

Abstract
Background
Stress is a generic term used to describe non-specific responses of the body to all kinds of challenges. A very large variability in the response can be observed across individuals, depending on numerous conditioning factors like genetics, early influences and life history. As a result, there is a wide range of individual vulnerability and resilience to stress, also called robustness. The importance of robustness-related traits in breeding strategies is increasing progressively towards the production of animals with a high level of production under a wide range of climatic conditions and management systems, together with a lower environmental impact and a high level of animal welfare. The present study aims at describing blood transcriptomic, hormonal, and metabolic responses of pigs to a systemic challenge using lipopolysaccharide (LPS). The objective is to analyze the individual variation of the biological responses in relation to the activity of the HPA axis measured by the levels of plasma cortisol after LPS and ACTH in 120 juvenile Large White (LW) pigs. The kinetics of the response was measured with biological variables and whole blood gene expression at 4 time points. A multilevel statistical analysis was used to take into account the longitudinal aspect of the data.
Results
Cortisol level reaches its peak 4 h after LPS injection. The characteristic changes of white blood cell count to LPS were observed, with a decrease of total count, maximal at t=+4 h, and the mirror changes in the respective proportions of lymphocytes and granulocytes. The lymphocytes / granulocytes ratio was maximal at t=+1 h. An integrative statistical approach was used and provided a set of candidate genes for kinetic studies and ongoing complementary studies focused on the LPS-stimulated inflammatory response.
Conclusions
The present study demonstrates the specific biomarkers indicative of an inflammation in swine. Furthermore, these stress responses persist for prolonged periods of time and at significant expression levels, making them good candidate markers for evaluating the efficacy of anti-inflammatory drugs.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-4363-5) contains supplementary material, which is available to authorized users.
Keywords: Stress, Hypothalamic-pituitary-adrenal (HPA) axis, Cortisol, Time-course, Systems biology, Microarray, Pig
Background
Over time, farms have evolved towards factory production units. This has led to a decline of the welfare of animals that becomes an important concern for consumers [1]. Moreover, this type of farming has led to the selection of animals with high production traits such as rapid growth, lean meat, or large litters. However, the strong selection focus on these characteristics is suspected to reduce functional traits, such as viability of the newborns or disease resistance. Consequently, the genetic potential of animals is usually not fully expressed in commercial conditions, due to the limiting influence of the environment. Robustness is a specific quality of an individual to express a high production potential in a wide variety of environmental conditions and is now a major specific breeding goal in the context of sustainable farm animal breeding. Various strategies are available to increase robustness, and we have suggested that the reinforcement of the neuroendocrine stress responses may favour the processes of adaptation and dampen the negative consequences of the environment [2]. The hypothalamic-pituitary-adrenocortical (HPA) axis is the main neuroendocrine system involved in adaptation to stress and is strongly influenced by genetic factors [3]. It is therefore a primary candidate for the selection of more robust animals [2].
In modern intensive livestock production, pigs are easily threatened by different types of inflammation. Immunological stress is a comprehensive process involving immunological, neurological, and endocrinological responses [4]. The reciprocal “subjugation” of the brain and the immune system via cytokines and stress hormones is now well demonstrated [5, 6]. The resulting balance has more recently been demonstrated at the level of blood cell transcriptome [7], with chronic stress increasing the expression of genes regulated by inflammatory mediators and decreasing those regulated by glucocorticoid hormones [8]. This approach has been used to evaluate the negative consequences of adverse environmental conditions, mostly in humans but also in farm animals (horses [9]). More recently, individual differences have also been described as related to personality dimensions in humans [10]. However the relationships with individual variations of HPA axis activity, including genetic factors, is still unexplored.
We have previously shown large variations in biological and transcriptomic responses to an ACTH stimulation test [11]. Indeed, the adrenal response to ACTH is a major source of variability of HPA axis function [12]. The present study aims at describing blood transcriptomic, hormonal, and metabolic responses of pigs to a systemic challenge using lipopolysaccharide (LPS), a major component of the outer membrane in gram-negative bacteria [13]. LPS provokes an acute inflammatory syndrome resulting eventually in all kinds of pathophysiological damages [14]. The objective is to analyze the individual variation of the biological responses in relation to the activity of the HPA axis. This was assessed through the level of cortisol released by LPS (this experiment) and also, in the same animals, through the level of cortisol released after an ACTH stimulation test (in an experiment previously published [11]).
Methods
Animals, treatment and blood sampling and biological analyses
The same 120 piglets (63 females and 57 males) as described in [11] were used for this study. In addition to the ACTH stimulation test, previously described, each animal was injected in the neck muscles with LPS at 8 weeks (E. coli serotype 055:B5, Sigma-Aldrich, Saint Quentin Fallavier, FR) at a dose of 15 μg/kg body weight. Injections occurred from 10:00-11:00 AM to avoid nycthemeral variations. Blood samples were collected before the injection (t=0) and 1 h (t=+1), 4 h (t=+4) and 24 h (t=+24) after injection with the same protocol as described in [11].
Cortisol, glucose, free fatty acid (FFA), blood cell counts (including: white cells count, proportion of lymphocytes, monocytes and granulocytes, red cells count, hematocrit, concentration of hemoglobin, red cells width and volume, platelets count and platelets width and volume) were obtained using the same protocol as in the previous study. Fifteen biological variables were measured on the 120 pigs in addition to birth and weaning weights. All these variables were preprocessed for outlier and missing value correction and to ensure normality as in the previous study.
Whole blood transcriptome
A subset of 30 females from 2 batches only was used to study pangenomic expression in whole blood cells at each time point (120 samples). Total RNA isolation and purification was done as described in [11].
Gene expression analysis was performed at the GeT-TRiX facility (GénoToul, Génopole Toulouse Midi-Pyrénées) using Agilent SurePrint G3 porcine microarray GPL16524 (Agilent, 8 ×60 K) following the manufacturer’s instructions (Agilent Technologies, Santa Clara, California). For each of 120 samples, Cyanine-3 (Cy3) labeled cRNA was prepared from 200 ng of total RNA using the One-Color Quick Amp Labeling kit (Agilent) according to the manufacturer’s instructions, followed by Agencourt RNAClean XP (Agencourt Bioscience Corporation, Beverly, Massachusetts). 600 ng of Cy3-labelled cRNA were hybridized on the microarray slides following the manufacturer’s instructions. Immediately after washing, the slides were scanned on Agilent G2505C Microarray Scanner using Agilent Scan Control A.8.5.1 software and fluorescence signal extracted using Agilent Feature Extraction software v10.10.1.1 with default parameters (grid 037880_D_F_20120213 and protocol GE1_1010_Sep10).
Hybridization protocol
Blood samples of 2 pigs at one time step each were of poor quality and thus not used. The same experimental design than the one described in [11] was used to secure the kinetics of the response for each individual and to prevent confounding effects between batch and array. After quality control and filtering, 27,837 probes were kept and log2 transformed. Technical biases and missing data were handled similarly than in the previous study.
Fluidigm Biomark RT-PCR
For validation of array data by Fluidigm technology 22 animals (88 samples) were kept to fit the technical constraints of this technique. Total RNA (1 μg) used in microarray experiments was reverse-transcribed as previously described [15]. Primer sequences for genes were designed using Primer3plus software (http://primer3plus.com) and are given in Additional file 1. The TFRC gene (transferrin receptor) and EPRS gene (glutamyl-prolyl-tRNA synthetase) were used as internal controls. Pre-amplified samples were analyzed with a 96 ×96 Dynamic Array™ IFC (Fluidigm) following the protocol defined by [16], with some modifications. All measurements were performed on the same plate. Each gene was tested twice for each sample. Four dilution points containing a pool of all samples were used to determine PCR efficiency. Data were analyzed using BioMark Gene Expression Data Analysis software (Fluidigm) to obtain Ct values. The Pfaffl method was applied to compute the relative expression of each gene [17]. Pearson correlations were computed to compare the expression values of microarray and quantitative real-time PCR. Quantitative RT-PCR data were also analyzed for time effect by ANOVA with repeated measurements for every gene.
Statistical analyses
All analyses were performed with the R software, version 3.2.2 [18]. They were designed so as to address two main questions: the first one is the study of the evolution over time of the different variables (plasma metabolites, cortisol and gene expression) after LPS injection. The second one is the study of the relation between the different variables and one of the most relevant measure of sensitivity of the adrenals, the cortisol level one hour after ACTH injection (data from [11], obtained on the same animals).
Longitudinal data analysis of the evolution over time of the different variables can be performed using different statistical methods. A very common approach is to fit curves (for instance splines as in [19, 20]) a as prior processing to the statistical analysis. However, four time steps are too few number of time points to obtain an accurate fit. The analysis was thus performed using two main approaches: the first one relies on linear models with the time as a factor covariate and the second one is based on a decomposition of sources of variations, as was already proven successful for repeated measurements analysis in [21] and for longitudinal data analysis in [11].
Statistical analysis of plasma metabolites and cortisol
First, all variables were subjected to a one-way ANOVA with repeated measures and the time step as a factor covariate. In order to control the false discovery rate (FDR) [22], p-values were adjusted using a Benjamini-Hochberg (BH) approach (Table 1). Variables with an adjusted p-value (FDR <0.05) were then subjected to 3 paired t-tests to assess the difference between t=0 and t=+1, between t=0 and t=+4 and between t=0 and t=+24. The full list of p-values was adjusted using a BH approach (Fig. 1).
Table 1.
Reference values (at t=0) for the biological variables, birth weight and weaning weight (n=120)
| Units | Min | Max | Mean | SEM | F | |
|---|---|---|---|---|---|---|
| Tympanic temperature | °C | 36.100 | 40.257 | 39.168 | 0.050 | 258.110 |
| White cells | log10(G/l) | 0.491 | 1.472 | 1.188 | 0.011 | 572.970 |
| Lymphocytes | % | 46.600 | 91.900 | 67.477 | 0.555 | 112.180 |
| Monocytes | % | 3.900 | 16.200 | 8.557 | 0.191 | 69.500 |
| Granulocytes | % | 2.500 | 35.600 | 22.608 | 0.527 | 78.210 |
| L/G ratio | 1.355 | 36.760 | 3.466 | 0.295 | 64.650 | |
| Red cells | T/l | 1.490 | 7.330 | 5.163 | 0.054 | 69.420 |
| Mean corpuscular volume | fl | 39.700 | 63.700 | 52.008 | 0.333 | 62.120 |
| Hematocrit | % | 6.800 | 37.400 | 26.828 | 0.299 | 78.990 |
| Hemoglobin | g/dl | 6.900 | 12.800 | 8.947 | 0.092 | 46.680 |
| Red blood cells distribution width | fl | 29.100 | 33.800 | 32.029 | 0.081 | 74.440 |
| Platelets | log10(G/l) | 2.330 | 2.998 | 2.667 | 0.011 | 227.400 |
| Mean platelet volume | fl | 7.600 | 13.000 | 9.682 | 0.102 | 71.210 |
| Platelet distribution width | % | 9.600 | 12.000 | 10.771 | 0.045 | 122.790 |
| Cortisol | log10(ng/ml) | 1.041 | 2.033 | 1.475 | 0.017 | 370.240 |
| Free fatty acids | 0.079 | 0.560 | 0.162 | 0.005 | 111.040 | |
| Glucose | mmol/l | 5.850 | 9.525 | 8.035 | 0.061 | 123.990 |
| Bilirubin | μmol/l | 4.660 | 13.000 | 8.523 | 0.190 | 178.610 |
| Birth weight | kg | 0.400 | 2.680 | 1.492 | 0.033 | |
| Weaning weight | kg | 5.460 | 16.564 | 9.486 | 0.174 |
Results of the ANOVA for time effect (F value): all variables varied significatively during the experiment, with an FDR <10−12, except for the weights (not tested because constant)
Fig. 1.
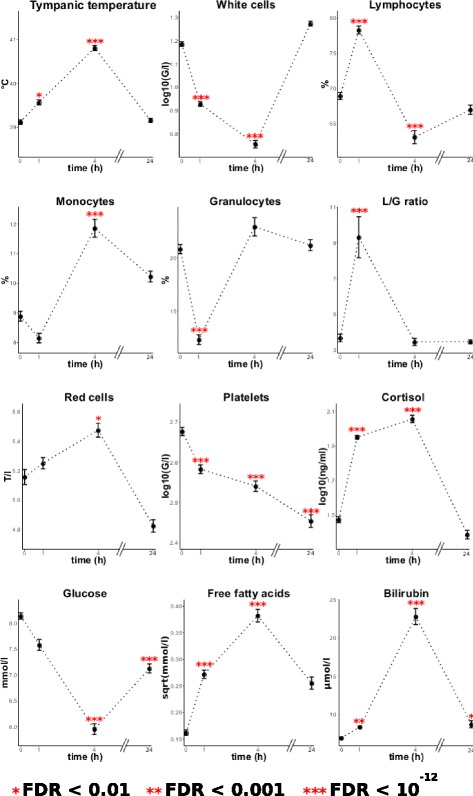
Mean evolution of the biological variables over time. Vertical bars correspond to + and - SEM at each point
In addition, the influence of sex on the biological variables was tested using a two-way ANOVA with repeated measures including sex as a covariate. p-values were adjusted using a BH approach.
Cortisol levels measured one hour after ACTH injection are the most relevant measure to assess the sensitivity of the adrenals to ACTH (data from [11]). Hence, correlations between biological variables at t∈{0,+1,+4,+24} and the level of cortisol one hour after ACTH injection were investigated using t-tests of the linear regression on ACTH level. p-values were adjusted using a BH approach.
Statistical analysis of the transcriptome
Differentially expressed probes (DEP)
The whole blood is composed of different types of white cells with distinct roles which express different kinds of transcripts [23]. It is thus likely that a modification in blood cell composition may influence the gene expression level without having cells actually express transcripts differently. As blood cell composition was found to vary over time after LPS injection, we used the (L/G) ratio as a covariate in our analyses.
Three different approaches were used to identify relevant probes. The first two are longitudinal analyses aiming, respectively, at identifying probes with an expression significantly varying from their basal levels after LPS injection and probes with a varying contribution of the L/G ratio to their expressions after LPS injection. The last analysis searched for probes correlated to the level of cortisol one hour after ACTH injection.
Firstly, we identified probes differentially expressed at each time step while taking blood cell composition into account. Blood cell composition was measured by the L/G ratio. Three models (one for each time step t ′ where t ′∈{+1,+4,+24}) were fitted to each probe using observations at t=0 and t=t ′.
| 1 |
with i=1,…n is animal i. exprit is the expression of the probe being studied for animal i at time step t (t∈{0,t ′}), μ 0 is the specific contribution of time step t=0, is the effect of time step t ′, is the effect of L/G ratio in this model and is an error term.
We then tested the contribution of time step t ′ against the null hypothesis H 0: . The full list of p-values was globally adjusted using a Bonferroni approach. As the Bonferroni approach exerts a more stringent control than the BH approach, it was used to obtain a narrowed list of the most significant probes. Probes with at least one adjusted p-value <0.01 were probes for which the expression adjusted by the L/G ratio was significantly different from the basal level. In the sequel, this list of genes will be referred to as (List1).
Secondly, we identified probes for which the L/G ratio effect is different according to the time step. To that aim, we compared a complete model, including all time step contributions and the L/G ratio effect according to the time step (Eq. (2)):
| 2 |
(with t∈{0,1,4,24} and β t is the interaction effect between time step t and the L/G ratio of individual i at time step t), to a reduced model, including only the average L/G ratio and all time step contributions (Eq. (3)):
| 3 |
An F-test was then performed to test the null hypothesis, H 0: β 0=β 1=β 4=β 24, against the alternate hypothesis, H 1: ∃t 1,t 2 such as . Multiple testing was handled by applying a BH approach (FDR <0.05). Probes for which the test was significant were probes for which the effect of L/G varied over time. In the sequel, this list of genes will be referred to as (List2).
Finally, we studied correlations between all probes and cortisol level when it reaches its peak in blood after LPS injection. Thus, Pearson correlations, ρ, were computed between DEP expression at each time step and cortisol level at t=+4. A correlation test was then performed to test the null hypothesis, H 0: ρ=0 against H 1: ρ≠0. Multiple testing was handled by using a BH approach (FDR <5%). This list of genes will be referred as (List3) in the sequel.
In addition, to link probes responding to a LPS injection with a measure of the HPA axis activity, we studied correlations between expression of all probes and the cortisol level at one hour after ACTH injection, as measured on the same pigs in [11].
All lists of DE probes were then annotated and duplicated probes were removed by keeping only DEP with the smallest FDR per annotated gene and all non-annotated genes. Remaining genes will be referred as differentially expressed genes (DEG) in the sequel.
Functional analysis
Enrichment analysis was performed using tools available at WEB-based GEne SeT AnaLysis Toolkit (WebGestalt) [24, 25]. Entrez gene IDs were used as unique gene identifiers. Target gene lists for main effects and interactions and a background gene set consisting of all 9530 genes were used to identify enrichment in GO, KEGG, Transcription Factor Target, Microarray Target, Protein Interaction Network Module, and Phenotype Analysis in WebGestalt using Fisher’s exact test and BH correction for multiple testing.
A pathway is an interconnected arrangement of processes, representing the functional roles of genes in the genome. The biological processes in which individual genes may participate were identified using the “Gene Ontology” database AmiGO (http://amigo.geneontology.org). The DEG were assembled into networks using Ingenuity Pathway Analysis (IPA Ⓒ) (http://www.ingenuity.com). This application includes algorithms that automatically identify the biological pathways and functions of selected genes. It is based on a large bibliographic database with various types of interaction already identified between pairs of genes. Every biological network extracted by IPA corresponds to the best possible arrangement of the genes, and are associated with a score derived from a p-value (right-tailed Fisher’s exact test, − log10-transformed).
Time course analyses
In the case of time course analyses, the approach previously described (applying a univariate linear model on each variable followed by multiple test correction) is common. However, this approach disregards the dependencies between genes and does not allow for a global view of the relationships between the repeated measurements in high dimensional data. The multilevel approach, already proven successful to investigate the relationships between repeated measurements in [21] was thus used and combined with multivariate data analysis methods.
The multilevel approach [21] is inspired by the mixed-model framework and uses a split-up variation of the (n T)×p matrix X that contains the observations of p variables (clinical biology variables or gene expressions) on n animals with T=4 times of measurements:
| 4 |
Similarly to what was performed in [11], multivariate approaches were performed on X w to bring out the most relevant correlations between variables in the dataset, independently from individual variations. First a multilevel PCA was performed on the biological variables to study the overall effect of LPS on plasma metabolites and cortisol over time. Then, a multilevel multiple factor analysis (MFA) [26] was used to investigate the overall relationships between clinical biology and transcriptomic data.
Results
Plasma cortisol, metabolites, and blood cell counts
Baseline values of biological variables and the global time effect, and birth and weaning weights are shown in Table 1. Figure 1 shows the evolution of the main variables over time.
Tympanic temperature peaked at t=+4 (40.8 °C vs 39.1 °C) and returned to basal levels at t=+24. A decrease of total count of white blood cell count was observed, maximal at t=+4 (5.70 vs 15.35 G/l) and the mirror changes in the respective proportions of lymphocytes and granulocytes. This indicated that the lymphocytes/granulocytes ratio (L/G) was a good measure to use in order to take into account these changes that result mainly from the redistribution of lymphocytes into the tissues [27]. The L/G ratio was maximal at t=+1 (9.32 vs 3.67) and back to basal levels at t=+4. The red blood cell count and associated measures (hematocrit and hemoglobin concentration) showed a biphasic change, with an initial increase, maximal at t=+4 (5.47 vs 5.16 T/l) and a subsequent long-lasting decrease (4.82 T/l at t=+24). The platelet count showed a steady decrease until at least t=+24 (284 vs 475 G/l). These measures were not influenced by sex, except the mean red cell volume and hematocrit that were slightly lower in males (FDR <0.05).
Cortisol levels peaked at t=+4 with a 3.83-fold increase (114.3 vs 29.8 ng/ml). Circulating glucose levels were reduced by 26.9% to 5.95 mmol/l at t=+4. The circulating concentration of free fatty acids increased from 0.026 to 0.146 mmol/l at t=+4. None of these biochemical measures was influenced by sex.
Overall effect of LPS on clinical biological variables
The overall effect of LPS over time was investigated with a multilevel PCA (Fig. 2). The first component of the multilevel PCA opposes the observations at t=0 (negative coordinates on this axis) to the observations at t=+4 (positive coordinates on this axis), this time step corresponding to the peak of LPS effect. The second component opposes the observations at t=+24 (positive coordinates on this axis) to the other observation times (negative coordinates on this axis). The representation of the variables shows that the first axis is mainly driven by an opposition between free fatty acids (FFA), bilirubin, temperature and cortisol (high measures at t=+4), and white cell count and glucose (low measures at t=+4). The second axis of the PCA is driven by L/G ratio and platelet count that are high at t=+1.
Fig. 2.
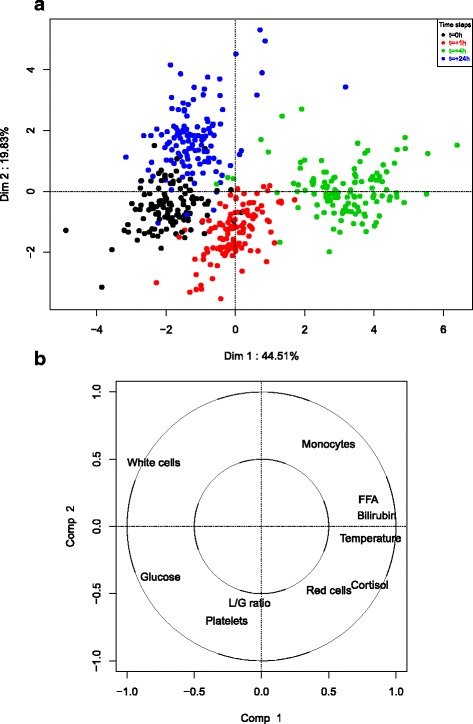
Multilevel PCA on the biological variables responding to LPS. Colors represent the time step; Black: t=0; Red: t=+1; Green: t=+4; Blue: t=+24; a: Projection of the individuals on dimensions 1 - 2; b: Projection of the variables on dimensions 1 - 2
No biological variable was found to be correlated to cortisol level one hour after ACTH injection.
Differentially expressed genes related to key immune functions
In our study, we used a comprehensive gene expression profiling by means of microarray analysis to identify clusters of genes differentially expressed in peripheral blood cells, taking into consideration the kinetic of the response with 4 time points (t∈{0,+1,+4,+24}). LPS induces dramatic changes in blood cell number and lymphocyte/granulocyte (L/G) ratio that introduces a confusion between time and cell type effects, and a major challenge for the interpretation of transcriptomic data. Therefore we based the interpretation of the results on three different lists of genes, (List1), (List2), and (List3).
All genes found to be differentially expressed after ACTH injection in our previous study [11] were also found in one of these three lists.
Analysis of each list of genes
The first list of genes (List1) consists of 9530 unique genes (22,794 transcripts, Additional files 2 and 3) for which the expression adjusted by the L/G ratio was significantly different from basal level. (List1) was submitted to gene ontology and enrichment analysis. These analyses showed 106 classes significant at FDR < 0.05. Due to the important number of DEG, generic classes were removed (such as morphogenesis, transcription, locomotion and others). Only genes that were well-known and well described in the literature were chosen to define a final selected list of 284 genes. These genes were grouped into 6 functional classes that were all found enriched for genes (List1) (Immunity and Inflammation, Chemotaxis, Apoptosis, Calcium ion transport, Metabolism, Hormonal Response).
The “immunity and inflammation” class (175 genes) is related to the inflammatory cascade after activation of leukocytes by LPS via TLR4 receptor (a receptor for bacterial lipopolysaccharide). TLR4 is a critical driver of immune responses to bacterial infections. Signals from TLR4 promote NF- κB and AP-1 activation, leading to inflammatory gene expression [28] (DEG for TLR4, TNF, JUNB, and NF-B pathway).
The “chemotaxis” class is composed of 59 genes. Among them ABHD2, ACADS, AIF1, ANXA7, ARPC1A, ARPC2, CD97, CHL1, CLIC1, CNTFR, COQ3, DGKD, DNASE2, GP1BA, GPI, HCLS1, HPS6, IL1RN, IL8RA, KAT5, LOC100523056, LSP1, MAN2B1, PARK7, PTPN6, SMAD7, SPG21, TMEM173, TMSB10, TMSB4X, TRDMT1, and TSPO genes are related to immune cell trafficking. This observation is in agreement with the observed blood cell redistribution.
The “apoptosis” class (33 genes) includes C5AR1, CCL24, CCR1, CCR3, CXCL13, IRG1, ALDOC, C3AR1, CADM1, CAPN3, HEXA, ID3, MAEA, PLAU, PRDX5, PROC, and CXCR2 genes related to apoptosis and inflammatory response, and TNFSF13B and NFKBIA involved in cell-activating factor signalling pathway.
Twelve genes (CD9, ANXA5, COMT, DDIT3, ADAM10, BAD, SOD2, ADRB2, CLN8, LTA, TGFBR1 and PTEN) form a “calcium ion transport” class.
The “metabolism” class includes four genes (EDN1, COFILIN, PLA2G4A, and CORO1A), and the “hormonal responses” class includes one gene (HMOX1).
The 284 remaining genes from the first list (List1) were clustered into 4 clusters using HAC (Fig. 3, Additional file 3).
Fig. 3.
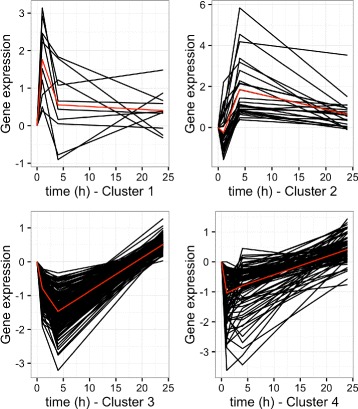
Black: Average evolution the genes in each of the clusters identified by the HAC on the 284 DEG identified in list (List1). Evolution of each gene is translated so that it is equal to 0 at t=0; Red: Average evolution over all genes in the cluster (cluster 1: 8 genes, cluster 2: 12 genes, cluster 3: 159 genes, cluster 4: 77 genes). 28 genes were unclassified
The second list of genes (List2), consists of 154 unique genes (209 transcripts, Additional file 4) for which the contribution of the L/G ratio to the expression varied over time steps. Among these genes, 132 genes were further assembled into six functional networks that notably revealed hematological system development and function, tissue morphology, cancer, organismal injury and abnormalities, reproductive system disease, cellular growth and proliferation. The average evolution of all these genes shows the same expression profile. This group of genes is characterized by genes decreasing with a peak of expression at t=+1 and stable between t=+4 and t=+24. According to this analysis, it is unlikely that genes for which the contribution of the L/G ratio to the expression varied over time steps are directly involved in the immune response to LPS injection.
The third list of genes (List3) consists of 116 unique genes (185 transcripts, Additional file 5) for which the expression was found to be correlated to the level of cortisol at t=+4. This time point was chosen as the peak of plasma cortisol concentration after LPS (Fig. 1). The most significant functions are: cellular function and maintenance – function of blood cells (30 genes); cellular movement, immune cell trafficking – leucocyte migration (35 genes); lymphoid tissue structure and development, tissue morphology – quantity of lymphatic system cells (34 genes); cellular function and maintenance – function of leucocytes (27 genes); hematological system development and function, tissue morphology – quantity of leucocytes (36 genes); cellular movement, hematological system development and function, immune cell trafficking – cell movement of leucocytes (32 genes); immunological disease – systemic autoimmune syndrome (39,374 genes) (Additional file 6).
Validation of differential expression by quantitative real-time PCR
Twenty-two DEG were selected for further examination by quantitative real-time PCR using the Fluidigm technique (Table 2). These genes were selected from the three studied lists ((List1)), (List 2), and (List 3)). Pearson correlations between the differences in expression measured by quantitative real-time PCR and microarray were greater than 0.70 for 7 genes (CHI3L1, MYLIP, LCK, SOD2, VAT1, COMT, and FAS). Lower correlations (between 0.5 and 0.6) were obtained for GALK2, VNN2, JAK2, KAT5, ADAM10, RARA, and ANXA7.
Table 2.
Correlations between quantitative real-time PCR with microarray expression for selected genes (n=22)
| Gene name | Gene description | List that provided the gene | Pearson correlation | p-value (Fluidigm) | Gene expression profile |
|---|---|---|---|---|---|
| CHI3L1 | chitinase 3 like 1 | (List 1) | 0.72 | 1.68e-07 | down at t=+1 |
| up at t=+24 | |||||
| CHIT1 | chitinase 1 | (List 1) | 0.68 | 0.00214 | down at t=+1 |
| up at t=+24 | |||||
| CLEC2D | C-type lectin domain family 2 member D | (List 2), (List 3) | 0.60 | 0.0019 | down at t=+1 |
| up at t=+24 | |||||
| GALK2 | galactokinase 2 | (List 1) | 0.50 | 9.52e-11 | down at t=+1 |
| up at t=+24 | |||||
| HSD17B11 | hydroxysteroid (17-beta) | (List 1) | 0.67 | 3.02e-11 | down at t=+1 |
| dehydrogenase 11 | up at t=+24 | ||||
| KAT5 | lysine acetyltransferase 5 | (List 1) | 0.53 | 5.37e-09 | down at t=+1 |
| up at t=+24 | |||||
| LCK | LCK proto-oncogene, Src family | (List 1) | 0.74 | 3.77e-10 | down at t=+1 |
| tyrosine kinase | up at t=+24 | ||||
| MSN | moesin | (List 1) | 0.61 | 5.68e-05 | down at t=+1 |
| up at t=+24 | |||||
| MYLIP | myosin regulatory light chain | (List 1) | 0.72 | 2.26e-10 | down at t=+1 |
| interacting protein | up at t=+24 | ||||
| RAB31 | RAB31, member RAS oncogene family | (List 1) | 0.65 | 0.00152 | down at t=+1 |
| up at t=+24 | |||||
| RARA | retinoic acid receptor alpha | (List 1) | 0.58 | 6.59e-05 | down at t=+1 |
| up at t=+24 | |||||
| SSH1 | slingshot protein phosphatase 1 | (List 1), (List 2) | 0.67 | 1.28e-11 | down at t=+1 |
| (List 3) | up at t=+24 | ||||
| VAT1 | vesicle amine transport 1 | (List 1) | 0.75 | 1.26e-12 | down at t=+1 |
| up at t=+24 | |||||
| VNN2 | vanin 2 | (List 1) | 0.52 | 0.74 | down at t=+1 |
| up at t=+24 | |||||
| CERS4 | ceramide synthase 4 | (List 2), (List 3) | 0.60 | 1.16e-06 | down at t=+1 |
| up at t=+4 | |||||
| FAS | Fas cell surface death receptor | (List 1), (List 2) | 0.83 | <2e-16 | down at t=+1 |
| (List 3) | up at t=+4 | ||||
| JAK2 | Janus kinase 2 | (List 2), (List 3) | 0.52 | 2.54e-10 | down at t=+1 |
| up at t=+4 | |||||
| ADAM10 | ADAM metallopeptidase domain 10 | (List 1) | 0.56 | 8.15e-05 | down at t=+4 |
| up at t=+24 | |||||
| ANXA7 | annexin A7 | (List 1) | 0.59 | 0.000417 | down at t=+4 |
| up at t=+24 | |||||
| COMT | catechol-O-methyltransferase | (List 1) | 0.75 | 1.64e-14 | down at t=+4 |
| up at t=+24 | |||||
| STMN1 | stathmin 1 | (List 1) | 0.69 | 3.24e-09 | down at t=+4 |
| up at t=+24 | |||||
| SOD2 | superoxide dismutase 2, | (List 1) | 0.74 | <2e-16 | up at t=+4 |
| mitochondrial | down at t=+24 |
p-value (Fluidigm) gives the time effect of quantitative real-time PCR data for every gene
Discussion
Plasma cortisol, metabolites, and blood cell counts
In pigs like in other species, LPS is responsible for the fever and inflammatory reaction induced by gram-negative bacterial infection, as shown by the increase in the circulating levels of pro-inflammatory cytokines and acute phase proteins, as well as the changes in white blood cell counts [27, 29–31], which explained the characteristic changes of white blood cell count to LPS observed in our study.
LPS also induces profound endocrine and metabolic changes and our results are consistent with previously published data in pigs [27, 29, 31]. A large increase in circulating levels of cortisol (and catecholamines, not measured here) has been described and these hormonal changes can be involved in the release of the mediators of inflammation [27, 31]. It was shown previously in mice that this hypoglycaemia cannot be explained by changes in insulin concentrations that are also reduced by LPS [32], but it could result from the increased glycolysis in muscles and immune cells, as well as from a reduced hepatic glucose production [33]. The increase of circulating concentration of free acids can result from the lipolytic action of catecholamines and cortisol that are massively released by LPS [11, 34] and from LPS-induced changes in hepatic and fat tissue lipid metabolism [35, 36]. A sharp increase in bilirubin concentrations was also measured at t=+4 (17.72 vs 2.14 μmol/l), reflecting the hepatic toxicity of LPS [37, 38].
Clustering of differentially expressed genes (List1)
The 284 remaining genes from the first list (List1) were clustered into 4 clusters using HAC. This clustering exhibited patterns related to different kinetics of their response.
The first cluster includes 8 genes up-regulated at t=+1 and related to immune cell tracking (ALOX12, JUNB, TNFAIP3, CCL20, CXCL5, NFKBIA, LTA, EDN1). Inflammation is a powerful protective mechanism which is coordinated and controlled by cytokines and chemokines and, as expected, we detected an increase in the expression level of members of the CXCL family. JUNB gene (a member of Jun family) also participates in the immune response; it is activated by the TLR signalling pathway [39] and can induce expression of interleukins [40–43]. Hormone activation of the glucocorticoid receptor in leukocytes results in a profound suppression of pro-inflammatory gene networks such as the NF- κB mediated transcription of pro-inflammatory cytokine genes and CXCL2 together with LTA were described by [44] as glucocorticoid-regulated genes. These findings show that wide variation in glucocorticoid sensitivity exists between individuals which may influence susceptibility to inflammatory diseases [11].
The second cluster includes 12 genes up-regulated at t=+4 and related to connective tissue disorders and inflammatory diseases (GYG1, PDXK, RETN, C3, IL27, TLR4, IL1RN, ICAM1, CXCL13, C3AR1, FAS, SOD2, and TLR4). Toll-like receptor 4 (TLR4) is essential for initiating the innate response to lipopolysaccharide from Gram-negative bacteria by acting as a signal-transducing receptor. As the pig industry faces a unique array of related pathogens, it is anticipated that the genotype of swine TLR4 could be of crucial importance in future strategies aimed at improving genetic resistance to infectious diseases [45].
The third cluster includes the genes down-regulated at t=+4. This cluster groups 159 DEG related to the inflammatory response. It is associated with functions linked to immunological disease, cancer, cell death and survival, immune cell tracking, and belongs to a series of twelve canonical pathways, including leukocyte extravasation signalling, NF- κB activation, and glucocorticoid receptor signalling.
The fourth cluster includes the genes down-regulated at t=+1. These genes are related to apoptosis, NF- κB, and death receptor signalling canonical pathways.
Comparison of all lists of genes
Figure 4 shows the overlap of the three lists of genes ((List1), (List2), and (List3)). Twenty two genes are common between the three analyses: ABHD2, C3, C3AR1, C5AR1, CAPN3, CCDC47, CD163, CXCL13, DBN1, DGAT2, FAS, GYG1, HMOX1, NFAM1, PDXK, SELL, SERPING1, SOD2, TLR4, TNFRSF1A, TNFSF13B and TXNIP. Identification of genes common to all three analyses, which are known in literature to have an important role in chemotaxis, apoptosis, calcium ion transport and metabolism, confirms their roles in immunity and inflammation in pigs. These genes could further serve in a panel of tissue prognosis indicators of porcine immune response. IPA analysis showed that these genes form two functional networks, NW1 and NW2 (Table 3). The first functional network (NW1, Fig. 5) is related to infectious diseases, cellular movement, hematological system development and function, cell-to-cell signalling and interaction. These genes form a node connected to ESR1. This gene encodes the estrogen receptor 1, a ligand-activated transcription factor. Estrogen receptors are also involved in pathological processes including breast cancer, endometrial cancer, and osteoporosis [46]. Among the genes that form these networks, two are particularly interesting, TLR4 and CD163. Toll-like receptor 4 (TLR4) signalling pathway is the essential member in TLRs family, which plays an important role in a variety of inflammatory reaction such as in diarrhoea and hydropsy of weaned piglets infected by pathogens [47]. The TLR4 gene was described as one of the important immunological factors influencing for example the development of mycoplasma pneumonia of swine [48]. TLR4 dysregulation promoted aberrant cytokine production in bacterial sepsis [49].
Fig. 4.
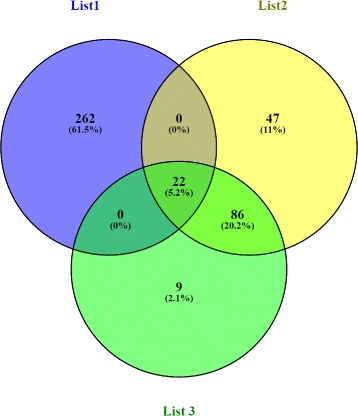
Venn diagram showing DEG common to all lists of genes ((List1), (List12 and (List3); 22 DEG)
Table 3.
Gene networks (NW) with commons DEG between the three lists of genes (in bold)
| NW | Genes in network | Genes in present study | Top diseases and functions |
|---|---|---|---|
| 1 | ABHD2 ∗, ALB, C3AR1, C5AR1, Calmodulin, CAPN3, CD163, DBN1, ESR1*, FAS, Gpcr, GPR158, IgG, ITPKA, NFAM1, PDXK, SELL, SERPING1, SMARCA4, SYK, TLR4, TNFR/Fas, TNFRSF1A, TREM1, YWHAZ | 13 | Infectious Diseases, Cellular Movement, Hematological System Development and Function, Cell-To-Cell Signaling and Interaction |
| 2 | 2-methoxyestradiol, ABHD2 ∗, C3, Cbp/p300, CCDC47, CDKN1A, CXCL13, DGAT2, EGFR, EP300, ESR1*, GYG1, HMOX1, NR3C1, PPARG, SOD2, STAT1, TNFSF13B, TXNIP | 10 | Cell Death and Survival, Cellular Development, Cellular Growth and Proliferation |
ABHD2 ∗ and ESR1 ∗ are common to both networks
Fig. 5.
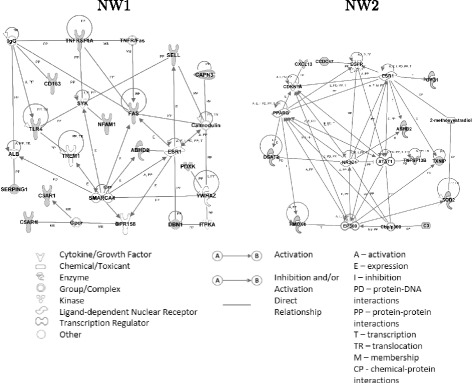
Functional networks formed by the 22 DEG common to (List1), (List2) and (List3). NW1: related to infectious diseases, cellular movement, hematological system development and function, cell-to-cell signalling and interaction. NW2: related to cell death and survival, cellular development, cellular growth and proliferation
The expression of porcine CD163 (a scavenger receptor belonging to a cysteine-rich superfamily) on monocytes/macrophages correlates with permissiveness to African swine fever infection [50]. CD163 is considered as the most important receptor for porcine reproductive and respiratory syndrome attachment and internalization [51]. Cell entry of simian hemorrhagic fever virus is also dependent on CD163 [52].
The second network (NW2) is related to cell death and survival, cellular development, cellular growth and proliferation. The NR3C1 (nuclear receptor subfamily 3, group C, member 1) gene encodes the glucocorticoid receptor, which can function both as a transcription factor that binds to glucocorticoid response elements in the promoters of glucocorticoid responsive genes, and as a regulator of other transcription factors. Signal transducer and activated transcription 1 (STAT1) has been identified as a point of convergence for the cross talk between the pro-inflammatory cytokine interferon γ (IFNγ) and the Toll-like receptor-4 (TLR4) ligand LPS in immune cells [53]. LPS activates STAT1 via the NF-κB pathway [54].
Several transcriptomic studies of immune and inflammatory responses have been published in pigs, however little is known about longitudinal changes. The peripheral blood transcriptome reflects variations in immunity traits and a few potential gene biomarkers were found for immunocompetence (RALGDS gene was shown for prediction of IL2; ALOX12 for phagocytosis; GNLY, KLRG1 and CX3CR1 for CD4-/CD8+ cell count) [55]. Zhou et al. [56] investigated the transcriptional responses of pig peripheral blood mononuclear cells following an experimental challenge with the intracellular protozoan Toxoplasma gondii. Zhao et al. [57] studied the response to the foot-and-mouth disease infection. Peripheral blood mononuclear cells transcriptome profiles were studied by Islam et al. [58] to identify potential candidate genes and functional networks controlling the innate and adaptive immune responses to the porcine reproductive and respiratory syndrome vaccine. The innate immune transcriptional network was found to be regulated by LCK, STAT3, ATP5B, UBB and RSP17 genes. The adaptive immune transcriptional response to the porcine reproductive and respiratory syndrome vaccine in peripheral blood mononuclear cells is related to TGFb1, IL7R, RAD21, SP1 and GZMB. Altogether these results show the value of gene expression studies to explore inflammatory and immune responses and the factors of their regulation.
Conclusion
We have presented here an integrative biological approach combining different statistical models and biological measures and taking into consideration the longitudinal aspect of the data. This analysis of biological data required the development of a methodology adapted to both the multi-dimensional and longitudinal data.
LPS stimulation was chosen because it is standard to study general inflammation processes in many species. Immunological stress is the status of animals challenged by bacteria or viruses. It is associated with immunological, neurological, and endocrinological responses [4]. A four time point kinetic was studied. It has been reported that time points earlier than 24 h are more relevant to decipher the onset of the response to stimulus as shown in kinetics studies in cow [59], pig [60], mouse [61] or human [62]. Moreover, kinetic studies have revealed that many genes return to their basal expression level by 24-48 h of stimulation, suggesting that homeostasis is restored at that time [59, 60]. Our results provide many candidate genes to test for kinetic studies and ongoing complementary studies focused on this topic. It is worth mentioning that the different responses to LPS are not influenced by the adrenal gland reactivity as measured by the cortisol response to ACTH.
In conclusion, we have demonstrated that there are specific biomarkers indicative of an LPS-stimulated inflammatory response. Furthermore, these responses persist for prolonged periods of time and at significant expression levels, making them good candidate markers for evaluating the efficacy of anti-inflammatory drugs. The majority of the genes identified have known roles in the inflammatory process. Subsequently, these biomarkers may serve collectively as an indication of inflammation in swine. The knowledge gained from this series of experiments may help in the development of a model for further studies.
Additional files
Sequences of oligonucleotide primers tested by quantitative RT-PCR analysis. Gene name: name of the gene; Gene description: informations on the gene’s molecular function; Forward Primer; Reverse Primer. (XLSX 11 kb)
List of 9,530 unique genes differentially expressed in list (List1). (XLSX 2360 kb)
List of 284 unique genes differentially expressed in list (List1) included in non-generic biological functions (n=30). Generic biological functions include functions such as morphogenesis, transcription, locomotion. Gene name: name of the gene; Gene description: informations on the gene’s molecular function; Cluster: cluster in which the gene is classified by HAC; Time point: time measurement where the gene is DE; Expression: whether the DEG is up or down-regulated. (XLSX 21.2 kb)
List of 154 unique genes differentially expressed in list (List2). (XLSX 35.1 kb)
List of 116 unique genes differentially expressed in list (List3). (XLSX 22.9 kb)
Biological functions enriched by differentially expressed genes in list (List3) (n=30). Categories: Category of the enriched function; Diseases or Function Annotation: name of the enriched function; FDR: FDR of the enrichment test; Genes: list of genes enriching the biological function; Genes: number of genes enriching the biological function. (XLSX 10 kb)
Acknowledgements
We thank the team of “le Magneraud” experimental station who took care of animal breeding, GeT-TRiX platform (Toulouse) for expression data production, GenPhySE INRA Toulouse (more specificaly Nathalie Iannuccelli, Katia Fève and Juliette Riquet) for experimental support, ANEXPLO platform (Toulouse) for biological assays, PEGASE INRA Saint-Gilles (more specifically Rafael Comte) for cortisol assay, Aurélie Ducan for hematology analysis, Aurélie Sécula-Tircazes for Fluidigm analysis and Magali San Cristobal and Pascal Martin for helpful discussions on the analyses.
Funding
This project received financial support from the French National Research Agency (contract SUSoSTRESS, ANR-12-ADAP-0008) which also funded the PhD thesis of VS together with the Région Midi-Pyrénées. DB was supported by the Embassy of France in Russia (Mechnikov Program). The funding bodies had no role in the design and conduct of the study, the collection, management, analysis, and interpretation of the data; or the preparation, review, or approval of the manuscript.
Availability of data and materials
The datasets generated and analysed during the current study are available in Gene Expression Omnibus (GEO repository, http://www.ncbi.nlm.nih.gov/geo/) through the accession number GSE107487.
Authors’ contributions
VS, CY and NVV developed and performed all the statistics. PM and ET developed and undertook the experimental design, performed experimentations and ensured the biological interpretation. LG, ET and LL ensured the experimental transcriptomic analysis. YL and CN provided the transcriptomic data set. VS, ET, DB and LL performed the transcriptomic related biology interpration. LG, DB, and AS performed the sampling, the gene expression study and the data management. YB was responsible of the animal management. VS, NVV, ET and PM wrote the paper and supervised all experimentations. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Animal breeding was taken care of at “le Magneraud” experimental station, INRA (Charente-Maritime, France). All animal use was performed under European Union and French legislation (directive 201063UE, décret 2013-118). The protocol and procedures were approved by the local (Poitou-Charentes) ethics committee (decision CE2013-1, 21012013).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-4363-5) contains supplementary material, which is available to authorized users.
Contributor Information
Elena Terenina, Email: elena.mormede@inra.fr.
Valérie Sautron, Email: valerie.sautron@gmail.com.
Caroline Ydier, Email: c.ydier@hotmail.fr.
Darya Bazovkina, Email: darya.bazovkina@gmail.com.
Amélie Sevin-Pujol, Email: amelie.sevin@orange.fr.
Laure Gress, Email: laure.gress@inra.fr.
Yannick Lippi, Email: yannick.lippi@inra.fr.
Claire Naylies, Email: claire.naylies@inra.fr.
Yvon Billon, Email: yvon.billon@inra.fr.
Laurence Liaubet, Email: laurence.liaubet@inra.fr.
Pierre Mormede, Email: pierre.mormede@inra.fr.
Nathalie Villa-Vialaneix, Email: nathalie.villa-vialaneix@inra.fr.
References
- 1.Tawse J. Consumer attitudes towards farm animals and their welfare: a pig production case study. Biosci Horizons. 2010;3(2):156–65. doi: 10.1093/biohorizons/hzq020. [DOI] [Google Scholar]
- 2.Mormede P, Terenina E. Molecular genetics of the adrenocortical axis and breeding for robustness. Domest Anim Endocrinol. 2012;43(2):116–31. doi: 10.1016/j.domaniend.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 3.Mormede P, Foury A, Barat P, Corcuff JB, Terenina E, Marissal-Arvy N, et al. Molecular genetics of hypothalamic-pituitary-adrenal axis activity and function. Ann N Y Acad Sci. 2011;1220(1):127–36. doi: 10.1111/j.1749-6632.2010.05902.x. [DOI] [PubMed] [Google Scholar]
- 4.Song C, Jiang J, Han X, Yu G, Pang Y. Effect of immunological stress to neuroendocrine and gene expression in different swine breeds. Mol Biol Rep. 2014;41(6):3569–76. doi: 10.1007/s11033-014-3219-1. [DOI] [PubMed] [Google Scholar]
- 5.Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9(1):46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pavlov VA, Tracey KJ. Neural circuitry and immunity. Immunol Res. 2015;63(1-3):38–57. doi: 10.1007/s12026-015-8718-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Irwin MR, Cole SW. Reciprocal regulation of the neural and innate immune systems. Nat Rev Immunol. 2011;11(9):625–32. doi: 10.1038/nri3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cole SW. Elevating the perspective on human stress genomics. Psychoneuroendocrinology. 2010;35(7):955–62. doi: 10.1016/j.psyneuen.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lansade L, Valenchon M, Foury A, Neveux C, Cole SW, Layé S et al. Behavioral and transcriptomic fingerprints of an enriched environment in horses (equus caballus) PloS ONE. 2014;9(12):e114384. doi: 10.1371/journal.pone.0114384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vedhara K, Gill S, Eldesouky L, Campbell BK, Arevalo JMG, Ma J, et al. Personality and gene expression: do individual differences exist in the leukocyte transcriptome? Psychoneuroendocrinology. 2015;52:72–82. doi: 10.1016/j.psyneuen.2014.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sautron V, Terenina E, Gress L, Lippi Y, Billon Y, Larzul C et al. Time course of the response to ACTH in pig: biological and transcriptomic study. BMC Genomics. 2015;16(961):PMC4650497. doi: 10.1186/s12864-015-2118-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larzul C, Terenina E, Foury A, Billon Y, Louveau I, Merlot E, et al. The cortisol response to ACTH in pigs, heritability and influence of corticosteroid-binding globulin. Animal. 2015;9(12):1929–34. doi: 10.1017/S1751731115001767. [DOI] [PubMed] [Google Scholar]
- 13.Hou X, Zhang J, Ahmad H, Zhang H, Xu Z, Wang T. Evaluation of antioxidant activities of ampelopsin and its protective effect in lipopolysaccharide-induced oxidative stress piglets. PloS ONE. 2014;9:e108314. doi: 10.1371/journal.pone.0108314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Westphal M, Stubbe H, Sielenkämper A, Borgulya R, Van Aken H, Ball C, et al. Terlipressin dose response in healthy and endotoxemic sheep: impact on cardiopulmonary performance and global oxygen transport. Intensive Care Med. 2003;29(2):301–8. doi: 10.1007/s00134-002-1546-5. [DOI] [PubMed] [Google Scholar]
- 15.Bonnet A, Bevilacqua C, Benne F, Bodin L, Cotinot C, Liaubet L, et al. Transcriptome profiling of sheep granulosa cells and oocytes during early follicular development obtained by laser capture microdissection. BMC Genomics. 2011;12(1):417. doi: 10.1186/1471-2164-12-417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spurgeon SL, Jones RC, Ramakrishnan R. High throughput gene expression measurement with real time PCR in a microfluidic dynamic array. PLoS ONE. 2008;3(466):e1662. doi: 10.1371/journal.pone.0001662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.R Core Team. R: A Language and Environment for Statistical Computing. Vienna; 2015. Available from: http://www.R-project.org.
- 19.Déjean S, Martin PGP, Baccini A, Besse P. Clustering time-series gene expression data using smoothing spline derivatives. EURASIP J Bioinforma Syst Biol. 2007;2007(1):70561. doi: 10.1155/2007/70561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Straube J. Gorse AD, PROOF Centre of Excellence Team. Huang BE, Lê Cao KA. A linear mixed model spline framework for analysing time course ‘omics’ data. PLoS ONE. 2015;10(8):e0134–540. doi: 10.1371/journal.pone.0134540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liquet B, Lê Cao KA, Hocini H, Thiébaut R. A novel approach for biomarker selection and the integration of repeated measures experiments from two assays. BMC Bioinformatics. 2012;13(1):325. doi: 10.1186/1471-2105-13-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B. 1995;57:289–300. [Google Scholar]
- 23.Gao Y, Flori L, Lecardonnel J, Esquerré D, Hu ZL, Teillaud A, et al. Transcriptome analysis of porcine PBMCs after in vitro stimulation by LPS or PMA/ionomycin using an expression array targeting the pig immune response. BMC Genomics. 2010;11:292. doi: 10.1186/1471-2164-11-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005;1(W33):W741—8. doi: 10.1093/nar/gki475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang J, Duncan D, Shi Z, Zhang B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res. 2013;41(W1):W77—83. doi: 10.1093/nar/gkt439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thurstone LL. Multiple factor analysis. Psychol Rev. 1931;38(5):406. doi: 10.1037/h0069792. [DOI] [Google Scholar]
- 27.Williams PN, Collier CT, Carroll JA, Welsh TH, Laurenz JC. Temporal pattern and effect of sex on lipopolysaccharide-induced stress hormone and cytokine response in pigs. Domest Anim Endocrinol. 2009;37(3):139–47. doi: 10.1016/j.domaniend.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 28.Rosadini CV, Kagan JC. Early innate immune responses to bacterial LPS. Curr Opin Immun. 2016;44:14–19. doi: 10.1016/j.coi.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Myers MJ, Farrell DE, Baker JD, Cope CV, Evock-Clover CM, Steele NC. Challenge differentially affects cytokine production and metabolic status of growing and finishing swine. Domest Anim Endocrinol. 1999;17(4):345–60. doi: 10.1016/S0739-7240(99)00057-0. [DOI] [PubMed] [Google Scholar]
- 30.Dänicke S, Brosig B, Kersten S, Kluess J, Kahlert S, Panther P, et al. The Fusarium toxin deoxynivalenol (DON) modulates the LPS induced acute phase reaction in pigs. Toxicol Lett. 2013;220(2):172–80. doi: 10.1016/j.toxlet.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 31.Llamas Moya S, Boyle L, Lynch P, Arkins S. Age-related changes in pro-inflammatory cytokines, acute phase proteins and cortisol concentrations in neonatal piglets. Neonatology. 2006;91(1):44–8. doi: 10.1159/000096970. [DOI] [PubMed] [Google Scholar]
- 32.Harizi H, Homo-Delarche F, Amrani A, Coulaud J, Mormede P. Marked genetic differences in the regulation of blood glucose under immune and restraint stress in mice reveals a wide range of corticosensitivity. J Neuroimmunol. 2007;189(1):59–68. doi: 10.1016/j.jneuroim.2007.06.019. [DOI] [PubMed] [Google Scholar]
- 33.Sun H, Huang Y, Yin C, Guo J, Zhao R, Yang X. Lipopolysaccharide markedly changes glucose metabolism and mitochondrial function in the longissimus muscle of pigs. Animal. 2016;10(7):1204–12. doi: 10.1017/S1751731116000100. [DOI] [PubMed] [Google Scholar]
- 34.Eigler N, Saccà L, Sherwin RS. Synergistic interactions of physiologic increments of glucagon, epinephrine, and cortisol in the dog: a model for stress-induced hyperglycemia. J Clin Investi. 1979;63(1):114. doi: 10.1172/JCI109264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo J, Liu Z, Sun H, Huang Y, Albrecht E, Zhao R, et al. Lipopolysaccharide challenge significantly influences lipid metabolism and proteome of white adipose tissue in growing pigs. Lipids Health Dis. 2015;14:68. doi: 10.1186/s12944-015-0067-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Z, Liu W, Huang Y, Guo J, Zhao R, Yang X. Lipopolysaccharide significantly influences the hepatic triglyceride metabolism in growing pigs. Lipids Health Dis. 2015;14:64. doi: 10.1186/s12944-015-0064-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stanek C, Reinhardt N, Diesing AK, Nossol C, Kahlert S, Panther P, et al. A chronic oral exposure of pigs with deoxynivalenol partially prevents the acute effects of lipopolysaccharides on hepatic histopathology and blood clinical chemistry. Toxicol Let. 2012;215(3):193–200. doi: 10.1016/j.toxlet.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 38.Trauner M, Fickert P, Stauber RE. Inflammation-induced cholestasis. J Gastroenterol Hepatol. 1999;14(10):946–59. doi: 10.1046/j.1440-1746.1999.01982.x. [DOI] [PubMed] [Google Scholar]
- 39.Liu W, Ouyang X, Yang J, Liu J, Li Q, Gu Y, et al. AP-1 activated by toll-like receptors regulates expression of IL-23 p19. J Biol Chem. 2009;284(36):24006–16. doi: 10.1074/jbc.M109.025528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rohrbach S, Engelhardt S, Lohse MJ, Werdan K, Holtz J, Muller-Werdan U. Activation of AP-1 contributes to the beta-adrenoceptor-mediated myocardial induction of interleukin-6. Mol Med. 2007;13(11/12):605–14. doi: 10.2119/2007-00071.Rohrbach. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kang SS, Woo SS, Im J, Yang JS, Yun CH, Ju HR, et al. Human placenta promotes IL-8 expression through activation of JNK/SAPK and transcription factors NF- κB and AP-1 in PMA-differentiated THP-1 cells. Int Immunopharmacol. 2007;7(11):1488–95. doi: 10.1016/j.intimp.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 42.Park J, Chung SW, Kim SH, Kim TS. Up-regulation of interleukin-4 production via NF-AT/AP-1 activation in T cells by biochanin A, a phytoestrogen and its metabolites. Toxicol Appl Pharmacol. 2006;212(3):188–99. doi: 10.1016/j.taap.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 43.Hess J, Angel P, Schorpp-Kistner M. AP-1 subunits: quarrel and harmony among siblings. J Cell Sci. 2004;117(25):5965–73. doi: 10.1242/jcs.01589. [DOI] [PubMed] [Google Scholar]
- 44.Donn R, Berry A, Stevens A, Farrow S, Betts J, Stevens R, et al. Use of gene expression profiling to identify a novel glucocorticoid sensitivity determining gene, BMPRII. FASEB J. 2007;21(2):402–14. doi: 10.1096/fj.06-7236com. [DOI] [PubMed] [Google Scholar]
- 45.Thomas AV, Broers AD, Vandegaart HF, Desmecht DJM. Genomic structure, promoter analysis and expression of the porcine (Sus scrofa) TLR4 gene. Mol Immunol. 2006;43(6):653–9. doi: 10.1016/j.molimm.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 46.Clarke R, Tyson JJ, Dixon JM. Endocrine resistance in breast cancer – an overview and update. Mol Cellular Endocrinol. 2015;418:22034. doi: 10.1016/j.mce.2015.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Werling D, Jungi TW. TOLL-like receptors linking innate and adaptive immune response. Vet Immunol Immunopathol. 2003;91(1):1–12. doi: 10.1016/S0165-2427(02)00228-3. [DOI] [PubMed] [Google Scholar]
- 48.Borjigin L, Shimazu T, Katayama Y, Li M, Satoh T, Watanabe K, et al. Immunogenic properties of Landrace pigs selected for resistance to mycoplasma pneumonia of swine. Animal Sci J. 2016;87(3):321–9. doi: 10.1111/asj.12440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tan Y, Kagan JC. A cross-disciplinary perspective on the innate immune responses to bacterial lipopolysaccharide. Mol Cell. 2014;54:212–23. doi: 10.1016/j.molcel.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lithgow P, Takamatsu H, Werling D, Dixon L, Chapman D. Correlation of cell surface marker expression with African swine fever virus infection. Vet Microbio. 2014;168(2):413–9. doi: 10.1016/j.vetmic.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Niu P, Shabir N, Khatun A, Seo BJ, Gu S, Lee SM, et al. Effect of polymorphisms in the GBP1, Mx1 and CD163 genes on host responses to PRRSV infection in pigs. Vet Microbiol. 2016;182:187–95. doi: 10.1016/j.vetmic.2015.11.010. [DOI] [PubMed] [Google Scholar]
- 52.Caì Y, Postnikova EN, Bernbaum JG, Yú S, Mazur S, Deiuliis NM, et al. Simian hemorrhagic fever virus cell entry is dependent on CD163 and uses a clathrin-mediated endocytosis-like pathway. J Virol. 2015;89(1):844–56. doi: 10.1128/JVI.02697-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sikorski K, Chmielewski S, Przybyl L, Heemann U, Wesoly J, Baumann M, et al. STAT1-mediated signal integration between IFN γ and LPS leads to increased EC and SMC activation and monocyte adhesion. Am J Physiol Cell Physiol. 2011;300(6):C1337—44. doi: 10.1152/ajpcell.00276.2010. [DOI] [PubMed] [Google Scholar]
- 54.Luu K, Greenhill CJ, Majoros A, Decker T, Jenkins BJ, Mansell A. STAT1 plays a role in TLR signal transduction and inflammatory responses. Immunol Cell Biol. 2014;92(9):761–9. doi: 10.1038/icb.2014.51. [DOI] [PubMed] [Google Scholar]
- 55.Mach N, Gao Y, Lemonnier G, Lecardonnel J, Oswald IP, Estellé J, et al. The peripheral blood transcriptome reflects variations in immunity traits in swine: towards the identification of biomarkers. BMC Genomics. 2013;17(14):894. doi: 10.1186/1471-2164-14-894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou CX, Zhou DH, Liu GX, Suo X, Zhu XQ. Transcriptomic analysis of porcine PBMCs infected with Toxoplasma gondii RH strain. Acta Tropica. 2016;154:82–8. doi: 10.1016/j.actatropica.2015.11.009. [DOI] [PubMed] [Google Scholar]
- 57.Zhao FR, Xie YL, Liu ZZ, Shao JJ, Li SF, Zhang YG, et al. Transcriptomic analysis of porcine PBMCs in response to FMDV infection. Acta Tropica. 2017;173:69–75. doi: 10.1016/j.actatropica.2017.05.009. [DOI] [PubMed] [Google Scholar]
- 58.Islam MA, Große-Brinkhaus C, Pröll MJ, Uddin MJ, Rony SA, Tesfaye D, et al. PBMC transcriptome profiles identifies potential candidate genes and functional networks controlling the innate and the adaptive immune response to PRRSV vaccine in Pietrain pig. PLoS ONE. 2017;12(3):e0171828. doi: 10.1371/journal.pone.0171828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Donaldson L, Vuocolo T, Gray C, Strandberg Y, Reverter A, McWilliam S, et al. Construction and validation of a bovine innate immune microarray. BMC Genomics. 2005;6:135. doi: 10.1186/1471-2164-6-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ledger TN, Pinton P, Bourges D, Roumi P, Salmon H, Oswald IP. Development of a macroarray to specifically analyze immunological gene expression in swine. Clin Diagn Lab Immunol. 2004;11(4):691–8. doi: 10.1128/CDLI.11.4.691-698.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wells CA, Ravasi T, Faulkner GJ, Carninci P, Okazaki Y, Hayashizaki Y, et al. Genetic control of the innate immune response. BMC Immunol. 2003;4:5. doi: 10.1186/1471-2172-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boldrick JC, Alizadeh AA, Diehn M, Dudoit S, Liu CL, Belcher CE, et al. Stereotyped and specific gene expression programs in human innate immune responses to bacteria. Proc Natl Acad Sci. 2002;99(2):972–7. doi: 10.1073/pnas.231625398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequences of oligonucleotide primers tested by quantitative RT-PCR analysis. Gene name: name of the gene; Gene description: informations on the gene’s molecular function; Forward Primer; Reverse Primer. (XLSX 11 kb)
List of 9,530 unique genes differentially expressed in list (List1). (XLSX 2360 kb)
List of 284 unique genes differentially expressed in list (List1) included in non-generic biological functions (n=30). Generic biological functions include functions such as morphogenesis, transcription, locomotion. Gene name: name of the gene; Gene description: informations on the gene’s molecular function; Cluster: cluster in which the gene is classified by HAC; Time point: time measurement where the gene is DE; Expression: whether the DEG is up or down-regulated. (XLSX 21.2 kb)
List of 154 unique genes differentially expressed in list (List2). (XLSX 35.1 kb)
List of 116 unique genes differentially expressed in list (List3). (XLSX 22.9 kb)
Biological functions enriched by differentially expressed genes in list (List3) (n=30). Categories: Category of the enriched function; Diseases or Function Annotation: name of the enriched function; FDR: FDR of the enrichment test; Genes: list of genes enriching the biological function; Genes: number of genes enriching the biological function. (XLSX 10 kb)
Data Availability Statement
The datasets generated and analysed during the current study are available in Gene Expression Omnibus (GEO repository, http://www.ncbi.nlm.nih.gov/geo/) through the accession number GSE107487.