

Abstract
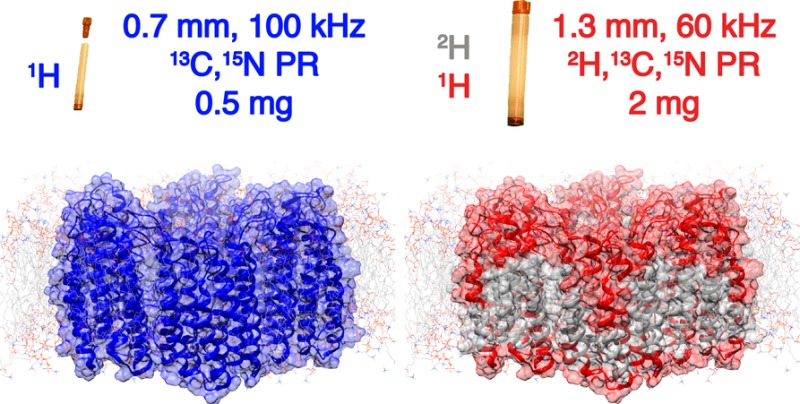
The structures and properties of membrane proteins in lipid bilayers are expected to closely resemble those in native cell-membrane environments, although they have been difficult to elucidate. By performing solid-state NMR measurements at very fast (100 kHz) magic-angle spinning rates and at high (23.5 T) magnetic field, severe sensitivity and resolution challenges are overcome, enabling the atomic-level characterization of membrane proteins in lipid environments. This is demonstrated by extensive 1H-based resonance assignments of the fully protonated heptahelical membrane protein proteorhodopsin, and the efficient identification of numerous 1H–1H dipolar interactions, which provide distance constraints, inter-residue proximities, relative orientations of secondary structural elements, and protein–cofactor interactions in the hydrophobic transmembrane regions. These results establish a general approach for high-resolution structural studies of membrane proteins in lipid environments via solid-state NMR.
Introduction
Atomic level characterization of membrane proteins in lipid bilayers is essential for understanding their functions, although extremely challenging. Membrane proteins in lipid environments generally lack long-range order, and tumble slowly in solutions, which respectively render scattering investigations infeasible and jeopardize liquid-state NMR investigations. Magic-angle spinning (MAS) solid-state NMR spectroscopy is a powerful tool that can reveal both structural and dynamical details of such systems,1 yet its application has been limited by low spectral sensitivity and resolution, as well as by the difficulty in obtaining large (∼20 mg) quantities of isotopically labeled proteins.
Many strategies have been employed to overcome the resolution and sensitivity issues that impede structural characterization of membrane proteins by MAS NMR. Proton detection is a powerful technique that exploits the high gyromagnetic ratio and abundance of proton nuclei to enhance the spectral sensitivity.2 However, despite encouraging proof-of-principle studies performed on fully protonated model systems,3 applications of 1H-detection to membrane proteins in native-like lipid environments have been hindered by the low 1H spectral resolution under moderate MAS rates (<40 kHz). Higher spectral resolution4 can be achieved in part by proton dilution strategies (typically perdeuteration and back-protonation at the exchangeable sites) to quench the 1H–1H dipolar couplings that broaden NMR signals.5 This strategy, however, is problematic during protein expression, due to anemic growth in deuterium oxide which sometimes is even incompatible with protein expression, as for example in mammalian cells. When feasible, it allows reintroduction of 1H species exclusively at sites that are exchangeable and accessible to solvent, which notably do not include the extensive hydrophobic transmembrane regions, thereby precluding their analyses by 1H-detected spectroscopy.6 Unfolding and refolding membrane proteins leads to the introduction of 1H species at the exchangeable sites of transmembrane regions, however such protocols are not general, and specific examples are rare.1l,5d,7 To address this in part, isotopic labeling strategies have been developed in which membrane proteins are expressed in H2O in the presence of deuterated 13C glucose, such that 1H/2H species are homogeneously distributed in both water-accessible and inaccessible regions.8 Nevertheless, in such cases the 1H/2H isotopomeric distributions often result in poorly resolved 13C resonances from side-chain moieties that are crucial for structure determination.
The advent of MAS NMR probes capable of spinning at rates of 100 kHz or greater has reduced the amount of sample required,2,5e,5f,9 and, most importantly, reduced the need for proton dilution.10 This has opened unprecedented opportunities for structural investigations of biosolids by using sensitive 1H-detected methods,10,11 with a dramatic reduction in homogeneous line broadening to improve the resolution of 1H resonances. However, even at the fastest (∼100 kHz) sample spinning rates and the highest magnetic fields (23.5 T) currently available, membrane proteins in lipid bilayers remain challenging to study by NMR (or other methods), because of their inherently heterogeneous lipid bilayer environments in which they are naturally diluted and which limit spectral resolution and signal sensitivity.
Transmembrane proteins, in particular, are incorporated into lipid bilayers and perform sensing, transport and enzymatic functions in support of cellular viability. One example is the green variant of proteorhodopsin, a light-activated H+-ion pump of 240 residues that in solution has an archetypical heptahelical transmembrane protein structure with a retinal cofactor.12 While the structure of monomeric proteorhodopsin in detergents has been determined by solution NMR (pdb code: 2L6X),13 the structure of the protein in native-like lipid environments remains unknown. This is complicated further by the tendency of proteorhodopsin molecules in bilayers to assemble into pentamers and hexamers, which are thought to mediate protein function.14 Conventional 13C-detected MAS NMR methods have enabled the extensive assignment of backbone and side-chain 13C and 15N resonances of proteorhodopsin oligomers in lipids.15 However, only a partial assignment of the solvent-exposed amide 1H resonances was possible with 1H-detected measurements on perdeuterated protein, due to incomplete solvent exchange.6
Here, we demonstrate very fast (100 kHz) MAS NMR to be a general approach for structural analyses of fully protonated membrane proteins in near-native lipid environments. Notably, we show that the use of fast 100 kHz MAS conditions expedites sequence-specific resonance assignments and facilitates the detection of 1H–1H proximities in hydrophobic transmembrane regions, which are essential features of protein structure and for their function.
Experimental Section
Sample Preparation
Expression of isotopically labeled proteorhodopsin was carried out as described by Ward et al.6 with a few differences. Following overnight growth of E. coli cells in the 25 mL culture, the cells were pelleted by centrifugation at ∼5000 rpm and resuspended in 75 mL of M9 minimal media with all labels present. Subsequently, the 75 mL culture was grown at 37 °C to an O.D.600 of 1.0–1.5 (approximately 6 h) and then added to 925 mL of M9 media (all labels present). Protein expression was induced at an O.D.600 of 0.8 by the addition of IPTG to a concentration of 1 mM and allowed to proceed for ∼24 h at room temperature without shaking. Protein purification was carried out using methods described previously14a with a few modifications. Cells were lysed by a freeze fracture step with three freeze–thaw cycles using liquid nitrogen in addition to probe tip sonication and incubation with DNase, lysozyme, and MgCl2. Then, the large cell fragments containing proteorhodopsin were pelleted by centrifugation at 5000 rpm and then washed with 250 mL of phosphate buffered solution (150 mM KCl and KH2PO4, pH ≈ 8.7) by repeatedly suspending the cell pellet in 40 mL of buffer, shaking the solution for 5 min, and pelleting cells by centrifugation. Subsequently, proteorhodopsin was extracted from lysed E. coli membranes by overnight incubation of the washed cell fragments in a phosphate buffered solution containing 4% (w/v) n-dodecyl-β,d-maltoside surfactant. Following the Ni-NTA resin binding, washing and elution steps,2 the optical purities of the proteorhodopsin samples, as measured by the ratio of absorbances at 280 to 520 nm, typically ranged between 1.8 and 2.2. The concentration of proteorhodopsin was estimated based on the absorbance at 520 nm, using an extinction coefficient of 49 000 M–1cm–1. Proteorhodopsin was reconstituted into 1,2-dimyristoyl-sn-glycero-3-phosphate (DMPA) and 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) liposomes using procedures described previously, except using a 10 mM HEPES buffer that was titrated to a pH 6.2 using dilute HCl.15b
NMR Spectroscopy
All experiments were carried out on a Bruker Avance III 1 GHz standard bore spectrometer operating at a static field of 23.4 T, equipped with a triple channel H, C, N, 0.7 mm probe, at a MAS rate ωr/2π of 100 kHz. Sample temperature was maintained at about 305 K using a Bruker cooling unit with regulated N2 gas directed at the rotor. The temperature of this gas measured just before reaching the sample was 280 K. Chemical shifts were referenced to adamantane (1H signal at 1.87 ppm).
The nonselective pulses were set to 1.1 μs at 227 kHz rf-field amplitude (1H), 5.5 μs at 45 kHz rf-field amplitude (15N) and 3.1 μs at 81 kHz rf-field amplitude (13C). The dipolar-based 15N,1H and 13C,1H CP-HSQC experiments (H)CH and (H)NH follow, with little modifications, those introduced by Rienstra and co-workers.3b,5a (H)NCAHA, (H)N(CO)CAHA, (H)CANH, (H)(CO)CA(CO)NH, and (H)CONH experiments were performed as described recently.5d,11b The irradiation schemes are displayed in Figure S1 of the Supporting Information, SI. The 1H–15N and 1H–13C cross-polarization (CP) were performed using a constant RF frequency applied to 15N and 13C of 40 kHz and a pulse linearly ramped from 90% to 100% of a maximum RF frequency of 130 kHz on 1H. The 13C–15N CP was performed using a constant RF frequency of 60 kHz on 13C and a 10% tangent ramp of 40 kHz on 15N for 10 ms. Low power WALTZ-16 decoupling of 10 kHz was applied for heteronuclear decoupling. Swept low-power TPPM (slTPPM)16 decoupling was used during 13C, 15N chemical shift evolution with a 1H RF frequency of 25 kHz and a pulse-length duration of 20 μs. DIPSI-2 of γB1/2π = 20 kHz was used for 13C decoupling during acquisition due to the presence of homonuclear 13C–13C J-couplings. Suppression of solvent signals4a was applied using the MISSISSIPPI scheme17 without the homospoil gradient for 200 ms. The interscan recycle delay was 1 s.
The (H)CCH experiment follows that reported recently.10,11b The composite 13C pulses of 25 kHz were applied for the TOCSY mixing for 15 ms. In the 3D (H)CHH experiment, 1H–1H RFDR recoupling16 was applied after the back-CP at a 1H RF frequency of 200 kHz, for 1.4 ms. No loss of water from the sample was observed during the acquisition of the spectra. Spectra were apodized in each dimension with 60° to 90° shifted squared sine-bells (“qsine 3” or “qsine 2” in Bruker Topspin), and zero-filled to at least twice the number of points in the indirect dimensions. Where line widths are reported, no apodization was applied for the reported frequencies. Acquisition and processing parameters specific for each data set are summarized in Tables S2. Spectra were processed with Topspin3.5, and their analysis was performed using Cara. The resonance assignments for 1H, 13C, and 15N nuclei are listed in Table S3.
Results and Discussion
A dipolar-mediated 2D 1H–15N correlation spectrum (Figure 1, blue) of fully protonated U–[13C,15N] proteorhodopsin acquired at 100 kHz MAS shows highly resolved signals from the amide moieties of the protein backbone. These correlations have an average proton line width of 190 Hz fwhm that is significantly narrower than in a spectrum acquired on an identical sample at 60 kHz MAS rates in a 1.3 mm probe (Figure S2). Surprisingly, these spectra show comparable signal sensitivities (Figure S3), despite the significantly lower sample quantity (∼0.5 mg, 0.7 mm rotor) at 100 kHz MAS, compared to 60 kHz MAS (∼2.0 mg, 1.3 mm rotor). Deuterated proteorhodopsin reprotonated at the amide sites by exchange in 100% protonated buffers, yields enhanced resolution in a 1H–15N correlation spectrum (Figure 1, red) acquired under conventional 60 kHz MAS rates, showing average line widths of 140 Hz fwhm. However, the spectrum acquired on the perdeuterated sample at 60 kHz MAS has far fewer cross-peaks (Figure 1, red) than that from the fully protonated (Figure 1, blue) protein at 100 kHz MAS. This reflects an incomplete reintroduction of HN species in perdeuterated proteorhodopsin, predominantly at residues in the hydrophobic transmembrane regions, which precludes their detection and structural analysis. By comparison, the ubiquity of 1H species in fully protonated proteorhodopsin allows the entire biomolecule to be probed by 1H-detected spectroscopy, in particular moieties on the aliphatic side-chains from which critical structural constraints are derived.
Figure 1.
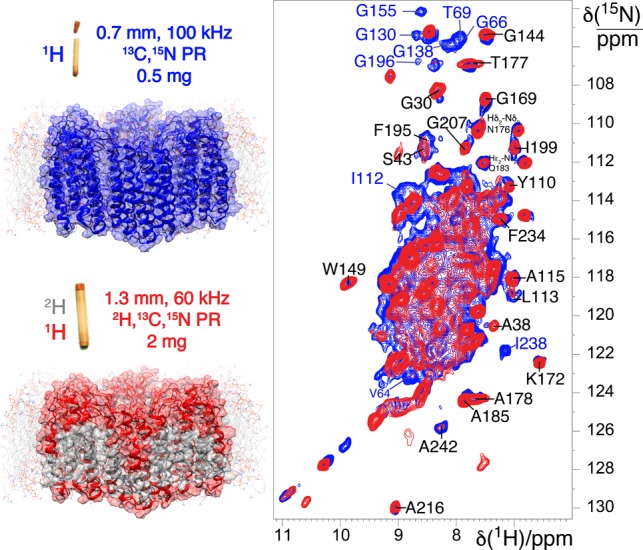
(A) Comparison of 2D 1H–15N CP-HSQC MAS NMR spectra acquired at 305 K on (blue trace) fully protonated U–[13C,15N] proteorhodopsin in DMPC:DMPA lipids at 100 kHz MAS, and (red trace) U–[2H,15N,13C] proteorhodopsin, reprotonated in 100% protonated buffer, in the same lipids at 60 kHz MAS and a field strength of 23.5 T. (B) Schematic diagrams of proteorhodopsin oligomers, modeled from the monomeric protein structure (pdb code: 2L6X, see SI), in which residues with 1HN species are highlighted in blue and red for the fully protonated and perdeuterated samples, respectively.
For example, the 2D 13C–1H CP-HSQC spectra of fully protonated proteorhodopsin at 60 kHz (Figure 2, left) and 100 kHz (Figure 2, right) MAS show correlated signals from 1H and 13C nuclei in the side-chains (top panel) and α positions (bottom panel). Substantially enhanced proton resolution is observed under the faster MAS conditions, as established by the larger number of fully resolved correlations that appear only in the spectrum recorded at 100 kHz; these include many 1Hα resonances labeled in Figure 2, bottom panels, as well as 1H methyl resonances (Figure 2, top panels). For several peaks resolved even at 60 kHz, the line widths are observed to be 50–100 Hz broader (Figure 2). The dramatic improvements in resolution enable the use of aliphatic side-chain protons as crucial reporters of protein structure. The significant increase in spectral resolution observed at 100 kHz MAS is surprising. While microcrystalline proteins, capsids, and fibrils often are homogeneous samples with rigid architectures that are amenable to MAS-averaging of homonuclear dipolar interactions, membrane proteins are less homogeneous, comparably flexible, and undergo a range of motions18 that could reduce the benefits of faster MAS rates in improving signal resolution.
Figure 2.
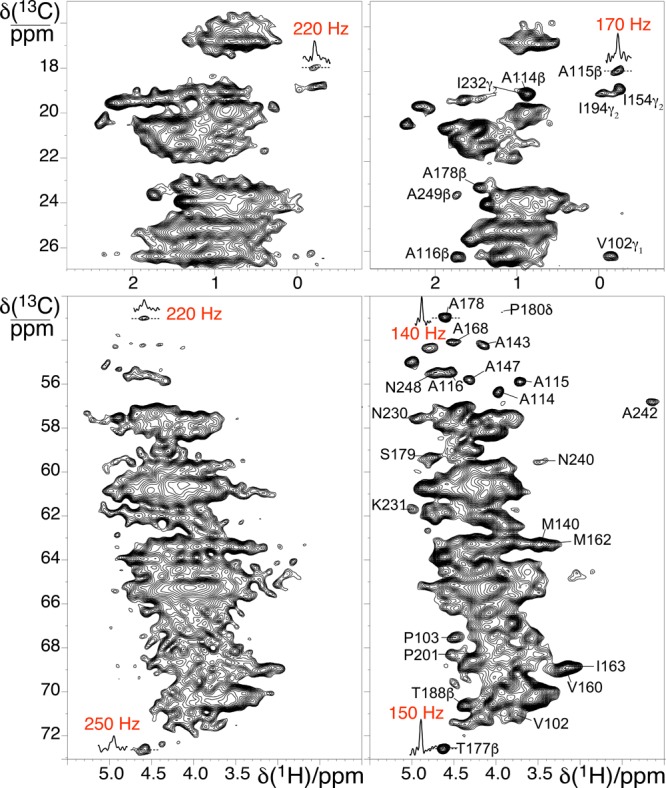
2D 1H–13C CP-HSQC MAS NMR spectra acquired at 305 K and 23.5 T on fully protonated U–[13C,15N] proteorhodopsin in lipids at MAS rates of 60 kHz (left) and 100 kHz (right). The side chain and alpha regions of the spectra are shown in the top and bottom panels, respectively.
Nevertheless, in spectra from fully protonated proteorhodopsin in lipids, the average 1H line width of 15N–1H correlations from amide moieties is ∼190 Hz fwhm at 100 kHz MAS, compared to ∼280 Hz fwhm at 60 kHz MAS. Greater resolution improvements are observed for aliphatic 1H signals, where average line widths are approximately 145 Hz fwhm at 100 kHz MAS, versus about 235 Hz fwhm estimated from the few resolved signals at 60 kHz. The bulk 1H coherence lifetimes were measured to be 2.5 ms on the fully protonated protein at 100 kHz MAS, which corresponds to residual homogeneous components of ∼125 Hz that suggest inhomogeneous line widths of 155 and 70 Hz for the 1HN and 1Hα signals, respectively. The larger inhomogeneous components for the 1HN species likely arise from a distribution of hydrogen bonding environments, consistent with the larger range of amide 1HN shifts reported generally for proteins in the BMRB. The substantial homogeneous broadening remaining even at 100 kHz MAS conditions indicates that further narrowed 1H line widths could be obtained for faster MAS rates and/or higher magnetic fields.2,19
Additionally, the 1H signal resolution of fully protonated proteorhodopsin at 100 kHz MAS is comparable to that obtained with state-of-the-art partial isotopic labeling schemes. These labeling strategies, including fractional deuteration,5h,20 isoleucine–leucine–valine labeling,1l,21 proton clouds,5g and stereospecific array isotopic labeling (SAIL),22 selectively introduce 1H side-chains into a deuterated protein matrix. Spectra of fully protonated proteorhodopsin in lipids at 100 kHz MAS show 20% higher 1H resolution than for the similar α-helical transmembrane K+ channel Kcsa in lipid bilayers labeled with an inverse fractional deuteration approach and using 60 kHz MAS rates.8 Importantly, while this labeling scheme yields 1H/2H isotopomers that can account for up to 0.3 ppm dispersions in 13C chemical shifts,23 such effects are negligible in fully protonated proteins probed using 100 kHz MAS.
To facilitate rapid and global sequence-specific resonance assignments, judicious selections of 3D correlation experiments are essential for high sensitivity, in addition to high spectral resolution. Between the two different types of protein backbone 13C species, the coherence lifetimes are longest for 13C′ species (T2′ = 21 ms, compared to 13Cα T2′ = 12.5 ms, see Table S1), yielding considerable sensitivity advantages for 3D NMR measurements that rely on evolution of 13C′ versus 13Cα coherences. Thus, for sequential resonance assignments of fully protonated proteorhodopsin, we chose the combination of two strategies that leverage the longer lifetimes of the 13C′ spins by using J-mediated 13C′-13Cα coherence transfers24 and detection of either HN5d or Hα resonances.11b These two approaches, respectively, use (H)CANH and (H)(CO)CA(CO)NH spectra to correlate signals from 1H–15N amide pairs to 13Cα resonances of adjacent residues, or use (H)NCAHA and (H)N(CO)CAHA to correlate the signals of 1Hα–13Cα pairs to intra and inter-residue 15N species.11b Sequential backbone assignments are achieved by simultaneously linking correlations of both 1H–15N or 1Hα–13Cα pairs through their mutual 13Cα or 15N chemical shifts established in the amide or α proton-detected spectra, respectively, as depicted in Figure 3. Here, representative portions of the four spectra that demonstrate sequential linking of amide and alpha pairs are reported. The choice of these pairs of experiments is motivated by the coherence lifetimes, which for membrane proteins are not as long as for microcrystalline samples. For comparison, while these 3D spectra were acquired in less than 2 weeks (Figure S1), 1HN-detected experiments that rely on the faster decaying 13Cα spins to enable 13C′- or 13Cβ-linking25 have lower transfer efficiencies and significantly longer acquisition times. The 1Hα–13Cα and 1H–15N pairs also have roughly equal sensitivities, and the narrow dispersion in the 1Hα dimension is offset by the narrow line width. This makes both types of spin pairs similarly useful in providing sequence-specific assignments. The backbone resonance assignments are further corroborated by analyses of the 13C–13C-1H TOCSY spectrum (Figure S4) that yields the assignment of the 1H and 13C side-chain resonances, thus enabling the identification of the amino acid types.
Figure 3.
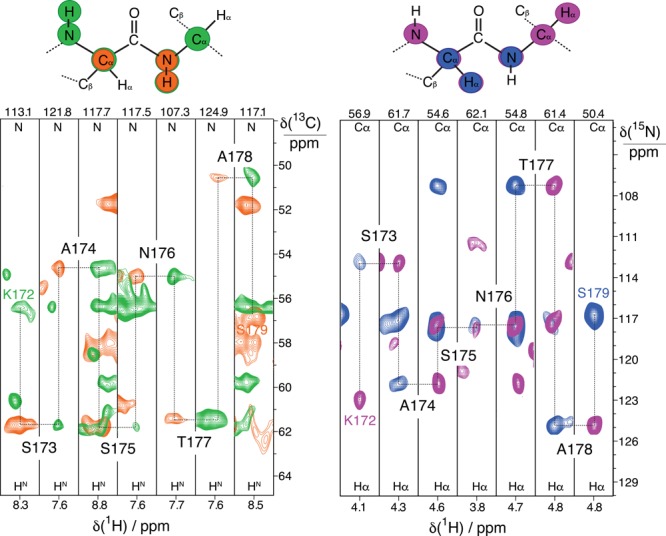
Sequential assignments of intensity correlations for residues 172–179 in fully protonated proteorhodopsin in lipids bilayers. 2D 1Hα–13Cα slices extracted from (H)CANH (green trace) and (H)(CO)CA(CO)NH (orange trace) spectra are shown in the left panel, and 2D 1H–15N slices from (H)NCAHA (magenta trace) and (H)N(CO)CAHA (blue trace) spectra in the right. The four spectra were acquired at 100 kHz MAS and 23.5 T.
Resonance assignments for extensive portions of the proteorhodopsin backbone and side-chains were made based on spectra acquired at 100 kHz MAS. Despite the high degeneracy of aliphatic residues (32 Leu, 32 Ala, 24 Gly, 21 Val, and 19 Ile residues) that account for 49% of the proteorhodopsin sequence and the typically low chemical shift dispersions for helical proteins, the backbone resonances of 146 residues were sequence specifically assigned (Figure S5). Continuous linkages through 5 of the 6 proline residues were identified from analyses of 1Hα-detected 3D NMR correlation spectra. These residues are distributed in the transmembrane helices and extra-membrane loop regions. Importantly, resonance assignments were established for 57% of the 1H and 13C moieties of the aliphatic side-chains. The backbone chemical shifts clearly identify the seven transmembrane helices, connected by interhelical loops, and one additional short helix located in the extracellular E–F loop, in agreement with the structure of proteorhodopsin in detergents (Figure S6).
Such extensive resonance assignments facilitate the identification of inter-residue 1H–1H proximities that yield detailed site-specific information on proteorhodopsin structure in lipids. Key insights into the intra- and interhelical proximities between side-chains are obtained from analyses of high-resolution radio frequency-driven-recoupling (RFDR) spectra. For example, the 3D H(H)CH RFDR spectrum (1.4 ms mixing time, Figure 4) acquired from fully protonated proteorhodopsin shows numerous cross-signals that can be assigned to specific 1H species using the resonance assignments established above. Subsequent analyses yield the identification of structural constraints, several of which are depicted schematically on the protein structure derived by solution NMR data, shown in Figure 4B. These include both intrahelical proximities, such as between the methyl 1H of M134 and 1Hα of G138 (Figure 4B, right, middle), and interhelical proximities, including the methyl 1Hs of A116 and V182 (Figure 4B, right, bottom). These internuclear contacts within the transmembrane region are a direct way to probe the relative orientations of secondary structural elements. Especially important are the 1H–1H proximities of the 1Hα of Gly residues and methyl groups, as these provide extremely useful structural constraints for α-helical proteins. In addition, the spectrum contains 1H–1H cross peaks between 1Hα of Gly155 and two methyl groups with 1H signals at 2.0 and 1.6 ppm, respectively, which are tentatively assigned to the retinal cofactor (Figure 4B, right, top). Such signals are valuable to establish the location, orientation and configuration of the chromophore in the transmembrane region of the protein, which is directly related to the protein functionality. In contrast, similar 3D spectra using perdeuterated and back-exchanged proteorhodopsin can only reveal 1HN–1HN contacts that primarily provide short- and medium-range intrahelical distance restraints. Importantly, much higher signal sensitivity and resolution were observed from the fully protonated sample at 100 kHz MAS versus an otherwise identical measurement at 60 kHz MAS on a 5-fold larger sample (Figure 4A).
Figure 4.
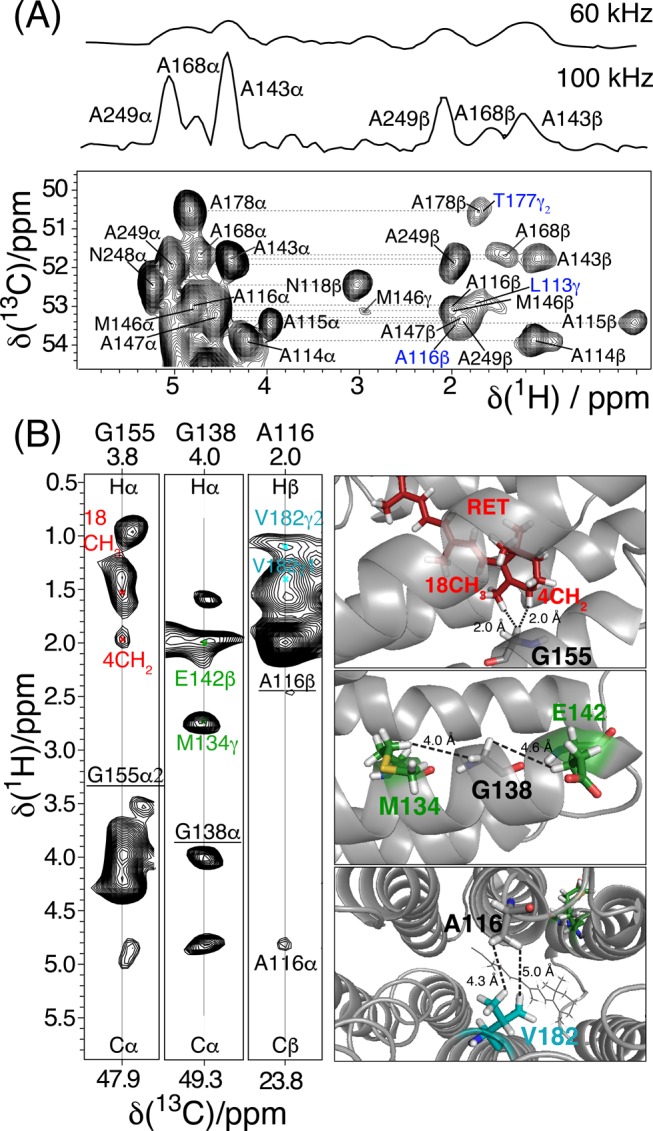
A) Alanine region of the 2D 1H–13C projection of a 3D H(H)CH RFDR spectrum acquired on U–[15N,13C] proteorhodopsin in lipids at 100 kHz MAS at 305 K and 23.5 T, using a 1.4 ms mixing time during which the RFDR rf-field was 200 kHz. Diagonal peaks are labeled in black and cross peaks in blue. Shown above the 2D projection are 1D 13C slices extracted at the A249 Cα–Hα position (∼51.9 ppm in the indirect dimension of the 2D projection) from the 3D RFDR spectrum acquired at 100 kHz and (up 60 kHz MAS. (B) 2D cross sections (left) of the 3D RFDR spectrum with 1H–1H correlations assigned to intra/interhelical and helix-retinal contacts cofactor, as depicted in the schematic 3D structure of the protein (right).
The structures and properties of fully protonated membrane proteins in lipid bilayers are expected to closely resemble those in native cell-membrane environments. Interestingly, all of the 1H–1H contacts between the transmembrane helices reported above can be explained on the basis of the structure of proteorhodopsin in micellar (diheptanoyl-phosphocholine, diC7PC) surfactant solution.13 In combination with the 13C chemical shift analysis above, this establishes that for the compositions and conditions investigated, the structures of proteorhodopsin in lipid bilayers and in micellar surfactant solution are very similar, and that even the position of the retinal cofactor within the transmembrane pocket is maintained. While in many cases solubilizing detergents have been observed to alter the structures or functionalities of membrane proteins,26 that is not the case here. NMR structural analyses of fully protonated membrane proteins in lipids enabled by fast MAS represent an essential step to validating the conclusions from solution NMR data in detergent micelles.
From the present data, the oligomeric state of proteorhodopsin in lipid bilayers cannot be concluded, since it is not possible to identify any intermonomer cross peak in the 3D H(H)CH RFDR spectrum reflecting the presence of pentamers and/or hexamers. Unambiguous detection of such cross-peaks is extremely challenging due to the partial side chain assignment, the signal degeneracy, and the sample heterogeneity in terms of oligomeric composition. In order to identify such contacts, different strategies aimed at decreasing the sample heterogeneity and the spectral overlap and increasing the signal sensitivity can be adopted, such as the expression of mutants that stabilize a single oligomeric form to increase the sample homogeneity, or the use of tailored labeling schemes to decrease the spectral crowding, the acquisition of selective proton–proton distance restraints to increase the signal-to-noise, or the acquisition of 4D spectra with increased heteronuclear dimensionality to improve the spectral resolution.
Conclusions
Extensive atomic-level structural insights on a fully protonated membrane protein in native-like lipid environments are provided by 1H-detected solid-state NMR spectra acquired under 100 kHz MAS conditions and at high (23.5 T) magnetic field. This approach yields highly resolved 1H resonances from moieties throughout the protein, including those from transmembrane amide sites that are generally inaccessible to chemical exchange with water, and that are therefore absent in spectra of perdeuterated samples. This enables the sequential assignments of the protein resonances, including the majority of the aliphatic 1H moieties, and notably the identification of long-range interhelical 1H–1H contacts between side-chains in transmembrane protein regions. To the best of our knowledge, this is the first report of long-range proximities between side-chain protons in a fully protonated membrane protein. Remarkably, this information was obtained with less than 0.5 mg of sample without the need for deuteration, thus circumventing a major roadblock to the structural characterization of membrane proteins by solid-state MAS NMR or other methods. This represents an important step toward the determination of membrane protein structures and their relationships to functional interactions in native-like lipid environments. The approach is expected to open opportunities to investigate a variety of complicated structure-dependent biochemical phenomena, including protein interactions in near-native environments or molecular recognition mechanisms that govern ligand binding to transmembrane receptors.
Acknowledgments
Financial support is acknowledged from the French CNRS (IR-RMN FR3050), from the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Programme (ERC-2015-CoG GA no. 648974), from the National Institutes of Health (R01GM116128), and from the USARO through the Institute for Collaborative Biotechnologies (grant W911NF-09-0001) and award W911NF-14-1-0617. L.B.A was supported by a MC incoming fellowships (REA grant agreement no. 624918 “MEM-MAS”).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b05269.
NMR coherence lifetimes and line widths. Diagrams with the NMR pulse sequence used. 2D 1H–15N CP–HSQC NMR spectra acquired for U–[15N, 13C] PR in a 1.3 mm probe at 60 kHz and in a 0.7 mm probe at 100 kHz. 13C–13C 2D projection of the 3D (H)CCH-TOCSY spectrum. Topological plot of PR with the assigned residues and prediction of the secondary structure based on the analysis of the backbone chemical shifts. Probe considerations. Table of fully assigned 1H, 13C, and 15N chemical shifts (PDF)
Author Present Address
∥ Max-Plank Institute for Biophysical Chemistry, Am Fassberg 11, Göttingen, Germany.
The authors declare no competing financial interest.
Supplementary Material
References
- a Lange A.; Giller K.; Hornig S.; Martin-Eauclaire M. F.; Pongs O.; Becker S.; Baldus M. Nature 2006, 440, 959–962. 10.1038/nature04649. [DOI] [PubMed] [Google Scholar]; b Xu J.; Durr U. H.; Im S. C.; Gan Z.; Waskell L.; Ramamoorthy A. Angew. Chem., Int. Ed. 2008, 47, 7864–7867. 10.1002/anie.200801338. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Etzkorn M.; Kneuper H.; Dunnwald P.; Vijayan V.; Kramer J.; Griesinger C.; Becker S.; Unden G.; Baldus M. Nat. Struct. Mol. Biol. 2008, 15, 1031–1039. 10.1038/nsmb.1493. [DOI] [PubMed] [Google Scholar]; d Bajaj V. S.; Mak-Jurkauskas M. L.; Belenky M.; Herzfeld J.; Griffin R. G. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 9244–9249. 10.1073/pnas.0900908106. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Cady S. D.; Schmidt-Rohr K.; Wang J.; Soto C. S.; Degrado W. F.; Hong M. Nature 2010, 463, 689–692. 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Sharma M.; Yi M.; Dong H.; Qin H.; Peterson E.; Busath D. D.; Zhou H. X.; Cross T. A. Science 2010, 330, 509–512. 10.1126/science.1191750. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Bhate M. P.; McDermott A. E. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 15265–15270. 10.1073/pnas.1211900109. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Park S. H.; Das B. B.; Casagrande F.; Tian Y.; Nothnagel H. J.; Chu M.; Kiefer H.; Maier K.; De Angelis A. A.; Marassi F. M.; Opella S. J. Nature 2012, 491, 779–783. 10.1038/nature11580. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Shahid S. A.; Bardiaux B.; Franks W. T.; Krabben L.; Habeck M.; van Rossum B. J.; Linke D. Nat. Methods 2012, 9, 1212–1217. 10.1038/nmeth.2248. [DOI] [PubMed] [Google Scholar]; j Wang S.; Munro R. A.; Shi L.; Kawamura I.; Okitsu T.; Wada A.; Kim S. Y.; Jung K. H.; Brown L. S.; Ladizhansky V. Nat. Methods 2013, 10, 1007–1012. 10.1038/nmeth.2635. [DOI] [PubMed] [Google Scholar]; k Wylie B. J.; Bhate M. P.; McDermott A. E. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 185–190. 10.1073/pnas.1319577110. [DOI] [PMC free article] [PubMed] [Google Scholar]; l Andreas L. B.; Reese M.; Eddy M. T.; Gelev V.; Ni Q. Z.; Miller E. A.; Emsley L.; Pintacuda G.; Chou J. J.; Griffin R. G. J. Am. Chem. Soc. 2015, 137, 14877–14886. 10.1021/jacs.5b04802. [DOI] [PMC free article] [PubMed] [Google Scholar]; m Becker-Baldus J.; Bamann C.; Saxena K.; Gustmann H.; Brown L. J.; Brown R. C.; Reiter C.; Bamberg E.; Wachtveitl J.; Schwalbe H.; Glaubitz C. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 9896–9901. 10.1073/pnas.1507713112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreas L. B.; Le Marchand T.; Jaudzems K.; Pintacuda G. J. Magn. Reson. 2015, 253, 36–49. 10.1016/j.jmr.2015.01.003. [DOI] [PubMed] [Google Scholar]
- a Samoson A.; Tuherm T.; Gan Z. Solid State Nucl. Magn. Reson. 2001, 20, 130–136. 10.1006/snmr.2001.0037. [DOI] [PubMed] [Google Scholar]; b Zhou D. H.; Shea J. J.; Nieuwkoop A. J.; Franks W. T.; Wylie B. J.; Mullen C.; Sandoz D.; Rienstra C. M. Angew. Chem., Int. Ed. 2007, 46, 8380–8383. 10.1002/anie.200702905. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Marchetti A.; Jehle S.; Felletti M.; Knight M. J.; Wang Y.; Xu Z. Q.; Park A. Y.; Otting G.; Lesage A.; Emsley L.; Dixon N. E.; Pintacuda G. Angew. Chem., Int. Ed. 2012, 51, 10756–10759. 10.1002/anie.201203124. [DOI] [PubMed] [Google Scholar]; d Vasa S. K.; Rovo P.; Giller K.; Becker S.; Linser R. Phys. Chem. Chem. Phys. 2016, 18, 8359–8363. 10.1039/C5CP06601H. [DOI] [PubMed] [Google Scholar]
- a Paulson E. K.; Morcombe C. R.; Gaponenko V.; Dancheck B.; Byrd R. A.; Zilm K. W. J. Am. Chem. Soc. 2003, 125, 15831–15836. 10.1021/ja037315+. [DOI] [PubMed] [Google Scholar]; b Chevelkov V.; Rehbein K.; Diehl A.; Reif B. Angew. Chem., Int. Ed. 2006, 45, 3878–3881. 10.1002/anie.200600328. [DOI] [PubMed] [Google Scholar]
- a Zhou D. H.; Shah G.; Cormos M.; Mullen C.; Sandoz D.; Rienstra C. M. J. Am. Chem. Soc. 2007, 129, 11791–11801. 10.1021/ja073462m. [DOI] [PubMed] [Google Scholar]; b Knight M. J.; Webber A. L.; Pell A. J.; Guerry P.; Barbet-Massin E.; Bertini I.; Felli I. C.; Gonnelli L.; Pierattelli R.; Emsley L.; Lesage A.; Herrmann T.; Pintacuda G. Angew. Chem., Int. Ed. 2011, 50, 11697–11701. 10.1002/anie.201106340. [DOI] [PubMed] [Google Scholar]; c Knight M. J.; Pell A. J.; Bertini I.; Felli I. C.; Gonnelli L.; Pierattelli R.; Herrmann T.; Emsley L.; Pintacuda G. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 11095–11100. 10.1073/pnas.1204515109. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Barbet-Massin E.; Pell A. J.; Retel J. S.; Andreas L. B.; Jaudzems K.; Franks W. T.; Nieuwkoop A. J.; Hiller M.; Higman V.; Guerry P.; Bertarello A.; Knight M. J.; Felletti M.; Le Marchand T.; Kotelovica S.; Akopjana I.; Tars K.; Stoppini M.; Bellotti V.; Bolognesi M.; Ricagno S.; Chou J. J.; Griffin R. G.; Oschkinat H.; Lesage A.; Emsley L.; Herrmann T.; Pintacuda G. J. Am. Chem. Soc. 2014, 136, 12489–12497. 10.1021/ja507382j. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Lamley J. M.; Iuga D.; Oster C.; Sass H. J.; Rogowski M.; Oss A.; Past J.; Reinhold A.; Grzesiek S.; Samoson A.; Lewandowski J. R. J. Am. Chem. Soc. 2014, 136, 16800–16806. 10.1021/ja5069992. [DOI] [PubMed] [Google Scholar]; f Agarwal V.; Penzel S.; Szekely K.; Cadalbert R.; Testori E.; Oss A.; Past J.; Samoson A.; Ernst M.; Bockmann A.; Meier B. H. Angew. Chem., Int. Ed. 2014, 53, 12253–12256. 10.1002/anie.201405730. [DOI] [PubMed] [Google Scholar]; g Sinnige T.; Daniels M.; Baldus M.; Weingarth M. J. Am. Chem. Soc. 2014, 136, 4452–4455. 10.1021/ja412870m. [DOI] [PubMed] [Google Scholar]; h Mance D.; Sinnige T.; Kaplan M.; Narasimhan S.; Daniels M.; Houben K.; Baldus M.; Weingarth M. Angew. Chem., Int. Ed. 2015, 54, 15799–15803. 10.1002/anie.201509170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward M. E.; Shi L.; Lake E.; Krishnamurthy S.; Hutchins H.; Brown L. S.; Ladizhansky V. J. Am. Chem. Soc. 2011, 133, 17434–17443. 10.1021/ja207137h. [DOI] [PubMed] [Google Scholar]
- a Zhou D. H.; Nieuwkoop A. J.; Berthold D. A.; Comellas G.; Sperling L. J.; Tang M.; Shah G. J.; Brea E. J.; Lemkau L. R.; Rienstra C. M. J. Biomol. NMR 2012, 54, 291–305. 10.1007/s10858-012-9672-z. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Eddy M. T.; Su Y.; Silvers R.; Andreas L.; Clark L.; Wagner G.; Pintacuda G.; Emsley L.; Griffin R. G. J. Biomol. NMR 2015, 61, 299–310. 10.1007/s10858-015-9903-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros-Silva J.; Mance D.; Daniels M.; Jekhmane S.; Houben K.; Baldus M.; Weingarth M. Angew. Chem., Int. Ed. 2016, 55, 13606–13610. 10.1002/anie.201606594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama Y. Solid State Nucl. Magn. Reson. 2016, 78, 24–36. 10.1016/j.ssnmr.2016.06.002. [DOI] [PubMed] [Google Scholar]
- Andreas L. B.; Jaudzems K.; Stanek J.; Lalli D.; Bertarello A.; Le Marchand T.; Cala-De Paepe D.; Kotelovica S.; Akopjana I.; Knott B.; Wegner S.; Engelke F.; Lesage A.; Emsley L.; Tars K.; Herrmann T.; Pintacuda G. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 9187–9192. 10.1073/pnas.1602248113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mroue K. H.; Nishiyama Y.; Kumar Pandey M.; Gong B.; McNerny E.; Kohn D. H.; Morris M. D.; Ramamoorthy A. Sci. Rep. 2015, 5, 11991. 10.1038/srep11991. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Stanek J.; Andreas L. B.; Jaudzems K.; Cala D.; Lalli D.; Bertarello A.; Schubeis T.; Akopjana I.; Kotelovica S.; Tars K.; Pica A.; Leone S.; Picone D.; Xu Z. Q.; Dixon N. E.; Martinez D.; Berbon M.; El Mammeri N.; Noubhani A.; Saupe S.; Habenstein B.; Loquet A.; Pintacuda G. Angew. Chem., Int. Ed. 2016, 55, 15504–15509. 10.1002/anie.201607084. [DOI] [PubMed] [Google Scholar]
- a Bamann C.; Bamberg E.; Wachtveitl J.; Glaubitz C. Biochim. Biophys. Acta, Bioenerg. 2014, 1837, 614–625. 10.1016/j.bbabio.2013.09.010. [DOI] [PubMed] [Google Scholar]; b Inoue K.; Kato Y.; Kandori H. Trends Microbiol. 2015, 23, 91–98. 10.1016/j.tim.2014.10.009. [DOI] [PubMed] [Google Scholar]
- Reckel S.; Gottstein D.; Stehle J.; Lohr F.; Verhoefen M. K.; Takeda M.; Silvers R.; Kainosho M.; Glaubitz C.; Wachtveitl J.; Bernhard F.; Schwalbe H.; Guntert P.; Dotsch V. Angew. Chem., Int. Ed. 2011, 50, 11942–11946. 10.1002/anie.201105648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Stone K. M.; Voska J.; Kinnebrew M.; Pavlova A.; Junk M. J.; Han S. Biophys. J. 2013, 104, 472–481. 10.1016/j.bpj.2012.11.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Edwards D. T.; Huber T.; Hussain S.; Stone K. M.; Kinnebrew M.; Kaminker I.; Matalon E.; Sherwin M. S.; Goldfarb D.; Han S. Structure 2014, 22, 1677–1686. 10.1016/j.str.2014.09.008. [DOI] [PubMed] [Google Scholar]; c Maciejko J.; Mehler M.; Kaur J.; Lieblein T.; Morgner N.; Ouari O.; Tordo P.; Becker-Baldus J.; Glaubitz C. J. Am. Chem. Soc. 2015, 137, 9032–9043. 10.1021/jacs.5b03606. [DOI] [PubMed] [Google Scholar]
- a Shi L.; Ahmed M. A. M.; Zhang W.; Whited G.; Brown L. S.; Ladizhansky V. J. Mol. Biol. 2009, 386, 1078–1093. 10.1016/j.jmb.2009.01.011. [DOI] [PubMed] [Google Scholar]; b Shi L.; Lake E. M.; Ahmed M. A.; Brown L. S.; Ladizhansky V. Biochim. Biophys. Acta, Biomembr. 2009, 1788, 2563–2574. 10.1016/j.bbamem.2009.09.011. [DOI] [PubMed] [Google Scholar]
- Bennett A. E.; Rienstra C. M.; Auger M.; Lakshmi K. V.; Griffin R. G. J. Chem. Phys. 1995, 103, 6951–6958. 10.1063/1.470372. [DOI] [Google Scholar]
- Zhou D. H.; Rienstra C. M. J. Magn. Reson. 2008, 192, 167–172. 10.1016/j.jmr.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Good D. B.; Wang S.; Ward M. E.; Struppe J.; Brown L. S.; Lewandowski J. R.; Ladizhansky V. J. Am. Chem. Soc. 2014, 136, 2833–2842. 10.1021/ja411633w. [DOI] [PubMed] [Google Scholar]; b Saurel O.; Iordanov I.; Nars G.; Demange P.; Le Marchand T.; Andreas L. B.; Pintacuda G.; Milon A. J. Am. Chem. Soc. 2017, 139, 1590–1597. 10.1021/jacs.6b11565. [DOI] [PubMed] [Google Scholar]
- Asami S.; Szekely K.; Schanda P.; Meier B. H.; Reif B. J. Biomol. NMR 2012, 54, 155–168. 10.1007/s10858-012-9659-9. [DOI] [PubMed] [Google Scholar]
- Asami S.; Schmieder P.; Reif B. J. Am. Chem. Soc. 2010, 132, 15133–15135. 10.1021/ja106170h. [DOI] [PubMed] [Google Scholar]
- Huber M.; Hiller S.; Schanda P.; Ernst M.; Bockmann A.; Verel R.; Meier B. H. ChemPhysChem 2011, 12, 915–918. 10.1002/cphc.201100062. [DOI] [PubMed] [Google Scholar]
- Wang S.; Parthasarathy S.; Nishiyama Y.; Endo Y.; Nemoto T.; Yamauchi K.; Asakura T.; Takeda M.; Terauchi T.; Kainosho M.; Ishii Y. PLoS One 2015, 10, e0122714. 10.1371/journal.pone.0122714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Venters R. A.; Farmer B. T. II; Fierke C. A.; Spicer L. D. J. Mol. Biol. 1996, 264, 1101–1116. 10.1006/jmbi.1996.0699. [DOI] [PubMed] [Google Scholar]; b Smith A. A.; Ravotti F.; Testori E.; Cadalbert R.; Ernst M.; Bockmann A.; Meier B. H. J. Biomol. NMR 2017, 67, 109–119. 10.1007/s10858-016-0087-0. [DOI] [PubMed] [Google Scholar]
- Barbet-Massin E.; Pell A. J.; Jaudzems K.; Franks W. T.; Retel J. S.; Kotelovica S.; Akopjana I.; Tars K.; Emsley L.; Oschkinat H.; Lesage A.; Pintacuda G. J. Biomol. NMR 2013, 56, 379–386. 10.1007/s10858-013-9757-3. [DOI] [PubMed] [Google Scholar]
- Barbet-Massin E.; Pell A. J.; Knight M. J.; Webber A. L.; Felli I. C.; Pierattelli R.; Emsley L.; Lesage A.; Pintacuda G. ChemPhysChem 2013, 14, 3131–3137. 10.1002/cphc.201201097. [DOI] [PubMed] [Google Scholar]
- Cross T. A.; Sharma M.; Yi M.; Zhou H. X. Trends Biochem. Sci. 2011, 36, 117–125. 10.1016/j.tibs.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.