with the average of 0.7464 and the variance of 0.006. To compare, we built a set of 100 randomized modules maintaining as same node numbers and degree distributions for each module as those at L6 by edge perturbations. In contrast, the clustering coefficients of the 100 randomized modules vary significantly from 0.01 to 0.65 with a lower average of 0.2080 and a larger variance of 0.022. There was additional significant difference between the two sets of modules regarding the clustering coefficients (P-value = 1.2e-81 by ANOVA). The above results show that the nodes (genes or miRNAs) of the modules at L6 are connected more closely than those of the randomized ones, which implies the modular characterization of the genes and/or miRNAs in functionality.
Figure 5.

Topological and functional analysis on the optimally partitioned bilayer modules. (A) The clustering coefficient distribution of the 100 modules at L6. Horizontal lines indicate the average cluster coefficients. (B) The statistics for GO functional enrichment of the 100 modules at L6. Inset pies show the percentage of enriched (color filled) and unenriched (blue filled) modules.
Secondly, based on the conclusion of our previous research (52) that the miRNAs in the same family or in the same cluster share a larger functional similarities than randomly selected miRNAs, we investigated whether the miRNAs in the same family or in the same cluster tend to be clustered in the same module. For the 81 miRNA families, 76 families (94%) have more than half of their member miRNAs been clustered in same modules, of which 50 families (62%) are completely clustered in same modules. For the 50 families, half of them are completely clustered in more than one module, with the maximum of 15 modules. These suggest that the miRNAs in the same family tend to participate in forming the same functional module and have pleiotropy.
For the 59 miRNA clusters, 50 clusters (85%) have more than half of their members been clustered in same modules, of which 39 clusters (66%) are completely clustered in same modules. This suggests a similar conclusion to miRNA family that the miRNAs located in the same cluster tend to be a same functional module. However, we also found a significant difference between them. That is only three of 59 miRNA clusters are completely clustered in more than one module, wherein only one cluster is completely in maximum 3 modules, another two are completely in 2 modules. It suggests that the miRNAs in the same cluster do not exhibit characterization of pleiotropy as the miRNA in the same family. We speculated on the reason for the difference is that miRNA regulates its target genes by base-pared binding the UTR (untranslated region) of the mRNA, so that the pleiotropy of a set of miRNAs depends more on whether they share the same seed sequences rather than the locations in the genome, because a miRNA family always shares a same seed sequence.
Functional enrichment analysis and a case study in soybean fatty acid synthesis
Enrichment analysis is a computational method that widely used to determine whether an a priori defined set of genes shows statistically significant, concordant differences between two biological states (e.g. Gene Ontology) (69). To estimate whether the genes are significantly enriched in a module extracted by pseudo-3D clustering algorithm, we performed functional enrichment analysis on 100 bilayer modules at 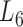 based on Gene ontology (GO) annotation. Except four modules (i.e. #1, #2, #7 and #11), the rest 96 modules are significantly enriched in three aspects of GO, i.e. biological process (BP), molecular function (MF) and cellular component (CC) (see Figure 5B). In details, 96 modules are enriched in BP, 89 modules in MF and 83 modules in CC, covering an average of 91% genes of each module (#34 module has the maximum coverage of 99.435%). The online database SoyFN (70) allows the retrieval of detailed enrichment results for all modules (http://nclab.hit.edu.cn/SoyFN/SoyModule.php).
based on Gene ontology (GO) annotation. Except four modules (i.e. #1, #2, #7 and #11), the rest 96 modules are significantly enriched in three aspects of GO, i.e. biological process (BP), molecular function (MF) and cellular component (CC) (see Figure 5B). In details, 96 modules are enriched in BP, 89 modules in MF and 83 modules in CC, covering an average of 91% genes of each module (#34 module has the maximum coverage of 99.435%). The online database SoyFN (70) allows the retrieval of detailed enrichment results for all modules (http://nclab.hit.edu.cn/SoyFN/SoyModule.php).
Soybean is the largest oilseed crop produced and consumed worldwide, accounting for 56% of the world oilseed production (SoyStats 2014, http://soystats.com). In this subsection, therefore, we take soybean oil (fat) synthesis as an example to investigate whether and/or how the bilayer modules produced by pseudo-3D clustering algorithm can characterize a real biological process. In order to get the genes and miRNAs involved in fat synthesis, we searched in four public databases and consulted seven literatures (Supplementary Methods, M3). As a result, we got 177 genes that encode 24 key enzymes and 102 miRNA types (see Supplementary Methods M3 for ‘miRNA type’ definition), which are also represented as Supplementary Figure S5 according to the three stages of fat synthesis.
We firstly analyzed the distribution of 177 genes encoding 23 key enzymes (except MCMT, EC:2.3.1.39) in the 100 bilayer modules at L6. The results are provided as a matching matrix shown in Supplementary Figure S6 and show that the enzyme encoding genes of glycolysis and fatty acid synthesis are significantly concentrated in the different functional modules, indicating that the genes in the modules partitioned by pseudo-3D clustering algorithm have a higher correlation in function. Exceptionally, the genes that encode the enzymes involved in fatty acid modification and TAG assembly (the third stage of the fat synthesis) are not matched to any modules. We think the possible reasons are: (i) compared with such basic metabolic processes as glycolysis, the research of this process was not thorough enough, accumulating less functional annotation data of the related genes; (ii) there are relatively less enzyme encoding genes involved in this process. We also found that different modules can gather the same enzyme encoding gene (set), which is the evidence that the functional modules identified by pseudo-3D clustering algorithm are overlapping.
Secondly, using the 177 genes and 102 miRNA types, we extracted a subnetwork regarding fat synthesis from the global soybean miRNA–gene bilayer network. As shown in Figure 6, the subnetwork contains 380 edges among 175 genes (gene–gene), 553 edges among 102 miRNA types (miRNA–miRNA) and 242 edges between 102 miRNAs and 68 their target genes (miRNA–gene). Supplementary file 3 provides the detailed information of the sub network, which can be recreated by imported into Cytoscape 3.3.0 (71). Supplementary Table S4 provides the detailed statistics of the global topological properties of this subnetwork.
Figure 6.

A graphic view of the miRNA–gene bilayer subnetwork for soybean fat synthesis. All abbreviations of the enzyme names are given in Supplementary Figure S5.
As apparent from Supplementary Table S4, in the miRNA–gene bilayer subnetwork for soybean fat synthesis, there are relatively sparse connections among genes with smaller clustering coefficient (0.333) and network density (0.025), but dense connections among miRNAs with larger clustering coefficient (0.528) and density (0.107). This indicates that the genes, as the enzyme encoding units, are relatively more homogeneous in function, while the miRNAs, as the post-transcriptional regulators, exhibit multifunctional and synergistic characteristics. Figure 6 shows that genes encoding the same enzyme (in red font) themselves constitute a spokewise module with one or two genes as the hub (red nodes). The hub genes play more critical roles in encoding corresponding enzymes and should be the preferential candidates for biological verification. Also the spokewise topology results in the small clustering coefficient and density. The spokewise modules are not completely independent. The modules encoding the same type of enzymes, such as the modules PFK, HK, PGK and PK, are connected to form a complete module network (the left of lower layer, Figure 6).
Application to human miRNA–gene bilayer network reveals the consistent robustness of pseudo-3D clustering
We further employed a more informative human bilayer network to better illustrate the usefulness and biological implications of pseudo-3D clustering algorithm. The human bilayer network was constructed based on the public available data sources in the context of disease (54,55,72) (Supplementary Methods M4) that contains much more biological information than that from an unpopular plant, soybean. Pseudo-3D clustering algorithm clustered this human bilayer network at 14 different hierarchies, with the 14th level (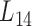 ) being the optimal partition based on the modularity measure (Supplementary Table S5). At
) being the optimal partition based on the modularity measure (Supplementary Table S5). At 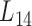 , the human miRNA–gene bilayer network is partitioned into 37 overlapping bilayer modules. L14 has an average of 696 genes and 158 miRNAs per module, with the average cohesiveness of 0.4404 and the average clustering coefficient of 0.4343. Supplementary File 2 (sheet 2) provides the summary statistics of the 37 modules at L14. In average, each miRNA targets 8.9 intra-module genes (i.e.
, the human miRNA–gene bilayer network is partitioned into 37 overlapping bilayer modules. L14 has an average of 696 genes and 158 miRNAs per module, with the average cohesiveness of 0.4404 and the average clustering coefficient of 0.4343. Supplementary File 2 (sheet 2) provides the summary statistics of the 37 modules at L14. In average, each miRNA targets 8.9 intra-module genes (i.e. 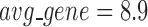 ), and each gene is targeted by 2.0 intra-module miRNAs (i.e.
), and each gene is targeted by 2.0 intra-module miRNAs (i.e. 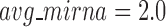 ).
).
To further illustrate how the pseudo-3D clustering algorithm groups the human bilayer network consistently with the functional categories, we first performed GO enrichment analysis (biological process) on the gene set of each module, separately, to investigate the enriched functions. The top ten enriched GO terms were merged to compare the functional similarity of pair-wise modules (Supplementary Methods M4 and Supplementary File 4). Then the hierarchical clustering was performed based on functional enrichment and pseudo-3D partitions to compare their consistency. We found that all modules were completely clustered into the same three clusters based on these two types of independent information (Supplementary Figure S7). A quantitative score, named consensus score, was defined to indicate this consistency (Supplementary Methods M4). The results shows that pseudo-3D clustering algorithm can produce a bilayer network partition with a significantly high consistency with the functional enrichment (consensus score = 1.0, P-value < 2.2e-16).
Additionally, enrichment analysis of human diseases on all 37 modules shows additional evidence indicating the robustness of pseudo-3D algorithm (Supplementary Methods M4). We found that only a small part of 37 modules significantly enriched in a sub group of diseases, therein most of these disease-related modules are extremely enriched in one or a limited diseases, such as module 31 (m31) is significantly related to breast cancer (Supplementary Figure S8A). Hierarchical clustering based on disease enrichment also shows a significantly high consistency with the pseudo-3D partitioned hierarchies (consensus score = 1.0, P-value < 2.2e-16) (Supplementary Figure S8B).
To sum up, the pseudo-3D clustering algorithm is demonstrated to successfully cluster the bilayer networks from both the plant and human into hierarchically organized and overlapping bilayer modules with high topological cohesiveness. Furthermore, functional enrichment analyses, as well as the biological evidence derived from database retrieval and literature collection, proved its excellent performance in identifying bilayer modules with functional consistency from a bilayer network. Additionally, the pseudo-3D clustering algorithm has been implemented as a runnable JAR file for public download and application to other types of bilayer networks. The manual and JAR file can be accessed at http://nclab.hit.edu.cn/SoyFN/SoyModule.php.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank the editors and reviewers for their valuable comments and suggestions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [61571163, 61532014, 61402132, 91335112 and 61271346]. Funding for open access charge: National Natural Science Foundation of China [61571163].
Conflict of interest statement. None declared.
REFERENCES
- 1. Lorenz D.M., Jeng A., Deem M.W.. The emergence of modularity in biological systems. Phys. Life Rev. 2011; 8:129–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hartwell L.H., Hopfield J.J., Leibler S., Murray A.W.. From molecular to modular cell biology. Nature. 1999; 402:C47–C52. [DOI] [PubMed] [Google Scholar]
- 3. Tornow S., Mewes H.W.. Functional modules by relating protein interaction networks and gene expression. Nucleic Acids Res. 2003; 31:6283–6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ravasz E., Somera A.L., Mongru D.A., Oltvai Z.N., Barabasi A.L.. Hierarchical organization of modularity in metabolic networks. Science. 2002; 297:1551–1555. [DOI] [PubMed] [Google Scholar]
- 5. Ihmels J., Friedlander G., Bergmann S., Sarig O., Ziv Y., Barkai N.. Revealing modular organization in the yeast transcriptional network. Nat. Genet. 2002; 31:370–377. [DOI] [PubMed] [Google Scholar]
- 6. Han J.D.J., Bertin N., Hao T., Goldberg D.S., Berriz G.F., Zhang L.V., Dupuy D., Walhout A.J.M., Cusick M.E., Roth F.P. et al. Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature. 2004; 430:88–93. [DOI] [PubMed] [Google Scholar]
- 7. Csermely P., Korcsmaros T., Kiss H.J.M., London G., Nussinov R.. Structure and dynamics of molecular networks: A novel paradigm of drug discovery A comprehensive review. Pharmacol. Ther. 2013; 138:333–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang Y.J., Xuan J.H., de los Reyes B.G., Clarke R., Ressom H.W.. Reverse engineering module networks by PSO-RNN hybrid modeling. BMC Genomics. 2009; 10(Suppl. 1):S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Segal E., Shapira M., Regev A., Pe'er D., Botstein D., Koller D., Friedman N.. Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nat. Genet. 2003; 34:166–176. [DOI] [PubMed] [Google Scholar]
- 10. Jaimovich A., Rinott R., Schuldiner M., Margalit H., Friedman N.. Modularity and directionality in genetic interaction maps. Bioinformatics. 2010; 26:i228–i236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maraziotis I.A., Dimitrakopoulou K., Bezerianos A.. An in silico method for detecting overlapping functional modules from composite biological networks. BMC Syst. Biol. 2008; 2:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Palla G., Derenyi I., Farkas I., Vicsek T.. Uncovering the overlapping community structure of complex networks in nature and society. Nature. 2005; 435:814–818. [DOI] [PubMed] [Google Scholar]
- 13. Guruharsha K.G., Rual J.F., Zhai B., Mintseris J., Vaidya P., Vaidya N., Beekman C., Wong C., Rhee D.Y., Cenaj O. et al. A protein complex network of drosophila melanogaster. Cell. 2011; 147:690–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brohee S., van Helden J.. Evaluation of clustering algorithms for protein-protein interaction networks. BMC Bioinformatics. 2006; 7:488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bader G.D., Hogue C.W.. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003; 4:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pu S.Y., Ronen K., Vlasblom J., Greenblatt J., Wodak S.J.. Local coherence in genetic interaction patterns reveals prevalent functional versatility. Bioinformatics. 2008; 24:2376–2383. [DOI] [PubMed] [Google Scholar]
- 17. Langfelder P., Horvath S.. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008; 9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Segal E., Friedman N., Koller D., Regev A.. A module map showing conditional activity of expression modules in cancer. Nat. Genet. 2004; 36:1090–1098. [DOI] [PubMed] [Google Scholar]
- 19. Jeon J., Jeong J.H., Baek J.H., Koo H.J., Park W.H., Yang J.S., Yu M.H., Kim S., Pak Y.K.. Network clustering revealed the systemic alterations of mitochondrial protein expression. PLoS Comput. Biol. 2011; 7:e1002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Georgii E., Dietmann S., Uno T., Pagel P., Tsuda K.. Enumeration of condition-dependent dense modules in protein interaction networks. Bioinformatics. 2009; 25:933–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhao S.W., Li S.. A co-module approach for elucidating drug-disease associations and revealing their molecular basis. Bioinformatics. 2012; 28:955–961. [DOI] [PubMed] [Google Scholar]
- 22. Kovacs I.A., Palotai R., Szalay M.S., Csermely P.. Community Landscapes: An integrative approach to determine overlapping network module hierarchy, identify key nodes and predict network dynamics. Plos One. 2010; 5:e12528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Freyre-Gonzalez J.A., Alonso-Pavon J.A., Trevino-Quintanilla L.G., Collado-Vides J.. Functional architecture of Escherichia coli: new insights provided by a natural decomposition approach. Genome Biol. 2008; 9:R154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Saez-Rodriguez J., Gayer S., Ginkel M., Gilles E.D.. Automatic decomposition of kinetic models of signaling networks minimizing the retroactivity among modules. Bioinformatics. 2008; 24:I213–I219. [DOI] [PubMed] [Google Scholar]
- 25. Ma H.W., Zhao X.M., Yuan Y.J., Zeng A.P.. Decomposition of metabolic network into functional modules based on the global connectivity structure of reaction graph. Bioinformatics. 2004; 20:1870–1876. [DOI] [PubMed] [Google Scholar]
- 26. Yoon J., Si Y.G., Nolan R., Lee K.. Modular decomposition of metabolic reaction networks based on flux analysis and pathway projection. Bioinformatics. 2007; 23:2433–2440. [DOI] [PubMed] [Google Scholar]
- 27. Chen J.C., Yuan B.. Detecting functional modules in the yeast protein-protein interaction network. Bioinformatics. 2006; 22:2283–2290. [DOI] [PubMed] [Google Scholar]
- 28. Luo F., Yang Y.F., Chen C.F., Chang R., Zhou J.Z., Scheuermann R.H.. Modular organization of protein interaction networks. Bioinformatics. 2007; 23:207–214. [DOI] [PubMed] [Google Scholar]
- 29. Ziv E., Middendorf M., Wiggins C.H.. Information-theoretic approach to network modularity. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2005; 71:046117. [DOI] [PubMed] [Google Scholar]
- 30. Newman M.E.J. Modularity and community structure in networks. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:8577–8582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nacher J.C., Schwartz J.M.. Modularity in protein complex and drug interactions reveals new polypharmacological properties. Plos One. 2012; 7:e30028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen Z.J., He Y., Rosa P., Germann J., Evans A.C.. Revealing modular architecture of human brain structural networks by using cortical thickness from MRI. Cereb. Cortex. 2008; 18:2374–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jia P.L., Zheng S.Y., Long J.R., Zheng W., Zhao Z.M.. dmGWAS: dense module searching for genome-wide association studies in protein-protein interaction networks. Bioinformatics. 2011; 27:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ulitsky I., Shamir R.. Identification of functional modules using network topology and high-throughput data. BMC Syst. Biol. 2007; 1:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ulitsky I., Shamir R.. Identifying functional modules using expression profiles and confidence-scored protein interactions. Bioinformatics. 2009; 25:1158–1164. [DOI] [PubMed] [Google Scholar]
- 36. Hou L., Wang L., Qian M.P., Li D., Tang C., Zhu Y.P., Deng M.H., Li F.T.. Modular analysis of the probabilistic genetic interaction network. Bioinformatics. 2011; 27:853–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ciriello G., Cerami E., Sander C., Schultz N.. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 2012; 22:398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang X.R., Zhou Y., Jin R., Chan C.. Reconstruct modular phenotype-specific gene networks by knowledge-driven matrix factorization. Bioinformatics. 2009; 25:2236–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li H., Sun Y., Zhan M.. The discovery of transcriptional modules by a two-stage matrix decomposition approach. Bioinformatics. 2007; 23:473–479. [DOI] [PubMed] [Google Scholar]
- 40. Zhang W.S., Edwards A., Fan W., Zhu D.X., Zhang K.. svdPPCS: an effective singular value decomposition-based method for conserved and divergent co-expression gene module identification. BMC Bioinformatics. 2010; 11:338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zou Q., Li J., Hong Q., Lin Z., Shi H., Wu Y., Ju Y.. Prediction of microRNA-disease associations based on social network analysis methods. BioMed Res. Int. 2015; 2015:810514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kelley R., Ideker T.. Systematic interpretation of genetic interactions using protein networks. Nat. Biotechnol. 2005; 23:561–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pinter R.Y., Rokhlenko O., Yeger-Lotem E., Ziv-Ukelson M.. Alignment of metabolic pathways. Bioinformatics. 2005; 21:3401–3408. [DOI] [PubMed] [Google Scholar]
- 44. Sharan R., Suthram S., Kelley R.M., Kuhn T., McCuine S., Uetz P., Sittler T., Karp R.M., Ideker T.. Conserved patterns of protein interaction in multiple species. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:1974–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ali W., Deane C.M.. Functionally guided alignment of protein interaction networks for module detection. Bioinformatics. 2009; 25:3166–3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen Y.Y., Wang Z., Wang Y.Y.. Spatiotemporal positioning of multipotent modules in diverse biological networks. Cell Mol. Life Sci. 2014; 71:2605–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bauer-Mehren A., Bundschus M., Rautschka M., Mayer M.A., Sanz F., Furlong L.I.. Gene-Disease Network Analysis reveals functional modules in mendelian, complex and environmental diseases. Plos One. 2011; 6:e20284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zeng X., Liao Y., Liu Y., Zou Q.. Prediction and validation of disease genes using HeteSim Scores. IEEE/ACM Trans. Comput. Biol. Bioinform. 2016; 99:1. [DOI] [PubMed] [Google Scholar]
- 49. Dhami P., Saffrey P., Bruce A.W., Dillon S.C., Chiang K., Bonhoure N., Koch C.M., Bye J., James K., Foad N.S. et al. Complex exon-intron marking by histone modifications is not determined solely by nucleosome distribution. Plos One. 2010; 5:e12339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yildirim M.A., Goh K.I., Cusick M.E., Barabasi A.L., Vidal M.. Drug-target network. Nat. Biotechnol. 2007; 25:1119–1126. [DOI] [PubMed] [Google Scholar]
- 51. Zou Q., Li J., Song L., Zeng X., Wang G.. Similarity computation strategies in the microRNA-disease network: A Survey. Brief. Funct. Genomic. 2016; 15:55–64. [DOI] [PubMed] [Google Scholar]
- 52. Xu Y., Guo M., Liu X., Wang C., Liu Y.. Inferring the soybean (Glycine max) microRNA functional network based on target gene network. Bioinformatics. 2013; 30:94–103. [DOI] [PubMed] [Google Scholar]
- 53. Xu Y.G., Guo M.Z., Zou Q., Liu X.Y., Wang C.Y., Liu Y.. System-level insights into the cellular interactome of a non-model organism: inferring, modelling and analysing functional gene network of soybean (Glycine max). Plos One. 2014; 9:e113907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zaman N., Li L., Jaramillo M.L., Sun Z., Tibiche C., Banville M., Collins C., Trifiro M., Paliouras M., Nantel A. et al. Signaling network assessment of mutations and copy number variations predict breast cancer subtype-specific drug targets. Cell Rep. 2013; 5:216–223. [DOI] [PubMed] [Google Scholar]
- 55. Bhajun R., Guyon L., Pitaval A., Sulpice E., Combe S., Obeid P., Haguet V., Ghorbel I., Lajaunie C., Gidrol X.. A statistically inferred microRNA network identifies breast cancer target miR-940 as an actin cytoskeleton regulator. Sci. Rep. 2015; 5:8336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mahanta P., Ahmed H., Bhattacharyya D., Kalita J.K.. Emerging Trends and Applications in Computer Science (NCETACS), 2011 2nd National Conference on. 2011; IEEE; 1–6. [Google Scholar]
- 57. Yoon S.R., De Micheli G.. Prediction of regulatory modules comprising microRNAs and target genes. Bioinformatics. 2005; 21:93–100. [DOI] [PubMed] [Google Scholar]
- 58. Pio G., Ceci M., D'Elia D., Loglisci C., Malerba D.. A novel biclustering algorithm for the discovery of meaningful biological correlations between microRNAs and their target genes. BMC Bioinformatics. 2013; 14(Suppl. 7):S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Peng X.X., Li Y., Walters K.A., Rosenzweig E.R., Lederer S.L., Aicher L.D., Proll S., Katze M.G.. Computational identification of hepatitis C virus associated microRNA-mRNA regulatory modules in human livers. BMC Genomics. 2009; 10:373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tran D.H., Satou K., Ho T.B.. Finding microRNA regulatory modules in human genome using rule induction. BMC Bioinformatics. 2008; 9(Suppl. 12):S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Joung J.G., Hwang K.B., Nam J.W., Kim S.J., Zhang B.T.. Discovery of microRNA-mRNA modules via population-based probabilistic learning. Bioinformatics. 2007; 23:1141–1147. [DOI] [PubMed] [Google Scholar]
- 62. Zhang S.H., Li Q.J., Liu J., Zhou X.J.. A novel computational framework for simultaneous integration of multiple types of genomic data to identify microRNA-gene regulatory modules. Bioinformatics. 2011; 27:I401–I409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Enright A.J., Van Dongen S., Ouzounis C.A.. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002; 30:1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nepusz T., Yu H.Y., Paccanaro A.. Detecting overlapping protein complexes in protein-protein interaction networks. Nat. Methods. 2012; 9:471–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rhrissorrakrai K., Gunsalus K.C.. MINE: Module Identification in Networks. BMC Bioinformatics. 2011; 12:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ravasz E., Barabasi A.L.. Hierarchical organization in complex networks. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2003; 67:026112. [DOI] [PubMed] [Google Scholar]
- 67. Barabasi A.L., Ravasz E., Oltvai Z.. Hierarchical organization of modularity in complex networks. Lect. Notes Phys. 2003; 625:46–65. [Google Scholar]
- 68. Hwang T., Atluri G., Xie M., Dey S., Hong C., Kumar V., Kuang R.. Co-clustering phenome-genome for phenotype classification and disease gene discovery. Nucleic Acids Res. 2012; 40:e146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Xu Y.G., Guo M.Z., Liu X.Y., Wang C.Y., Liu Y.. SoyFN: a knowledge database of soybean functional networks. Database (Oxford). 2014; 2014:bau019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Smoot M.E., Ono K., Ruscheinski J., Wang P.-L., Ideker T.. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011; 27:431–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ru Y., Kechris K.J., Tabakoff B., Hoffman P., Radcliffe R.A., Bowler R., Mahaffey S., Rossi S., Calin G.A., Bemis L. et al. The multiMiR R package and database: integration of microRNA-target interactions along with their disease and drug associations. Nucleic Acids Res. 2014; 42:e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.