

Abstract
Background
Bat-borne virus surveillance is necessary for determining inter-species transmission risks and is important due to the wide-range of bat species which may harbour potential pathogens. This study aimed to monitor coronaviruses (CoVs) and paramyxoviruses (PMVs) in bats roosting in northwest Italian regions. Our investigation was focused on CoVs and PMVs due to their proven ability to switch host and their zoonotic potential. Here we provide the phylogenetic characterization of the highly conserved polymerase gene fragments.
Results
Family-wide PCR screenings were used to test 302 bats belonging to 19 different bat species. Thirty-eight animals from 12 locations were confirmed as PCR positive, with an overall detection rate of 12.6% [95% CI: 9.3–16.8]. CoV RNA was found in 36 bats belonging to eight species, while PMV RNA in three Pipistrellus spp. Phylogenetic characterization have been obtained for 15 alpha- CoVs, 5 beta-CoVs and three PMVs; moreover one P. pipistrellus resulted co-infected with both CoV and PMV. A divergent alpha-CoV clade from Myotis nattereri SpA is also described. The compact cluster of beta-CoVs from R. ferrumequinum roosts expands the current viral sequence database, specifically for this species in Europe. To our knowledge this is the first report of CoVs in Plecotus auritus and M. oxygnathus, and of PMVs in P. kuhlii.
Conclusions
This study identified alpha and beta-CoVs in new bat species and in previously unsurveyed Italian regions. To our knowledge this represents the first and unique report of PMVs in Italy. The 23 new bat genetic sequences presented will expand the current molecular bat-borne virus databases. Considering the amount of novel bat-borne PMVs associated with the emergence of zoonotic infections in animals and humans in the last years, the definition of viral diversity within European bat species is needed. Performing surveillance studies within a specific geographic area can provide awareness of viral burden where bats roost in close proximity to spillover hosts, and form the basis for the appropriate control measures against potential threats for public health and optimal management of bats and their habitats.
Keywords: Bat-borne viruses, Coronavirus, Emerging viruses, Genetic characterization, Paramyxovirus, Surveillance
Background
Bats (order Chiroptera) represent at least one-fifth of existing mammals, consisting of over 1300 known species of which at least 44 are present in Europe [1] and 34 in Italy [2]. Species diversity is expected to increase as some taxa, i.e. Myotis nattereri complex, are in the processes of being defined as cryptic species using molecular approaches rather than using morphological characteristics [3]. Bats are grouped into two suborders: the fruit-eating megabats (Megachiroptera), or flying foxes consisting of the single family Pteropodidae, and the echolocating insectivorous microbats (Microchiroptera) comprising 16 bat families [4].
Bat borne viruses are arousing increased interest since viral infections in bats have been associated with zoonotic disease outbreaks in humans and domestic animals, including livestock. Rabies virus, Hendra and Nipah viruses, Severe Acute Respiratory Syndrome (SARS) and Middle East Respiratory Syndrome (MERS) coronaviruses, as well as Filoviruses exemplify the role of bats in spreading viruses [5–7].
In the last fifteen years, at least two widespread outbreaks have been caused by novel coronaviruses jumping the species barrier, SARS in 2002–2003 and MERS starting from the Arabian Peninsula since 2012 [6, 7]. Genetic similarities between the viral sequences detected during outbreaks and CoV sequences in bats suggest the viruses originated in flying mammals and presumably passed to humans through a previous adaptation in intermediate hosts, i.e. civet cats and dromedaries [8]. Coronaviruses (family Coronaviridae, subfamily Coronavirinae) are divided into four main genera: Alphacoronavirus (alpha-CoV) and Betacoronavirus (beta-CoV) found mainly in mammals, Gammacoronavirus detected in birds and marine mammals and Deltacoronavirus found mainly in birds. Several alpha and beta-CoVs have been described worldwide in different bat species (e.g. [9–17]). From the first report in China, Rhinolophus species have been specifically associated with SARS-like CoVs [18–20], belonging to the lineage b of beta-CoV genus. Further investigations are needed to clarify the origin of all mammalian coronaviruses, assumed to be from viral ancestors residing in bats [21], untill the recent discovery of a new and highly divergent CoV (i.e. WESV) from house shrews in China [22].
As of 2010, the circulation of CoV in Italian bat population has been notified in only few published studies: SARS-like beta-CoVs have been identified in Rhinolophus species [23] and CoVs sequences are available only for Italian Pipistrellus kuhlii, Hypsugo savii, Nyctalus noctula, Epseticus serotinus, Myotis blythii and R. hipposideros species from fecal samples [24, 25]. Despite the rapid accumulation of bat CoV sequences in the last decade, any viral isolation trial, on different mammalian and bat cell lines failed till 2013, when the first isolation of SARS-like CoV from bat fecal samples succeeded in China [26].
On the list of emerging zoonoses there is a broad diversity of bat-borne paramyxoviruses (PMV), belonging to the wide Paramyxoviridae family, as the emergent Nipah virus and Hendra virus (Henipaviruses) and rubulaviruses (e.g. Menangle virus, Tioman virus and Tuhoko virus 1, 2 and 3) (e.g. [27–29] and references therein). Detection and isolation of paramyxoviruses from tissues and urine have been obtained mainly from flying foxes of the genus Pteropus in Africa, Asia, and South America (e.g. [27, 30, 31]) and in Australia (e.g. [32–34]), but also microbat species not previously indicated as PMV reservoirs tested positive for PMV RNA in Africa and Europe [27, 35–37]. Moreover, the ever-increasing attention paid to bat-associated pathogens, has led to the discovery of numerous novel and yet unclassified PMV, revealing an unexpected genetic diversity in the Paramyxovirinae sub-family [36]. PMV identification has been reported in only few studies in insectivorous bats in Europe from Germany, Bulgaria, Romania and Luxembourg, with none of the novel viruses closely related with highly or human pathogenic paramyxoviruses [16, 17, 27, 36].
Following the increasing need of surveillance for bat-borne viruses and the wide range of bat species potentially representing reservoirs for known or unknown pathogens, this study aimed to estimate the viral diversity and distribution in the bat population resident in Northwest Italy. Our investigation was focused on coronaviruses and paramyxoviruses due to their proved ability to switch host and their zoonotic potential. Here we provide the phylogenetic characterization of viral polymerase gene fragments, which are highly conserved within the viral families under investigation.
Methods
Sites and sample collection
Since all bat species in Europe are protected under the Habitats Directive of the European Union [38] and the Agreement on the Conservation of Populations of European Bats [39], samples collection and bat species identification were performed by expert chiropterologists authorized by the Italian Ministry of Environment (authorization number DPN/2010/0011879 and 000882/PNM/08052014).
Bats were captured, during the three years of surveillance (2013–2016) in the Northwestern Italian regions of Piedmont and Liguria, following ethical and safety recommendations [40]. Samplings were conducted from mid-June to October, a period that approximately corresponds to the pregnancy, lactation, dispersion and mating activity of European bats. To minimize animal disturbance, bats were caught soon after parturition with nylon mist-nets of mesh size of 16 to 19 mm positioned at 10–20 m from the reproductive and temporary roost along flight paths towards foraging and drinking areas. During autumn catches were focused particularly at swarming sites in caves where individuals from different colonies meet to mate [41]. All nets were checked every 10 min and captured bats were removed carefully from nets as soon as possible to minimize injury, drowning, strangulation, or stress and individually placed into disposable cloth bags awaiting species identification, collection of biometric data and biological samples.
Species identification was carried out according to Dietz & Kiefer [1] and individual details such as age, class, sex, reproductive status, forearm length, and body mass were recorded. Saliva and urine drops, when present, were collected directly on the animal by swabbing, while feces were recovered, when present, from the cotton bag. All bats were released in the same place of capture after minimal manipulations and were not tagged.
Based on the results of the first two years of surveillance, an increase in feces collection was performed in 2016 setting up random, non-invasive feces samplings underneath single- species reproductive roosts. Briefly, plastic films were left on the ground under different areas of each reproductive colony, then 15 min later single fresh droppings were collected with clean disposable forks, placed in 1 ml of buffered peptone water and kept at 4 °C till analyses. Dead animals in good post-mortem conditions were also collected and stored at −20 °C for further analyses.
RNA extraction and cDNA synthesis
Swabs and feces were maintained in 1 ml of UTM™ Viral Transport Medium (Catalog Number: 360C; Copan Diagnostics, Corona, California) and stored at −20 °C. Before any further analyses took place, the presence of the rabies virus antigen was investigated on dead animals by direct immunofluorescent staining in a BSL3 Laboratory, after necropsy. Once rabies infection has been excluded, samples underwent a pre-treatment before being submitted to automatic nucleic acid purification with magnetic beads.
Pre-treatment for tissues involved the preparation of a tissues pool composed by heart, lung, spleen and intestine from individual animals. The pools were homogenized at a ratio of 1:10 w/V in 1 ml of DEPC-treated PBS in a TissueLyser (Qiagen, Hilden, Germany). Tissue homogenates were then clarified at 13,000×g for 10 min at 4 °C, then 200 μl of tissues pool supernatant were incubated at 56 °C for 10 min with 180 μl of ATL buffer and 20 μl of Qiagen protease provided by the EZ1 Virus Mini Kit v2.0 (Qiagen, Hilden, Germany).To avoid any biosafety risk, the pre-treatment for swabs (saliva and urine) and feces suspensions involved the direct inactivation of 200 μl of each suspension in 200 μl of ATL buffer under a BSL3 hood. Nucleic acid purification (RNA/DNA) was finally accomplished on the EZ1 Advanced XL Instrument using an amount of 400 μl as sample input and a final elution volume of 60 μl of RNase-DNase free water, following the manufacturer’s guidelines. RNA was stored at −80 °C until amplification protocols were performed.
cDNA was synthetized from 5 μl of each RNA/DNA sample with the Transcriptor First Strand cDNA Synthesis Kit (Roche Diagnostics, Mannheim, Germany), according to manifacturer’s instructions.
Coronavirus detection
For coronavirus detection, 2 μl of cDNA were amplified with an end-point PCR assay targeting a conserved RNA-dependent RNA polymerase (RdRp) gene fragment (537 bp), as described by Poon et al. [42]. The amplification was set up in a 25 μl reaction mixture containing 0.2 mM deoxynucleoside triphosphates, 1.5 mM MgCl2, 0.2 μM of IN-6 and IN-7 primer and 1 U of Platinum Taq Polymerase (Invitrogen, Carlsbad, CA). The cycling conditions were 94 °C for 2 min, 40 cycles at 94 °C for 1 min, 48 °C for 1 min, 72 °C for 1 min and final elongation step at 72 °C for 7 min.The annealing temperature of primer was modified from 58 °C to 48 °C.
Upon amplification, 20 μl of PCR products were run in 1.5% agarose gel electrophoresis and visualized by GelGreen Nucleic Acid Gel Stain (Biotium) staining; bands of the expected size were excised from the gel for sequencing.
Paramyxovirus detection
For paramyxovirus detection, a broadly reactive seminested PCR assay specific for the RNA polymerase (L)- gene (538 bp) of the Paramyxovirinae subfamily was applied. 0 2 μl of cDNAs were amplified using the PAR primers designed by Tong et al. [43] and the protocol optimized with Taguchi method by Kurth et al. (36). Briefly for first round, the final concentration of the 25 μl reaction mix was: 0.1 mM deoxynucleoside triphosphates, 10 mM MgCl2, 0.12 μM of PAR F1 and PAR R primers and 1.25 U of Platinum Taq Polymerase (Invitrogen, Carlsbad, CA). The cycling conditions were 94 °C for 2 min, 40 cycles at 94 °C for 15 s, 50 °C for 30 s, 72 °C for 30 s and a final elongation step at 72 °C for 7 min. Then 1 μl first round PCR product was used in the second round with the same concentrations except for the MgCl2, set up at 1 mM and the use of PAR F2 and PAR R primers, cycling parameters were identical to the first round.
PCR products (20 μl) were run and recovered from a 1.5% agarose gel, as described before.
Sequencing and phylogenetic analysis
Amplicons were purified by gel extraction with the QIAquick Gel Extraction kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions. After elution, nucleic acid quantification of the recovered DNA was done using Thermo Scientific Nanodrop spectrofotometer and submitted for direct sequencing to BMR Genomics, Padua, Italy. The obtained chromatograms were manually checked for unclear base calls and edited using Geneious R7.1.7 software (Geneious, Auckland, New Zealand).
The sequences were aligned using Muscle (implemented in Geneious software) and the alignment was used to evaluate the best evolutionary model (Modeltest ver 3.7) and to draw a bayesian phylogenetic tree (MrBayes ver. 3.1.2). Consensus tree was created after at least 1 million of heuristic search generations and after eliminating the first 25% of evaluated tree topologies (burnin = 25%).
Biomolecular species identification
A total genomic DNA extraction was performed only for PCR positive individuals starting from the original swab suspensions using the QIAmp DNA Mini kit (Qiagen, Hilden, Germany) and following the manufacturer protocol. To confirm species identification by genetic determinations, the complete mitochondrial Cytochrome b gene (Cytb) was amplified as in Puechmaille et al. [3]. PCR products were submitted for direct sequencing to BMR Genomics, Padua, Italy. The obtained chromatograms were manually checked for unclear base calls and edited using Geneious R7.1.7 software (Geneious, Auckland, New Zealand). Species identification was conducted by comparing the obtained sequences to on-line available reference sequences (BLAST alignment, NCBI web site).
Results
Samples collection
Starting from June 2013 till October 2016 a total of 302 animals (35 dead; 267 live) belonging to 19 bat species were collected during 49 capture sessions in 38 locations of Piedmont and five of Liguria regions. Collection of saliva, urine and feces from the same animal was not possible for each of the 267 live bats handled, leading to the final collection of 123 oral swabs (37%), 49 urine swabs (15%) and 158 fecal drops (48%). Sex definition was determined for 195 bats: 117 males and 78 females; the additional 107 single fecal droppings collected in 2016 under 4 different monospecific colonies were considered as non-assigned individual samples. All captured species are listed in Table 1.
Table 1.
Sampled bat species and CoV and PMV prevalences detected
| Genus | Species | n°sampled (n° pos) | CoV detection; n/N (%) | PMV detection; n/N (%) |
|---|---|---|---|---|
| Pipistrellus | Pipistrellus kuhlii | 56 (4) | 2/56; 3.6% | 2/56; 3.6% |
| Pipistrellus pipistrellus | 20 (5) | 4/20; 20% | 1/20; 5% | |
| Pipistrellus nathusii | 2 | |||
| Myotis | Myotis myotis | 43 (4) | 4/43; 9.3% | |
| Myotis brandtii | 1 | |||
| Myotis bechsteinii | 1 | |||
| Myotis nattereri | 22 (3) | 3/22; 13,6% | ||
| Myotis daubentonii | 24 (2) | 2/24; 8.3% | ||
| Myotis emarginatus | 29 | |||
| Myotis oxygnathus | 23 (2) | 2/23; 8.7% | ||
| Myotis mistacinus | 3 | |||
| Hypsugo | Hypsugo savii | 5 | ||
| Plecotus | Plecotus auritus | 14 (1) | 1/14; 7.1% | |
| Plecotus austriacus | 1 | |||
| Plecotus macrobullaris | 1 | |||
| Barbastella barbastellus | 17 | |||
| Nyctalus | Nyctalus leisleri | 1 | ||
| Rhinolophus | Rhinolophus ferrumequinum | 38 (18) | 18/38; 47.4% | |
| Rhinolophus hipposideros | 1 | |||
| Total | 302 (39) | 36/302; 12% [95% CI: 9.6–17] | 3/302; 1% [95% CI: 0.3–3,1] |
95% Confidence Interval (95% CI) is expressed only for CoV and PMV overall rates
No animal captured during the active surveillance showed signs of disease. During necropsies, no macroscopic lesions referring to infectious diseases were observed, and all the examined bats were negative in the rabies virus antigen IF test.
Coronavirus and paramyxovirus detection
CoV and PMV positive sample types included feces (33/158; 21%) and urine swabs (6/49; 12.2%). None of the tissue pools from dead bats or oral swabs were PCR positive. A significantly greater percentage of female bats, 11.5% (9/78), were PCR-positive than males, 4.3% (5/117), (p = 0.05).
Coronavirus and/or paramyxovirus RNA was found in 38 animals belonging to eight bat species (Table 1). Specifically, CoV RNA was detected in 36 bats from 12 sampling sites in Piedmont and one in Liguria, while PMV RNA in three animals from three sampling sites in Piedmont; a map showing the positive sites is presented in Fig. 1. In our sample set, the detection rate of CoV was 12% (36/302; 95% confidence interval [CI] = 9.6–17) ranging between 3.6% for P. kuhlii, despite representing the most abundant species in our sample, and 47.4% for R. ferrumequinum.
Fig. 1.
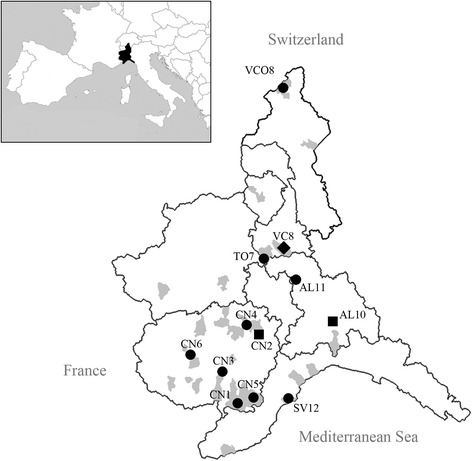
Map of Piedmont and Liguria sites where a CoV or PMV sequence was detected. Circles represent CoV positive sites; squares identify PMV positive sites and diamonds represent the site positive for both CoV and PMV. Sites are identified according to a code formed by the province abbreviation and progressive numbers, i.e. in Piedmont, for Cuneo province CN1: Ormea, CN2: Rodello, CN3: Pianfei, CN4: Santa Vittoria d’Alba, CN5: Garessio CN6: Villar San Costanzo; for Torino province TO7: Verrua Savoia; for Vercelli province VC8: Trino; for Verbano-Cusio-Ossola province VCO9: Baceno; for Alessandria province AL10: Tassarolo, AL11: Vignale Monferrato; in Liguria, for Savona province SV12: Finale Ligure. Sampled municipalities that were found negative are reported in grey.
Phylogenetic analysis was performed on 20 unique sequences obtained from 36 samples that yielded a PCR product of the expected size after the CoV PCR screening. The positive samples were collected from: M. nattereri (n = 3), M. myotis (n = 2), M. oxygnathus (n = 1), P. kuhlii (n = 1), P. pipistrellus (n = 3), P. auritus (n = 1) and R. ferrumequinum (n = 9). Any new sequences identified were submitted to GenBank and the accession numbers assigned are given in Table 2. The PMV strains were detected in three different provinces from two P. kuhlii at CN2 and AL10 sites and one P. pipistrellus at VC8 site; moreover, phylogenetic analysis based on the L-gene fragment was possible for all the three strains retrieved in this study. Interestingly, one P. pipistrellus from VC8 site was coinfected by both CoV and PMV as PCR positive results were obtained from the same urine sample. Details of positive sequenced samples are displayed in Table 2.
Table 2.
CoV and PMV positive samples for which a sequence is available
| Species | ID | Sample type | Capture date | Site | Settinga | Sex/ageb | CoV sequence (AN)/CoV genus | PMV sequence (AN) |
|---|---|---|---|---|---|---|---|---|
| Myotis nattereri | 560 | Feces | 31/08/13 | CN1 | T roost | M/ad | Mnat560_IT_13 (KY780381)/alpha |
|
| 562 | Urine | F/juv | Mnat562_IT_13 (KY780382)/alpha |
|||||
| 1021 | Feces | 16/08/14 | TO7 | R roost | F/juv | Mnat1021_IT_14 (KY780387)/alpha |
||
| Pipistrellus pipistrellus | 1015 | Urine | 05/08/14 | VC8 | R roost | F/ad | Ppip1015C_IT_14 (KY780385)/alpha |
Ppip1015P_IT_14 (KY780403) |
| 1016 | Feces | F/juv | Ppip1016_IT_14 (KY780386)/alpha |
|||||
| 1000 | Feces | 11/08/14 | VC9 | Fora-ging | M/ad | Ppip1000_IT_14 (KY780384)/alpha |
||
| Pipistrellus kuhlii | 600 | Feces | 19/08/14 | CN2 | R roost | F/ad | Pkuh600_IT_14 (KY780401) | |
| 605 | Feces | F/ad | Pkuh605_IT_14 (KY780383)/alpha |
|||||
| 621 | Urine | 06/08/14 | AL10 | R roost | F/ad | Pkuh621_IT_14 (KY780402) | ||
| Myotis myotis | 4658 | Feces | 15/08/16 | CN4 | R roost | Mmyo4658_IT_16 (KY780397)/alpha |
||
| 4663 | Feces | Mmyo4663_IT_16 (KY780398)/alpha |
||||||
| Myotis oxygnathus | 4235 | Feces | 06/07/16 | SV12 | R roost | Moxy4235_IT_16 (KY780395)/alpha |
||
| Plecotus auritus | 4241 | Feces | 20/09/16 | CN5 | Swar-ming | M/ad | Paur4241_IT_16 (KY780396)/beta |
|
| Rhinolophus ferrumequinum | 4009 | Feces | 04/07/16 | CN6 | R roost | Rfer4009_IT_16 (KY780388)/alpha |
||
| 4011 | Feces | Rfer4011_IT_16 (KY780389)/alpha |
||||||
| 4015 | Feces | Rfer4015_IT_16 (KY780390)/alpha |
||||||
| 4019 | Feces | Rfer4019_IT_16 (KY780391)/beta |
||||||
| 4024 | Feces | Rfer4024_IT_16 (KY780392)/alpha |
||||||
| 4025 | Feces | Rfer4025_IT_16 (KY780393)/alpha |
||||||
| 4027 | Feces | Rfer4027_IT_16 (KY780394)/beta |
||||||
| 4674 | Feces | 13/07/16 | AL11 | R roost | Rfer4674_IT_16 (KY780399)/beta |
|||
| 4675 | Feces | Rfer4675_IT_16 (KY780400)/beta |
ID: Identification number corresponds to the progressive and unique number assigned to each analyzed sample. Site codes are displayed in Fig. 1
athe setting where bats were caught, R roost: reproductive roost; T roost: temporary roost
bage definitions are juv: juvenile and ad: adult
CoV phylogeny
RdRp phylogeny is presented in Fig. 2 and shows that 15 CoV strains from this study clustered in the alphacoronavirus genus and 5 in the beta-coronavirus genus.
Fig. 2.
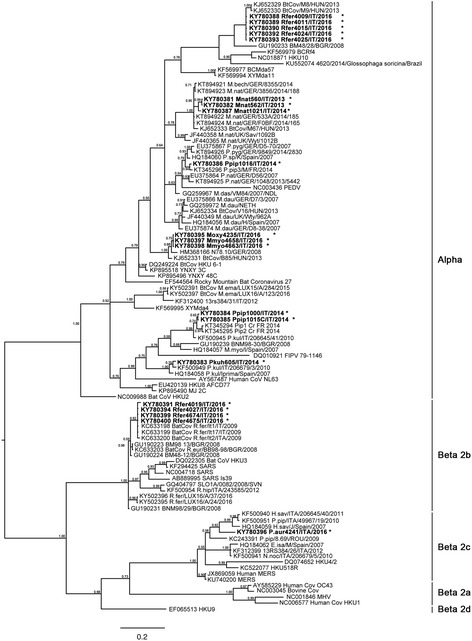
Bayesian phylogenetic tree of alpha and beta-CoVs derived from bats and other species. Representative RdRp sequences were extracted from GenBank and the alignment carried out on 372 nucleotides for a total of 101 sequences: twenty original and 81 available from Genbank, among the alpha and beta-CoVs genera. Members of betacoronaviruses are separated into four lineages, 2a, 2b, 2c and 2d. Posterior probability values of the clades are reported above branches. The CoVs name in the tree is composed by the sequence GenBank accession number plus the name of the strain. Our 20 new sequences are reported in bold and labeled with a star (*)
As shown in Fig. 2, the three M. nattereri alpha-CoV strains (560, 562 site CN1 and 1021 site TO7) cluster with nucleotide similarities ranging from 94 to 96% within a CoV clade composed of three M. nattereri and one M. bechstenii from Germany (AN: KT94921–924) and another M. nattereri from Hungary (AN: KJ652333), but show only an 86% identity with M. nattereri CoVs strains from UK 2009.
Genetic species determination based on the Cyt B gene fragment of 837 bp for these M. nattereri species showed a 99% sequence identity with a French M. nattereri isolate (AN: JF412408) named “MspA Mnat22 cytochrome b gene” was highlighted.
Based on this finding, our new CoVs strains belong to the M. nattereri SpA, a putative new species within the M. nattereri species complex.
Three alpha-CoV strains found in feces samples of three bats belonging to the Myotis genus show 100% identity to each other (4235 from M. oxygnathus, site SV12 and 4658 and 4663 from M. myotis, site CN4) and form a divergent clade. When compared to other CoVs, this clade showed the highest identity (~97%) with two M. myotis CoV strains, from Germany and Hungary (AN: HM368166 and KJ652331).
Two P. pipistrellus CoV sequences (1000 site VCO9 and 1015 site VC8) cluster together with two P. pipistrellus strains (Pip1, Pip2) from the same species detected in France in 2014 (AN: KT345294–95) and one P. pipistrellus strain from Italy (AN: KF500945); interestingly the third P. pipistrellus CoV (1016 site VC8) is ~27% divergent from the others and clusters near Pip3 CoV strain from France (AN: KT345296).
The P. kuhlii CoV sequence clusters (605 site CN2) with a similarity of ~97%, within a clade of two P. kuhlii strains from Italy 2007 (AN: KF500949) and Spain (AN: HQ184058).
Five R. ferrumequinum alpha-CoV sequences (4009, 4011, 4015, 4024, 4025 site CN6) found in fecal droppings from the same monospecific roost, showed 100% identity with each other clustering within the clade formed by the only three R. ferrumequinum alpha-CoV sequences detected in Europe so far, 3% divergent from the ones from Hungary (AN: KJ652329–30) and 13% from the Bulgarian one (AN:GU190233).
Among the beta-CoV group (lineage b) four R. ferrumequinum CoV strains (4019, 4027 site CN6 and 4674, 4675 site AL11) cluster together with other three Italian beta-CoV sequences from the same species (AN: KC33198–200). Interestingly, the 4027 sequence is 100% identical with 4674 and 4675, although originating from two R. ferrumequinum roosts located at 130 km distance.
One novel beta-CoV sequence from Plecotus auritus (AN: KY780396) clusters separately in the beta-CoV group (lineage c) showing only a ~88% similarity with two H. savii CoV strains one from Spain (AN: HQ184059) and one from Italy (AN: KF500940) and a P. pipistrellus strain from Italy (AN: KF500951). It’s divergence from a MERS CoV strain isolated in 2014 from a camel (AN: KU740200) is 14%. Phylogenetic analyses of this short fragment show that CoVs cluster based on the relatedness of host species.
PMV phylogeny
PMV phylogeny based on representative L-gene sequences available from GenBank is presented in Fig. 3.
Fig. 3.
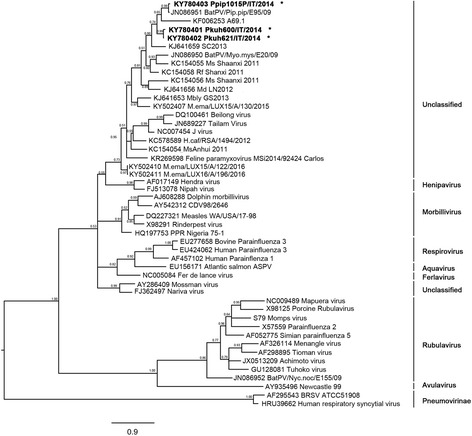
Bayesian phylogenetic tree of the Paramixoviridae family. The tree is built on a L-gene fragment of 393 nucleotides on a total of 48 taxa: three original sequences and 45 sequences. Available L-gene sequences, representative of the seven currently known and unclassified genera of the Paramixovirinae sub-family, together with two strains from the Pneumovirinae sub-familiy were extracted from GenBank. Posterior probability values of the clades are reported above branches. The samples name in the tree is composed by the GenBank accession number plus the name of the strain. New obtained sequences are sequences are reported in bold and labeled with a star (*)
The new PMV strains were detected in three different locations from one P. pipistrellus at VC8 site and two P. kuhlii bats at CN2 and AL10 sites (80 km distance).
Our 1015 P. pipistrellus strain revealed a 97% nucleotide identity with the E95 PMV strain (AN: JN086951) detected in the same bat species in Germany in 2009, but is more than 23% divergent from any other known PMV sequence.
The two PMV sequences from P. kuhlii (600 site CN2 and 621 site AL10) are 97% similar to each other and cluster separately from previously known PMV sequences (18–20% divergence) in the L-gene fragment phylogenetic tree.
To our knowledge, the P. kuhlii species was never previously implicated as paramyxovirus host.
Discussion
Recently, emerging disease surveillance programs have intensified to investigate the role of bats in the evolution and spillover of zoonotic pathogens from wildlife. Our study involved three years of active and passive surveillance to characterize the viral diversity of the Northwestern Italian bat population. Using viral family-wide PCRs we identified and phylogenetically characterized 20 new CoVs and 3 PMVs strains. To date, studies on bat CoVs phylogeny are mainly based on datasets of short sequences (i.e. 440 bp) (e.g. [9, 10, 13, 14, 24, 25, 44]) due to the difficulties of obtaining isolates and good quality viral RNA from bats, but ideally long sequence fragments would be beneficial to infer more reliable phylogenies.
The high prevalence of positive fecal samples (21%) in our study is in concordance with other studies, which identified feces as the best sample type for CoVs detection in bats [9, 18]. Rather than collecting samples from individually caught bats, which is time consuming and labor intensive, collecting single fecal droppings under mono-species roosts turned out to be a reliable and non-invasive method for virological surveillance of bat roosts during their reproductive period. Moreover, urine is confirmed as the most suitable and appropriate sample types for detection of paramyxoviruses in bat populations [33], considering that 2 out of the 3 PMV positive samples from our study were urine swabs.
In 2013–2014 coronavirus circulation was identified in at least four species-specific reproductive roosts of Piedmont: TO7 site for M. nattereri, VC8 site for P. pipistrellus, CN2 and AL10 for P. kuhlii. Unfortunately, attempts to re-test the same roosts in 2016 failed since the VC8 colony moved due to the effect of human disturbance (i.e. robbery of the copper roof cover used as refuge by P. pipistrellus bats), and the other three colonies located in private buildings were inaccessible due to logistical reasons. The likelihood of roost disturbance should be taken into account when putting in place bat surveillance plans to enable a steady follow up of the colonies over time.
Bats social behavior could explain the significantly higher infection rate detected in our study for female bats all sampled in August near maternity roosts. Previous studies documented higher virus detection rates in females and juveniles captured near maternity roosts in summer, supporting the hypothesis that virus amplification occurs mainly in reproductive roosts [11, 45].
The identification of the same CoV strains (100% identical) in different roosts of the same bat species (i.e. R. ferrumequinum and P. pipistrellus) located also at over 100 km distance, seems to confirm that most bat-CoVs appear species-specific and thus more closely associate with the host species than the sampling location [11, 15, 20]. Interestingly, we identified a divergent alpha-CoV lineage in M. nattereri SpA, representing a cryptic lineage within the Myotis nattereri species complex in the Mediterranean region. The lineage is known to be present in Italy, however no information is available for Germany and Hungary [46]. Following the host-virus coevolution theory based on their close phylogenetic concordance [47], the small divergence (from 3.5 to 5%) between our M. nattereri SpA CoV strains and the German or Hungarian M. nattereri ones could indicate that they all reside in the M. nattereri SpA host, considering that molecular species identification for those specimens is lacking.
The detection of identical alpha-CoV sequences in two different species belonging to the Myotis genus (M. oxygnathus and M. myotis) from two distinct roosts (sites SV12 and CN4) 90 km apart could be due to the expansion and overlapping of habitats and foraging areas of Myotis spp. through the Maritime Alpine chain and valleys. To our knowledge this is the first report of CoV in the M. oxygnathus species.
The compact cluster of almost identical beta-CoV (lineage b) strains from two separate R. ferrumequinum roosts gives further indications that the Rhinolophus genus may represent the specific host for SARS-like CoVs and gives an important contribution in terms of available beta-CoV sequences from this species in Europe. To our knowledge, this is the first report of CoV in the P. auritus species. This sequence clusters separately within the beta-CoV (lineage c) showing a 14% divergence with a MERS strain identified from a camel in Egypt.
The detection of highly divergent alpha-CoV strains within one P. pipistrellus reproductive roost, the circulation of both alpha and beta-CoVs within one R. ferrumequinum roost and the co-infection of P. pipistrellus with both CoV and PMV provide further evidence that bats are able to carry more than one virus. While infection with multiple CoVs in the same species/bat/colony is well known, and has been previously reported in China [26, 48, 49] and Europe [19], apart from metagenomic studies notably biased towards the identification of sequences from dsDNA viruses, to our knowledge the coinfection of different ssRNA viral families in the same animal was so far reported only in one study in Europe from P. pygmaeus in Hungary [44]. In the specific, the coinfection with two ssRNA viral families within the same host may be explicable in the light of the IFN inhibition used by paramyxoviruses to circumvent host’ innate immune response [50]. This mechanism, known as IFN antagonism, may be exploited by other viruses able to escape the adaptive immunity, e.g. CoVs, to be introduced and proliferate in the same host, as observed in mallards [51].
By the increased viral surveillance, a considerable number of novel paramyxoviruses has been discovered in pteropoid and non-pteropoid species, but to date the number of bat PMV sequences for Europe is very scarce and only from few bat species [16, 17, 27, 36]. The three new PMV strains, two in P. kuhlii and one in P. pipistrellus species, couldn’t be classified within any of the current seven known PMV genera, but cluster in the crowded, unassigned PMV clade, which comprises several bat derived strains. Our report represents the first identification of PMVs in the P. kuhlii species worldwide. The two sequences, retrieved from two roosts located 90 km apart, are divergent from previously known PMV clusters, which may indicate a stronger association to the host species rather than the geographic area also for paramyxoviruses. This viral tropism is also strongly supported by the high similarity of our P. pipistrellus sequence to that of the one other E95 PMV sequence retrieved in Germany from the same bat species. In support of this hypothesis, a study on renal tissues from African bats underlined how paramyxovirus divergence in pteroid and non-pteroid bats correlates with bat taxonomy, suggesting a strong association with bat genera [37]. Because the L-gene fragment used as genetic marker in the aforementioned study is not overlapping with the sequence we used, we couldn’t phylogenetically compare them. Nevertheless, given the high similarity our P. pipistrellus sequence shows with the E95 PMV strain, our findings support this association. Moreover, an extensive collection of urine samples from the colony would be necessary to facilitate PMVs isolation, which remains a critical requirement for full genome and pathogenic characterization of the strains detected.
Conclusions
Compared to previous studies published in Italy [24, 25], we detected alpha and beta-CoVs in not previously surveyed Italian regions and in new bat species; moreover, this report represents the first and novel identification of PMVs in Italy. The 23 new bat genetic sequences will fill gaps and expand the current molecular bat-borne virus databases.
Considering the amount of novel bat-borne PMVs associated with the emergence of zoonotic infections in animals and humans in the last years define the virus diversity within European bat species is needed. Performing surveillance studies within a specific geographic area can provide awareness of viral burden where bat roosts are in close proximity to spillover hosts, and can form the basis for the appropriate control measures to curb potential threats for public health and optimal management of bats and their habitats.
Acknowledgements
We thank Carla Lo Vecchio for the assistance with the laboratory work and Mara Calvini for dead animals’ collection.
Funding
Financial support for this study and its publication was provided by the Italian Ministry of Health in the context of Ricerca Sanitaria Corrente 2013 (Code: IZS PLV 09/13 RC).
Availability of data and materials
Data generated or analyzed during this study are included in this paper and can be made available by the corresponding author upon a reasonable request. Sequence data obtained in this study are deposited in GenBank database: full sequence name and GenBank accession no are indicated in Table 2.
Authors’ contributions
MLM, SR1. and RO participated in the conception and coordination of the study; RT and PC performed all the captured and bat species identification in the field; FR and AL performed the experiments and FR wrote the paper; LB and KE analyzed the genetic data; SZ and AD performed the necropsies; AK and SR2 contributed reagents, materials and analysis tools. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Samples collection and bat species identification were performed by expert chiropterologists authorized by the Italian Ministry of Environment (authorization number DPN/2010/0011879 and 000882/PNM/08052014). All field operations were performed in compliance with the Habitats Directive of the European Union [41] and the Agreement on the Conservation of Populations of European Bats [45], and respecting ethical and safety recommendations [46].
Consent for publication
Not applicable
Competing interests
The authors declare no competing interests. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Francesca Rizzo, Phone: +39-011-268-6272, Email: francesca.rizzo@izsto.it.
Kathryn M. Edenborough, Email: EdenboroughK@rki.de
Roberto Toffoli, Email: rtoffoli@iol.it.
Paola Culasso, Email: chirosphera@gmail.com.
Simona Zoppi, Email: simona.zoppi@izsto.it.
Alessandro Dondo, Email: alessandro.dondo@izsto.it.
Serena Robetto, Email: serena.robetto@izsto.it.
Sergio Rosati, Email: sergio.rosati@unito.it.
Angelika Lander, Email: LanderA@rki.de.
Andreas Kurth, Email: KurthA@rki.de.
Riccardo Orusa, Email: riccardo.orusa@izsto.it.
Luigi Bertolotti, Email: luigi.bertolotti@unito.it.
Maria Lucia Mandola, Email: marialucia.mandola@izsto.it.
References
- 1.Dietz C, Kiefer A. Bats of Britain and Europe. London: Bloomsbury Publishing; 2016. [Google Scholar]
- 2.Lanza B. Mammalia V Chiroptera. In: Calderini editors. Fauna d’Italia, vol XLVII. Milano: Calderini de Il Sole 24; 2012.
- 3.Puechmaille SJ, Allegrini B, Boston ES, Dubourg-Savage MJ, Evin A, Knochel A, et al. Genetic analyses reveal further cryptic lineages within the Myotis Nattereri species complex. Mammalian Biology-Zeitschrift für Säugetierkunde. 2012;77:224–228. doi: 10.1016/j.mambio.2011.11.004. [DOI] [Google Scholar]
- 4.Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T. Bats: important reservoir hosts of emerging viruses. Clin Microbiol Rev. 2006;19:531–545. doi: 10.1128/CMR.00017-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wong S, Lau S, Woo P, Yuen KY. Bats as a continuing source of emerging infections in humans. Rev Med Virol. 2007;17:67–91. doi: 10.1002/rmv.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peiris JS, Lai ST, Poon LLM, Guan Y, Yam LYC, Lim W. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Memish ZA, Mishra N, Olival KJ, Fagbo SF, Kapoor V, Epstein JH, et al. Middle East respiratory syndrome coronavirus in bats. Saudi Arabia Emerg Infect Dis. 2013;19:1819–1823. doi: 10.3201/eid1911.131172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu B, Ge X, Wang LF, Shi Z. Bat origin of human coronaviruses. Virol J. 2015;12:221. doi: 10.1186/s12985-015-0422-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang XC, Zhang JX, Zhang SY, Wang P, Fan XH, Li LF, et al. Prevalence and genetic diversity of coronavirus in bats from China. J Virol. 2006;80:7481–7490. doi: 10.1128/JVI.00697-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dominguez SR, O’Shea TJ, Oko LM, Holmes KV. Detection of group 1 coronaviruses in bats in North America. Emerg Infect Dis. 2007;13:1295–1300. doi: 10.3201/eid1309.070491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gloza-Rausch F, Ipsen A, Seebens A, Gottsche M, Panning M, Drexler JF, et al. Detection and prevalence patterns of group I coronaviruses in bats northern Germany. Emerg Infect Dis. 2008;14:626–631. doi: 10.3201/eid1404.071439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Falcon A, Vazquez-Moron S, Casas I, Aznar C, Ruiz G, Pozo F, et al. Detection of alpha and betacoronaviruses in multiple Iberian bat species. Arch Virol. 2011;156:1883–1890. doi: 10.1007/s00705-011-1057-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anthony SJ, Ojeda-Flores R, Rico-Chávez O, Navarrete-Macias I, Zambrana-Torrelio CM, Rostal MK, et al. Coronaviruses in bats from Mexico. J Gen Virol. 2013;94:1028–1038. doi: 10.1099/vir.0.049759-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goffard A, Demanche C, Arthur L, Pinçon C, Michaux J, Dubuisson J. Alphacoronaviruses detected in french bats are phylogeographically linked to coronaviruses of european bats. Viruses. 2015;7:6279–6290. doi: 10.3390/v7122937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Asano KM, Hora AS, Scheffer KC, Fahl WO, Iamamoto K, Mori E, et al. Alphacoronavirus in urban Molossidae and Phyllostomidae bats Brazil. Virol J. 2016;13:110. doi: 10.1186/s12985-016-0569-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer K, Zeus V, Kwasnitschka L, Kerth G, Haase M, Groschup MH, et al. Insectivorous bats carry host specific astroviruses and coronaviruses across different regions in Germany. Infect Genet Evol. 2016;37:108–116. doi: 10.1016/j.meegid.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pauly M, Pir JB, Loesch C, Sausy A, Snoeck CJ, Hübschen JM, Muller CP. Novel Alphacoronaviruses and paramyxoviruses Cocirculate with type 1 and severe acute respiratory system (SARS)-related Betacoronaviruses in Synanthropic bats of Luxembourg. Appl Environ Microbiol. 2017;83:e01326–e01317. doi: 10.1128/AEM.01326-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lau SK, Woo PC, Li KS, Huang Y, Tsoi HW, Wong BH, et al. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc Natl Acad Sci U S A. 2005;102:14040–14045. doi: 10.1073/pnas.0506735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drexler JF, Gloza-Rausch F, Glende J, Corman VM, Muth D, Goettsche M, et al. Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J Virol. 2010;84:11336–11349. doi: 10.1128/JVI.00650-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balboni A, Battilani M, Prosperi S. The SARS-like coronaviruses: the role of bats and evolutionary relationships with SARS coronavirus. Microbiologica-quarterly journal of. Microbiol Sci. 2012;35:1. [PubMed] [Google Scholar]
- 21.Woo PC, Lau SK, Lam CS, Lau CC, Tsang AK, Lau JH, et al. Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J Virol. 2012;86:3995–4008. doi: 10.1128/JVI.06540-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang W, Lin X-D, Liao Y, Guan X-Q, Guo W-P, Xing J-G, et al. Discovery of a highly divergent coronavirus in the Asian house shrew from China illuminates the origin of the alphacoronaviruses. J Virol. 2017;91:e00764–e00717. doi: 10.1128/JVI.00764-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balboni A, Palladini A, Bogliani G, Battilani M. Detection of a virus related to betacoronaviruses in Italian greater horseshoe bats. Epidemiol Infect. 2011;139:216–219. doi: 10.1017/S0950268810001147. [DOI] [PubMed] [Google Scholar]
- 24.Lelli D, Papetti A, Sabelli C, Rosti E, Moreno A, Boniotti MB. Detection of coronaviruses in bats of various species in Italy. Viruses. 2013;5:2679–2689. doi: 10.3390/v5112679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Benedictis P, Marciano S, Scaravelli D, Priori P, Zecchin B, Capua I, et al. Alpha and lineage C betaCoV infections in Italian bats. Virus Genes. 2014;48:366–371. doi: 10.1007/s11262-013-1008-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ge XY, Li JL, Yang XL, Chmura AA, Zhu G, Epstein JH, et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature. 2013;503:535–538. doi: 10.1038/nature12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drexler JF, Corman VM, Müller MA, Maganga GD, Vallo P, Binger T, et al. Bats host major mammalian paramyxoviruses. Nat Commun. 2012;3:796. doi: 10.1038/ncomms1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Edwards S, Marsh GA. Henipaviruses: bat-borne paramyxoviruses. Microbiol Aust. 2017;38:4–7. [Google Scholar]
- 29.Baker KS, Todd S, Marsh GA, Crameri G, Barr J, Kamins AO, et al. Novel, potentially zoonotic paramyxoviruses from the African straw-colored fruit bat Eidolon Helvum. J Virol. 2013;87:1348–1358. doi: 10.1128/JVI.01202-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drexler JF. CormanVM, Gloza-Rausch F, Seebens a, Annan a, Ipsen a, et al. Henipavirus RNA in African bats. PLoS One. 2009;4:e6367. doi: 10.1371/journal.pone.0006367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chua KB, Koh CL, Hooi PS, Wee KF, Khong JH, Chua BH, et al. Isolation of Nipah virus from Malaysian island flying-foxes. Microbes Infect. 2002;4:145–151. doi: 10.1016/S1286-4579(01)01522-2. [DOI] [PubMed] [Google Scholar]
- 32.Halpin K, Young PL, Field HE, Mackenzie JS. Isolation of Hendra virus from pteropid bats: a natural reservoir of Hendra virus. J Gen Virol. 2000;81:1927–1932. doi: 10.1099/0022-1317-81-8-1927. [DOI] [PubMed] [Google Scholar]
- 33.Barr JA, Smith C, Marsh GA, Field H, Wang LF. Evidence of bat origin for Menangle virus a zoonotic paramyxovirus first isolated from diseased pigs. J Gen Virol. 2012;93:2590–2594. doi: 10.1099/vir.0.045385-0. [DOI] [PubMed] [Google Scholar]
- 34.Barr J, Smith C, Smith I, de Jong C, Todd S, Melville D, et al. Isolation of multiple novel paramyxoviruses from pteropid bat urine. J Gen Virol. 2015;96:24–29. doi: 10.1099/vir.0.068106-0. [DOI] [PubMed] [Google Scholar]
- 35.Wilkinson DA, Temmam S, Lebarbenchon C, Lagadec E, Chotte J, Guillebaud J, et al. Identification of novel paramyxoviruses in insectivorous bats of the Southwest Indian Ocean. Virus Res. 2012;170:159–163. doi: 10.1016/j.virusres.2012.08.022. [DOI] [PubMed] [Google Scholar]
- 36.Kurth A, Kohl C, Brinkmann A, Ebinger A, Harper JA, Wang LF, et al. Novel paramyxoviruses in free-ranging European bats. PLoS One. 2012;7:e38688. doi: 10.1371/journal.pone.0038688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mortlock M, Kuzmin IV, Weyer J, Gilbert AT, Agwanda B, Rupprecht CE, et al. Novel paramyxoviruses in bats from sub-Saharan Africa 2007–2012. Emerg Infect Dis. 2015;21:1840–1843. doi: 10.3201/eid2110.140368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.The Council of the European Communities Council directive 92/43/EEC of 21 may 1992 on the conservation of natural habitats and of wild fauna and flora. Off J Eur Union. 1992;206:7–50. [Google Scholar]
- 39.UNEP/EUROBATS Agreement on the Conservation of Populations of European Bats. EUROBATS, London. 1991. http://www.eurobats.org/official_documents/agreement_text. Accessed on 25 February 2013.
- 40.Kunz TH, Parsons S. Ecological and behavioural methods for the study of bats. 2. Baltimore: Johns Hopkins University Press; 2009. [Google Scholar]
- 41.Veith M, Beer N, Kiefer A, Johannesen J, Seitz A. The role of swarming sites for maintaining gene flow in the brown long-eared bat (Plecotus Auritus) Heredity. 2004;93:342–349. doi: 10.1038/sj.hdy.6800509. [DOI] [PubMed] [Google Scholar]
- 42.Poon LL, Chu DK, Chan KH, Wong OK, Ellis TM, Leung YH, et al. Identification of a novel coronavirus in bats. J Virol. 2005;79:2001–2009. doi: 10.1128/JVI.79.4.2001-2009.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tong S, Chern SW, Li Y, Pallansch MA, Anderson LJ. Sensitive and broadly reactive reverse transcription-PCR assays to detect novel paramyxoviruses. J Clin Microbiol. 2008;46:2652–2658. doi: 10.1128/JCM.00192-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kemenesi G, Dallos B, Görföl T, Boldogh S, Estók P, Kurucz K, et al. Molecular survey of RNA viruses in Hungarian bats: discovering novel astroviruses coronaviruses and caliciviruses. Vector Borne Zoonotic Dis. 2014;14:846–855. doi: 10.1089/vbz.2014.1637. [DOI] [PubMed] [Google Scholar]
- 45.Plowright RK, Field HE, Smith C, Divljan A, Palmer C, Tabor G, et al. Reproduction and nutritional stress are risk factors for Hendra virus infection in little red flying foxes (Pteropus Scapulatus) Proc R Soc Lond B Biol Sci. 2008;275:861–869. doi: 10.1098/rspb.2007.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salicini I, Ibáñez C, Juste J. Deep differentiation between and within Mediterranean glacial refugia in a flying mammal the Myotis Nattereri bat complex. J Biogeogr. 2013;40:1182–1193. doi: 10.1111/jbi.12062. [DOI] [Google Scholar]
- 47.Cui J, Han N, Streicker D, Li G, Tang X, Shi Z, et al. Evolutionary relationships between bat coronaviruses and their hosts. Emerg Infect Dis. 2007;13:1526–1532. doi: 10.3201/eid1310.070448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lau SK, Poon RW, Wong BH, Wang M, Huang Y, Xu H, et al. Coexistence of different genotypes in the same bat and serological characterization of Rousettus bat coronavirus HKU9 belonging to a novel Betacoronavirus subgroup. J Virol. 2010;84:11385–11394. doi: 10.1128/JVI.01121-10. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 49.Yuan J, Hon CC, Li Y, Wang D, Xu G, Zhang H, et al. Intraspecies diversity of SARS-like coronaviruses in Rhinolophus Sinicus and its implications for the origin of SARS coronaviruses in humans. J Gen Virol. 2010;91:1058–1062. doi: 10.1099/vir.0.016378-0. [DOI] [PubMed] [Google Scholar]
- 50.Parks GD, Alexander-Miller MA. Paramyxovirus activation and inhibition of innate immune responses. J Mol Biol. 2013;425:4872–4892. doi: 10.1016/j.jmb.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wille M, Avril A, Tolf C, Schager A, Larsson S, Borg O, et al. Temporal dynamics diversity and interplay in three components of the virodiversity of a mallard population: influenza a virus avian paramyxovirus and avian coronavirus. Infect Genet Evol. 2015;29:129–137. doi: 10.1016/j.meegid.2014.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data generated or analyzed during this study are included in this paper and can be made available by the corresponding author upon a reasonable request. Sequence data obtained in this study are deposited in GenBank database: full sequence name and GenBank accession no are indicated in Table 2.