

Abstract
The A2A adenosine receptor (A2AAR) is a G protein-coupled receptor that is pharmacologically targeted for the treatment of inflammation, sepsis, cancer, neuro-degeneration, and Parkinson’s disease. Recently, we applied long-timescale molecular dynamics simulations on two ligand-free receptor conformations, starting from the agonist-bound (PDB ID:3QAK) and antagonist-bound (PDB ID:3EML) X-ray structures. This analysis revealed four distinct conformers of the A2AAR: the active, intermediate 1, intermediate 2, and inactive. In this study, we apply the fragment-based mapping algorithm, FTMap, on these receptor conformations to uncover five non-orthosteric sites on the A2AAR. Two sites that are identified in the active conformation are located in the intracellular region of the transmembrane helices (TM) 3/TM4 and the G protein-binding site in the intracellular region between TM2/TM3/TM6/TM7. Three sites are identified in the intermediate 1 and intermediate 2 conformations, annexing a site in the lipid interface of TM5/TM6. Five sites are identified in the inactive conformation, comprising of a site in the intracellular region of TM1/TM7, and in the extracellular region of TM3/TM4 of the A2AAR. We postulate that these sites on the A2AAR be screened for allosteric modulators for the treatment of inflammatory and neurological diseases.
Keywords: G protein-coupled receptors, GPCR, A2A Adenosine Receptor, Allostery, Fragment Mapping, FTMap
Graphical Abstract
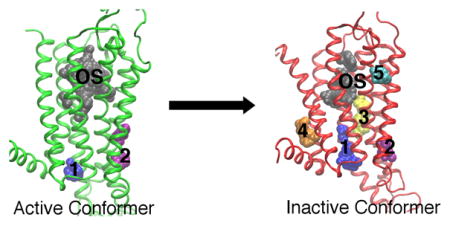
We uncover five non-orthosteric sites on the A2A adenosine receptor that can be screened for allosteric modulators for the treatment of inflammatory and neurological diseases. Site one is in the intracellular region of the transmembrane helices (TM) 3/TM4, and site two is the G protein-binding site between TM2/TM3/TM6/TM7. Site three is on the lipid interface of TM5/TM6, site four is in the intracellular region of TM1/TM7, and site five is in the extracellular region of TM3/TM4.
Introduction
The adenosine receptors are a class of four G protein-coupled receptors, A1, A2A, A2B, and A3, whose endogenous ligand is the purine adenosine [1–3]. The A2A adenosine receptor (A2AAR) is expressed in leukocytes, such as T cells, monocytes, macrophages, and natural killer cells (NKT) [2–4], and in the brain, specifically the GABAergic neurons in the basal ganglia that are responsible for voluntary movement [4, 5]. The receptor couples to the Gs protein in the periphery and the Golf protein in the brain [2, 4]. The G protein stimulates the adenylyl cyclase pathway, causing a surge in cAMP, and the activation of the protein kinase A (PKA) signaling cascade [1–3, 5].
In leukocytes, the activation of PKA by the A2AAR produces anti-inflammatory effects [2, 3]. Furthermore, a knockout of the A2AAR in mice causes an accumulation of pro-inflammatory cytokines and leads to tissue damage [6]. When the A2AAR is activated on T cells, the adenylyl cyclase/cAMP/PKA pathway leads to the inhibition of NF-κB, a pro-inflammatory transcription factor [3]. Additionally, the pathway activates anti-inflammatory transcription factors, which leads to the decrease in pro-inflammatory cytokines [2]. Because of its role in the immune response, A2AAR agonists are being developed and studied for the treatment of sepsis, which is caused by hyper-inflammation and characterized by multiple organ failure, and multiple sclerosis, where leukocytes attack the myelin on neurons and cause lesions in the brain [1, 2, 7]. Moreover, the anti-inflammatory role of the A2AAR in the immune system is protective to tumors, suggesting that antagonists of the receptor can be developed to treat cancer [8].
Antagonists of the A2AAR is a treatment option for Parkinson’s disease. Parkinson’s disease is a motor disorder caused by the degradation of the dopamine pathway. In the GABAergic neurons, the activation of the A2AAR acts counter to the D2 dopamine signaling pathway, and suppresses dopamine release [9].
Overall, A2AAR antagonists can treat Parkinson’s disease [9] and cancer [8]. A2AAR agonists can treat sepsis [2] and inflammation [1, 7]. Allosteric modulators bind to sites spatially different from the orthosteric binding site [10, 11]. Because allosteric regions are not as conserved as the orthosteric ones across receptor subtypes, modulators that bind to non-orthosteric sites can maintain subtype specificity. Allosteric modulators alter the association or disassociation rate of the endogenous ligand [10]. They include positive allosteric modulators (PAM), which increase the effect of the orthosteric ligand and negative allosteric modulators (NAM), which decrease the effect of the orthosteric ligand [10].
The structure of the A2AAR consists of 7 transmembrane helices (TM1-TM7) connected by three intracellular loops (ICL1-ICL3) and three extracellular loops (ECL1-ECL3) [12–21]. The receptor has been crystallized in complex with agonists, including adenosine [12–15], antagonists [16–21], and coupled to a G protein fragment [15]. The A2AAR has also been crystallized with interacting lipids [13, 14, 16, 19–21] and sodium ions [20, 21].
Recently, we applied molecular dynamics (MD) to the ligand–free (apo) form of two conformations of the A2AAR X-ray structures, agonist-bound (PDB ID: 3QAK) [13] and antagonist-bound (PDB ID: 3EML) [16]. We uncovered the deactivation process of the A2AAR from the active conformation of 3QAK to the inactive crystal structure 3EML [22]. We identified four distinct conformations of the receptor, the active, intermediate 1, intermediate 2, and inactive, which were characterized particularly by the motions of the Y2887.53 residue in the NPxxY motif. The number ‘288’ denotes the residue number, ‘7’ denotes the transmembrane helix number, and ‘53’ represents the location of the residue in the transmembrane helix relative to the most conserved residue (denoted 50) [23]. In the active conformation, the χ1 angle of Y2887.53 remained in the trans conformation and the side-chain faced the intracellular region of the receptor. In the intermediate 1 conformation, Y2887.53 interacted with residues in TM2, TM6, and TM7. In the intermediate 2 conformation, the χ1 angle of Y2887.53 switched from trans to gauche conformation, causing a break in the interactions between TM2 and TM6. Finally, in the simulation of the inactive A2AAR, a salt bridge was formed between the D1013.50 and E2286.30 residues, or the ‘ionic-lock’ [22].
In this study, we obtain representative conformations of the A2AAR from the previous MD simulations. We identify non-orthosteric sites on the active, intermediate 1, intermediate 2, and inactive conformers, and 20 A2AAR crystal structures using FTMap [24]. FTMap is fragment-based mapping algorithm, which screens the surface of the receptor with small probes, such as ethanol and isopropanol, and identifies hot spots based on regions of the receptor where multiple probes bind at low energy. This tool was previously used to identify five non-orthosteric sites on the β2 adrenergic receptor (β2AR), four non-orthosteric sites on the β1 adrenergic receptor (β1AR) [25], and seven non-orthosteric sites on the M2 muscarinic receptor [26]. These receptors are all membrane-bound proteins and the sites identified spanned the extracellular, intracellular and lipid interface regions. We classify two non-orthosteric sites on the active conformations, three on the intermediate 1 and intermediate 2 conformations, and five non-orthosteric sites on the inactive conformations of the A2AAR.
Materials and Methods
Simulations
In a previous study [22], we performed two simulations on the ligand-free A2AAR starting from the agonist-bound (PDB ID: 3QAK) [13] and the antagonist-bound (PDB ID:3EML) [16] X-ray structures. 3QAK was crystallized in complex with the agonist UK-432097 at a resolution of 2.71 Å [13]. 3EML was crystallized in complex with the antagonist ZM241385 at a resolution of 2.6 Å [16]. The ligands were removed from each crystal structure. In this study, the term “agonist-bound apo” denotes the ligand-free simulation starting from the 3QAK X-ray structure. The term “antagonist-bound apo” denotes the ligand-free simulation starting from the 3EML X-ray structure. The agonist-bound apo and antagonist-bound apo simulations were performed using all-atom MD simulations on the Anton supercomputer [27] for 1.57 μs and 1.75 μs, respectively, as previously described in Caliman, AD, et al [22].
FTMap
FTMap is a fragment-based site mapping algorithm that identifies potential binding sites on a receptor. The program docks 16 molecules, ethanol, isopropanol, isobutanol, acetone, acetaldehyde, dimethyl ether, cyclohexane, ethane, acetonitrile, urea, methylamine, phenol, benzaldehyde, benzene, acetamide, and N,N-dimethylformamide, to the surface of the receptor [24]. To use this algorithm, PDB files are uploaded to the online server, http://FTMap.bu.edu. The output of FTMap is a PDB file with the coordinates of the receptor and the probes found in low-energy hot spots. Hot spots are probe clusters distributed different sites of the receptor. The hot spots are identified and ranked by the FTMap protocol [24, 28]. All hot spots identified on the A2AAR were analyzed.
Clustering of Receptor Conformations
In the agonist-bound apo simulation, the receptor transitioned from the active to the intermediate 1 conformation at 250 ns. The receptor transitioned from intermediate 1 to intermediate 2 at 1.25 μs. The antagonist-bound apo simulation remained in the inactive conformation for the entire 1.75 μs [22]. The receptor frames were aligned on the backbone atoms of the transmembrane helices, and RMSD-based clustering of the receptor conformations was performed with a 1.5 Å cutoff to identify representative structures of the active, intermediate 1, intermediate 2, and inactive conformations. There were a total of 16 receptor clusters obtained from the agonist-bound apo simulation and 14 receptor clusters from the antagonist-bound apo simulation. Three receptor clusters were found in the active conformer. Nine clusters were found in the intermediate 1 conformer. Four receptor clusters were found in the intermediate 2 conformer, and 14 receptor clusters were found in the inactive conformer [22]. All 30 receptor clusters were submitted to the FTMap server for analysis.
Crystal Structures
Twenty crystal structures were analyzed using the FTMap server. The antagonist-bound crystal structures included are shown in Table 1 and the agonist-bound crystal structures are shown in Table 2. The crystal structures were prepared with Schrödinger’s Protein Preparation Wizard, where missing side chains, residues, and loops were added [29].
Table 1.
Antagonist-bound X-ray structures used for site-mapping. The PDB ID, ligand name, and resolution, interesting features, and references are shown.
| PDB ID | Ligand | Resolution in Å | Interesting Features | Reference |
|---|---|---|---|---|
| 3EML | ZMA | 2.6 | Lipid Interactions | [16] |
| 3PWH | ZMA | 3.3 | [17] | |
| 3REY | XAC | 3.31 | [17] | |
| 3RFM | Caffeine | 3.6 | [17] | |
| 3UZA | T4G | 3.27 | [18] | |
| 3UZC | T4E | 3.34 | [18] | |
| 3VG9 | ZMA | 2.7 | Lipid Interactions | [19] |
| 4EIY | ZMA | 1.8 | Lipid Interactions, Sodium | [20] |
| 5IU4 | ZMA | 1.72 | Lipid Interactions, Sodium | [21] |
| 5IU7 | 6DY | 1.9 | Lipid Interactions, Sodium | [21] |
| 5IUA | 6DX | 2.2 | Lipid Interactions, Sodium | [21] |
| 5IUB | 6DV | 2.1 | Lipid Interactions, Sodium | [21] |
| 5IU8 | 6DZ | 2 | Lipid Interactions, Sodium | [21] |
Table 2.
Agonist-bound X-ray structures used for site-mapping. The PDB ID, ligand name, and resolution, interesting features, and references are shown.
| PDB ID | Ligand | Resolution in Å | Interesting Features | Reference |
|---|---|---|---|---|
| 2YDO | Adenosine | 3.0 | [12] | |
| 2YDV | NECA-agonist | 2.6 | [12] | |
| 3QAK | UKA-agonist | 2.71 | Lipid Interactions | [13] |
| 4UG2 –Subunit A | NGI -agonist | 2.6 | Lipid Interactions, Two Subunits Crystallized | [14] |
| 4UG2 – Subunit B | NGI -agonist | 2.6 | Lipid Interactions, Two Subunits Crystallized | [14] |
| 5G53 – Subunit A | NECA + G protein | 3.4 | Two Subunits Crystallized | [15] |
| 5G53 – Subunit B | NECA + G protein | 3.4 | Two Subunits Crystallized | [15] |
Probe Occupancy Analysis
The total number of probes within 5 Å of the Cα atom of each residue was calculated for every receptor cluster and X-ray structure. Because these probes are small, if any atom on the probe was within 5 Å of the Cα atom a residue, it was included in the probe occupancy. The probe occupancy designates the probability that a probe was found within 5 Å of a residue. The probe occupancy was calculated by:
| (1) |
where Pj is the number of probes within 5 Å of a residue i in receptor cluster j, Nc is the total number of receptor clusters, Wj is the number of simulation frames that receptor cluster j represents, Nres is the total number of residues, and P is the total number of possible probes that could interact with that residue, or
| (2) |
where 16 is the total number of probes and T is the total number of simulation frames. When the probe occupancy is calculated for all antagonist or agonist-bound crystal structures, the weight, Wj is 1, and T is the number of antagonist or agonist-bound crystal structures, respectively.
The binding site residues plotted in the probe occupancy figures include I662.64, V843.32, L853.33, T883.36, Q893.37, I923.40, L167ECL2, F168ECL2, E169ECL2, M1775.38, N1815.42, W2466.48, L2496.51, H2506.52, N2536.55, T2566.58, H264, L2676.32, M2707.35, Y2717.36, I2747.39, S2777.42, and H2787.43 (Fig. S1 and Fig. 1).
Figure 1. Probe Occupancy Per Residue during the Molecular Dynamic Simulations.

Probe occupancies for the agonist-bound apo simulation [A], and the antagonist-bound apo simulation [B] are shown. Binding site residues are shown in red, non-orthosteric residues are shown in blue, and black boxes represent the transmembrane regions. ICL1 is located in between TM1 and TM2. ECL1 is located between TM2 and TM3. ICL2 is located between TM3 and TM4. ECL2 is located between TM4 and TM5. ICL3 is located between TM5 and TM6, and ECL3 is located between TM6 and TM7.
Center-of-Mass
If the distance between the center-of-mass for one hot spot was within 5 Å of the center-of-mass of another hot spot, they were considered a part of one larger site, and identified as a non-orthosteric site.
Figures 2–7, Supp. Figure 3, and Supp. Figures 5–7 were generated with VMD [30].
Figure 2. The Hot Spots on the A2AAR.

The hot spots for probe-binding in the active (green) [A], intermediate 1 (cyan) [B], intermediate 2 (orange) [C], and inactive (red) [D] conformers are shown. The orthosteric site (OS) is shown in gray, site 1 in purple, site 2 in blue, site 3 yellow, site 4 in orange, and site 5 in cyan.
Figure 7. Hot spot 5: ‘The Extracellular Cleft’.

is located between TM3 (gray), and TM4 (orange) helices. The probes are shown as cyan spheres [A]. The key interacting residues are shown as bonds [B], and the probes that bind to this site are shown as cyan bonds.
Results and Discussion
Probe Distribution in X-ray Structures
The overall distribution of probes in a receptor obtained from the FTMap analysis can help us to identify specific residues within the receptor with a propensity for interacting with drug fragments. The probe occupancy for the simulation starting agonist-bound (PDB ID: 3QAK) and simulation starting antagonist-bound (PDB ID: 3EML) X-ray structures were calculated (Fig. S1A and Fig. S1C). Additionally, the probe occupancy for all antagonist-bound and all agonist-bound X-ray structures as listed in Table 1 and Table 2, respectively, were calculated (Fig. S1B and Fig. S1D).
In the simulation starting agonist-bound X-ray structure, probes interact with residues in the TM, ICL1, and ECL2 regions (Fig. S1A). In all agonist-bound X-ray structures, the distribution of the probes to the receptor is identical to the distribution of the simulation starting X-ray structure, but the number of residues in TM1, ICL1, TM3, ECL2, and TM6 within 5 Å of a probe increases (Fig. S1B). In the simulation starting antagonist-bound X-ray structure, probes interact with the TM, ECL1, ECL2, and ICL2 regions of the receptor (Fig. S1C). In the antagonist-bound X-ray structures, there are fewer residues within 5 Å of a probe in TM1, while there are more residues in the ICL1, ECL1, and TM7 regions that are within 5 Å of a probe (Fig. S1D).
Given that there are only slight differences in the probe distribution between the X-ray structures (Fig. S1), and that X-ray structures represent static snapshots of the receptor, an FTMap analysis on MD structures will provide a more complete description of the probe distribution, and reveal transient sites in different conformers of the receptor.
Residues with the Highest Probe Occupancies
In the MD sampled representative structures of the receptor, probes interact primarily with the transmembrane regions, with residues with the highest probe occupancies being found in the TM2, TM3, ECL2, TM5, and TM7 regions in the agonist-bound apo simulation (Fig. 1A), and in the TM2, TM3, TM6, and TM7 regions in the antagonist-bound apo simulation (Fig. 1B). In the X-ray structures, the residues with the highest probe occupancies are A632.61, S672.65, A973.45, L167ECL2, and F168ECL2 (Fig. S1 and Table S1). The residues with the highest probe occupancies in both simulations are T883.36 and D522.50 (Fig. S1, Fig. 1, Table S1, and Table S2). Another residue with high probe occupancies in both simulations is S913.39.
Residue T883.36 is located in the sodium ion binding site (Fig. S2) [31] and the orthosteric binding site in the X-ray structures of the agonist-bound A2AAR [12–14]. The probe occupancy surrounding T883.36 is higher in the agonist-bound X-ray structures (0.38) compared with the antagonist-bound X-ray structures (0.03) (Table S1). In the agonist-bound apo simulation, the probe occupancy for T883.36 is 0.99, and the probe occupancy in the active conformer is 1.0 (Table S1 and Table S2). This is consistent with previous findings that mutating T883.36 affects agonist-binding, but not antagonist binding [12, 13]. The probe occupancy in the intermediate 1 conformer is 0.93, 0.87 in the intermediate 2 conformer, and 0.96 in the antagonist-bound apo simulation or the inactive conformer (Table S1 and Table S2). The high probe occupancies found in the intermediate 1, intermediate 2, and inactive conformers compared to the X-ray structures is due to the entrance of a sodium ion into the sodium ion binding pocket during both simulations (Fig. S2).
The probe occupancy of D522.50 is 0.86 in the agonist-bound apo simulation and 0.99 in the antagonist-bound apo simulation (Table S1). D522.50 is a conserved residue located in the sodium ion-binding site [20, 21, 31]. A sodium ion is not found in either the simulation starting agonist-bound X-ray structure [13] or the simulation starting antagonist-bound X-ray structure [16]. Appropriately, the probe occupancy surrounding D522.50 is 0.0 in the simulation starting agonist-bound and antagonist-bound X-ray structures, as well as in most A2AAR X-ray structures (Table S1). It has been suggested that sodium binding to D2.50 triggers deactivation of the A2AAR and other GPCRs [20, 31, 32]. In the agonist-bound apo simulation, a sodium ion enters the site at 405 ns (Fig. S2B), after the receptor has transitioned from the active state to intermediate 1 [22]. Thus, the probe occupancy for D522.50 in the active conformer is 0.28 and is 1 in the intermediate 1 conformer (Table S2). In the antagonist-bound apo simulation, a sodium ion enters the site after 34 ns (Fig. S2C).
Residue S913.39 interacts with a sodium ion [20, 31], which enters the sodium binding site in both simulations (Fig. S2). S913.39 has a probe occupancy of 0.44 in the agonist-bound apo simulation and 0.42 in the antagonist bound apo simulation, compared to 0.0 in the X-ray structures (Table S1). Surprisingly, the probe occupancy is higher in the active conformer (0.85) where a sodium ion does not bind, compared to the intermediate 1 (0.43), intermediate 2 (0.17), and inactive conformers (0.42) (Table S2). This suggests the modulators could bind to some residues of the sodium ion binding site in the active or inactive conformations.
Higher Probe Occupancies Observed in MD Simulations of the A2AAR
Residues C1284.49, F933.41, and V2837.48 have higher probe occupancies in the agonist-bound X-ray structures compared to the antagonist- bound X-ray structures (Fig. S1B, Fig. S1D, and Table S1). The TM5, ECL2, and residue S2817.46 have higher probe occupancies in the agonist-bound apo simulation structures compared to the antagonist-bound apo simulation structures (Fig. 1A and Table S1). TM5 is more flexible in the agonist-bound apo simulation than in the agonist-bound apo simulation [22]. Additionally, during receptor deactivation, residue Y1975.58 rotates the side chain towards the lipid interface, while in the antagonist-bound apo simulation, Y1975.58 faces the helical bundle [22]. Residue S2817.46 is also a part of the sodium ion binding pocket [20, 31] (Fig. S2). S2817.46 has a probe occupancy of 0.57 in the agonist-bound apo simulation and 0.09 in the antagonist-bound apo simulation, compared to 0.0 in the starting X-ray structures (Table S1). The probe occupancy is 0.63 in the active and intermediate 1 conformers, and 0.18 in the intermediate 2 conformer (Table S2).
TM2, TM3, A813.29, and D1013.49 have higher probe occupancies in the antagonist-bound X-ray structures than in the agonist-bound X-ray structures (Fig. S1D and Fig. S1B). TM4 and TM6 (including residue W2466.48), and residue L853.33 have higher probe occupancies in the antagonist-bound apo simulation, compared to the agonist-bound simulation. W6.48 is located in the conserved CWxP site, a rotameric trigger for GPCR activation [33], and residue in the sodium ion binding site (Fig. S2A) [31]. W2466.48 is also a key residue identified in the intrinsic water pathway in A2AAR [34]. During the antagonist-bound apo simulation, intracellular water molecules enter into the receptor and interact with W2466.48 (Fig. S4B). W2466.48 has a probe occupancy of 0.44 in the antagonist-bound apo simulation and 0.11 in the agonist-bound apo simulation (Table S1). The probe occupancy is 0.01 in the active conformer, 0.1 in the intermediate 1 conformer, and 0.14 in the intermediate 2 conformer (Table S2).
The probe occupancy surrounding L853.33 is 0.31 in the agonist-bound apo simulation (0.54 in the active, 0.15 in the intermediate 1, and 0.63 in the intermediate 2) and 0.74 in the antagonist-bound apo simulation. L853.33 is a residue in the orthosteric binding site in the agonist-bound and antagonist-bound X-ray structures [13, 16]. However, the probe occupancy in the simulation starting agonist-bound X-ray structure is much higher (0.34) than in the simulation starting antagonist-bound (0.0) X-ray structure (Table S1). The probe occupancy for residue L853.33 is 0.43 in the all agonist-bound and 0.03 in all antagonist-bound X-ray structures (Table S1).
Hot Spots of Probe Molecules Identified in the A2AAR during Deactivation
Different conformers of the A2AAR were identified from the long time-scale molecular dynamics simulations: the active, intermediate 1, intermediate 2, and inactive [22]. FTMap analysis revealed sites and relevant key residues on each of these conformers (Table 3 and Fig. 2). The inactive conformer has more non-orthosteric sites compared with the active conformer (Fig. 2), consistent with findings on the simulation starting agonist and antagonist-bound X-ray structures (Fig. S3).
Table 3.
Non-orthosteric sites in the A2AAR conformers are listed. The site number, location, transmembrane helices and residues are shown.
| Site Number | Location | Regions | Residues |
|---|---|---|---|
| 1 | Intracellular Crevice | TM3/TM4/TM5 | I3.40, F3.41, L3.44, A3.45, D3.49 (TM3) I4.45, I4.48, C4.49 (TM4) Y112 (ICL1) C5.46, P5.50 (TM5) |
| 2 | G Protein-Coupling Site | TM2/TM3/TM6/TM7 | N39 (ICL1) T2.39, N2.40 (TM2) D3.49, R3.50 (TM3) H6.32, S6.36, F6.44 (TM6) Y7.53 (TM7) I292 (C-Term) |
| 3 | The Lipid Interface | TM5/TM6 | P5.50, M5.54 (TM5) V6.41, F6.44, W6.48 (TM6) |
| 4 | C-Terminus Cleft | TM1/TM7 | L1.45, G1.49, L1.52 (TM1) V7.47, P7.50, F7.51 (TM7) |
| 5 | Extracellular Cleft | TM3/TM4 | C3.30, F3.31, V3.34 (TM3) L4.58, G4.57, F4.54 (TM4) |
Each conformer and the X-ray structures have an orthosteric site (Fig. 2 and Fig. S3). Residues in the orthosteric site include I662.64, V843.32, L853.33, T883.36, Q893.37, I923.40, L167ECL2, F168ECL2, E169ECL2, M1775.38, N1815.42, W2466.48, L2496.51, H2506.52, N2536.55, T2566.58, H264ECL3, L2677.32, M2707.35, Y2717.36, I2747.39, S2777.42, and H2787.43. This site is smaller in the inactive conformer and the simulation starting antagonist-bound X-ray structure than in the active conformer and simulation starting agonist-bound X-ray structure (Fig. 2A, Fig. 2D, and Fig. S3).
Non-orthosteric site 1 is located in the intracellular crevice between TM3, TM4, and TM5 (Fig. 2 and Fig. 3). Non-orthosteric site 2 is located in the G protein-coupling site of TM2, TM3, TM6, and TM7 (Fig. 2 and Fig. 4). These sites are found in the active, intermediate 1, intermediate 2, and inactive conformers (Fig. 2), as well as the simulation starting agonist-bound and antagonist-bound X-ray structures (Fig. S3). Non-orthosteric site 3 is located on the lipid interface of TM5 and TM6 (Fig. S3 and Fig. 5). This site is found in the intermediate 1, intermediate 2, and inactive conformers (Fig. 2B, Fig. 2C, and Fig. 2D), and in the simulation starting antagonist-bound X-ray structures (Fig. S3B). Non-orthosteric site 4 is located on the intracellular end of TM1 and TM7, near the C-terminus (Fig. 2D and Fig. 6). It is found in the inactive conformer (Fig. 2D), the simulation starting agonist-bound X-ray, and the simulation starting antagonist-bound X-ray structures (Fig. S3). Non-orthosteric site 5 is located between the extracellular ends of TM3 and TM4 (Fig. 2D and Fig. 7). This site is found in the inactive conformer (Fig. 2D).
Figure 3. Hot spot 1: ‘The Intracellular Crevice’.

is located in the intracellular region between TM3 (gray), TM4 (orange) and TM5 (tan). The probes in the active [A] and the intermediate 1 conformers [C] are shown as purple spheres, with the numbers ‘1.1’, ‘1.2’ and ‘1.3’ denoting different regions of the pocket. The key interacting residues for the active clusters [B] and intermediate 1 conformers [D] are shown as bonds, and the probes that bind to this site are shown as purple bonds.
Figure 4. Hot spot 2: ‘The G Protein-Coupling Site’.

is located between TM2 (green), TM3 (gray), TM6 (purple), and TM7 (red). The probes in the active [A] and inactive [C] conformers are shown as blue spheres. The key interacting residues for active conformers [B] and inactive conformers [D] are shown as bonds, and the probes that bind to this site are shown as blue bonds.
Figure 5. Hot spot 3: ‘The Lipid interface’.

is located between TM5 (tan) and TM6 (purple). The probes are shown as yellow spheres [A]. The key interacting residues are shown as bonds [B], and the probes that bind to this site are shown as yellow bonds.
Figure 6. Hot spot 4: ‘The C-Terminus Cleft’.

is located between TM1 (blue), TM7 (red), and TM8 (red). The probes are shown as orange spheres [A]. The key interacting residues are shown as bonds [B], and the probes that bind to this site are shown as orange bonds.
The key residues in each of these sites are listed in Table 3.
Site 1: The Intracellular Crevice
The “intracellular crevice” is located in between TM3, TM4, and TM5. The three regions in this site are denoted 1.1, 1.2, and 1.3 (Fig. 3), and correspond to the different probe clusters in the receptor. Site 1.1 is positioned closest to the intracellular region and involves residues Y112ICL2, D1013.49 and A973.45, and I1244.45 (Fig. 3A and Fig. 3B). D3.49 is a conserved residue of the DRY motif and Y112ICL2 is a key residue in the G protein-coupling site [15]. This region of the intracellular crevice is located in the active, intermediate 2, and inactive conformers (Fig. 2A, Fig. 2C, and Fig. 2D), and the simulation starting antagonist-bound X-ray structure (Fig. S3B). The aromatic and hydrophobic probes that interact with this region are cyclohexane and benzene.
Site 1.2 is present in every structure (Fig. 2 and Fig. S3) and encompasses residues I923.40, C1855.46, and P1895.50 (Fig. 3D). Hydrophobic cyclohexane, benzene, and phenol, and hydrophilic isobutanol interact with this site. Site 1.3 is only located in the intermediate 1 conformer (Fig. 2B), and comprises of residues I923.40, C1855.46, and P1895.50 (Fig. 3D). Probes in this region include benzaldehyde, benzene, cyclohexane, acetone, and phenol. Site 1.1 and 1.2 are separated by I1244.45 and A973.45, while site 1.2 and 1.3 are separated by F933.41 (Fig. 3B and Fig. 3D).
The intracellular crevice site is also found in the β1AR, β2AR, denoted sites 3 and 5 in Ref. [25], and the M2 muscarinic receptor, denoted site 4 in Ref. [26]. In the M2 receptor, there are two components to this site, 4.1 and 4.2. Site 4.1, located closest to the intracellular region, can be found in the inactive and intermediate 2 conformers. In ref. [26], the conformers of the M2 receptor include the inactive, intermediate 1, intermediate 2 and the active, where the intermediate 1 is found between inactive and intermediate 2, and the intermediate 2 is found between intermediate 1 and active. Site 4.2 is identified in the intermediate 1 and active states of the M2 receptor.
Site 2: G Protein-Coupling Site
The “G protein-coupling site” is located between the intracellular ends of the TM2, TM3, TM6, and TM7 (Fig. 4). Allosteric modulators designed to bind to this site could prevent or enhance the binding of the G protein, attenuating the signaling cascade. In the active conformer, residues encompassing the G protein-coupling site include H2306.32, S2346.36, Y2887.53, R1023.50, and I292C-TERM, and the probes that interact with this site are cyclohexane, N, N-dimethylformamide, and phenol (Fig. 4B). In the inactive conformer, the interacting residues are D1013.49, R1023.50, T412.39, F2426.44, I2877.52, Y2887.53, N422.40, and N39ICL1 (Fig. 4D).
Probes found in this site include methylamine, acetone, cyclohexane, ethanol, urea, benzene, N, N-dimethylformamide, acetamide, and benzaldehyde. The G protein coupling site is smaller in the active conformer and the simulation starting agonist-bound structure (Fig. 2A, Fig. S3A, and Fig. 4A), than in the intermediate 1, intermediate 2 and inactive conformers, and simulation starting antagonist-bound X-ray structure (Fig. 2B, Fig. 2C, Fig. 2D, Fig. S3B, and Fig. 4C). In the antagonist-bound apo simulation, an internal water channel floods the intracellular region of the receptor (Fig. S4B). In the agonist-bound apo simulation, fewer water molecules enter the receptor (Fig. S4A).
The residues in this site are key to deactivation of the A2AAR [22]. Residue Y2887.53 is oriented toward the intracellular region of the receptor in the active conformation. In the intermediate 1, Y2887.53 forms hydrogen bonds with N422.40 and S2346.36. Next, the side chain of Y2887.53 switches from the gauche to the trans conformation. In the inactive conformation, a salt bridge is formed between R1023.50 and E2286.30. Additionally, F2426.44 is a residue in the sodium ion binding site [35]. R3.50 is a key residue in the G protein binding site [15].
This G protein-coupling site is found in the β1AR, β2AR, denoted site 4 in ref. [25], and the M2 receptor, denoted site 7 in ref. [26]. In the M2 receptor, this site is identified only in the active state.
Site 3: The Lipid Interface
The “lipid interface” site is located on the lipid exposed region of TM5 and TM6 (Fig. 5). It is present in the intermediate 1, intermediate 2, and inactive conformers (Fig. 2B, Fig. 2C, and Fig. 2D), and the simulation starting antagonist-bound X-ray structure of the A2AAR (Fig. S3B). The residues P1895.50, W2466.48, F2426.44, M1935.54, and V2396.41 interact with probes, such as acetone, benzene, ethane, acetaldehyde, acetamide, isobutanol, isopropanol, urea, ethanol, dimethyl ether, and acetonitrile (Fig. 5B).
The conformations of P1895.50 and F2426.44 determine whether this site is present in the conformers (Fig. 5 and Fig. S5). In the active conformer, the side chains of key residues in this site, P1895.50 and F2426.44, face towards each other. The distance between the Cα atoms of these two residues is smaller than 10 Å (Fig. S5B). When the receptor transitions to the intermediate 1 conformation in the agonist-bound apo simulation, the distance between P1895.50 and F2426.44 increases to an average of 12 Å (Fig. S5B). This distances increases to 15 Å in the intermediate 2 conformer (Fig. S5B). In the antagonist-bound apo simulation, the distance between these two residues is consistently above 15 Å (Fig. S5C).
While this site is not present in the β1AR or β2AR receptors [25], it is present in the inactive M2 muscarinic receptor, denoted site 2 in ref. [26]. Allosteric modulators designed to bind to site 3 could lock the protein in the inactive or intermediate conformers, by interacting with residues P1895.50, F2426.44, and W2466.48 (the “toggle switch” for GPCR activation) [33].
Site 4: The C-Terminus Cleft and Site 5: Extracellular Cleft
The “C-terminus cleft” is located in the intracellular end of TM1 and TM7 (Fig. 6). It is present in the inactive conformer (Fig. 2D) and the simulation starting antagonist-bound X-ray structure (Fig. S3B). The residues located in the C-terminus cleft include V2827.47, F2867.51, P2857.50, L261.52, G231.49, L221.48, and L191.45. These residues interact with benzene, cyclohexane, phenol, N, N-dimethylformamide, acetone, cyclohexane, isobutanol, and benzaldehyde (Fig. 6B).
This cleft is caused by a tilt in TM1 in the inactive conformer relative to the active conformer (Fig. S6). The side chains of residues V2827.47 and L191.45 move towards each other with a distance of 4.9 Å, while the side chain of residues F2867.51 and L261.52 move away from each other with a distance of 10.0 Å, forming a cleft (Fig. S6).
The “extracellular cleft” is located between the extracellular ends of TM3 and TM4 (Fig. 7). It is present in the inactive conformer (Fig. 2D). The residues located in the extracellular cleft site include F833.31, C823.30, L1374.58, G1364.57, F1334.54, and V863.34 (Fig. 7B). This site interacts with acetone, benzene, dimethyl ether, ethane, and phenol.
The extracellular cleft is only present in the inactive conformer of the A2AAR (Fig. 2D), but in the M2 receptor, the extracellular cleft is found in intermediate 2 and active conformers, denoted as site 3.2 in ref [26].
Conclusion
The A2AAR is a GPCR that plays a key role in the immune and neurological systems [3, 9]. In the immune system, the receptor is anti-inflammatory, and activation of the A2AAR leads to an inhibition of pro-inflammatory cytokines and a secretion of anti-inflammatory cytokines [2, 3, 6]. In the neurological disorders, such as Parkinson’s Disease, A2AAR activation acts counter to the D2 dopamine receptor [9]. Given these roles, A2AAR agonists are being developed for the treatment of immunological diseases, such as sepsis, and A2AAR antagonists are being developed for the treatment of PD [5].
Allosteric modulators bind to regions outside of the orthosteric site. Some of these modulators do not have activity on their own, but either increase or decrease the activity of the endogenous ligand [10]. In this study, we applied standard FTMap, a fragment mapping technique, to analyze representative receptor structures obtained from previous agonist-bound apo and antagonist-bound apo MD simulations [22] and the 20 X-ray structures of the A2AAR to identify allosteric sites on the receptor. FTMap identified non-orthosteric sites on this membrane receptor distributed in the lipid interface, the extracellular and intracellular surfaces. Many ligands do not bind to the lipid interface of the receptor, but there are known ligands that can partition into the lipid bilayer [36].
First, we calculated the probe occupancy per residue. In the MD simulation representative clusters and the X-ray structures, probes mostly interacted with residues in the TM regions. Many residues with high probe occupancies, I923.40, S2817.46, T883.36, D522.50, L853.33, and W2466.48, are involved in sodium binding. Sodium cannot bind to the active state of the receptor. Previous A2AAR studies have shown that a sodium ion can bind to when the receptor is in complex with an antagonist, but cannot bind when the receptor is bound to an agonist [31]. Additionally, studies of the M3 muscarinic receptor reveal that sodium binding locks the receptor in the inactive conformation [32]. The propensity of these probes to bind to residues of the sodium ion binding site suggest that negative allosteric modulators could be developed to target this region of the receptor.
In the agonist-bound apo and antagonist-bound apo MD simulations, four key receptor conformers, active, intermediate 1, intermediate 2, and inactive, were identified during deactivation of the A2AAR [22]. We identified five non-orthosteric sites on the A2AAR that can be targeted for designing non-orthosteric modulators. Overall, the inactive conformer and antagonist-bound X-ray structures exhibited more non-orthosteric sites than the active conformer and agonist-bound X-ray structures.
The intracellular crevice, present on all conformers, is located between the intracellular ends of TM3/TM4/TM5. This site is also present in the β1AR, β2AR, and M2 muscarinic receptor. Key interacting residues include D1013.49 of the conserved DRY motif. The G protein-coupling site, present on all conformers, is located in the intracellular mouth of TM2/TM3/TM6/TM7. This site is also present in the β1AR, β2AR, and M2 muscarinic receptor. Key interacting residues include Y2887.53 of the NPxxY motif, and R1023.50 of the conserved DRY motif. This site is larger in the inactive conformer than in the active conformer due the intracellular influx of water molecules. Given the role of two key residues involved in GPCR activation, Y2887.53 and R1023.50 of the ionic lock, either PAMS or NAMs could be developed to bind this site.
The lipid interface site, present on the intermediate 1, intermediate 2 and inactive conformers, is located on the lipid interface between TM5/TM6. This site is present in the M2 muscarinic receptor, and includes key residues including W2466.48. The C-terminus cleft is located in the intracellular end of TM1/TM7. The extracellular cleft is present on the extracellular region of TM3/TM4. These sites are not found in the active conformer, and thus could be targeted for designing novel NAMs.
Overall, these sites provide an array of available non-orthosteric sites on the active, intermediate 1, intermediate 2, and inactive A2AAR conformers. These sites, including the sites found on the lipid interface, can be screened for novel PAMs for the treatment of sepsis and novel NAMs for the treatment of PD and cancer.
Supplementary Material
Table S1. Probe Occupancies of Key Residues. Probe Occupancies of Key Residues
Table S2. Probe Occupancies of Key Residues by Deactivation Conformation.
Figure S1. Probe Occupancy for A2AAR X-ray Structures.
Figure S2. Sodium Ion Binding Site.
Figure S3. Receptor Hotspots in Starting Structures.
Figure S4. Internal water channel.
Figure S5. Conformation of P1895.50 and F2426.44 in Site 3.
Figure S6. Conformation of TM1 and TM7 in Site 4.
Acknowledgments
Computing time was provided on the Anton supercomputer, courtesy of D.E. Shaw Research. Additional support is provided by the National Science Foundation (NSF grant: MCB1020765), National Institutes of Health (NIH grant: GM31749), Howard Hughes Medical Institute, National Biomedical Computation Resource (NBCR), and the National Institute of Health Training Grant (NIH Training Grant: T32GM007752).
Footnotes
Conflict of Interest
The authors declare that there are no conflicts of interests.
References
- 1.Thiel M, Caldwell CC, Sitkovsky MV. The critical role of adenosine A2A receptors in downregulation of inflammation and immunity in the pathogenesis of infectious diseases. Microbes and Infection. 2003;5(6):515–526. doi: 10.1016/s1286-4579(03)00068-6. [DOI] [PubMed] [Google Scholar]
- 2.Sivak KV, et al. Adenosine A2A receptor as a drug target for treatment of sepsis. Molecular Biology. 2016;50(2):200–212. doi: 10.7868/S0026898416020233. [DOI] [PubMed] [Google Scholar]
- 3.Cekic C, Linden J. Purinergic regulation of the immune system. Nature Reviews Immunology. 2016;16(3):177–192. doi: 10.1038/nri.2016.4. [DOI] [PubMed] [Google Scholar]
- 4.Chen JF, Eltzschig HK, Fredholm BB. Adenosine receptors as drug targets — what are the challenges? Nature Reviews Drug Discovery. 2013;12(4):265–286. doi: 10.1038/nrd3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pinna A. Adenosine A2A receptor antagonists in Parkinson’s disease: progress in clinical trials from the newly approved istradefylline to drugs in early development and those already discontinued. CNS Drugs. 2014;28(5):455–474. doi: 10.1007/s40263-014-0161-7. [DOI] [PubMed] [Google Scholar]
- 6.Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflamation and protetion from tissue damage. Nature. 2001;414(6866):916–920. doi: 10.1038/414916a. [DOI] [PubMed] [Google Scholar]
- 7.Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nature Reviews Drug Discovery. 2006;5(3):247–264. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ohta A, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(35):13132–13137. doi: 10.1073/pnas.0605251103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morelli M, Carta AR, Jenner P. Adenosine A2A receptors and Parkinson’s Disease. In: Wilson CN, Mustafa SJ, editors. Adenosine Receptors in Health and Disease. Springer; Berlin Heidelberg: 2009. pp. 589–615. [DOI] [PubMed] [Google Scholar]
- 10.Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nature Reviews Drug Discovery. 2009;8(1):41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goblyos A, Ijzerman AP. Allosteric modulation of adenosine receptors. Biochimica et Biophysica Acta. 2011;1808(5):1309–1318. doi: 10.1016/j.bbamem.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 12.Lebon G, et al. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011;474(7352):521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu F, et al. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332(6027):322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lebon G, et al. Molecular Determinants of CGS21680 Binding to the Human Adenosine A2A Receptor. Molecular Pharmacology. 2015;87(6):907–915. doi: 10.1124/mol.114.097360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carpenter B, et al. Structure of the adenosine A(2A) receptor bound to an engineered G protein. Nature. 2016;536(7614):104–7. doi: 10.1038/nature18966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaakola VP, et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322(5905):1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doré Andrew S, et al. Structure of the adenosine A2A receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure. 2011;19(9):1283–1293. doi: 10.1016/j.str.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Congreve M, et al. Discovery of 1,2,4-triazine derivatives as adenosine A2A antagonists using structure based drug design. Journal of Medicinal Chemistry. 2012;55(5):1898–1903. doi: 10.1021/jm201376w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hino T, et al. G-protein-coupled receptor inactivation by an allosteric inverse-agonist antibody. Nature. 2012;482(7384):237–240. doi: 10.1038/nature10750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu W, et al. Structural Basis for Allosteric Regulation of GPCRs by Sodium Ions. Science. 2012;337(6091):232–236. doi: 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Segala E, et al. Controlling the Dissociation of Ligands from the Adenosine A2A Receptor through Modulation of Salt Bridge Strength. Journal of Medicinal Chemistry. 2016;59(13):6470–6479. doi: 10.1021/acs.jmedchem.6b00653. [DOI] [PubMed] [Google Scholar]
- 22.Caliman AD, et al. Investigation of the conformational dynamics of the apo A2A adenosine receptor. Protein Science. 2015;24(6):1004–1012. doi: 10.1002/pro.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-dunction relations in G protein-coupled receptor. Methods in Neurosciences. 1995;25:366–428. [Google Scholar]
- 24.Brenke R, et al. Fragment-based identification of druggable ‘hot spots’ of proteins using Fourier domain correlation techniques. Bioinformatics. 2009;25(5):621–627. doi: 10.1093/bioinformatics/btp036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ivetac A, McCammon JA. Mapping the druggable allosteric space of G-protein coupled receptors: a fragment-based molecular dynamics approach. Chemical Biology & Drug Design. 2010;76(3):201–217. doi: 10.1111/j.1747-0285.2010.01012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miao Y, Nichols SE, McCammon JA. Mapping of allosteric druggable sites in activation-associated conformers of the M2 muscarinic receptor. Chemical Biology & Drug Design. 2014;83(2):237–246. doi: 10.1111/cbdd.12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaw DE, et al. Millisecond-scale molecular dynamics simulations on Anton. Proceedings of the 2009 ACM/IEEE Conference on Supercomputing (SC09); Portland, OR: ACM Press; 2009. [Google Scholar]
- 28.Kozakov D, et al. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat Protoc. 2015;10(5):733–55. doi: 10.1038/nprot.2015.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jacobson MP, et al. A hierarchical approach to all-atom protein loop prediction. Proteins. 2004;55(2):351–367. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 30.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. The Journal of Molecular Graphics and Modelling. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 31.Gutierrez-de-Teran H, et al. The role of a sodium ion binding site in the allosteric modulation of the A(2A) adenosine G protein-coupled receptor. Structure. 2013;21(12):2175–2185. doi: 10.1016/j.str.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miao Y, Caliman AD, McCammon JA. Allosteric effects of sodium ion binding on activation of the m3 muscarinic g-protein-coupled receptor. Biophysical Journal. 2015;108(7):1796–1806. doi: 10.1016/j.bpj.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olivella M, Caltabianco G, Cordomí A. The role of Cysteine 6.47 in class A GPCRs. BMC Structural Biology. 2013;13(3):1–10. doi: 10.1186/1472-6807-13-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuan S, et al. W246(6.48) Opens a Gate for a Continuous Intrinsic Water Pathway during Activation of the Adenosine A2A Receptor. Angewandte Chemie International Edition. 2015;54(2):556–559. doi: 10.1002/anie.201409679. [DOI] [PubMed] [Google Scholar]
- 35.Katritch V, et al. Allosteric sodium in class A GPCR signaling. Trends in Biochemical Sciences. 2014;39(5):233–244. doi: 10.1016/j.tibs.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pyne NJ, Pyne S. Sphingosine 1-Phosphate Receptor 1 Signaling in Mammalian Cells. Molecules. 2017;22(3) doi: 10.3390/molecules22030344. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Probe Occupancies of Key Residues. Probe Occupancies of Key Residues
Table S2. Probe Occupancies of Key Residues by Deactivation Conformation.
Figure S1. Probe Occupancy for A2AAR X-ray Structures.
Figure S2. Sodium Ion Binding Site.
Figure S3. Receptor Hotspots in Starting Structures.
Figure S4. Internal water channel.
Figure S5. Conformation of P1895.50 and F2426.44 in Site 3.
Figure S6. Conformation of TM1 and TM7 in Site 4.