

Abstract
Heparanase, the sole heparan sulfate degrading endoglycosidase, regulates multiple biological activities that enhance tumor growth, metastasis, angiogenesis and inflammation. Heparanase accomplishes this by degrading heparan sulfate and thereby regulating the bioavailability of heparin-binding proteins, priming the tumor microenvironment, mediating tumor-host crosstalk and inducing gene transcription, signaling pathways, exosome formation and autophagy that together promote tumor cell performance and chemoresistance. In contrast, heparanase-2, a close homolog of heparanase, lacks enzymatic activity, inhibits heparanase activity and regulates selected genes that promote normal differentiation, ER-stress, tumor fibrosis and apoptosis, together resulting in tumor suppression. The emerging premise is that heparanase is a master regulator of the aggressive phenotype of cancer, while heparanase-2 functions as a tumor suppressor.
Keywords: Heparanase, heparanase-2, tumor microenvironment, exosomes, chemoresistance, tumor suppressor
Heparanase-1 vs. heparanase-2
Cloning and expression of mammalian heparanase-1 (= heparanase, see Glossary) elicited a broad interest in the enzyme and prompted extensive basic and clinical research, focusing on the involvement of heparanase in cancer, inflammation and kidney dysfunction. Heparanase expression is increased in multiple types of cancers, and this elevation is associated with aggressive disease and poor prognosis. The enzyme is highly implicated in the crosstalk between cells and their microenvironment, a consequence of cleavage of heparan sulfate (HS) (see Glossary) and remodeling of the extracellular matrix (ECM) underlying epithelial and endothelial cells. Despite important and unexpected advances in basic and translational features of the heparanase enzyme its multiple modes of action and significance in health and disease are underemphasized. Even more so are recent developments in the biology of heparanase-2 (Hpa2, see Glossary), a close homolog of heparanase that lacks heparanase enzymatic activity. It appears that Hpa2 not only inhibits heparanase activity but also regulates selected genes that affect tumor vascularity, tumor fibrosis, cell differentiation, ER-stress and apoptosis, together resulting in tumor suppression. Several excellent reviews present basic and translational aspects related to the involvement of heparanase in cancer and inflammation [1–5]. In contrast, research on heparanase-2 (Hpa2) is in its infancy and has not been reviewed. The present review addresses recent developments in the heparanase field, highlighting the multiple pro-tumorigenic functions of heparanase, the emerging role of Hpa2 as a tumor suppressor, and the related clinical implications.
Heparanase-1
Biochemical features
Unlike synthesis and modification of heparan sulfate (HS) chains that require the activity of an array of enzymes, intra-chain cleavage of HS is carried out by one enzyme, heparanase (= heparanase-1; endo-β-D-glucuronidase), an endoglycosidase that cleaves the HS side chains of HS proteoglycans (see Glossary) into fragments of 10–20 sugar units [6]. Heparanase is initially translated as a preproenzyme containing a signal peptide. Cleavage of the signal sequence yields a latent 65-kDa pro-heparanase, which must undergo further processing for activity. Proteolytic removal by cathepsin L [7] of a 6-kDa linker liberates an N-terminal 8-kDa subunit and a C-terminal 50-kDa subunit, which remain associated as a noncovalent heterodimer in mature active heparanase [8]. For structural information, see Box 1.
Box. 1. Heparanase crystal structure and mode of action.
Heparanase is a member of the glycoside hydrolase 79 (GH79) family of carbohydrate-processing enzymes [69, 88]. It catalyzes hydrolysis of internal glucuronic acid (GlcUA) β1–4 N-sulfoglucosamine (GlcNS) linkages in HS, with net retention of anomeric configuration. The GH79 family belongs to the larger GH-A clan, which is characterized by a (β/α)8 domain containing the catalytic site [89]. The crystal structure of human heparanase reveals how an endo-acting binding cleft is exposed by proteolytic activation of latent pro-heparanase [69]. The cleft contains residues Glu343 and Glu225, which have been identified as the catalytic nucleophile and acid-base of heparanase required for retaining the catalytic mechanism [88]. In accordance with the negatively charged nature of its HS substrate, the heparanase binding cleft is lined by basic side chains contributed by Arg35, Lys158, Lys159, Lys161, Lys231, Arg272, Arg273 and Arg303 [69]. The positioning of the 8-kDa and 50-kDa-subunits indicates that the excised Ser110–Gln157 linker of pro-heparanase lie very near or even within the active site cleft of the (β/α)8 domain [7, 69]. This positioning hinders incoming HS substrates and is consistent with a steric-block mechanism for pro-heparanase inactivation by its own linker [7]. The crystal structure confirmed that sulfation is key for heparanase interaction with HS and that the recognized cleavage site is a trisaccharide accommodated in the heparanase binding cleft [69]. The recently resolved structure of pro-heparanase is similar to that of mature heparanase, with the same (β/α)8 and β-sheet domains clearly discernible. Unexpectedly, the 6-kDa linker does not entirely block access to the catalytic residues of pro-heparanase, but instead contributes to the formation of an exo-glycosidase-like ‘binding pocket’ that can accommodate smaller molecules [70]. Thus, small and likely soluble HS substrates are possibly accessible to the active site even of the latent enzyme.
Cellular uptake, exosome formation, autophagy and chemoresistance
A number of studies have shown that secreted or exogenously added latent heparanase rapidly interacts with HS side chains of syndecans (see Glossary), followed by internalization and processing into a highly active enzyme [9] (Fig. 1), a process that may be used by cancer cells to increase their own levels of heparanase. Heparanase uptake is a pre-requisite for the delivery of latent heparanase to lysosomes and its subsequent proteolytic processing and activation (Fig. 1). While syndecan-1 is regarded as the primary receptor for heparanase endocytosis, low density lipoprotein receptor-related protein (LRP) and the cation-independent mannose-6-phosphate receptor (CIMPR) have been identified as cell surface heparanase-binding proteins [10, 11] that contribute to heparanase uptake. Heparanase resides primarily within endocytic vesicles, assuming a polar, peri-nuclear localization and co-localizing with lysosomal markers (Fig. 1). Residence and accumulation of heparanase in lysosomes indicate that the enzyme may function in the normal physiology of this organelle. In a search for such function, we identified a role of lysosomal heparanase in augmenting autophagy [12] (Fig. 2b, c) (see Glossary), a physiological process required to remove unfolded proteins and damaged organelles, thus maintaining cellular homeostasis [13]. This implies that heparanase function is not limited to the extracellular milieu but can operate inside the cell as well [14]. Thus, heparanase inhibitors (i.e., aspirin and other small molecules) [15] should ideally be able also to penetrate cells and target heparanase within lysosomes.
Figure 1.

Schematic presentation of heparanase and Hpa2 biosynthesis and trafficking. Pre-pro-heparanase is first targeted to the ER lumen via its own signal peptide (1). The 65 kDa pro-heparanase is then shuttled to the Golgi apparatus, and is subsequently secreted via vesicles that bud from the Golgi (2). Once secreted, heparanase rapidly interacts with syndecans (3), followed by rapid endocytosis of the heparanase-syndecan complex that accumulates in late endosomes (4). Heparanase uptake is inhibited by heparin or Hpa2, resulting in extracellular accumulation of the latent enzyme (9). Conversion of endosomes to lysosomes results in heparanase processing and activation (primarily by cathepsin L) (5). Typically, heparanase appears in perinuclear lysosomes (merge: heparanase-green; lysotracker-red) (6). Lysosomal heparanase may translocate to the nucleus, where it affect gene transcription, and/or can get secreted via secretory lysosomes and exosomes. Similar to heparanase, Hpa2 is first targeted to the ER lumen (1), secreted via vesicles that bud from the Golgi (2) and interacts with syndecan on the cell surface (7). Unlike heparanase, Hpa2 is retained on the cell surface (merge: Hpa2-green; syndecan1-red) for a relatively long period followed by a decline at later time points (>4 hours), possibly due to proteolysis (7), or release from the cell surface by shedding of syndecan (8). Accumulation of Hpa2 in the extracellular compartment is enhanced by heparin or anti-Hpa2 monoclonal antibody (1c7) (10).
Figure 2.

Heparanase uptake is mediated by syndecan and promotes autophagy. A. Heparanase uptake is mediated by syndecan cytoplasmic tail. Heparanase was added (1μg/ml, 1h) to U87 glioma cells transfected with control empty vector (Vo; left panels), syndecan-1 (WT; middle panels) or syndecan-1 lacking its cytoplasmic tail (Delta; right panels). Cells were fixed with cold methanol and subjected to immunofluorescent staining applying anti-heparanase mouse monoclonal antibody (upper panels, green). Merged images with rat anti-syndecan staining (red) are shown in the lower panels. Note retention of heparanase at the cell membrane, co-localizing with syndecan lacking the entire cytoplasmic tail, and increased heparanase-positive endocytic vesicles in cells over expressing wild type (WT) syndecan-1. B. Heparanase co-localizes with LC3-II. Heparanase (1 μg/ml) was added exogenously to HeLa cells stably expressing GFP-LC3 gene construct for 24h. Cells were then deprived of amino acids in the presence of chloroquine for 3 h, fixed with methanol and subjected to immunofluorescent staining applying anti-heparanase (middle panel, red) antibody. Co-localization of heparanase and GFP-LC3 appears yellow (lower panel). C. Electron microscopy. Pancreas tissues from control (WT) and transgenic mice over expressing heparanase (Hpa-Tg) were fixed and processed for electron microscopy. Shown are representative images at x10,000 magnification. Note a substantial increase in the number and size of autophagosomes in heparanase-transgenic mice.
To examine the role of syndecan-1 cytoplasmic domain in heparanase processing [16], cells were transfected with a full length mouse syndecan-1 or deletion constructs lacking the entire cytoplasmic domain (delta) (Fig. 2a), the conserved (C1 or C2) or variable (V) regions of syndecan. It was found that the C2 and V regions of the cytoplasmic tail of syndecan mediate heparanase uptake (Fig. 2a). Internalization of the latent enzyme is followed by proteolytic processing. There is no indication for direct interaction between the cytoplasmic domain of syndecan and heparanase, nor for a role of syndecan signaling in heparanase processing. Furthermore, syntenin, known to interact with syndecan C2 domain, and α actinin are essential for heparanase processing [16]. Interestingly, syndecans and syntenin, via interaction with ALIX, have been implicated in regulating the biogenesis of exosomes [17] (see Glossary) and heparanase was found to promote the secretion of exosomes and alter their composition and biological function [18]. Further investigation indicated that heparanase activates the syndecan-syntenin-ALIX pathway establishing a linear axis that regulates exosome formation [19]. Heparanase was also found to be heavily loaded into exosomes as cargo. Because of the known role of heparanase in promoting metastasis and angiogenesis, delivery of the enzyme via exosomes possibly play a role in establishing niches to which tumor cells eventually home and grow. Emerging data indicate that exosomes can act as barriers to anti-cancer therapy by interacting with tumor cells and enhancing their chemoresistance [20] (see Glossary). This was shown to occur in myeloma cells, where treatment with anti-myeloma drugs stimulated a burst of exosome secretion and those exosomes (referred to as chemoexosomes) were loaded with heparanase (Fig. 3) [21]. Chemoexosomes were capable of docking with other myeloma cells or macrophages and transferring heparanase to those cells (Fig. 3). This enhances the shedding of syndecan-1 by the recipient myeloma cells and likely also augments their resistance to killing by the drugs. Chemoexosomes stimulate macrophage migration and upregulate secretion of TNF-α (Fig. 3), a cytokine known to support myeloma growth. Analysis of the chemoexosomes revealed that heparanase was present on the exosome surface. Intact heparanase-containing chemoexosomes are capable of releasing HS fragments from the extracellular matrix indicating that once released by tumor cells these exosomes can remodel their surrounding matrix and perhaps contribute to tumor and host cell migration (Fig. 3). Together, heparanase helps drive exosome secretion, alters exosome composition and facilitates production and docking of exosomes that impact both tumor and host cell behavior thereby promoting tumor progression and chemoresistance [18] [21].
Figure 3.

Exosomes released following chemotherapy facilitate tumor progression. Treatment of myeloma cells with bortezomib causes a burst of exosome secretion. These exosomes (referred to as chemoexosomes) carry an abundance of heparanase bound to the heparan sulfate of syndecan-1 that is present on the exosome surface. Chemoexosomes can transfer their heparanase to other tumor cells thereby enhancing their chemoresistance. Heparanase also stimulates syndecan-1 shedding by those cells. Shed syndecan-1 facilitates tumor metastasis and angiogenesis by enhancing both myeloma and endothelial cell migration via activation of the VEGFR-2 receptor [87]. Chemoexosomes may also have VEGF bound on their surface that aids in driving angiogenesis. Chemoexosomes can also dock with macrophages, stimulating their migration and enhancing their expression of TNF-α. Because heparanase is present on the exosome surface, it is also available to degrade heparan sulfate within the ECM which may liberate tumor-supporting growth factors and facilitate tumor and host cell migration.
One protein with multiple functions
Even though the straightforward function of heparanase is to loosen the ECM, the role of heparanase extends much beyond tissue invasion. For example, both enzymatically active and inactive heparanase promotes signal transduction, including Akt, STAT, Src, Erk, HGF-, IGF- and EGF-receptor signaling [22–24], highlighting the significance of non-enzymatic activities of heparanase in tumor progression [25, 26]. This is best exemplified in a recent study in which the regulatory elements of the mouse mammary tumor virus (MMTV) were used to direct expression of heparanase and its C-domain (8c) to the mammary gland epithelium of transgenic mice [27]. Mammary gland branching morphogenesis was increased in MMTV-heparanase and MMTV-8c mice, associating with increased Akt, Stat5 and Src phosphorylation [27]. Furthermore, the growth of tumors generated by mouse breast cancer cells and the resulting lung metastases are enhanced in MMTV-heparanase mice, thus supporting the notion that heparanase contributed by the tumor microenvironment (Box 2) plays a decisive role in tumorigenesis [28]. Remarkably, MMTV-8c mice develop spontaneous tumors in their mammary gland. Although this occurs at low rates and requires long latency, it evidently demonstrates the pro-tumorigenic capacity of heparanase signaling [27]. Heparanase also regulates the transcription of pro-angiogenic (i.e., VEGF-A, VEGF-C, COX-2, MMP-9), pro-thrombotic (i.e., tissue factor), pro-inflammatory (i.e., TNFα, IL-1, IL-6), pro-fibrotic (i.e., TGFβ), mitogenic (i.e., HGF), osteolyic (RANKL) and other genes [5, 29], further expanding its functional repertoire and mode of action [22–24].
Box. 2. Targeting the tumor microenvironment.
The tumor microenvironment (TME) is the cellular and a-cellular environment in which the tumor exists, including surrounding blood vessels, immune cells, fibroblasts, bone marrow-derived inflammatory cells, lymphocytes, signaling molecules and the extracellular matrix (ECM). Traditionally, the ECM network of proteins, glycoproteins and proteoglycans was regarded as an inert scaffold providing a structural framework for cells to form tissues and organs. Subsequently it was realized that the ECM plays an active role in the control of cell survival, proliferation and differentiation. The tumor and the surrounding microenvironment are closely related and interact constantly to support the survival, growth and evolution of cancerous cells. An active interplay exists between cells and the ECM where cells synthesize matrix components which in turn dictate and regulate cell shape and function. Moreover, the ECM serves as a reservoir for numerous bioactive molecules (i.e., growth factors, cytokines, chemokines, enzymes) that bind primarily to heparan sulfate (HS) proteoglycans (HSPGs) and together orchestrate cellular responses to both normal and pathological situations. Cleavage of HSPGs ultimately releases these proteins and converts them into bioavailable and bioactive mediators, ensuring rapid tissue response to local or systemic cues. This function of HS provides the cell with a rapidly accessible reservoir, precluding the need for de novo synthesis when the requirement for a particular protein is increased. Heparanase, secreted by tumor cells, platelets, activated endothelial and immune cells, primes the microenvironment by releasing HS-bound factors and extracellular signals that promote tumor growth, angiogenesis, peripheral immune tolerance and formation of a metastatic niche. Thus, the tumor microenvironment plays multifaceted roles during cancer progression and can promote or inhibit tumor development, depending on local and systemic conditions. Briefly, tumor cells can change the nature of the microenvironment, and conversely, the microenvironment can affect how a tumor grows and spreads. Interestingly, a strategy by which tumor cells gain drug resistance is by modifying the tumor microenvironment. Understanding the mechanisms by which the tumor microenvironment functions would greatly facilitate the development of therapeutic interventions that target the tumor niche [90]
Compelling evidences tie heparanase with all steps of tumor formation including tumor initiation, growth, metastasis, and chemoresistance [12, 30–34]. Mechanistic studies on heparanase action have focused primarily on its expression by tumor cells and revealed that heparanase promotes an aggressive tumor behavior via multiple mechanisms [3, 4]. Yet, non-tumor (host) cells including T lymphocytes, B lymphocytes, neutrophils, monocyte/macrophages (see Glossary), endothelial cells, osteoclasts and fibroblasts can also upregulate heparanase expression upon activation and thereby contribute not only to cancer progression and chemoresistance [5, 12, 30–34], but also to acute and chronic inflammation [35–37], autoimmunity [36, 38], atherosclerosis [39], tissue fibrosis [40], kidney dysfunction [41–44], ocular surface dysfunction [45], viral infection [46], diabetes [47] and diabetic complications [48, 49]. These functions dynamically impact multiple pathological pathways, but at the same time, may fulfill some normal functions associated, for example, with vesicular traffic, exosome formation, lysosomal-based secretion, stress response, and heparan sulfate turnover [50–52]. Noteworthy, heparanase appears to activate cells of the innate immune system and soluble HS fragments generated by heparanase trigger the expression and secretion of pro-inflammatory cytokines through toll-like receptors (TLR) [1, 53–55]. While heparanase up-regulation by tumor cells is well documented, the pro-tumorigenic function of heparanase mediated by the host has not been sufficiently emphasized. The significance of heparanase residing in the tumor microenvironment (Box 2) has been demonstrated by showing that tumors are more aggressive when developed in heparanase over-expressing transgenic mice (Hpa-Tg, MMTV-heparanase), while smaller tumors develops in Hpa-KO mice [56, 57]. Also, heparanase-neutralizing antibodies attenuate the growth of lymphoma cells that do not express heparanase, implying that neutralization of heparanase contributed by the tumor microenvironment may be sufficient to restrain tumor growth [28]. As a direct result of these and other studies, heparanase was advanced from being an obscure enzyme with a poorly understood function to a highly promising drug target, offering new treatment strategies for various cancers and other diseases.
Cells of the immune system
Heparanase is critically required for activation and function of macrophages, an important constituent of the tumor microenvironment that may accelerate or suppress tumor progression [56, 58] (Box 2). Macrophages isolated from heparanase knockout (Hpa-KO) mice express lower levels of cytokines (i.e., TNF-α, IL1-β) and exhibit lower motility and phagocytic capacities as compared to wild type (WT) macrophages [56]. Intriguingly, inoculating WT monocytes together with Lewis lung carcinoma (LLC) cells into Hpa-KO mice resulted in a nearly complete inhibition of tumor growth. In striking contrast, inoculating LLC cells together with monocytes isolated from Hpa-KO mice did not affect tumor growth. Mechanistically, we identified a linear cascade by which heparanase activates Erk, p38 and JNK signaling in macrophages, leading to increased c-Fos levels and induction of cytokine expression [56]. These results identify heparanase as a key mediator of macrophage activation and function in tumorigenesis and cross-talk with the tumor microenvironment, paving the way for the development of heparanase-based treatment modalities that either stimulate or inhibit macrophage activation and function in a broad range of human diseases, including cancer, inflammation, kidney failure, diabetes and atherosclerosis.
Natural killer (NK) cells are highly efficient at preventing cancer metastasis but are infrequently found in the core of primary tumors. NK cell invasion into primary tumors and recruitment to the site of metastasis strictly depend on the presence of heparanase [59]. In fact, mice lacking heparanase specifically in NK cells were highly tumor prone when challenged with a carcinogen or when inoculated with metastatic melanoma, prostate carcinoma, or mammary carcinoma cells [59]. Moreover, cytokine and immune checkpoint blockade immunotherapy for metastases was compromised when NK cells lacked heparanase [59]. This should be considered when systemically treating cancer patients with heparanase inhibitors, since the potential adverse effect on NK cell infiltration might limit the antitumor activity of the inhibitors. These findings advocate a more selective targeting of heparanase in tumor cells that would avoid the potentially adverse effect of reducing effector T cell or NK cell infiltration into tumors. In line with this thought, heparanase pro-invasion function has been exploited in humans by engineering CAR T cells overexpressing heparanase [60]. Such T cells showed improved ECM degradation in vitro and efficient infiltration into solid tumors in vivo, ultimately leading to improved antitumor activity [60]. Thus, under certain conditions, heparanase may exert effects that may expedite tumor cell killing.
Clinical considerations
Numerous clinical association studies have consistently demonstrated that upregulation of heparanase expression correlates with increased tumor size, tumor angiogenesis, enhanced metastasis and poor prognosis [23, 61]. In contrast, knockdown of heparanase or treatments of tumor-bearing mice with heparanase-inhibiting compounds, markedly attenuate tumor progression [3, 5, 37, 62, 63] further underscoring the potential of anti-heparanase therapy for multiple types of cancer. In addition, recent studies revealed that high levels of heparanase in the tumor metastases predict poor prognosis in stage IVc melanoma patients [64]. This result implies that heparanase not only enhances tumor cells dissemination but also promotes the growth and aggressiveness of the resulting metastases. Interestingly, in breast cancer patients, heparanase expression in the metastatic lesion does not always reflect its expression in the primary tumor. Accordingly, in some cases the primary lesion was stained positive for heparanase while the metastasis stained negative, and vice versa (unpublished). Thus, precision medicine approaches applying heparanase inhibitors must examine heparanase levels in the tumor metastases. Although heparanase expression is noted to be induced in all major types of cancer (carcinomas, sarcomas and hematological malignancies) [37], the molecular mechanisms that regulate heparanase expression are largely unknown. A few recent publications describe the involvement of microRNAs in induction and repression of heparanase expression in human tumors [65, 66].
Heparanase neutralizing monoclonal antibodies block myeloma and lymphoma tumor growth and dissemination [28]; this is attributable to a combined effect on the tumor cells and/or cells of the tumor microenvironment [28]. In fact, substantial impact of heparanase on tumor progression is related to its function in mediating tumor-host crosstalk, priming the tumor microenvironment to better support tumor seeding, growth and chemoresistance. Heparanase is upregulated in response to chemotherapy in cancer patients [33] and the surviving cells acquire chemoresistance [34], attributed, at least in part, to autophagy [12] (Fig. 2b, c). Consequently, heparanase inhibitors used in tandem with chemotherapeutic drugs overcome initial chemoresistance [34], providing a strong rationale for applying anti-heparanase therapy in combination with conventional anti-cancer drugs. Heparin-mimetics (i.e., Muparfostat, Roneparstat, Necuparanib, PG545) that inhibit heparanase enzymatic activity are being evaluated in clinical trials for various types of cancer and appear to be well tolerated [67]. Heparanase neutralizing monoclonal antibodies are being examined in pre-clinical studies [28], and heparanase-inhibiting small molecules are being developed, based on the recently resolved crystal structure of the heparanase protein [68–70]. While clinical trials focus on cancer patients, the same inhibitors are likely to be applied in other indications as well. For example, the efficacy of heparanase inhibitors has been demonstrated in preclinical models of sepsis [71], autoimmune diabetes [72], diabetic nephropathy [48], pancreatitis [73], viral infection [46], acute kidney injury [74] and other kidney diseases [44].
Heparanase 2 (Hpa2)
Biochemical features
McKenzie and colleagues reported the cloning of heparanase homolog termed heparanase 2 (Hpa2) [75]. The full-length HPSE2 gene consists of 2353 bp encoding a protein of 592 amino acids; Alignment of the coding region of heparanase and Hpa2 reveals an overall identity of 40% and sequence resemblance of 59%, including conservation of residues critical for heparanase enzymatic activity (Glu225 and Glu343). The segment corresponding to the linker region and cleavage sites of pro-heparanase are not conserved in Hpa2. Indeed, Hpa2 does not undergo proteolytic processing and hence lacks intrinsic HS-degrading activity, the hallmark of heparanase [76]. It is appealing to examine whether enzymatic activity can be restored following excision of a putative linker segment from the Hpa2 molecule. In addition to the full length Hpa2 protein (Hpa2c), several variants have been identified as a result of alternative splicing of the HPSE2 transcript, including Hpa2a (480 aa) and Hpa2b (534 aa) [75]. Notably, only Hpa2c is secreted, likely due to extra glycosylation sites that are lost in the splice variants [76]. Hpa2 retains the capacity to bind heparin/HS [76] and, importantly, exhibits even higher affinity towards heparin/HS than heparanase, thus competing for HS binding and inhibiting heparanase enzymatic activity (Fig. 4, key figure) [76]. Moreover, co-immunoprecipitation studies revealed physical association between Hpa2 and heparanase proteins [76], providing additional explanation for the inhibition of heparanase enzymatic activity by Hpa2. Notably, exogenously added Hpa2 binds to HS and clusters syndecan-1 and -4. Surprisingly, however, unlike heparanase, it fails to get internalized but remains on the cell surface for relatively long periods of time (Fig. 1) and does not undergo proteolytic processing [76].
Figure 4.
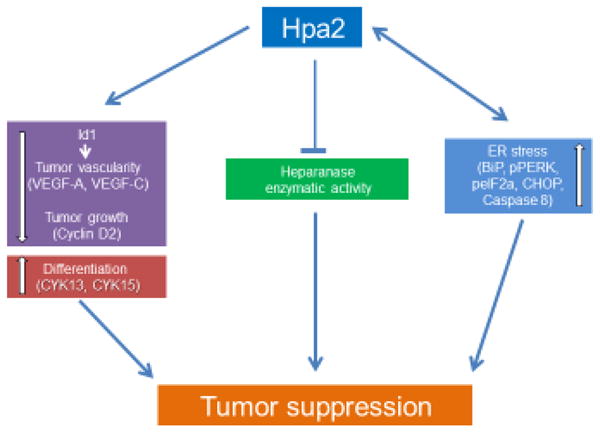
Hpa2 promotes tumor suppression via down regulation of Id1, inhibition of heparanase enzymatic activity, and stimulation of ER-stress. Down-regulation of the transcription factor Id1 reduces vascular (VEGF-A) and lymphatic (VEGF-C) tumor angiogenesis and growth (cyclin D2) and promotes cell differentiation (i.e., cytokeratin 13 & 15). Hpa2 also inhibits heparanase enzymatic activity thereby attenuating cell invasion and release of HS-bound growth and angiogenesis promoting factors. Hpa2 enhances ER-stress (BiP, pPERK, peIF2α, CHOP, cleaved caspase 8) through interaction with HS. Moreover, Hpa2 levels are elevated in response to hypoxia and ER-stress (i.e., thapsigargin), generating a vicious cycle that contributes to tumor suppression.
Hpa2 and urofacial syndrome
While the role played by Hpa2 in healthy tissues is unknown, genetic studies revealed that Hpa2 is critically important in urofacial syndrome (UFS) [77, 78], a rare autosomal recessive disease with severe dysfunctional urination including urinary incontinence (leading to renal failure) and peripheral neuropathies (i.e., facial abnormalities) [79]. The genetic basis for this condition involves mutation of the Hpa2 gene [77, 78]. The biallelic mutations in HPSE2 mostly result in frame-shifts that lead to an early stop codon and a truncated Hpa2 protein, resulting in a loss-of-function phenotype [77, 78]. In order to reveal the function of Hpa2 in the urinary tract, Stuart et al [80] and Guo et al [81] independently generated a Hpa2 mutant mouse. Interestingly, Hpa2 mutant mice display a distended bladder (megacystis) phenotype and abnormal voiding behaviour similar to that found in UFS patients, indicating that Hpa2 is necessary for peripheral neural control of bladder function [80, 81]. Further analysis of the Hpa2-KO mutant mice suggests involvement of Hpa2 in tissue remodelling and fibrosis, apparently via the TGFβ pathway [80]. Notably, homozygous Hpa2 mutant mice exhibited a growth retardation phenotype and poor weight gain after birth, and most mice have subsequently died [80, 81]. Given the lethal phenotype of the mutant mice it is conceivable that Hpa2 also plays a significant role in the function of organs other than the bladder in a manner that is yet to be defined. Unlike heparanase, Hpa2 is readily detected in normal epithelium (i.e., breast, bladder, oral cavity) and its expression levels are markedly reduced in the adjacent tumor tissue [82]. Importantly, we have demonstrated anti-tumoral and anti-angiogenic effects of Hpa2 [82, 83], providing a solid rationale for the notion that Hpa2 functions as a tumor suppressor (see Glossary) (Fig. 4).
Head & neck carcinoma
In head & neck cancer patients, Hpa2 expression correlates with prolonged time to disease recurrence and inversely correlates with tumor cell dissemination to regional lymph nodes [76]. In an attempt to reveal molecular parameters responsible for this function, Hpa2 was over-expressed in FaDu pharyngeal carcinoma cells. Tumor xenografts produced by cells over expressing Hpa2 were significantly smaller than xenografts produced by control cells. These tumors exhibited a marked decrease in lymphatic and blood vessels associated with reduced VEGF-A and VEGF-C expression (Fig. 4) [83]. In addition, levels of collagen and lysyl oxidase (LOX), an enzyme that is strongly implicated in collagen deposition and tissue fibrosis, were markedly increased in xenografts produced by Hpa2 overexpressing cells [82, 83], in agreement with the notion that LOX functions as a tumor suppressor [84], apparently via ECM cross-linking, collagen deposition and inhibition of tumor angiogenesis. Moreover, expression of differentiation markers (i.e., cytokeratins 13, 15) was also increased [83], implying increased cell differentiation and lower tumor grade (Fig. 4). Notably, Hpa2 over-expression resulted in reduced expression of Id-1 (Fig. 4), a pro-angiogenic transcription factor [85, 86]. This result was confirmed by the ability to rescue tumor growth of Hpa2 transfected cells by over expression of Id-1 gene construct [83].
Bladder cancer
Unlike head and neck tissues, Hpa2 is expressed in normal human bladder transitional epithelium and its levels are decreased substantially in bladder cancer, an expression pattern typical of a tumor suppressor. Remarkably, tumors that retain high levels of Hpa2 were diagnosed as low grade and low stage, suggesting that Hpa2 is required to preserve cell differentiation and halt cell migration and proliferation [82]. Notably, migration of bladder carcinoma cells was attenuated markedly by exogenous addition of purified Hpa2, and over expression of Hpa2 resulted in smaller tumors that were diagnosed as low grade and were abundantly decorated with stromal cells and collagen, correlating with a marked increase in Lox [82]. The association between Hpa2 and Lox was further confirmed clinically [82]. Importantly, endogenous heparanase enzyme activity was not altered in Hpa2 over-expressing cells, indicating heparanase-independent function of Hpa2 in bladder [82] and head and neck [83] tumor growth regulation. This and a pro-adhesive function even in the presence of heparin clearly suggest that Hpa2 exerts HS-, and heparanase-independent tumor suppressing function.
Pancreatic carcinoma
In the normal pancreas, Hpa2 staining is detected in beta islets whereas the duct epithelium is stained negative. Notably, Hpa2 expression is increased substantially in some pancreatic ductal adenocarcinomas associating inversely with tumor grade. In orthotopic model of pancreatic carcinoma, over-expression of Hpa2 resulted in tumors that were 8-folds smaller, associating with reduced blood vessel density, elevation in tumor fibrosis and Lox expression, and decreased levels of phospho-Met, phospho-STAT3, and phospho-Akt (unpublished). Notably, Hpa2 tumors exhibited higher degree of cell differentiation, indicated by elevated levels of cytokeratin 18 and E-cadherin. Further analyses revealed increased immunostaining of molecular determinants typical of ER-stress (i.e., BiP, phospho-PERK, CHOP, cleaved caspase-8) in Hpa2 tumors (Fig. 4). In fact, exogenous addition of Hpa2 to Panc01 cells stimulated ER-stress, and this effect was attenuated by antibody that blocks Hpa2 interaction with HS or by heparin, clearly implying that enhanced ER-stress by Hpa2 is HS-dependent (unpublished) (Fig. 4). Notably, Hpa2 expression was induced by ER-stress, thus establishing a loop that fuels itself and enhances ER-stress, likely leading to apoptotic cell death, together suggesting that Hpa2 functions as a tumor suppressor (Fig. 4). Induction of Hpa2 by ER-stress as well as hypoxia may explain why Hpa2 expression is increased in pancreatic and head and neck tumors compared with normal ductal epithelium. Mechanisms that mediate Hpa2 down-regulation as occurs in bladder cancer are yet to be characterized.
Concluding Remarks and Future Perspectives
As investigation of heparanase advances, new roles for the enzyme in signal transduction, gene regulation, exosome formation and function, autophagy, activation of innate immune cells and chemo-resistance, are emerging and thus widening the impact of this enzyme. Unraveling these and other aspects of heparanase biology is ongoing and is critical to our understanding of its multiple functions in health and disease. Notably, the enzymatic and non-enzymatic functions of heparanase are not tumor specific and hence cannot be ascribed to a specific type of cancer (a table listing various malignancies affected by heparanase is presented in ref. #37). Central to some of the downstream effects of heparanase is the enzyme’s ability to regulate gene transcription. At the molecular level, nuclear heparanase appears, among other effects, to regulate histone 3 lysine 4 (H3K4) methylation by influencing the recruitment of demethylases to transcriptionally active genes [29]. Yet our understanding of heparanase nuclear accessibility and mode of action is far from being complete (see Outstanding Questions). An important challenge in the field rests on structure-based rational development of clinically effective inhibitors (heparin mimics, neutralizing antibodies, small molecules) of heparanase that will be applied to treat cancer, inflammation and other diseases. Remarkably, heparanase inhibitors were effective even when the xenografted tumor cells were devoid of heparanase, emphasizing the significance of heparanase contributed by cells residing in the tumor microenvironment (Box 2). It appears that targeting the tumor microenvironment by heparanase inhibitors enhances the antitumor activity of approved therapies, further providing a strong rationale for applying anti-heparanase therapy in combination with conventional anti-cancer drugs. Collectively, the emerging premise is that heparanase expressed by tumor cells and cells of the tumor microenvironment is a dominant regulator of the aggressive phenotype of cancer, an important contributor to the poor outcome of cancer patients and a prime target for therapy.
Outstanding Questions Box.
Is there a heparanase/Hpa2 specific cell surface signaling receptor?
How does heparanase activate macrophages and other cells of the immune system?
Does host heparanase play a significant role in establishing a metastatic niche?
What is the role of Hpa2 in normal physiology (conditional Hpa2-KO mice)?
How does nuclear heparanase and Hpa2 affect gene transcription?
What are the structural features of Hpa2 and its mode of action as a tumor suppressor?
Can heparanase inhibitors be applied for indications other than cancer such as diabetes, diabetic nephropathy, fibrosis, autoimmunity, chronic and acute inflammatory disorders?
Unlike heparanase, the role played by Hpa2 in normal physiology and pathological disorders is still largely obscure. Genetic studies revealed that Hpa2 is critically important in urofacial syndrome (UFS), a rare autosomal recessive disease with severe dysfunctional urination and peripheral neuropathies. It appears that Hpa2 is necessary for peripheral neural control of bladder function. Given the lethal phenotype of Hpa2 mutant mice, it is conceivable that Hpa2 plays a significant role in the function of organs other than the bladder in a manner that is yet to be defined (see Outstanding Questions). In some normal epithelium Hpa2 is localized mainly to the cell nucleus but appears diffused within the cytoplasm of tumor cells suggesting a role in maintaining traits of normal epithelium, possibly associating with the regulation of gene transcription and cell differentiation. Hpa2 appears to attenuate tumor growth via gene transcription, decreasing the expression of pro-angiogenic mediators and inducing the manifestation of genes involved in tumor suppression and cell differentiation. Yet the molecular mechanisms that underlie these properties are obscure (see Outstanding Questions). Together, Hpa2 functions in heparanase activity- and HS- dependent and independent manners, and regulates the expression of selected genes that affect tumor vascularity, tumor fibrosis, cell differentiation, ER-stress and apoptosis of cancer cells, resulting in tumor suppression. Clearly, further research is needed (i.e., generation and characterization of conditional Hpa2 knockout and transgenic mice) in order to appreciate the scope of Hpa2 normal functions, mode of action in UFS and related complications and role as a tumor suppressor.
In summary, heparanase exerts strong pro-tumorigenic properties, promoting all aspects of tumor development (tumor initiation, growth, and metastasis) and chemo-resistance. The enzyme is expressed by tumor cells and cells of the tumor microenvironment and functions extracellularly to remodel the ECM as well as inside the cell, augmenting autophagy. Heparanase promotes the secretion of exosomes and alter their composition and biological function. Heparanase is also heavily loaded in exosomes as cargo, and delivery of the enzyme via exosomes plays a role in chemo-resistance and the establishment of niches to which tumor cells eventually home and grow. In striking contrast, Hpa2 suppresses tumor growth and vascularity, maintaining cell differentiation. Thus, unlike many enzyme families (i.e., MMPs, cathepsins), heparanase and its only close homolog Hpa2 exert opposing biological properties.
Trends Box.
Compelling evidence tie heparanase enzymatic and non-enzymatic activities with tumor initiation, growth, metastasis, and chemoresistance.
Studies emphasize the impact of host heparanase on tumor progression, supporting the clinical use of anti-heparanase therapy in combination with conventional anti-cancer drugs.
The heparanase-syndecan-syntenin axis regulates exosome formation and hence provides a mechanism for the crosstalk between tumor cells, the host and the tumor microenvironment.
Heparanase-inhibiting compounds are being examined in cancer patients.
Hpa2 inhibits heparanase activity and regulates the expression of selected genes that affect tumor angiogenesis, tumor fibrosis, cell differentiation, ER-stress and apoptosis of cancer cells, together resulting in tumor suppression.
Heparanase-1 and Hpa2 have multiple functions in health and disease in a context dependent manner.
The crystal structure of heparanase has been solved paving the way for rational design of heparanase inhibiting small molecules and monoclonal antibodies.
Take-home message.
Heparanase exerts strong pro-tumorigenic properties, promoting all aspects of tumor development (tumor initiation, growth, and metastasis) and chemo-resistance.
Heparanase is expressed by tumor cells and cells of the tumor microenvironment and functions extracellularly to remodel the ECM as well as inside the cell, augmenting autophagy.
Heparanase promotes the secretion of exosomes and alter their composition and biological function. Heparanase is also heavily loaded in exosomes as cargo, and delivery of the enzyme via exosomes plays a role in chemo-resistance and establishing niches to which tumor cells eventually home and grow.
In striking contrast, Hpa2 suppresses tumor growth and vascularity, maintaining cell differentiation. Thus, unlike many enzyme families (i.e., MMPs, cathepsins), heparanase and its only close homolog Hpa2 exert opposing biological properties.
Acknowledgments
This work was supported by grants from the National Institutes of Health CA138340 and CA211752 (RDS), the United States - Israel Binational Science Foundation (jointly to RDS and IV), the Israel Science Foundation (601/14 and 2277/15) and the Israel Cancer Research Fund (ICRF). Israel Vlodavsky is a Research Professor of the ICRF. We gratefully acknowledge the contribution, motivation, and assistance of the research teams in the Technion Integrated Cancer Center (TICC) and the UAB Comprehensive Cancer Center. We apologize for not citing several relevant articles, due to space limitation.
Glossary
- Autophagy
Autophagy is the natural, regulated, destructive mechanism of the cell that disassembles unnecessary or dysfunctional components. Autophagy allows the orderly degradation and recycling of cellular components. In macroautophagy, targeted cytoplasmic constituents are isolated from the rest of the cell within a double-membraned vesicle known as an autophagosome. The autophagosome eventually fuses with lysosomes and the contents are degraded and recycled. In disease, autophagy has been seen as an adaptive response to stress, which promotes survival, whereas in other cases it appears to promote cell death and morbidity. In the extreme case of starvation, the breakdown of cellular components promotes cellular survival by maintaining cellular energy levels. Autophagy is induced by starvation and stress, including chemotherapy, thereby promoting cancer cells survival and chemoresistance by providing their metabolic needs
- Chemoresistance
Chemotherapy is a category of cancer treatment that uses one or more anti-cancer drugs (chemotherapeutic agents) as part of a standardized chemotherapy regimen. Resistance is a major cause of treatment failure of chemotherapeutic drugs. There are several causes of resistance in cancer, one of which is the presence of small pumps, known as p-glycoprotein, on the surface of cancer cells that actively move chemotherapy from inside the cell to the outside, thereby protecting themselves from chemotherapeutics
- Exosomes
Exosomes are cell-derived vesicles (30–150 nm in diameter) that are present in eukaryotic fluids, including blood, urine, and cultured medium of cell cultures. Exosomes are released from cells when multivesicular bodies fuse with the plasma membrane. Once released exosomes can interact with target cells transferring molecules (proteins, mRNA, miRNAs) thereby impacting key processes such as coagulation, intercellular signaling, and waste management. Tumor-derived exosomes can travel through the body and impact resident cells at locations distal to the tumor thereby aiding in preparation of the pre-metastatic niche and influencing metastatic organotropism. Exosomes can be used for prognosis, for therapy, and as biomarkers for health and disease
- Glycosaminoglycan (GAG) and heparan sulfate (HS)
GAGs are polymers of disaccharide units. In heparan sulfate (HS), uronic acid (glucuronic acid or iduronic acid) and glucosamine (N-acetyl or N-sulfamino) repeats comprise its basic structure. These GAG chains are long, linear carbohydrate polymers that are negatively charged under physiological conditions due to sulfate and uronic acid groups. Despite the seemingly simple single repeating structural motif, these sugar polymers show a great deal of structural diversity generated by complex pattern of deacetylation, sulfation, and epimerization. Embryos that lack HS die during gastrulation. HSPGs function is not limited to developmental processes but play key roles in numerous biological settings, including cytoskeleton organization, cell-cell and cell-ECM interactions. HSPGs exert their multiple functional repertoires via distinct mechanisms that combine structural, biochemical and regulatory aspects. By interacting with other macromolecules such as laminin, fibronectin, and collagens I and IV, HSPGs contribute to the structural integrity, self-assembly and insolubility of the ECM and basement membranes, thus intimately modulating cell-ECM interactions
- Heparanase-1 (heparanase)
an endo-β-glucuronidase that cleaves HS side chains of HSPGs thereby releasing saccharide products of appreciable size (7–4 kDa) that can associate with protein ligands and modulate their biological potency. Heparanase-mediated breakdown of HS is not indiscriminate but instead is restricted to a small subset of glucuronic acids, thus reflecting a requirement for specific N- and O-sulfation patterns on neighboring sugars. Heparanase cleaves HS utilizing a hydrolase mechanism as opposed to bacterial heparinase III (heparitinase) which cleaves HS more extensively by a β-eliminase mechanism. The heparanase gene (HPSE) encodes a 61.2-kDa latent pro-enzyme that is post-translationally cleaved into 8-kDA and 50-kDa subunits that non-covalently associate to form the active heparanase heterodimer. Notably, lysosomes harbor HS degrading enzymes other than heparanase. In fact, lack of lysosomal enzymes that degrade HS is causally associated with lysosomal storage diseases, but unlike heparanase, these enzymes are exoglycosidases
- Heparanase-2 (Hpa2)
Heparanase-2 is a protein found based on sequence similarity to heparanase. In humans it is encoded by the HPSE2 gene mapped to chromosome 10 (unlike heparanase that was mapped to chromosome 4). It is associated with urofacial syndrome, other neuropathies and tumor suppression
- Macrophages
Macrophages are a type of white blood cell that engulfs and digests cellular debris, foreign substances, microbes and cancer cells in a process called phagocytosis. Macrophages are found in essentially all tissues where they patrol for potential pathogens by amoeboid movement. They take various forms throughout the body (e.g., histiocytes, Kupffer cells, alveolar macrophages, microglia, and others), but all are part of the mononuclear phagocyte system. Besides phagocytosis, they play a critical role in both innate and adaptive immunity. Beyond increasing inflammation and stimulating the immune system, macrophages also play an important anti-inflammatory role and can decrease immune reactions through the release of cytokines. Macrophages that support inflammation are referred to as M1 macrophages, whereas those that decrease inflammation and encourage tissue repair and cancer progression are called M2 macrophages
- Proteoglycans
consist of a core protein with one or more covalently attached GAG side chain(s). The major cell membrane HSPGs are the transmembrane syndecans and the glycosylphosphatidylinositol (GPI)-anchored glypicans. In the ECM (particularly basement membranes) perlecan, agrin and collagen XVIII core proteins are the main HS-bearing species
- Syndecan
Syndecans are single transmembrane domain proteins that act as co-receptors, especially for G protein-coupled receptors. These core proteins carry three to five heparan sulfate and chondroitin sulfate chains, which allow the interaction with a large variety of ligands including fibroblast growth factors, vascular endothelial growth factor, fibronectin and antithrombin-1. In some cases, a ternary complex composed of a ligand (i.e., bFGF), its high-affinity receptor and HSPG is required for most efficient signaling
- Tumor suppressor
Tumor suppressor gene, or anti-oncogene, is a gene that protects a cell from one step on the path to cancer. When this gene mutates to cause a loss or reduction in its function, the cell can progress to cancer, usually in combination with other genetic changes. Proteins encoded by tumor-suppressor genes have a repressive effect on the regulation of the cell cycle and/or promote apoptosis. These genes are often down-regulated in cancer
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hermano E, et al. Heparanase enzyme in chronic inflammatory bowel disease and colon cancer. Cell Mol Life Sci. 2012;69(15):2501–13. doi: 10.1007/s00018-012-0930-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li JP, Vlodavsky I. Heparin, heparan sulfate and heparanase in inflammatory reactions. Thromb Haemost. 2009;102(5):823–8. doi: 10.1160/TH09-02-0091. [DOI] [PubMed] [Google Scholar]
- 3.Rivara S, et al. Heparanase: a rainbow pharmacological target associated to multiple pathologies including rare diseases. Future Med Chem. 2016;8(6):647–80. doi: 10.4155/fmc-2016-0012. [DOI] [PubMed] [Google Scholar]
- 4.Sanderson RD, et al. Heparanase regulation of cancer, autophagy and inflammation: new mechanisms and targets for therapy. FEBS J. 2017;284(1):42–55. doi: 10.1111/febs.13932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vlodavsky I, et al. Heparanase: From basic research to therapeutic applications in cancer and inflammation. Drug resistance updates. 2016;29:54–75. doi: 10.1016/j.drup.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vlodavsky I, et al. Mammalian heparanase: gene cloning, expression and function in tumor progression and metastasis. Nat Med. 1999;5(7):793–802. doi: 10.1038/10518. [DOI] [PubMed] [Google Scholar]
- 7.Abboud-Jarrous G, et al. Cathepsin L is responsible for processing and activation of proheparanase through multiple cleavages of a linker segment. J Biol Chem. 2008;283(26):18167–76. doi: 10.1074/jbc.M801327200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levy-Adam F, et al. Heterodimer formation is essential for heparanase enzymatic activity. Biochem Biophys Res Commun. 2003;308(4):885–91. doi: 10.1016/s0006-291x(03)01478-5. [DOI] [PubMed] [Google Scholar]
- 9.Gingis-Velitski S, et al. Heparanase uptake is mediated by cell membrane heparan sulfate proteoglycans. J Biol Chem. 2004;279(42):44084–92. doi: 10.1074/jbc.M402131200. [DOI] [PubMed] [Google Scholar]
- 10.Vreys V, et al. Cellular uptake of mammalian heparanase precursor involves low density lipoprotein receptor-related proteins, mannose 6-phosphate receptors, and heparan sulfate proteoglycans. J Biol Chem. 2005;280(39):33141–48. doi: 10.1074/jbc.M503007200. [DOI] [PubMed] [Google Scholar]
- 11.Wood RJ, Hulett MD. Cell surface-expressed cation-independent mannose 6-phosphate receptor (CD222) binds enzymatically active heparanase independently of mannose 6-phosphate to promote extracellular matrix degradation. J Biol Chem. 2008;283(7):4165–76. doi: 10.1074/jbc.M708723200. [DOI] [PubMed] [Google Scholar]
- 12.Shteingauz A, et al. Heparanase enhances tumor growth and chemoresistance by promoting autophagy. Cancer Res. 2015;75(18):3946–57. doi: 10.1158/0008-5472.CAN-15-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosenfeldt MT, Ryan KM. The multiple roles of autophagy in cancer. Carcinogenesis. 2011;32(7):955–63. doi: 10.1093/carcin/bgr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ilan N, et al. Function from within: Autophagy induction by HPSE/heparanase-new possibilities for intervention. Autophagy. 2015;11(12):2387–2389. doi: 10.1080/15548627.2015.1115174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dai XY, et al. Aspirin inhibits cancer metastasis and angiogenesis via targeting heparanase. Clin Cancer Res. 2017 doi: 10.1158/1078-0432.CCR-17-0242. [DOI] [PubMed] [Google Scholar]
- 16.Shteingauz A, et al. Processing of heparanase is mediated by syndecan-1 cytoplasmic domain and involves syntenin and alpha-actinin. Cellular and molecular life sciences: CMLS. 2014;71(22):4457–70. doi: 10.1007/s00018-014-1629-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baietti MF, et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat Cell Biol. 2012;14(7):677–85. doi: 10.1038/ncb2502. [DOI] [PubMed] [Google Scholar]
- 18.Thompson CA, et al. Heparanase regulates secretion, composition, and function of tumor cell-derived exosomes. J Biol Chem. 2013;288(14):10093–9. doi: 10.1074/jbc.C112.444562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roucourt B, et al. Heparanase activates the syndecan-syntenin-ALIX exosome pathway. Cell Res. 2015;25(4):412–28. doi: 10.1038/cr.2015.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Au Yeung CL, et al. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nature Comm. 2016;7:11150. doi: 10.1038/ncomms11150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bandari SK, et al. Chemotherapy induces secretion of exosomes loaded with heparanase that degrades extracellular matrix and impacts tumor and host cell behavior. Matrix Biol. 2017 doi: 10.1016/j.matbio.2017.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barash U, et al. A novel human heparanase splice variant, T5, endowed with protumorigenic characteristics. FASEB J. 2010;24(4):1239–48. doi: 10.1096/fj.09-147074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ilan N, et al. Regulation, function and clinical significance of heparanase in cancer metastasis and angiogenesis. Int J Biochem Cell Biol. 2006;38(12):2018–39. doi: 10.1016/j.biocel.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 24.Sanderson RD, Iozzo RV. Targeting heparanase for cancer therapy at the tumor-matrix interface. Matrix Biol. 2012;31(5):283–4. doi: 10.1016/j.matbio.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 25.Fux L, et al. Structure-function approach identifies a COOH-terminal domain that mediates heparanase signaling. Cancer Res. 2009;69(5):1758–67. doi: 10.1158/0008-5472.CAN-08-1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fux L, et al. Heparanase: busy at the cell surface. Trends Biochem Sci. 2009;34(10):511–9. doi: 10.1016/j.tibs.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boyango I, et al. Targeting heparanase to the mammary epithelium enhances mammary gland development and promotes tumor growth and metastasis. Matrix Biol. 2017 doi: 10.1016/j.matbio.2017.08.005. [DOI] [PubMed] [Google Scholar]
- 28.Weissmann M, et al. Heparanase-neutralizing antibodies attenuate lymphoma tumor growth and metastasis. Proc Natl Acad Sci U S A. 2016;113(3):704–9. doi: 10.1073/pnas.1519453113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He YQ, et al. The endoglycosidase heparanase enters the nucleus of T lymphocytes and modulates H3 methylation at actively transcribed genes via the interplay with key chromatin modifying enzymes. Transcription. 2012;3(3):130–45. doi: 10.4161/trns.19998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arvatz G, et al. Post-transcriptional regulation of heparanase gene expression by a 3′ AU-rich element. Faseb J. 2011;24(12):4969–76. doi: 10.1096/fj.10-156372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barash U, et al. Heparanase enhances myeloma progression via CXCL10 downregulation. Leukemia. 2014;28(11):2178–87. doi: 10.1038/leu.2014.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boyango I, et al. Heparanase cooperates with Ras to drive breast and skin tumorigenesis. Cancer Res. 2014;74:4504–4514. doi: 10.1158/0008-5472.CAN-13-2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramani VC, et al. Chemotherapy induces expression and release of heparanase leading to changes associated with an aggressive tumor phenotype. Matrix Biol. 2016;55:22–34. doi: 10.1016/j.matbio.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramani VC, et al. Targeting heparanase overcomes chemoresistance and diminishes relapse in myeloma. Oncotarget. 2016;7:1598–607. doi: 10.18632/oncotarget.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goldberg R, et al. Versatile role of heparanase in inflammation. Matrix biology: journal of the International Society for Matrix Biology. 2013;32(5):234–40. doi: 10.1016/j.matbio.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li RW, et al. Dramatic regulation of heparanase activity and angiogenesis gene expression in synovium from patients with rheumatoid arthritis. Arthritis Rheum. 2008;58(6):1590–600. doi: 10.1002/art.23489. [DOI] [PubMed] [Google Scholar]
- 37.Vlodavsky I, et al. Significance of heparanase in cancer and inflammation. Cancer Microenviron. 2012;5(2):115–32. doi: 10.1007/s12307-011-0082-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Mestre AM, et al. Expression of the heparan sulfate-degrading enzyme heparanase is induced in infiltrating CD4+ T cells in experimental autoimmune encephalomyelitis and regulated at the level of transcription by early growth response gene 1. J Leukoc Biol. 2007;82(5):1289–300. doi: 10.1189/jlb.0507315. [DOI] [PubMed] [Google Scholar]
- 39.Vlodavsky I, et al. Involvement of heparanase in atherosclerosis and other vessel wall pathologies. Matrix Biol. 2013;32(5):241–51. doi: 10.1016/j.matbio.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Secchi MF, et al. Recent data concerning heparanase: focus on fibrosis, inflammation and cancer. Biomol Concepts. 2015;6(5–6):415–21. doi: 10.1515/bmc-2015-0021. [DOI] [PubMed] [Google Scholar]
- 41.Garsen M, et al. Heparanase is essential for the development of acute experimental glomerulonephritis. Am J Pathol. 2016;186(4):805–15. doi: 10.1016/j.ajpath.2015.12.008. [DOI] [PubMed] [Google Scholar]
- 42.Garsen M, et al. Endothelin-1 Induces Proteinuria by Heparanase-Mediated Disruption of the Glomerular Glycocalyx. J Am Soc Nephrol, JASN. 2016;27(12):3545–3551. doi: 10.1681/ASN.2015091070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van den Hoven MJ, et al. Heparanase in glomerular diseases. Kidney Int. 2007;72(5):543–8. doi: 10.1038/sj.ki.5002337. [DOI] [PubMed] [Google Scholar]
- 44.Rabelink TJ, et al. Heparanase: roles in cell survival, extracellular matrix remodelling and the development of kidney disease. Nature Rev Nephrol. 2017;13(4):201–212. doi: 10.1038/nrneph.2017.6. [DOI] [PubMed] [Google Scholar]
- 45.McKown RL, et al. Lacritin and other new proteins of the lacrimal functional unit. Exp Eye Res. 2009;88(5):848–58. doi: 10.1016/j.exer.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Agelidis AM, et al. Viral Activation of heparanase drives pathogenesis of herpes simplex virus-1. Cell reports. 2017;20(2):439–450. doi: 10.1016/j.celrep.2017.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parish CR, et al. Unexpected new roles for heparanase in Type 1 diabetes and immune gene regulation. Matrix Biol. 2013;32(5):228–33. doi: 10.1016/j.matbio.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 48.Gil N, et al. Heparanase is essential for the development of diabetic nephropathy in mice. Diabetes. 2012;61(1):208–16. doi: 10.2337/db11-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang X, et al. Heparanase expression correlates with poor survival in oral mucosal melanoma. Medical Oncol. 2013;30(3):633. doi: 10.1007/s12032-013-0633-5. [DOI] [PubMed] [Google Scholar]
- 50.David G, Zimmermann P. Heparanase tailors syndecan for exosome production. Mol & Cell Oncol. 2016;3(3):e1047556. doi: 10.1080/23723556.2015.1047556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Escobar Galvis ML, et al. Transgenic or tumor-induced expression of heparanase upregulates sulfation of heparan sulfate. Nat Chem Biol. 2007;3(12):773–8. doi: 10.1038/nchembio.2007.41. [DOI] [PubMed] [Google Scholar]
- 52.Wang A, et al. Heparin interaction with a receptor on hyperglycemic dividing cells prevents intracellular hyaluronan synthesis and autophagy responses in models of type 1 diabetes. Matrix Biol. 2015;48:36–41. doi: 10.1016/j.matbio.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blich M, et al. Macrophage activation by heparanase is mediated by TLR-2 and TLR-4 and associates with plaque progression. Arterioscler Thromb Vasc Biol. 2013;33:e56–65. doi: 10.1161/ATVBAHA.112.254961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brunn GJ, et al. Conditional signaling by Toll-like receptor 4. Faseb J. 2005;19(7):872–4. doi: 10.1096/fj.04-3211fje. [DOI] [PubMed] [Google Scholar]
- 55.Goodall KJ, et al. Soluble heparan sulfate fragments generated by heparanase trigger the release of pro-inflammatory cytokines through TLR-4. PLoS One. 2014;9(10):e109596. doi: 10.1371/journal.pone.0109596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gutter-Kapon L, et al. Heparanase is required for activation and function of macrophages. Proc Nat Aca Sci USA. 2016;113(48):E7808–E7817. doi: 10.1073/pnas.1611380113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kundu S, et al. Heparanase Promotes Glioma Progression and Is Inversely Correlated with Patient Survival. Mol Cancer Res, MCR. 2016;14(12):1243–1253. doi: 10.1158/1541-7786.MCR-16-0223. [DOI] [PubMed] [Google Scholar]
- 58.Hermano E, et al. Macrophage polarization in pancreatic carcinoma: role of heparanase enzyme. J Nat Cancer Inst. 2014;106(12) doi: 10.1093/jnci/dju332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Putz EM, et al. NK cell heparanase controls tumor invasion and immune surveillance. J Clin Invest. 2017;127(7):2777–2788. doi: 10.1172/JCI92958. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 60.Caruana I, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. 2015;21(5):524–9. doi: 10.1038/nm.3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vreys V, David G. Mammalian heparanase: what is the message? J Cell Mol Med. 2007;11(3):427–52. doi: 10.1111/j.1582-4934.2007.00039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hammond E, et al. The role of heparanase and sulfatases in the modification of heparan sulfate proteoglycans within the tumor microenvironment and opportunities for novel cancer therapeutics. Frontiers in Oncol. 2014;4:195. doi: 10.3389/fonc.2014.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vlodavsky I, et al. Heparanase: structure, biological functions, and inhibition by heparin-derived mimetics of heparan sulfate. Curr Pharm Des. 2007;13(20):2057–2073. doi: 10.2174/138161207781039742. [DOI] [PubMed] [Google Scholar]
- 64.Vornicova O, et al. The prognostic significance of heparanase expression in metastatic melanoma. Oncotarget. 2016;7(46):74678–74685. doi: 10.18632/oncotarget.12492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang L, et al. MicroRNA-1258 suppresses breast cancer brain metastasis by targeting heparanase. Cancer Res. 2011;71(3):645–54. doi: 10.1158/0008-5472.CAN-10-1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Qu H, et al. miRNA-558 promotes tumorigenesis and aggressiveness of neuroblastoma cells through activating the transcription of heparanase. Human Mol Genet. 2015;24(9):2539–51. doi: 10.1093/hmg/ddv018. [DOI] [PubMed] [Google Scholar]
- 67.Dredge K, et al. An open-label, multi-center phase I study of the safety and tolerability of the novel immunomodulatory agent PG545 in subjects with advanced solid tumors. J Clin Oncol. 2017;35(suppl):2017. abstr 3083. [Google Scholar]
- 68.Bohlmann L, et al. Functional and structural characterization of a heparanase. Nature Chem Biol. 2015;11(12):955–7. doi: 10.1038/nchembio.1956. [DOI] [PubMed] [Google Scholar]
- 69.Wu L, et al. Structural characterization of human heparanase reveals insights into substrate recognition. Nature Struct & Mol Biol. 2015;22(12):1016–22. doi: 10.1038/nsmb.3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu L, et al. Activity-based probes for functional interrogation of retaining beta-glucuronidases. Nature Chem Biol. 2017;13(8):867–873. doi: 10.1038/nchembio.2395. [DOI] [PubMed] [Google Scholar]
- 71.Schmidt EP, et al. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat Med. 2012;18(8):1217–23. doi: 10.1038/nm.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Simeonovic CJ, et al. Heparanase and autoimmune diabetes. Frontiers Immunol. 2013;4:471. doi: 10.3389/fimmu.2013.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Khamaysi I, et al. The role of heparanase in the pathogenesis of acute pancreatitis: A potential therapeutic target. Sci Reports. 2017;7(1):715. doi: 10.1038/s41598-017-00715-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Abassi Z, et al. Involvement of heparanase in the pathogenesis of acute kidney injury: nephroprotective effect of PG545. Oncotarget. 2017;8(21):34191–34204. doi: 10.18632/oncotarget.16573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McKenzie E, et al. Cloning and expression profiling of Hpa2, a novel mammalian heparanase family member. Biochem Biophys Res Commun. 2000;276(3):1170–7. doi: 10.1006/bbrc.2000.3586. [DOI] [PubMed] [Google Scholar]
- 76.Levy-Adam F, et al. Heparanase 2 interacts with heparan sulfate with high affinity and inhibits heparanase activity. J Biol Chem. 2010;285(36):28010–9. doi: 10.1074/jbc.M110.116384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Daly SB, et al. Mutations in HPSE2 cause urofacial syndrome. Am J Hum Genet. 2010;86(6):963–9. doi: 10.1016/j.ajhg.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pang J, et al. Loss-of-function mutations in HPSE2 cause the autosomal recessive urofacial syndrome. Am J Hum Genet. 2010;86(6):957–62. doi: 10.1016/j.ajhg.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Woolf AS, et al. Urofacial syndrome: a genetic and congenital disease of aberrant urinary bladder innervation. Pediatric nephrology. 2014;29(4):513–8. doi: 10.1007/s00467-013-2552-2. [DOI] [PubMed] [Google Scholar]
- 80.Stuart HM, et al. Urinary tract effects of HPSE2 mutations. J Am Soc Nephrol, JASN. 2015;26(4):797–804. doi: 10.1681/ASN.2013090961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Guo C, et al. A mouse model of urofacial syndrome with dysfunctional urination. Human Mol Genet. 2015;24(7):1991–9. doi: 10.1093/hmg/ddu613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gross-Cohen M, et al. Heparanase 2 expression inversely correlates with bladder carcinoma grade and stage. Oncotarget. 2016;7(16):22556–65. doi: 10.18632/oncotarget.8003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gross-Cohen M, et al. Heparanase 2 attenuates head and neck tumor vascularity and growth. Cancer Res. 2016;76(9):2791–2801. doi: 10.1158/0008-5472.CAN-15-1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Siddikuzzaman, et al. Lysyl oxidase: a potential target for cancer therapy. Inflammopharmacology. 2011;19(3):117–29. doi: 10.1007/s10787-010-0073-1. [DOI] [PubMed] [Google Scholar]
- 85.Benezra R, et al. The Id proteins and angiogenesis. Oncogene. 2001;20(58):8334–41. doi: 10.1038/sj.onc.1205160. [DOI] [PubMed] [Google Scholar]
- 86.Sikder HA, et al. Id proteins in cell growth and tumorigenesis. Cancer cell. 2003;3(6):525–30. doi: 10.1016/s1535-6108(03)00141-7. [DOI] [PubMed] [Google Scholar]
- 87.Jung O, et al. Heparanase-induced shedding of syndecan-1/CD138 in myeloma and endothelial cells activates VEGFR2 and an invasive phenotype: prevention by novel synstatins. Oncogenesis. 2016;5:e202. doi: 10.1038/oncsis.2016.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hulett MD, et al. Identification of active-site residues of the pro-metastatic endoglycosidase heparanase. Biochemistry. 2000;39(51):15659–67. doi: 10.1021/bi002080p. [DOI] [PubMed] [Google Scholar]
- 89.Cantarel BL, et al. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nuc Acids Res. 2009;37(Database issue):D233–8. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Werb Z, Lu P. The Role of Stroma in Tumor Development. Cancer J. 2015;21(4):250–3. doi: 10.1097/PPO.0000000000000127. [DOI] [PMC free article] [PubMed] [Google Scholar]