

Abstract
TIEG knockout (KO) mice exhibit a female-specific osteopenic phenotype and altered expression of TIEG in humans is associated with osteoporosis. Gene expression profiling studies identified sclerostin as one of the most highly up-regulated transcripts in the long bones of TIEG KO mice relative to WT littermates suggesting that TIEG may regulate SOST expression. TIEG was shown to substantially suppress SOST promoter activity and the regulatory elements through which TIEG functions were identified using promoter deletion and chromatin immunoprecipitation assays. Knockdown of TIEG in IDG-SW3 osteocyte cells using shRNA and CRISPR-Cas9 technology resulted in increased SOST expression and delayed mineralization, mimicking the results obtained from TIEG KO mouse bones. Given that TIEG is an estrogen regulated gene, and since changes in the hormonal milieu affect SOST expression, we performed ovariectomy (OVX) and estrogen replacement therapy (ERT) studies in WT and TIEG KO mice followed by miRNA and mRNA sequencing of cortical and trabecular compartments of femurs. SOST expression levels were considerably higher in cortical bone compared to trabecular bone. In cortical bone, SOST expression was increased following OVX only in WT mice and was suppressed following ERT in both genotypes. In contrast, SOST expression in trabecular bone was decreased following OVX and significantly increased following ERT. Interestingly, a number of miRNAs that are predicted to target sclerostin exhibited inverse expression levels in response to OVX and ERT. These data implicate important roles for TIEG and estrogen-regulated miRNAs in modulating SOST expression in bone.
Key terms: TIEG, KLF10, SOST, Sclerostin, Estrogen, Bone, Osteocyte
Introduction
Krüppel-like factors (KLFs) are zinc-finger containing transcription factors that are known to regulate diverse cellular functions including cell proliferation, apoptosis and differentiation (McConnell and Yang, 2010). Besides these cellular functions, they are also known to act as transcriptional co-activators and co-repressors in multiple cell and tissue types (McConnell and Yang, 2010). One member of this transcription factor family is TGFβ inducible early gene-1 (TIEG), also known as KLF10. This gene was originally discovered as an early response gene following TGFβ treatment in human osteoblasts (Subramaniam et al., 1995). Following its discovery, extensive work by our laboratory and others has identified a multitude of functions for TIEG in various cell types (Hawse et al., 2008b; Subramaniam et al., 2010b). Overexpression of TIEG has been shown to mimic the effects of TGFβ treatment in cancer cell lines by suppressing cell proliferation and inducing apoptosis (Hefferan et al., 2000a; Ribeiro et al., 1999; Tachibana et al., 1997). TIEG is known to regulate TGFβ signaling by inhibiting the expression of a negative regulator (Smad-7) and inducing the expression of a positive regulator (Smad-2), ultimately resulting in enhanced TGFβ signaling (Johnsen et al., 2002a; Johnsen et al., 2002b; Johnsen et al., 2002c).
Through the development of TIEG KO mice, we demonstrated that loss of TIEG expression in osteoblasts results in defective mineralization and decreased support of osteoclast differentiation in vitro (Subramaniam et al., 2005). To identify the cause of these defects, we also demonstrated that TIEG KO osteoblasts express decreased levels of Runx2, osterix, alkaline phosphatase and osteocalcin (Hawse et al., 2011; Subramaniam et al., 2005; Subramaniam et al., 2016) and increased levels of OPG (Subramaniam et al., 2010a). In osteoclasts, TIEG is known to decrease NFATc1 activation thereby suppressing AKT and MEK/ERK signaling ultimately resulting in reduced osteoclast survival (Cicek et al., 2011). At the tissue level, TIEG KO mice exhibit a gender specific (female only) osteopenic phenotype characterized by low bone mineral content and density throughout the skeleton (Bensamoun et al., 2006; Hawse et al., 2008a). Recently, using an ovariectomized (OVX) and estrogen replacement therapy (ERT) mouse model system, we demonstrated that TIEG plays an essential role in mediating estrogen (E2) signaling in bone(Hawse et al., 2014). In the human skeleton, TIEG is also known to play important roles in skeletal homeostasis as its mis-regulation and mutation are associated with osteoporosis in humans (Hopwood et al., 2009; Yerges et al., 2010).
Recently, we have demonstrated that TIEG KO mice exhibit decreased canonical Wnt pathway activity in bone (Subramaniam et al., 2017). Wnt signaling is well known to play essential roles in skeletal development and homeostasis (Baron and Kneissel, 2013). A major regulator of Wnt signaling in bone is sclerostin. Sclerostin is a secreted glycoprotein produced mainly by the osteocytes which functions to inhibit Wnt pathway activity by acting as a competitive inhibitor for Wnt ligand binding to LRP5 and LRP6 receptors (Bonewald, 2011). Mutations in, or deletion of, the SOST gene are known to cause a high bone mass phenotype in mice and humans (Balemans et al., 2001; Brunkow et al., 2001; Li et al., 2008). For these reasons, pharmaceutical companies have placed an emphasis on targeting sclerostin for the treatment of bone disorders including osteoporosis (McClung et al., 2014).
In the present report, we provide evidence that TIEG directly regulates the expression of SOST in the mouse skeleton. Through the use of gene expression profiling studies, we demonstrate that SOST mRNA levels are substantially elevated in the femurs of female TIEG KO mice relative to WT littermates. Concordantly, serum sclerostin protein levels are also significantly increased in TIEG KO mice. Through the use of transient transfection and chromatin immunoprecipitation assays, we have shown that TIEG binds to and suppresses SOST promoter activity. shRNA mediated suppression of TIEG, as well as CRISPR/Cas9-mediated disruption of the TIEG gene, in IDG-SW3 (SW3) osteocyte cells was shown to result in elevated expression of sclerostin during osteocyte differentiation. These effects were accompanied by a delay in the mineralization of SW3 cells. Through the use of RNA sequencing, we demonstrate that sclerostin expression levels are dramatically higher in cortical bone compared to trabecular bone compartments and provide the first evidence that sclerostin is differentially regulated by estrogen in these two types of bone. Finally, we have identified candidate miRNAs that may in part mediate sclerostin expression in bone since their expression levels inversely correlate with sclerostin mRNA levels. Overall, the results presented in this study implicate an important role for TIEG in regulating SOST gene expression which may in part explain the osteopenic phenotype observed in TIEG KO mice.
Methods
Mice and Study design
Generation of TIEG KO mice and characterization of their bone phenotype has been described previously (Bensamoun et al., 2006; Hawse et al., 2008a; Hawse et al., 2014; Subramaniam et al., 2005). For this study, littermate animals from heterozygous breeding pairs were utilized and sacrificed for analysis at approximately 2 months of age. For the ovariectomy and estrogen replacement studies, surgeries were conducted on 27 female wild-type and 27 TIEG KO mice at 4–5 weeks of age as previously described (Hawse et al., 2014). Mice were randomized to undergo sham surgeries (9 mice per genotype) or ovariectomy surgeries (18 mice per genotype). Immediately following surgeries, either 30 day slow releasing placebo or 17β-estradiol (1.88μg) pellets (Innovative Research of America, Sarasota, FL) were implanted subcutaneously between the shoulder blades. All mice used in this study were maintained in a temperature controlled room (22 ± 2°C) with light/dark cycle of 12 hours. Animals had free access to water and were fed with standard laboratory chow ad libitum. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Mayo Clinic Institutional Animal Care and Use Committee (Permit Number: A9615).
RNA isolation from bone tissues
For the studies presented in Figure 1, total RNA was extracted from femurs isolated from 10 WT female and 10 TIEG KO female mice. Briefly, mice were sacrificed at 2 months of age and femurs were dissected from both legs and cleaned of all muscle and tendon tissues. Bone marrow was flushed from femurs using a tuberculin syringe and PBS and entire bones were sheared in Trizol using an Ultra-Turrax homogenizer prior to extraction of total RNA. For the studies presented in Figures 5 and 6, nine WT and nine TIEG KO female mice from each of the surgical and treatment groups listed above were utilized. For these animals, femur epiphyses were trimmed at both ends and bone marrow was extracted by centrifugation. Trabecular and cortical bone compartments were separated as described previously (Kelly et al., 2014), sheared in Trizol and processed for total RNA isolation using a miRNeasy Mini Kit (Qiagen, Germantown, MD).
cDNA synthesis and Real-Time PCR analyses
For all of the animal studies, equal amounts of total RNA were pooled from 3 individual mice per genotype/surgical/treatment group to generate three replicate samples. RNA was reverse transcribed using the iScript™ cDNA Synthesis Kit (Bio-Rad, Hercules, CA) and real-time PCR was performed in triplicate using a Bio-Rad iCycler and a PerfeCTa™ SYBR Green Fast Mix™ for iQ real-time PCR kit (Quanta Biosciences, Gaithersburg, MD) as specified by the manufacturer. Cycling conditions were as follows: 95°C for 30 seconds followed by 40 cycles of 95°C for 3 seconds and 60°C for 30 seconds. Melt curves were generated to ensure amplification of a single PCR product. Quantitation of the PCR results was calculated based on the threshold cycle (Ct) following normalization to β-tubulin. All PCR primers were designed using Primer3 software (http://frodo.wi.mit.edu/primer3/) and were purchased from Integrated DNA Technologies (Coralville, IA). Primer sequences are listed in Supplemental Table 1.
Measurement of serum sclerostin levels
At the time of sacrifice, serum was isolated from terminal blood draws collected from 10 WT and 10 TIEG KO female mice. Serum sclerostin protein levels were determined using an ELISA kit from ALPCO (Salem, NH) following the manufacturer’s instructions. All assays were performed in duplicate and group averages were calculated.
SOST promoter constructs, transient transfection and luciferase assays
The 2 Kb mouse SOST promoter luciferase reporter vector used in this study was a kind gift from Dr. de Crombrugghe (MD Anderson) and its development was described previously (Zhou et al., 2010). Promoter deletion constructs (-1.95Kb and -1.8Kb) were generated by PCR amplification using the 2 Kb promoter construct as a template and subsequently cloned into the pGL3-basic luciferase reporter construct. The sequence integrity of all constructs was confirmed by Sanger sequencing. SOST promoter reporter constructs were transiently transfected into U2OS osteosarcoma cells using Fugene-6 (Roche, Indianapolis, IN) with either an empty pcDNA4-TO expression vector or a pcDNA4-TO mouse flag-tagged TIEG expression vector as previously described(Subramaniam et al., 2016). Twenty four hours following transfection, cells were lysed in passive lysis buffer (Promega, Madison, WI), lysates were quantitated for protein content and equal amounts of protein were used to measure luciferase activity using Luciferase Assay Reagent (Promega) and a Glomax-Dual Luminometer (Promega) (Hawse et al., 2011; Hawse et al., 2008c).
Chromatin Immunoprecipitation (ChIP) Assays
U2OS cells were plated at a density of approximately 50% in 100 mm tissue culture plates and transfected in triplicate with 5 μg of SOST promoter reporter constructs and 5 μg of a mouse flag-tagged TIEG expression vector. Following incubation for 24 hours, ChIP assays were performed as previously described (Subramaniam et al., 2017). Immunoprecipitations were carried out using 1 μg of either mouse IgG or M2 Flag antibody (Sigma). Inputs were generated in an identical fashion except for the exclusion of the antibody immunoprecipitation step. Quantitative Real-Time PCR was conducted in triplicate on all samples and representative data sets are shown. Quantitative PCR values were calculated based on the threshold cycle (Ct), normalized to input controls and compared to IgG immunoprecipitated samples. Primers flanking the putative TIEG binding sites are listed in Supplemental Table 1.
shRNA mediated suppression of TIEG in SW3 osteocyte cells
A pool of transduction ready shRNA lentiviral particles targeting mouse TIEG was purchased from Santa Cruz Biotechnology (sc-45464). SW3 osteocyte cells (a kind gift from Dr. Lynda Bonewald’s laboratory, Indiana University) were plated in 6 well tissue culture plates at a density of 50% and infected with the TIEG shRNA lentivirus following the manufacturer’s protocol. Forty eight hours following infection, cells were trypsinized and plated in 100mm plates with puromycin (1 μg/mL) supplemented cell culture medium to eliminate uninfected cells. Individual clonal cell lines were harvested, expanded and characterized for TIEG mRNA expression using RT-PCR as described above. Two clonal cell lines with significant suppression of TIEG expression (clones #3 and #36) were used for determination of SOST expression and mineralization capacity.
CRISPR-Cas9 mediated disruption of TIEG in SW3 osteocyte cells
An all-in-one CRISPR-Cas9 mouse TIEG gRNA expression construct (MTN278655) was purchased from GeneCopoeia (Rockville, MD). The expression vector was amplified in bacteria and 10 μg was subsequently transfected into SW3 osteocyte cells using Fugene-6 as described above. Forty eight hours following transfection, cells were trypsinized and plated at low density in 100 mm plates and cultured in G418 (300 μg/mL) containing cell culture medium to eliminate non-transfected cells. Clonal cell lines were expanded and examined for successful TIEG disruption using the IndelCheck™ CRISPR/TALEN insertion or deletion detection system (GeneCopoeia) following the manufacturer’s protocol. Two clonal cell lines with successful TIEG disruption (clones #6 and #8) were used for determination of SOST expression and mineralization capacity.
SOST expression and mineralization assays
The parental SW3 osteocyte cells, the TIEG shRNA clones #3 and #36 and the CRISPR clones #6 and #8 were plated in 12 well tissue culture plates at 30% confluence in αMEM containing 10% FBS (Gibco, Waltham, MA) and 1x antibiotic and antimycotic solution. Cells were maintained at 34°C in a 5% CO2 humidified incubator. When cells reached 100% confluence, the medium was replaced with differentiation medium containing 50 mg/L ascorbic acid and 10 mM β-glycerophosphate and placed at 37°C. Differentiation medium was replaced every third day throughout the differentiation time period. At indicated time points, cells were fixed in formalin and processed for Alizarin Red staining as previously described (Subramaniam et al., 2005). At these same time points, total RNA was also isolated from cells using Trizol reagent for analysis of TIEG and sclerostin mRNA expression as described above. Primers used for amplification of TIEG and sclerostin are listed in Supplemental Table 1.
miRNA and RNA sequencing
Following Trizol extraction, total RNA was isolated from trabecular and cortical bone compartments of WT and TIEG KO mice using a miRNeasy Mini kit (Qiagen) following the manufacturer’s protocol. The samples were sorted into 6 independent groups as follows. Three replicate RNA samples were generated for each genotype and treatment group by combining equal amounts of RNA isolated from trabecular or cortical bone compartments. All samples were tested for RNA integrity before submitting for sequencing. mRNA and miRNA sequencing was performed by the Mayo Clinic Molecular Biology Sequencing Core Facility using standard procedures and all data analysis was conducted by the Mayo Clinic Bioinformatics Core Facility (Kalari et al., 2014; Liao et al., 2014; Robinson et al., 2010; Trapnell et al., 2009; Wang et al., 2012).
Statistics
For all real-time PCR, ELISA, luciferase assays, chromatin immunoprecipitation assays and sequencing studies, a two-sided Student’s t-test was utilized. P-values < 0.05 were considered to be statistically significant. Data presented are mean of triplicate experiments +/− standard error. Calculations were conducted using Excel.
Results
Altered expression of SOST in TIEG KO mice
We have previously demonstrated that loss of TIEG expression in mice results in impaired canonical Wnt pathway activity in the skeleton (Subramaniam et al., 2017). Through gene expression profiling of WT and TIEG KO calvarial osteoblasts, we have identified differences in the expression levels of multiple Wnt ligands and their receptors between these two genotypes (Subramaniam et al., 2017). However, we were unable to detect sclerostin mRNA even in fully differentiated WT calvarial osteoblasts. To determine if altered expression of SOST may contribute to decreased canonical Wnt signaling in TIEG KO mice, we examined the expression levels of SOST in the long bones of WT and TIEG KO mice following removal of the marrow compartment. As shown in Figure 1A, SOST expression was increased by approximately 12-fold in the long bones of TIEG KO mice compared to WT littermates. These changes were confirmed at the protein level where serum sclerostin levels were significantly increased in TIEG KO animals (Figure 1B) suggesting that TIEG may directly regulate the expression of SOST in bone.
Figure 1.
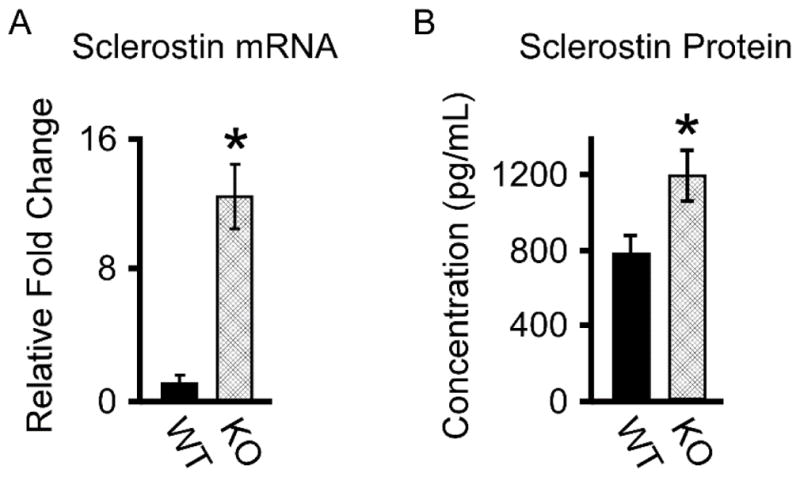
Sclerostin mRNA and protein levels in WT and TIEG KO mice. A). RT-PCR analysis of sclerostin mRNA levels in whole femurs devoid of marrow isolated from WT and TIEG KO mice. B). ELISA analysis of sclerostin protein levels in the serum of WT and TIEG KO mice. * Denotes significance at the P<0.05 level between WT and KO mice.
TIEG directly regulates SOST promoter activity
Given the observation that deletion of TIEG results in an increase in SOST expression, we sought to determine if SOST gene expression was under the control of TIEG. Transcription factor binding site analysis of the SOST promoter identified two putative TIEG binding sites (Figure 2A). To determine if TIEG could regulate SOST promoter activity, we performed transient transfection assays using a -2 Kb fragment of the SOST gene promoter in the presence and absence of TIEG over expression in U2OS cells. These assays revealed that over-expression of TIEG resulted in a greater than 90% reduction in SOST (2Kb) promoter activity (Figure 2B). Five-prime (5′)-deletion analysis suggested the involvement of both putative TIEG binding sites in mediating TIEG’s ability to suppress SOST promoter activity. Specifically, deletion of the distal TIEG binding site only partially relieved the inhibitory effects of TIEG (Figure 2C and D). However, deletion of both binding sites completely ablated the ability of TIEG to suppress SOST promoter activity (Figure 2C and D). Chromatin immunoprecipitation (ChIP) followed by PCR demonstrated that TIEG protein associates with the SOST promoter, effects that are completely abolished following deletion of both putative TIEG binding sites (Figure 2E). These results reveal that TIEG is able to regulate SOST gene expression by binding to and repressing SOST promoter activity.
Figure 2.

TIEG regulation of the SOST promoter. A). Identification of putative TIEG binding sites in the proximal promoter of the mouse SOST gene. B). Luciferase assays indicating activity of the SOST promoter following transfection into U2OS cells in the presence and absence of a TIEG expression construct. Depiction (C) and luciferase activity (D) of SOST promoter deletion constructs in U2OS cells in the presence and absence of a TIEG expression construct. E). Transient ChIP-PCR analysis indicating TIEG association with indicated SOST promoter constructs. * Denotes significance at the P<0.05 level between controls and indicated conditions.
Suppression of TIEG in SW3 osteocyte cells enhances SOST expression
Since deletion of TIEG in mice results in decreased Wnt pathway activity in bone that is concomitant with elevated SOST expression levels, we sought to mimic this effect in vitro using the SW3 osteocyte cell line. As a first step, SW3 clonal cell lines were developed in which TIEG expression was suppressed using a TIEG specific shRNA lentiviral construct. Multiple clonal cell lines in which TIEG expression was significantly repressed were identified and two representative cell lines (#’s 3 and 36) were chosen for further analysis (Figure 3A). To induce SOST gene expression, parental and clonal cell lines were differentiated in vitro for 29 days using ascorbic acid and β-glycerophosphate containing medium. Following differentiation, cell lines in which TIEG was suppressed exhibited significant increases in SOST mRNA levels relative to the parental cell line (Figure 3B). Suppression of TIEG expression also resulted in decreased mineralization capacity of SW3 osteocyte cells (Figure 3C).
Figure 3.

shRNA mediated knockdown of TIEG in SW3 cells. RT-PCR analysis of TIEG (A) and sclerostin (B) mRNA levels in parental SW3 osteocyte cells and two TIEG knockdown clonal cell lines (#’s 3 and 36). C). Alizarin red staining of parental SW3 osteocyte cells and two TIEG knockdown clonal cell lines depicting delayed mineralization following suppression of TIEG expression. * Denotes significance at the P<0.05 level between parental SW3 cells and indicated TIEG shRNA cell lines.
To validate these findings, we also developed SW3 cell lines in which the TIEG gene was disrupted using a CRISPR/Cas9 based approach. Multiple cell lines were identified and two representative clones (#’s 6 and 8) were chosen for further analysis (Figure 4A). Parental SW3 cells and the TIEG CRISPR clones were differentiated in vitro for 14 and 31 days and sclerostin mRNA levels were examined. As shown in Figure 4B, SOST expression was significantly increased at both time points in the TIEG CRISPR clones relative to the parental cell line. As was observed via shRNA mediated suppression of TIEG expression, genetic disruption of the TIEG gene also resulted in decreased mineralization capacity (Figure 4C). These in vitro results mimic the in vivo findings of elevated SOST expression in the long bones of TIEG KO mice and further demonstrate an important role for TIEG in regulating SOST expression in osteocyte cells.
Figure 4.

CRISPR mediated disruption of the TIEG gene in SW3 cells. A). PCR analysis depicting successful disruption of the TIEG gene via indel confirmation of two clonal cell lines (#’s 6 and 8) compared to parental cells. B). RT-PCR analysis of sclerostin mRNA levels in parental SW3 osteocyte cells and two TIEG CRISPR clones during the course of differentiation. C). Alizarin red staining of parental SW3 osteocyte cells and two TIEG CRISPR clonal cell lines depicting delayed mineralization following disruption of the TIEG gene. * Denotes significance at the P<0.05 level between parental SW3 cells and indicated TIEG CRISPR clonal cell lines.
Bone compartment-specific responses of SOST following OVX and ERT
We have previously demonstrated that global TIEG KO mice exhibit a female specific osteopenic bone phenotype as well as a muted skeletal response to OVX and ERT (Hawse et al., 2008a; Hawse et al., 2014). Further, sclerostin mRNA and protein are known to be elevated in post-menopausal women (Mirza et al., 2010), sclerostin expression exhibits an inverse relationship with free estrogen index (Drake and Khosla, 2017) and its expression decreases in response to short term ERT (Modder et al., 2011). In light of these past findings, and our present studies demonstrating that TIEG directly regulates SOST gene expression, we sought to determine if TIEG is involved in mediating the estrogen regulation of SOST expression. RNA sequencing was performed on trabecular and cortical bone compartments of WT and TIEG KO mice following sham operation, OVX + placebo and OVX + ERT. Interestingly, sclerostin mRNA levels were found to be significantly increased in cortical bone of TIEG KO mice relative to WT controls, but were actually slightly decreased in trabecular bone (Figure 5A and C). Following OVX, a significant increase in SOST expression was observed in cortical bone of WT mice, but not TIEG KO mice (Figure 5A). In trabecular bone, SOST expression actually decreased following OVX in both genotypes (Figure 5C). As expected, ERT resulted in decreased expression of SOST in the cortical bone of WT and TIEG KO mice, but substantially increased sclerostin mRNA levels in trabecular bone (Figure 5A and C). Overall, sclerostin mRNA levels were substantially higher in cortical bone relative to trabecular bone of both WT (approximately 20-fold) and TIEG KO (approximately 80-fold) mice (Figure 5A and C). RNA sequencing results were confirmed using RT-PCR (Figure 5B and D). These results demonstrate that SOST expression is dramatically higher in cortical bone compared to trabecular bone, that OVX and ERT have bone compartment specific effects on SOST expression and that TIEG does not appear to play a role in mediating estrogenic effects on the SOST gene.
Figure 5.

SOST expression levels in cortical vs. trabecular bone compartments of femurs isolated from WT and TIEG KO mice following sham, ovariectomy (OVX) and OVX + estrogen replacement therapy (OVX E2). Sclerostin mRNA levels in cortical bone as detected by mRNAseq (A) and RT-PCR confirmation (B) in indicated mouse groups. Sclerostin mRNA levels in trabecular bone as detected by mRNAseq (C) and RT-PCR confirmation (D) in indicated mouse groups. Bars denote significance at the P<0.05 level between indicated mouse groups.
Potential involvement of miRNAs in the estrogen regulation of SOST
Concurrent with the mRNA sequencing studies, we also performed microRNA (miRNA) sequencing on trabecular and cortical bone of WT and TIEG KO mice following sham, OVX and OVX + ERT to determine if altered miRNA expression profiles may impact SOST expression levels. As a first step, we utilized 4 different software platforms (miRSearch, miRDB, TargetScan and DIANA-microT) to identify miRNAs that are predicted to target sclerostin. Each of these programs identified multiple miRNAs, 11 of which were identified by at least 2 different programs (Figure 6A). The expression levels of these 11 miRNAs were culled from the miRNA sequencing data which demonstrated that only 3 of these miRNAs (miR-676-5p, -218-5p and -324-3p) were expressed in bone (Figure 6B). Interestingly, all three of these miRNAs were more highly expressed in trabecular bone compared to cortical bone, patterns that inversely correlate with SOST expression levels. miR-218-5p and -324-3p exhibited decreased expression in cortical bone of TIEG KO mice relative to WT littermates (Figure 6C), patterns of expression that are inverse of sclerostin mRNA levels in the same bone samples. Following OVX, only miR-324-3p exhibited decreased expression in the cortical bone of WT mice, but was unaffected in TIEG KO animals (Figure 6C). No consistent changes in the expression levels of these three miRs were identified in trabecular bone of WT or TIEG KO mice following OVX (Figure 6C). However, in response to ERT, the expression levels of miR-676-5p and -324-3p increased in the cortical bone of WT mice while miR-676-5p and -218-5p increased in TIEG KO mice (Figure 6C). In trabecular bone, the expression levels of miR-676-5p decreased and miR-218-5p increased in response to ERT (Figure 6C). Overall, these data suggest that specific miRs may play important roles in modulating SOST expression in specific bone compartments following OVX and ERT. However, further studies are necessary to confirm direct involvement of the identified miRNAs.
Figure 6.

Expression levels of miRNAs predicted to target sclerostin in cortical vs. trabecular bone compartments of femurs isolated from WT and TIEG KO mice following sham, ovariectomy (OVX) and OVX + estrogen replacement therapy (OVX E2). A). Venn diagram indicating the number and overlap of miRs predicted to target sclerostin using indicated software programs. B). Average counts per million (CPM) for all miRs predicted to target sclerostin by at least 2 of the 4 indicated software programs in cortical and trabecular bone compartments as determined by microRNAseq. C). Expression levels of sclerostin-targeting miRs in cortical and trabecular bone compartments of femurs isolated from indicated mouse groups as determined by microRNAseq. Bars denote significance at the P<0.05 level between indicated mouse groups.
Discussion
In this present study we have demonstrated that TIEG KO mice exhibit increased expression of SOST at the mRNA level in long bones, an effect that is reflected at the protein level in serum. We also show that TIEG down regulates SOST promoter activity by binding to its proximal promoter region and suppressing transcriptional activity. Using the SW3 mouse osteocyte cell line, we provide evidence that suppression of TIEG expression results in increased SOST expression during differentiation. Furthermore, decreased TIEG expression in SW3 cells resulted in delayed mineralization. Using an OVX induced mouse model of osteoporosis, as well as ERT, we provide evidence that sclerostin mRNA levels are differentially regulated in a bone compartment specific manner. Specifically, sclerostin levels increase following OVX and decrease in response to ERT in cortical bone, but exhibit opposite expression patterns in trabecular bone. Finally, we have identified 3 different miRNAs that are expressed in mouse bone and are predicted to target sclerostin that may be involved in modulating the expression levels of sclerostin in response to OVX and ERT.
Canonical Wnt signaling is essential for normal development and maintenance of the skeleton. Recently, we have demonstrated that TIEG plays an important role in regulating canonical Wnt signaling in bone (Subramaniam et al., 2017). In that study, we provide evidence that TIEG enhances Wnt signaling through two mechanisms, one by suppressing GSK-3β activity and inducing β-catenin nuclear localization and two by serving as a co-activator for Lef1 and β-catenin transcriptional activity (Subramaniam et al., 2017). In the present report, we provide an additional yet unidentified role for TIEG in regulating canonical Wnt pathway activity by suppressing sclerostin expression. Sclerostin is a secreted glycoprotein mainly produced by osteocytes in bone that functions as potent inhibitor of Wnt signaling (Bonewald, 2011). For these reasons, sclerostin antibody therapy is currently being used to treat osteoporosis and other skeletal disorders (Ishibashi et al., 2017; MacNabb et al., 2016).
It is interesting to note that TIEG, a transcription factor originally discovered in human osteoblasts (Subramaniam et al., 1995), is now known to regulate multiple important signaling pathways in the skeleton including TGFβ (Johnsen et al., 2002a; Johnsen et al., 2002b; Johnsen et al., 2002c), BMP (Hefferan et al., 2000b; Yadav et al., 2016) and Wnt (Subramaniam et al., 2017). With regard to bone, BMP, like Wnt signaling, is essential for bone development. We have previously demonstrated that deletion of TIEG inhibits the expression levels of Runx2 and osterix in osteoblasts cells and impairs the responsiveness of these two critical bone genes to both TGFβ and BMP signaling (Hawse et al., 2011; Subramaniam et al., 2016). Recently, Yadav and colleagues have demonstrated that TIEG is sufficient for the promotion of osteogenesis even in the absence of BMP signaling (Yadav et al., 2016), further suggesting that TIEG is a critical downstream regulator of this signaling pathway in bone. These past findings, our recent report (Subramaniam et al., 2017) and the present study demonstrating a direct role for TIEG in regulating SOST expression suggest that TIEG may also serve to mediate cross-talk between these pathways in the skeleton. Indeed, it is known that the TGFβ/BMP and Wnt signaling pathways are evolutionarily conserved and functionally integrated with regard to bone development and homeostasis (Guo and Wang, 2009; Itasaki and Hoppler, 2010). The relevance of TIEG in the skeleton is further supported by two studies demonstrating that mutations in the TIEG gene (Yerges et al., 2010), or alterations in TIEG expression (Hopwood et al., 2009), are associated with osteoporosis in humans.
In addition to modulating the TGFβ and BMP pathways, TIEG is also involved in regulating estrogen signaling in bone. We have previously shown that TIEG is an estrogen regulated gene (Hawse et al., 2008c), and that loss of TIEG expression diminishes the effects of OVX and ERT on the mouse skeleton (Hawse et al., 2014). A number of studies have suggested that sex steroids, including estrogen, can impact SOST expression and Wnt pathway activity in bone (Drake and Khosla, 2017). Specifically, post-menopausal women are known to have higher circulating sclerostin protein levels (Clarke and Drake, 2013; Modder et al., 2011) and treatment of post-menopausal women with estrogen is known to suppress sclerostin expression (Modder et al., 2011). A similar study by Chung and colleagues also showed that treatment of post-menopausal women with raloxifene, a selective estrogen receptor modulator, resulted in reduced sclerostin protein levels (Chung et al., 2012). In mice, two independent studies have reported conflicting results regarding the expression patterns of SOST following OVX and ERT. Studies by Kim and colleagues showed increased levels of sclerostin protein following OVX in the femur using IHC, an effect that was prevented with ERT (Kim et al., 2012). This observation is similar to that which has been reported in humans as mentioned above. Conversely, a second report by Jasterzebski and colleagues reported that sclerostin levels decrease in response to OVX in the mouse (Jastrzebski et al., 2013). However, they did not perform ERT in their study.
Our data presented here may provide a basis for some of the discrepancies that have been reported regarding SOST expression patterns following OVX and ERT in mice. We are the first to demonstrate bone compartment specific expression patterns of sclerostin in the mouse skeleton. In agreement with the humans studies and the report by Kim et al., we observed increased levels of sclerostin mRNA following OVX in cortical bone of mouse femurs. This increased expression was blocked by ERT. Interestingly, these patterns of sclerostin expression were opposite in trabecular bone of the femur where decreased levels of sclerostin mRNA were observed following OVX and increased expression was observed in response to ERT. The basis for the differential expression of SOST between these two bone compartments is not known but may relate to differences in the cellular composition, the expression levels of ERα and/or ERβ and the bone turnover rate. Further studies are warranted to confirm these findings and to elucidate the mechanistic basis and biological relevance for these differential responses.
MicroRNAs are well known to play important roles in a multitude of biological processes including bone development and maintenance. A number of specific miRNAs have also been shown to regulate canonical Wnt signaling in bone cells (Wang and Xu, 2010; Zhang et al., 2011). To determine if miRs may be involved in modulating the expression of SOST in cortical and trabecular bone following OVX and/or ERT, we examined the expression levels of specific miRs predicted to target sclerostin. Of the 11 miRs identified to potentially target sclerostin using target prediction software, only 3 were shown to be expressed in either cortical or trabecular bone using microRNA sequencing. Interestingly, all three of these miRs (676-5p, 218-5p and 324-3p) were expressed at approximately 2-fold higher levels in trabecular bone relative to cortical bone regardless of mouse genotype. These patterns are inversely correlated with the expression levels of SOST in these same bone samples. In cortical bone, where ERT was shown to suppress sclerostin mRNA levels, these three miRs exhibited increased expression in at least one of the two mouse genotypes. While these data are correlative in nature and do not prove that these miRs are involved in the modulation of sclerostin levels in mice, miR-218-5p has previously been shown to directly target the 3′ UTR of sclerostin in osteoblasts (Hassan et al., 2012) further supporting its potential involvement in the observations described here.
In summary, these data demonstrate that loss of TIEG expression in mice results in increased expression of sclerostin mRNA and protein. TIEG is shown to directly repress SOST expression in bone cell by interacting with 2 TIEG binding sites located in the proximal promoter of the SOST gene. Through the use of RNA sequencing of cortical and trabecular bone compartments, we have for the first time demonstrated that sclerostin mRNA levels substantially differ between cortical and trabecular bone and reveal that the expression patterns of SOST in response to OVX and ERT are completely opposite of one another in these two bone compartments. Finally, we have identified 3 miRs whose expression levels inversely correlate with sclerostin mRNA levels in cortical and trabecular bone that may also be involved in modulating sclerostin expression in response to changes in the hormonal milieu.
Supplementary Material
Acknowledgments
Contract grant sponsor: The National Institutes of Health, National Institute of Dental and Craniofacial Research; Contract grant number (JRH and MS): R01 DE14036.
Contract grant sponsor: The National Institutes of Health, National Institute of Arthritis and Musculoskeletal and Skin Diseases; Contract grant number (DGM): R01 AR068275.
Contract grant sponsor: The Eisenberg Foundation; Contract grant number (JRH and MS): None.
The authors would like to thank Dr. Thomas C. Spelsberg for his excellent career mentorship, support and helpful suggestions. The work presented here was supported by the National Institutes of Health under award numbers R01 DE14036 (JRH and MS) and R01 AR068275 (DGM) as well as a special gift from the Eisenberg Foundation (JRH and MS).
References
- Balemans W, Ebeling M, Patel N, Van Hul E, Olson P, Dioszegi M, Lacza C, Wuyts W, Van Den Ende J, Willems P, Paes-Alves AF, Hill S, Bueno M, Ramos FJ, Tacconi P, Dikkers FG, Stratakis C, Lindpaintner K, Vickery B, Foernzler D, Van Hul W. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST) Hum Mol Genet. 2001;10(5):537–543. doi: 10.1093/hmg/10.5.537. [DOI] [PubMed] [Google Scholar]
- Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. 2013;19(2):179–192. doi: 10.1038/nm.3074. [DOI] [PubMed] [Google Scholar]
- Bensamoun SF, Hawse JR, Subramaniam M, Ilharreborde B, Bassillais A, Benhamou CL, Fraser DG, Oursler MJ, Amadio PC, An KN, Spelsberg TC. TGFbeta inducible early gene-1 knockout mice display defects in bone strength and microarchitecture. Bone. 2006;39(6):1244–1251. doi: 10.1016/j.bone.2006.05.021. [DOI] [PubMed] [Google Scholar]
- Bonewald LF. The amazing osteocyte. J Bone Miner Res. 2011;26(2):229–238. doi: 10.1002/jbmr.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunkow ME, Gardner JC, Van Ness J, Paeper BW, Kovacevich BR, Proll S, Skonier JE, Zhao L, Sabo PJ, Fu Y, Alisch RS, Gillett L, Colbert T, Tacconi P, Galas D, Hamersma H, Beighton P, Mulligan J. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. 2001;68(3):577–589. doi: 10.1086/318811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung YE, Lee SH, Lee SY, Kim SY, Kim HH, Mirza FS, Lee SK, Lorenzo JA, Kim GS, Koh JM. Long-term treatment with raloxifene, but not bisphosphonates, reduces circulating sclerostin levels in postmenopausal women. Osteoporos Int. 2012;23(4):1235–1243. doi: 10.1007/s00198-011-1675-1. [DOI] [PubMed] [Google Scholar]
- Cicek M, Vrabel A, Sturchio C, Pederson L, Hawse JR, Subramaniam M, Spelsberg TC, Oursler MJ. TGF-β inducible early gene 1 regulates osteoclast differentiation and survival by mediating the NFATc1, AKT, and MEK/ERK signaling pathways. PLoS One. 2011;6(3) doi: 10.1371/journal.pone.0017522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke BL, Drake MT. Clinical utility of serum sclerostin measurements. Bonekey Rep. 2013;2:361. doi: 10.1038/bonekey.2013.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake MT, Khosla S. Hormonal and systemic regulation of sclerostin. Bone. 2017;96:8–17. doi: 10.1016/j.bone.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Wang XF. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 2009;19(1):71–88. doi: 10.1038/cr.2008.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan MQ, Maeda Y, Taipaleenmaki H, Zhang W, Jafferji M, Gordon JA, Li Z, Croce CM, van Wijnen AJ, Stein JL, Stein GS, Lian JB. miR-218 directs a Wnt signaling circuit to promote differentiation of osteoblasts and osteomimicry of metastatic cancer cells. J Biol Chem. 2012;287(50):42084–42092. doi: 10.1074/jbc.M112.377515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawse JR, Cicek M, Grygo SB, Bruinsma ES, Rajamannan NM, van Wijnen AJ, Lian JB, Stein GS, Oursler MJ, Subramaniam M, Spelsberg TC. TIEG1/KLF10 modulates Runx2 expression and activity in osteoblasts. PLoS One. 2011;6(4):e19429. doi: 10.1371/journal.pone.0019429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawse JR, Iwaniec UT, Bensamoun SF, Monroe DG, Peters KD, Ilharreborde B, Rajamannan NM, Oursler MJ, Turner RT, Spelsberg TC, Subramaniam M. TIEG-null mice display an osteopenic gender-specific phenotype. Bone. 2008a;42(6):1025–1031. doi: 10.1016/j.bone.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawse JR, Pitel KS, Cicek M, Philbrick KA, Gingery A, Peters KD, Syed FA, Ingle JN, Suman VJ, Iwaniec UT, Turner RT, Spelsberg TC, Subramaniam M. TGFbeta inducible early gene-1 plays an important role in mediating estrogen signaling in the skeleton. J Bone Miner Res. 2014;29(5):1206–1216. doi: 10.1002/jbmr.2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawse JR, Subramaniam M, Ingle JN, Oursler MJ, Rajamannan NM, Spelsberg TC. Estrogen-TGFbeta cross-talk in bone and other cell types: role of TIEG, Runx2, and other transcription factors. J Cell Biochem. 2008b;103(2):383–392. doi: 10.1002/jcb.21425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawse JR, Subramaniam M, Monroe DG, Hemmingsen AH, Ingle JN, Khosla S, Oursler MJ, Spelsberg TC. Estrogen receptor beta isoform-specific induction of transforming growth factor beta-inducible early gene-1 in human osteoblast cells: an essential role for the activation function 1 domain. Mol Endocrinol. 2008c;22(7):1579–1595. doi: 10.1210/me.2007-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefferan TE, Reinholz GG, Rickard DJ, Johnsen SA, Waters KM, Subramaniam M, Spelsberg TC. Overexpression of a nuclear protein, TIEG, mimics transforming growth factor-beta action in human osteoblast cells. J Biol Chem. 2000a;275(27):20255–20259. doi: 10.1074/jbc.C000135200. [DOI] [PubMed] [Google Scholar]
- Hefferan TE, Subramaniam M, Khosla S, Riggs BL, Spelsberg TC. Cytokine-specific induction of the TGF-beta inducible early gene (TIEG): regulation by specific members of the TGF-beta family. J Cell Biochem. 2000b;78(3):380–390. doi: 10.1002/1097-4644(20000901)78:3<380::aid-jcb4>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Hopwood B, Tsykin A, Findlay DM, Fazzalari NL. Gene expression profile of the bone microenvironment in human fragility fracture bone. Bone. 2009;44(1):87–101. doi: 10.1016/j.bone.2008.08.120. [DOI] [PubMed] [Google Scholar]
- Ishibashi H, Crittenden DB, Miyauchi A, Libanati C, Maddox J, Fan M, Chen L, Grauer A. Romosozumab increases bone mineral density in postmenopausal Japanese women with osteoporosis: A phase 2 study. Bone. 2017;103:209–215. doi: 10.1016/j.bone.2017.07.005. [DOI] [PubMed] [Google Scholar]
- Itasaki N, Hoppler S. Crosstalk between Wnt and bone morphogenic protein signaling: a turbulent relationship. Dev Dyn. 2010;239(1):16–33. doi: 10.1002/dvdy.22009. [DOI] [PubMed] [Google Scholar]
- Jastrzebski S, Kalinowski J, Stolina M, Mirza F, Torreggiani E, Kalajzic I, Won HY, Lee SK, Lorenzo J. Changes in bone sclerostin levels in mice after ovariectomy vary independently of changes in serum sclerostin levels. J Bone Miner Res. 2013;28(3):618–626. doi: 10.1002/jbmr.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsen SA, Subramaniam M, Janknecht R, Spelsberg TC. TGFbeta inducible early gene enhances TGFbeta/Smad-dependent transcriptional responses. Oncogene. 2002a;21(37):5783–5790. doi: 10.1038/sj.onc.1205681. [DOI] [PubMed] [Google Scholar]
- Johnsen SA, Subramaniam M, Katagiri T, Janknecht R, Spelsberg TC. Transcriptional regulation of Smad2 is required for enhancement of TGFbeta/Smad signaling by TGFbeta inducible early gene. J Cell Biochem. 2002b;87(2):233–241. doi: 10.1002/jcb.10299. [DOI] [PubMed] [Google Scholar]
- Johnsen SA, Subramaniam M, Monroe DG, Janknecht R, Spelsberg TC. Modulation of transforming growth factor beta (TGFbeta)/Smad transcriptional responses through targeted degradation of TGFbeta-inducible early gene-1 by human seven in absentia homologue. J Biol Chem. 2002c;277(34):30754–30759. doi: 10.1074/jbc.M204812200. [DOI] [PubMed] [Google Scholar]
- Kalari KR, Nair AA, Bhavsar JD, O’Brien DR, Davila JI, Bockol MA, Nie J, Tang X, Baheti S, Doughty JB, Middha S, Sicotte H, Thompson AE, Asmann YW, Kocher JP. MAP-RSeq: Mayo Analysis Pipeline for RNA sequencing. BMC Bioinformatics. 2014;15:224. doi: 10.1186/1471-2105-15-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly NH, Schimenti JC, Patrick Ross F, van der Meulen MC. A method for isolating high quality RNA from mouse cortical and cancellous bone. Bone. 2014;68:1–5. doi: 10.1016/j.bone.2014.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BJ, Bae SJ, Lee SY, Lee YS, Baek JE, Park SY, Lee SH, Koh JM, Kim GS. TNF-alpha mediates the stimulation of sclerostin expression in an estrogen-deficient condition. Biochem Biophys Res Commun. 2012;424(1):170–175. doi: 10.1016/j.bbrc.2012.06.100. [DOI] [PubMed] [Google Scholar]
- Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D’Agostin D, Kurahara C, Gao Y, Cao J, Gong J, Asuncion F, Barrero M, Warmington K, Dwyer D, Stolina M, Morony S, Sarosi I, Kostenuik PJ, Lacey DL, Simonet WS, Ke HZ, Paszty C. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23(6):860–869. doi: 10.1359/jbmr.080216. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- MacNabb C, Patton D, Hayes JS. Sclerostin Antibody Therapy for the Treatment of Osteoporosis: Clinical Prospects and Challenges. J Osteoporos. 2016;2016:6217286. doi: 10.1155/2016/6217286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClung MR, Grauer A, Boonen S, Bolognese MA, Brown JP, Diez-Perez A, Langdahl BL, Reginster JY, Zanchetta JR, Wasserman SM, Katz L, Maddox J, Yang YC, Libanati C, Bone HG. Romosozumab in postmenopausal women with low bone mineral density. N Engl J Med. 2014;370(5):412–420. doi: 10.1056/NEJMoa1305224. [DOI] [PubMed] [Google Scholar]
- McConnell BB, Yang VW. Mammalian Kruppel-like factors in health and diseases. Physiol Rev. 2010;90(4):1337–1381. doi: 10.1152/physrev.00058.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza FS, Padhi ID, Raisz LG, Lorenzo JA. Serum sclerostin levels negatively correlate with parathyroid hormone levels and free estrogen index in postmenopausal women. J Clin Endocrinol Metab. 2010;95(4):1991–1997. doi: 10.1210/jc.2009-2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modder UI, Clowes JA, Hoey K, Peterson JM, McCready L, Oursler MJ, Riggs BL, Khosla S. Regulation of circulating sclerostin levels by sex steroids in women and in men. J Bone Miner Res. 2011;26(1):27–34. doi: 10.1002/jbmr.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro A, Bronk SF, Roberts PJ, Urrutia R, Gores GJ. The transforming growth factor beta(1)-inducible transcription factor TIEG1, mediates apoptosis through oxidative stress. Hepatology. 1999;30(6):1490–1497. doi: 10.1002/hep.510300620. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam M, Cicek M, Pitel KS, Bruinsma ES, Nelson Holte MH, Withers SG, Rajamannan NM, Secreto FJ, Venuprasad K, Hawse JR. TIEG1 modulates beta-catenin sub-cellular localization and enhances Wnt signaling in bone. Nucleic Acids Res. 2017 doi: 10.1093/nar/gkx118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam M, Gorny G, Johnsen SA, Monroe DG, Evans GL, Fraser DG, Rickard DJ, Rasmussen K, van Deursen JM, Turner RT, Oursler MJ, Spelsberg TC. TIEG1 null mouse-derived osteoblasts are defective in mineralization and in support of osteoclast differentiation in vitro. Mol Cell Biol. 2005;25(3):1191–1199. doi: 10.1128/MCB.25.3.1191-1199.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam M, Harris SA, Oursler MJ, Rasmussen K, Riggs BL, Spelsberg TC. Identification of a novel TGF-beta-regulated gene encoding a putative zinc finger protein in human osteoblasts. Nucleic Acids Res. 1995;23(23):4907–4912. doi: 10.1093/nar/23.23.4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam M, Hawse JR, Bruinsma ES, Grygo SB, Cicek M, Oursler MJ, Spelsberg TC. TGFbeta inducible early gene-1 directly binds to, and represses, the OPG promoter in osteoblasts. Biochem Biophys Res Commun. 2010a;392(1):72–76. doi: 10.1016/j.bbrc.2009.12.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam M, Hawse JR, Rajamannan NM, Ingle JN, Spelsberg TC. Functional role of KLF10 in multiple disease processes. BioFactors (Oxford, England) 2010b;36(1):8–18. doi: 10.1002/biof.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam M, Pitel KS, Withers SG, Drissi H, Hawse JR. TIEG1 enhances Osterix expression and mediates its induction by TGFbeta and BMP2 in osteoblasts. Biochem Biophys Res Commun. 2016;470(3):528–533. doi: 10.1016/j.bbrc.2016.01.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana I, Imoto M, Adjei PN, Gores GJ, Subramaniam M, Spelsberg TC, Urrutia R. Overexpression of the TGFbeta-regulated zinc finger encoding gene, TIEG, induces apoptosis in pancreatic epithelial cells. J Clin Invest. 1997;99(10):2365–2374. doi: 10.1172/JCI119418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wang S, Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics. 2012;28(16):2184–2185. doi: 10.1093/bioinformatics/bts356. [DOI] [PubMed] [Google Scholar]
- Wang T, Xu Z. miR-27 promotes osteoblast differentiation by modulating Wnt signaling. Biochem Biophys Res Commun. 2010;402(2):186–189. doi: 10.1016/j.bbrc.2010.08.031. [DOI] [PubMed] [Google Scholar]
- Yadav PS, Khan MP, Prashar P, Duggal S, Rath SK, Chattopadhyay N, Bandyopadhyay A. Characterization of BMP signaling dependent osteogenesis using a BMP depletable avianized bone marrow stromal cell line (TVA-BMSC) Bone. 2016;91:39–52. doi: 10.1016/j.bone.2016.07.010. [DOI] [PubMed] [Google Scholar]
- Yerges LM, Klei L, Cauley JA, Roeder K, Kammerer CM, Ensrud KE, Nestlerode CS, Lewis C, Lang TF, Barrett-Connor E, Moffett SP, Hoffman AR, Ferrell RE, Orwoll ES, Zmuda JM. Candidate gene analysis of femoral neck trabecular and cortical volumetric bone mineral density in older men. J Bone Miner Res. 2010;25(2):330–338. doi: 10.1359/jbmr.090729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Tu Q, Bonewald LF, He X, Stein G, Lian J, Chen J. Effects of miR-335–5p in modulating osteogenic differentiation by specifically downregulating Wnt antagonist DKK1. J Bone Miner Res. 2011;26(8):1953–1963. doi: 10.1002/jbmr.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Zhang Z, Feng JQ, Dusevich VM, Sinha K, Zhang H, Darnay BG, de Crombrugghe B. Multiple functions of Osterix are required for bone growth and homeostasis in postnatal mice. Proc Natl Acad Sci U S A. 2010;107(29):12919–12924. doi: 10.1073/pnas.0912855107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.