

Abstract
Brucellosis is a commonly diagnosed zoonosis that causes infertility and abortion in cattle, it is acquired from handling of infected animals or consuming contaminated milk or milk products. In Colombia, it belongs to the official notifiable disease list, despite its relevance little is known about the origin, epidemiology and the genetic constituents of the strains circulating in dairy farms. Here we present the draft genome of B. abortus Ba Col-B012, an isolate obtained from a female Holstein belonging to a dairy farm in Nariño, Colombia. This genome comprises 3,234,714 bp and 3018 predicted protein-encoding genes. Using comparative genomics and phylogenetic analysis, we found that the strain Ba Col-B012 clustered with known biovar 4 variants. The analysis of the core genes allowed the identification of polymorphisms only present in biovar 4 genomes, these regions are proposed as possible targets for identification by PCR. The sequencing of B. abortus Ba Col-B012 genome provides important insights to improve the diagnosis and the epidemiology of this disease and represents the first report of the biovar 4 in Colombia.
Introduction
The brucellosis is one of the most important zoonotic diseases that causes infertility and abortion in cattle. In livestock, brucellosis is mainly caused by 10.1601/nm.1382, a Gram-negative coccobacillus that behaves as a facultative intracellular pathogen. There are up to eight variants of this species that differ on their physiological characteristics and are classified as biovars. However, some of these biovars differ only slightly and their status as true variants is unresolved. Some biovars have a wide geographic distribution; 10.1601/nm.1382 biovar1 and biovar2 are found around the world, while others as the biovar 5 are mainly distributed in Europe [1]. In South America, recent studies have identified several biovars, for instance, a survey of a 30-year 10.1601/nm.1382 collection from Brazil, found biovars 1, 2, and 3 [2], while in Ecuador, biovar 1 and 4 have been reported [3]. However, there still is a lack of sufficient information to establish biovar presence and distribution in other countries of the continent. In Colombia, even though there are regions with high prevalence and isolation of 10.1601/nm.1382 [4, 5], there are no reports on the identification of their corresponding biovars.
The genome presented here belongs to a larger collection of pathogens isolated as part of a monitoring program to identify the principal infectious agents related to infertility and abortion in cattle present in the southern part of Colombia [6]. During this survey, 12 10.1601/nm.1382 strains were isolated from dairy farms (Nariño, Colombia). Recently some of these strains were typified using AMOS-ERY-PCR [7] and MLVA methods [8], and a representative isolate was chosen for sequencing. Here we present the draft quality genome of the strain, 10.1601/nm.1382 Ba Col-B012, this genome contributes to a better understanding of the genomic constituents of local isolates and to the identification of virulence factors and conserved genes that code for immunogenic proteins that can eventually be used in the development of vaccines and new serological tests.
Organism information
Classification and features
10.1601/nm.1382 is a non-motil, Gram-negative short bacillus measuring about 0.6 to 1.5 μm by 0.5–0.7 μm (Fig. 1). The 10.1601/nm.1382 species belong to the family 10.1601/nm.1379, class 10.1601/nm.809 and phylum 10.1601/nm.808. Colonies are smooth, small, round, convex, and non-pigmented, on 10.1601/nm.1380 agar small colorless punctate colonies, appear within 48 to 72 h at 37 °C. Even though they are aerobes, providing a CO2 atmosphere may enhance growth.
Fig. 1.
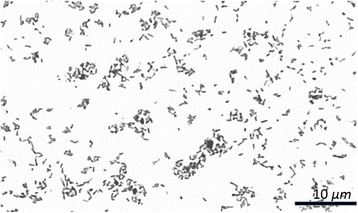
Photomicrograph of cells of B. abortus strain Ba Col-B012. Cells were grown on trypticase soy agar and brain infusion agar supplemented with 5% horse serum, this media was incubated at 37 °C for 48 h
The 10.1601/nm.1382 Ba Col-B012 strain was obtained from a female Holstein with an episode of abortion. The sample was taken from vaginal fluids with a swab and isolation was done on trypticase soy agar and brain infusion agar supplemented with 5% Horse serum, this media was incubated at 37 °C for 72 to 96 h, with a 5% CO2 atmosphere. Small transparent colonies were obtained with regular edges. Isolates were characterized by being non-motile and positive for the urease and oxidase tests and for the agglutination test by using polyclonal anti-10.1601/nm.1382 antibody (Difco). A summary of the classification and general features of 10.1601/nm.1382 strain Ba Col-B012 is presented in Table 1.
Table 1.
Classification and general features of B. abortus strain Ba Col-B012
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Classification | Domain Bacteria | TAS [25] | |
| Phylum Proteobacteria | TAS [25, 26] | ||
| Class Proteobacteria alfa | TAS [27] | ||
| Order “Rhizobiales” | TAS [28] | ||
| Family Brucellaceae | TAS [29, 30] | ||
| Genus Brucella | TAS [30, 31] | ||
| Species Brucella abortus | TAS [30, 31] | ||
| strain: Col-B012 | IDA | ||
| Gram stain | Negative | TAS [31] | |
| Cell shape | Coccobacilli | TAS [31] | |
| Motility | Non-motile | TAS [31] | |
| Sporulation | Non-sporulating | TAS [31] | |
| Temperature range | 20–40 °C | IDA | |
| Optimum temperature | 37 °C | TAS [31] | |
| pH range; Optimum | 6.6–7.4 | TAS [31] | |
| Carbon source | d-glucose, d-ribose, l-malate, dl-lactate | TAS [32] | |
| MIGS-6 | Habitat | Holstein cattle | TAS [6] |
| MIGS-6.3 | Salinity | – | NAS |
| MIGS-22 | Oxygen requirement | Facultative | TAS [31] |
| MIGS-15 | Biotic relationship | Host-associated | TAS [6] |
| MIGS-14 | Pathogenicity | Pathogenic | NAS |
| MIGS-23 | Isolation | IDA | |
| MIGS-4 | Geographic location | Nariño, Colombia | IDA |
| MIGS-5 | Sample collection | June, 1997 | IDA |
| MIGS-4.1 | Latitude | 00° 52’ N | IDA |
| MIGS-4.2 | Longitude | −77° 39’ W | IDA |
| MIGS-4.4 | Altitude | 2900 m a.s.I | IDA |
aEvidence codes - IDA: Inferred from Direct Assay; TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [12/17]
Genome sequencing and information
Genome project history
10.1601/nm.1382 strain Ba Col-B012 was isolated as part of a monitoring program to identify the principal infectious agents related to infertility and abortion in cattle present in the southern part of Colombia [6]. The main objective for sequencing 10.1601/nm.1382 genomes is to explore the genomic constituents of the local isolates and to identify virulence factors, polymorphic regions, and immunogenic proteins that can be eventually be used in the development of vaccines and new serological and molecular tests. A summary of the project information is shown in Table 2.
Table 2.
Project information
| MIGS ID | Property | Term |
|---|---|---|
| MIGS 31 | Finishing quality | Improved high-quality draft |
| MIGS-28 | Libraries used | One Illumina paired-end |
| MIGS 29 | Sequencing platforms | Illumina HiScan SQ |
| MIGS 31.2 | Fold coverage | 50 × Illumina |
| MIGS 30 | Assemblers | Newbler 2.0.01.14 |
| MIGS 32 | Gene calling method | GeneMarkS+, Glimmer, Prodigal |
| Locus Tag | LODQ01 | |
| Genbank ID | LODQ01000000. | |
| GenBank Date of Release | 01/09/2017 | |
| GOLD ID | _ | |
| BIOPROJECT | PRJNA305302 | |
| Project relevance | Host-associated |
Growth conditions and genomic DNA preparation
10.1601/nm.1382 strain Ba Col-B012 strain was grown on trypticase soy agar and brain infusion agar supplemented with 5% horse serum, this media was incubated at 37 °C for 72 h. Genomic DNA extraction was done with the CTBA-Phenol Chloroform method couple to ethanol precipitation [9]. DNA was quantified using the dsDNA HS (High Sensitivity) kit on a Qubit™ (Life Technologies), a greater than 30 ng/μl DNA concentration was obtained. Quality and purity of DNA was determined by spectrophotometry (Nanodrop® 2000 Thermo Fisher Scientific) obtaining a 260/280 and 260/230 ratio equal to 2.
Genome sequencing and assembly
Whole-genome sequencing of the 10.1601/nm.1382 strain Ba Col-B012 strain was performed by employing the Illumina HiScan SQ (Molecular Biology Lab, Corpoica). Libraries were generated using the Sure Select Strand Agilent Sample Preparation, once the DNA concentration was determined library amplification was done with the TruSeq PE Cluster Kit v3, (Illumina), using Cbot (Illumina). For de novo assembly, we used 3,956,238 paired-end Illumina reads (150 bp) and the Newbler v 2.0.01.14 software. The assembly resulted in 233 contigs with total genome length of 3227,565 bp and with 50× average coverage.
Genome annotation
Gene prediction was conducted with GeneMarkS+ [10], and PRODIGAL [11] and annotation was done automatically using the NCBI Prokaryotic Genome Annotation Pipeline. The annotation was corrected manually using the data from different databases (Swiss-Prot [12] and RAST [13]). We use LipoP v 1.0 [14] for finding genes with signal peptides and with transmembrane helices.
Genome properties
The genome statistics are provided in Table 3. The assembly resulted in 233 contigs with total genome length of 3227,565 bp and with 50× average coverage. The N50 contig size is 22,624 and a maximum contig size of 106,301 bp and a G + C content of 57.28 mol%. These values are similar to those reported for the genomes NC_006932.1, NZ_CP007709.1 and NZ_CP007705.1 of 10.1601/nm.1382 at NCBI. Using our annotation pipeline, it was possible to identify 3227 predicted genes of which 3018 were putatively protein-encoding, 166 pseudogenes, 42 tRNAs and 1 ncRNA. For the majority of the protein-encoding genes (78.12%) a function could be assigned. The distribution of these genes into COG functional categories [15] is shown in Table 4. This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession LODQ00000000. The version described in this paper is version LODQ01000000.
Table 3.
Genome statistics of B. abortus strain Ba Col-B012
| Attribute | Value | % of Total |
|---|---|---|
| Genome size (bp) | 3,234,714 | 100.00 |
| DNA coding (bp) | 2,685,762 | 83.02 |
| DNA G + C (bp) | 1,472,070 | 45.50 |
| DNA scaffolds | 243 | 100.00 |
| Total genes | 3227 | 100.00 |
| Protein coding genes | 3018 | 93.52 |
| RNA genes | 42 | 1.30 |
| Pseudo genes | 166 | 5.14 |
| Genes in internal clusters | 164 | 5.43 |
| Genes with function prediction | 2408 | 74.62 |
| Genes assigned to COGs | 2521 | 78.12 |
| Genes with Pfam domains | 2631 | 81.53 |
| Genes with signal peptides | 380 | 11.77 |
| Genes with transmembrane helices | 422 | 13.07 |
| CRISPR repeats | 0 | 0 |
Table 4.
Number of genes associated with general COG functional categories
| Code | Value | %age | Description |
|---|---|---|---|
| J | 160 | 5.30 | Translation, ribosomal structure and biogenesis |
| A | 0 | 0 | RNA processing and modification |
| K | 193 | 6.39 | Transcription |
| L | 117 | 3.87 | Replication, recombination and repair |
| B | 1 | 0.03 | Chromatin structure and dynamics |
| D | 28 | 0.92 | Cell cycle control, Cell division, chromosome partitioning |
| V | 50 | 1.65 | Defense mechanisms |
| T | 79 | 2.61 | Signal transduction mechanisms |
| M | 137 | 4.53 | Cell wall/membrane biogenesis |
| N | 30 | 0.99 | Cell motility |
| U | 23 | 0.76 | Intracellular trafficking and secretion |
| O | 98 | 3.24 | Posttranslational modification, protein turnover, chaperones |
| C | 169 | 5.59 | Energy production and conversion |
| G | 177 | 5.86 | Carbohydrate transport and metabolism |
| E | 307 | 10.17 | Amino acid transport and metabolism |
| F | 73 | 2.41 | Nucleotide transport and metabolism |
| H | 107 | 3.54 | Coenzyme transport and metabolism |
| I | 93 | 3.08 | Lipid transport and metabolism |
| P | 200 | 6.62 | Inorganic ion transport and metabolism |
| Q | 36 | 1.19 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 0 | 0 | General function prediction only |
| S | 481 | 15.93 | Function unknown |
| – | 497 | 16.46 | Not in COGs |
The total is based on the total number of protein coding genes (3018) in the genome
Insights from genome sequences
Genomes used in this study
A total of 28 10.1601/nm.1382 genomes were downloaded from the NCBI database of complete and draft bacterial genomes, even though there are many more genomes in the database, only those with identified biovar were used for further analyses. The genomes and their GeneBank accession numbers are listed in Table 5. The genes used in the analysis were predicted from the genomes using PRODIGAL with the default settings [11].
Table 5.
Genomes and accession numbers used in this study
| Biovar | Strain name | Genome assembly number |
|---|---|---|
| 1 | Brucella abortus biovar 1 NI435a | GCA_000245835.1 |
| 1 | Brucella abortus biovar 1 NI486 | GCA_000245855.1 |
| 1 | Brucella abortus biovar 1 NI474 | GCA_000245875.1 |
| 1 | Brucella abortus biovar 1 NI488 | GCA_000245895.1 |
| 1 | Brucella abortus biovar 1 NI010 | GCA_000245915.1 |
| 1 | Brucella abortus biovar 1 NI016 | GCA_000245935.1 |
| 1 | Brucella abortus biovar 1 NI021 | GCA_000245955.1 |
| 1 | Brucella abortus biovar 1 NI259 | GCA_000245975.1 |
| 1 | Brucella abortus biovar 1 str 134 | GCA_000298635.1 |
| 1 | Brucella abortus biovar 1 76–1413 | GCA_000413495.1 |
| 1 | Brucella abortus biovar 1 84–0928 | GCA_000413575.1 |
| 1 | Brucella abortus biovar 1 90–0742 | GCA_000413655.1 |
| 1 | Brucella abortus biovar 1 94–1313 | GCA_000413735.1 |
| 1 | Brucella abortus biovar 1 01–0648 | GCA_000413755.1 |
| 1 | Brucella abortus biovar 1 01–0585 | GCA_000413775.1 |
| 1 | Brucella abortus biovar 1 01–0065 | GCA_000413795.1 |
| 1 | Brucella abortus biovar 1 B10–0018 | GCA_000413815.1 |
| 1 | Brucella abortus biovar 1 B10–0091 | GCA_000413955.1 |
| 1 | Brucella abortus biovar 1 89–0363 | GCA_000413975.1 |
| 1 | Brucella abortus biovar 1 87–2211 | GCA_000413995.1 |
| 1 | Brucella abortus biovar 1 82–2330 | GCA_000414015.1 |
| 1 | Brucella abortus biovar 1 80–1399 | GCA_000478665.1 |
| 2 | Brucella abortus biovar 2 82–3893 | GCA_000413555.1 |
| 2 | Brucella abortus biovar 2 90–0737 | GCA_000413695.1 |
| 2 | Brucella abortus biovar 2 90–1280 | GCA_000413715.1 |
| 4 | Brucella abortus biovar 4 68-3396P | GCA_000413535.1 |
| 4 | Brucella abortus biovar 4 90–0775 | GCA_000413675.1 |
| 4 | Brucella abortus biovar 4292 | GCA_000157695.1 |
Genomic differences between 10.1601/nm.1382 BA col-B012 and the type strain 10.1601/nm.1382 2308
The comparative genomic analysis between 10.1601/nm.1382 strain BA Col-B012 and the type strain, 10.1601/nm.1382 2308, shows that both genomes shared 3015 genes, most of these genes are identical (2862 genes with 100%). Within this set of genes there are around 12 genes that are divergent with a nucleotide identity ranging from 77 to 94%, (Additional file 1: Table S1) among the genes are an ABC transporter permease, benzoate transporter, an alpha/beta hydrolase, a 5-hydroxymethyluracil DNA glycosylase, several hypothetical proteins and a hemolysin D gene (HlyD). Hemolysin D is part of the membrane transporter of the HlyA, a pore-forming toxin that affects the membrane of the host [16]. We also identified 16 genes present in strain BA Col-B012 that were not found in the type strain (Additional file 1: Table S2). Most genes in this set are hypothetical proteins, transporters and transcriptional regulators. These differences show that strain BA Col-B012 differs from the type strain 2308. In order to elucidate if these differences are related to the biovar classification a comparative genomic analysis with more strain was done in the next section.
The evolutionary distance and phylogenetic relationship of 10.1601/nm.1382 strain Ba col-B012
A phylogenomic approach was done to establish the evolutionary relationship of 10.1601/nm.1382 strain Ba Col-B012 and to evaluate whether biovars are congruent with true genetic groupings. The phylogenetic analysis was done by concatenating the alignment of orthologues genes shared by all strains. In order to identify a set of orthologous genes, an in-house PERL script that incorporates the reciprocal best match approach was used [17]. In brief, the predicted genes of strain Ba Col-B012 were searched using the blastn algorithm [18] against the genomic sequences of each of the remaining genomes. The best match for each query gene (genes with higher than 70% identity and alignment coverage) was extracted and searched against the complete gene complement of the Ba Col-B012 strain to identify reciprocal best matches. The reciprocal best match genes were denoted as orthologues, 3139 orthologous genes were shared among all strains, from these 2169 were identical among all strains (100% nucleotide identity). Average nucleotide identity (ANI) was quantified using the nucleotide identity of orthologues between the strain Ba Col-B012 and the other genomes, this is a measurement of genomic divergence that is used in modern taxonomy as the gold standard to delimitate new species [19, 20]. The ANI values between the Ba Col-B012 and the rest of the strains were higher than 99.6 % (Additional file 1), these high identity reflect the close evolutionary relationship between the 10.1601/nm.1382 strains that make difficult the identification of biovar variants. Despite the close relationship between all genomes, strain Ba Col-B012 showed a closest affiliation with biovar 4 strains (99.88%).
In order to corroborate the affiliation of Ba Col-B012 to biovar 4, the phylogenetic relationship of shared polymorphic genes, around 2961 genes, was inferred using the Neighbor Joining algorithm with the Jukes-Cantor distance and 1000 bootstraps (Fig. 2). As shown before by the ANI analyses, strain Ba Col-B012 was more closely related to the biovar 4 strains clustering in the same clade with a 100 bootstrap value. This represents the first confirmed report of a biovar 4 strain in Colombia, and may suggest a possible transfer from Ecuador which is the country that delimits with the Nariño region and where biovar 4 has been reported [3].
Fig. 2.

Evolutionary relationships of B. abortus strain Ba Col-B012. The evolutionary history was inferred using the Neighbor-Joining method [33]. The bootstrap consensus tree inferred from 1000 replicates [34] is taken to represent the evolutionary history of the taxa analyzed [35]. All positions containing gaps and missing data were eliminated. There were a total of 2,632,124 positions in the final dataset. Evolutionary analyses were conducted in MEGA6 [36]
Used of polymorphic regions in the identification of 10.1601/nm.1382 Biovar 4 and its potential for diagnosis and vaccination
Current identification of biovars is based on standard microbiological methods and molecular approaches like MLVA analysis. MLVA is particularly a high discriminatory method useful in epidemiological studies and in the identification of genetic variability of strains [21]. However, this methodology is not always conclusive. In order to complement the current methods of diagnosis with PCR-based amplification and sequencing, orthologous regions that could be used to differentiate biovar 4 genomes from others were identified. We found around 42 genes with polymorphism that differentiate biovar 4 genomes from the rest. Most genes have only one single nucleotide polymorphism (SNP), from this set almost half of the SNPs are non-synonymous. From all evaluated genes, only one hypothetical gene has two polymorphisms that are synonymous (set 12). We also found two genes that have insertion-deletions and three genes that are shorter than the biovar 1 counterpart due to the presence of an early stop codon (See Table 6 for a description of genes and differences). All gene set described in the analysis are provided in the Additional files section.
Table 6.
Analysis of polymorphic genes that differentiate biovar 4 from other genomes
| Set | Annotation | Type | Description |
|---|---|---|---|
| 0 | The major facilitator superfamily (MFS) is a class of membrane transport proteins | Syn | T-G (pos 87)| |
| 1 | Hypothetical protein | Syn | T-G (pos 283) |
| 2 | Multiple antibiotic resistance transporter | NonSyn | C-T (pos 424), P-S (pos 142) |
| 3 | Calcium/calmodulin dependent protein kinase II | Syn | C-A (pos 95) |
| 4 | Peptidase Do | Syn | C-A(pos 829) |
| 5 | 30S ribosomal protein S14 | Syn | C-A(pos 124) |
| 6 | Hypothetical protein | NonSyn | G-T (pos 186), Q-H (pos 62) |
| 7 | Excinuclease ABC subunit B | Stop | STOP codon |
| 8 | Hypothetical protein similar with BA14K family domain | In/Del | IN/DEL 12 nuc (pos 327) |
| 9 | Flagellar basal body rod protein FlgC | NonSyn | G-A (pos 55), A-T (pos 19) |
| 10 | Dipeptide ABC transporter permease DppC | NonSyn | T-C (pos 478), S-P (pos 160) |
| 11 | Na(+)/H(+) antiporter NhaA | Stop | IN/DEL-ORF G?- (pos 1917) |
| 12 | Hypothetical protein | Syn | G-A (pos 609), C-T (pos 633) |
| 13 | DNA-3-methyladenine glycosylase | Syn | C-T (pos 483) |
| 14 | Mannosyltransferase | NonSyn | A-C(pos 980), K-N (pos 349) |
| 15 | Hypothetical protein | NonSyn | C-T(pos 229), T-I (pos75) |
| 16 | Class II fumarate hydratase | NonSyn | C-T(pos 1323), A-V (pos 441) |
| 17 | Hypothetical protein | Syn | G-C(pos 250) |
| 18 | Acyl carrier protein | Syn | C-T (pos 260) |
| 19 | Tyrosine--tRNA ligase | Syn | C-G(pos1107) |
| 20 | Glycosyl transferase | NonSyn | C-T(pos 35), V-A(pos12) |
| 21 | D-alanyl-D-alanine carboxypeptidase | NonSyn | A-G(pos 451), T-A (pos 151) |
| 22 | Malic enzyme | Syn | T-C (pos723) |
| 23 | X-Pro dipeptidase | T-C(pos 280), F-L (pos 94) | |
| 24 | Putative multidrug efflux transporter protein | Stop | G-T(pos 229), E-STOP |
| 25 | D-ribose ABC transporter substrate-binding protein | NonSyn | C-T(pos 396), A-V(pos 132) |
| 26 | NAD-dependent dehydratase | NonSyn | A-G(pos196), M-V (pos 66) |
| 27 | Phosphogluconate dehydratase | NonSyn | C-T(pos620), A-V(pos 207) |
| 28 | Hypothetical protein | Syn | C-T (pos628) |
| 29 | Hypothetical protein | NonSyn | A-G(pos235), T-A (pos 79) |
| 30 | Glutamine synthetase | Syn | G-A(pos 1306) |
| 31 | N-formylglutamate amidohydrolase | Syn | A-G (pos 541) |
| 32 | Hypothetical protein | NonSyn | A-T(pos 36), K-M(pos12) |
| 33 | Branched-chain amino acid ABC transporter, ATP-binding/permease protein | NonSyn | A-G(pos452), N-S(pos 151) |
| 34 | DNA topoisomerase | NonSyn | C-A(pos 1827), |
| R-S(pos 609) | |||
| 35 | Aspartate carbamoyltransferase | Syn | A-G(pos 540) |
| 36 | 8-amino-7-oxononanoate synthase | NonSyn | A-G(pos 991), R-G(pos 331) |
| 37 | Secretion protein HlyD family protein-hemolysin secretion protein D | NonSyn | G-A(pos 415), V-I(pos 139) |
| 38 | Tetracycline resistance protein TetB | NonSyn | T-G(pos 765), F-L(pos 225) |
| 39 | Mannose-1-phosphate guanylyltransferase/mannose-6-phosphate isomerase | NonSyn | G-T(pos 590), F-C(pos 197) |
| 40 | ABC transporter permease | In/Del | Large Insertion of up to 43 aa |
| 41 | Aminobutyraldehyde dehydrogenase | Syn | T-C (pos 342) |
Position are relative to the gene set alignment. Alignments are provided as Additional file 2
In order to design primers for genetic markers for biovar 4, we focused on orthologues amplifiable by PCR (<400 bp) that have large INDELs or genes with synonymous polymorphisms, this guarantees that the observed changes are not under selection. We identified six genes that met this criteria, these were: hypothetical protein similar with BA14K family domain (gene set 8), hypothetical protein (gene set 12), 10.1601/strainfinder?urlappend=%3Fid%3DDNA+3-methyladenine glycosylase (gene set 13), tyrosine--tRNA ligase (gene set 19), glutamine synthetase (gene set 30), and ABC transporter permease (gene set 40). Based on these genes, we designed sets of primers that amplify the polymorphic regions and therefore can be used for the identification. Table 7 summarizes the designed primers and their predicted PCR conditions.
Table 7.
Designed primer sets to differentiate biovar 4 from others
| Set | F. pos (bp) | R. pos (bp) | Forward primer | Reverse primer | Tm (°C) |
|---|---|---|---|---|---|
| 8 | 281 | 435 | AGCCACGCACGACCTATATC | GCCCGAGCAATACTGATACC | 60 |
| 12 | 478 | 877 | GAAGCCGATCAGCAATTCAC | AAAGCAGGATCGCCACATAG | 60 |
| 13 | 178 | 552 | GGATTGTCGTGGCTTACGAT | GAAGGCATAGACCGTGGTTG | 60 |
| 19 | 962 | 1218 | ACGCAAGACCTTTGAAGACG | GAGCGACAGCTTGATGAGG | 60 |
| 30 | 923 | 1322 | CGCCTTACATCAATTCCTACAA | CGGTCATATTCGATCTGTTCC | 59 |
| 40 | 22 | 598 | ATTCTCGATCCGCATTTCAT | AGAGGCCGGAGAGAATAAGC | 60 |
Position of primers is relative to the gene set alignment
Comparative genome analysis of 10.1601/nm.1382 strains is a powerful tool for the identification of allele variants/polymorphism that modulate virulence. Interestingly, among the identified polymorphic genes, two genes have been associated with pathogenicity and immune response, a hypothetical protein similar with BA14K family domain (Table 6, gene set 8) and a gene coding for the subunit B of the exonuclease ABC (Table 6, gene set 7). The domain 10.1601/strainfinder?urlappend=%3Fid%3DBAL+14K had been demonstrated to induce a strong immunoreactivity in mice, with a Th1 response and induction of IL-12 secretion [22]. While changes in the subunit B of exonuclease ABC have been associated with minor virulence changes between attenuated and virulent 10.1601/nm.1380 strains [23]. It is also worth mentioning that several other sets of genes identified as polymorphic might also display immunogenic reactivity, as their coding proteins are located in the membrane at the interphase with the environment, for instance, several transporters in 10.1601/nm.1382 have been used to produce in vivo-induced antigens [24]. These genes are potential targets for future vaccination and diagnosis.
Conclusions
The genome of 10.1601/nm.1382 Ba Col-B012 contributes to the better understanding of the distribution and origin of zoonotic pathogens in Colombia and South America. A better representation of biovar genomes can be used to elucidate the correspondence between evolutionary relationship and phenotypic characteristics. The phylogenomic relationship between strain Ba Col-B012 and the examined genomes shows that biovar 4 strains form a distinctive clade with high bootstrap support. This pattern is not observed for other biovars, for example, strain 90–0737 and strain B10–0018, which cluster in the same clade, are classified into different biovar groups. The clear clustering of biovar 4 genomes reflects a common ancestor of the group and suggests the existence of allele differences that might be associated with the phenotypic and pathogenic characteristics of the group. Finally, the identification of biovar 4 distinctive genomic region allowed us to design sets of primers that coupled with sequencing could be incorporated into current methods of identification to distinguish biovar 4 strains from others. The 10.1601/nm.1382 Ba Col-B012 genome provides important insights to improve the diagnosis and the epidemiology of this disease and represents the first report of the biovar 4 in Colombia.
Additional files
Genes that differentiate Brucella abortus strain Ba Col-B012 from the type strain. (DOCX 56 kb)
Alignment of polymorphic genes, sets 0–41. (ZIP 58 kb)
Acknowledgements
We thank Yolanda Gomez Vargas, Johan Bernal Morales for their contribution in the DNA extraction, preparation of the genomic libraries and sequencing.
Funding
This study was funded by the Colombian Ministry of Agriculture (Ministerio de Agricultura y Desarrollo Rural de Colombia).
Abbreviations
- AAI
Average Amino Acid Identity
- MLVA
Multiple-Locus Variable number tandem repeat Analysis.
Authors’ contributions
AC and RP conceived of the study and participated in its design and coordination. IN, RP, JLR, SJ and LT collaborated in the isolation, conservation and characterization of the strains, as well as de DNA extraction. AC and MP analysis of them and drafted the manuscript. AC performed the phylogenetic and orthologous gene analysis, respectively. AC and MP participated in genome assembly, annotation and analysis. All authors contributed in improving the quality of the manuscript and approved the final version.
Competing interests
The authors declare that they have no competing interests.
This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s40793-017-0299-2) contains supplementary material, which is available to authorized users.
References
- 1.Garin-Bastuji B. Brucelloses bovine, ovine et caprine: Contrôle et prevention. Le Point vétérinaire: revue d'enseignement post-universitaie et de formation permanente. 1993;25:15–22. [Google Scholar]
- 2.Minharro S, Silva Mol J, Dorneles E, Pauletti R, Neubauer H, Melzer F, Poester F, Dasso M, Pinheiro E, Soares Filho P, Santos R, Heinemann M, Lage A. Biotyping and genotyping (MLVA16) of Brucella abortus isolated from cattle in Brazil, 1977 to 2008. PLoS One. 2013; 10.1371/journal.pone.0081152. [DOI] [PMC free article] [PubMed]
- 3.Rodriguez-Hidalgo R, Contreras-Zamora J, Benitez-Ortiz W, Guerrero-Viracocha K, Salcan-Guaman H, Minda E, Ron Garrido L. Circulating strains of Brucella abortus in cattle in Santo Domingo de Los Tsáchilas Province – Ecuador. Frontiers of. Public Health. 2015;3:45. doi: 10.3389/fpubh.2015.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rivera DY, Rueda OE, Calderon CP, Marino OC, Gall D, Nielsen K. Comparative evaluation of the indirect enzyme-linked immunosorbant assay in milk for the detection of cattle infected with Brucella abortus, in herds located in the province of Cundinamarca, Colombia. Revue Scientifique et Technique (International Office of Epizootics) 2003;22(3):1065–1075. [PubMed] [Google Scholar]
- 5.Griffiths IB, Gallego MI, De Leon LS. Levels of some reproductive diseases in the dairy cattle of Colombia. Trop Anim Health Prod. 1984;16(4):219–223. doi: 10.1007/BF02265325. [DOI] [PubMed] [Google Scholar]
- 6.González Cardona HG, Patiño Burbano RE: Principales agentes infectocontagiosos del aborto e infertilidad en el ganado lechero de Nariño y Alto Putumayo.1999.http://bibliotecadigital.agronet.gov.co/bitstream/11348/3879/1/20061127144049_Agentes%20aborto%20infertilidad%20ganado%20lechero.pdf. Accessed 12 November 2016.
- 7.Ocampo-Sosa A, Agüero-Balbín J, García-Lobo J. Development of a new PCR assay to identify Brucella abortus biovars 5, 6 and 9 and the new subgroup 3b of biovar 3. Vet Microbiol. 2005;110:41–51. doi: 10.1016/j.vetmic.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 8.Bricker BJ, Ewalt DR, Halling SM. Brucella 'HOOF-Prints': strain typing by multi-locus analysis of variable number tandem repeats (VNTRs) BMC Microbiol. 2003;3(15):1–13. doi: 10.1186/1471-2180-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Associates and John Wiley & Sons. Current Protocols in Molecular Biology. Vol I. Greene Publishing. 1997; 2.4.2–2.4.3.
- 10.Besemer J, Lomsadze A, Borodovsky M. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001;29:2607–2618. doi: 10.1093/nar/29.12.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hyatt D, Chen G, LoCascio P, Land M, Larimer F, Hauser L. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gasteiger E, Jung E, Bairoch A. SWISS-PROT: connecting biomolecular knowledge via a protein database. Current Issues Mol Biol. 2001;3:47–55. [PubMed] [Google Scholar]
- 13.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Juncker AS, Willenbrock H, Von Heijne G, Brunak S, Nielsen H, Krogh A. Prediction of lipoprotein signal peptides in gram-negative bacteria. Protein Sci. 2003;12:1652–1662. doi: 10.1110/ps.0303703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lenders MH, Beer T, Smits SH, Schmitt L. Vivo quantification of the secretion rates of the hemolysin a type I secretion system. Sci Rep. 2016;6 [DOI] [PMC free article] [PubMed]
- 17.Konstantinidis K, Serres M, Romine M, Rodrigues J, Auchtung J, McCue L, Lipton M, Obraztsova A, Giometti C, Nealson K, Fredrickson J, Tiedje J. Comparative systems biology across an evolutionary gradient within the Shewanella genus. Proc Natl Acad Sci U S A. 2009;106:15909–15914. doi: 10.1073/pnas.0902000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Altschul SF, Gish W, Miller W, Myers EW. Lipman DJ. Basic local alignment search tool. 1990;21:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 19.Konstantinidis K, Tiedje J. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci U S A. 2005;102:2567–2572. doi: 10.1073/pnas.0409727102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richter M, Rossello-Mora R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A. 2009;106:19126–19131. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tiller R, De B, Boshra M, Huynh L, Van Ert M, Wagner D, Klena J, Mohsen T, El-Shafie S, Keim P, Hoffmaster A, Wilkins P, Pimentel G. Comparison of two multiple-locus variable-number tandem-repeat analysis methods for molecular strain typing of human Brucella melitensis isolates from the middle east. J Clin Microbiol. 2009;47:2226–2231. doi: 10.1128/JCM.02362-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chirhart-Gilleland RL, Kovach ME, Elzer PH, Jennings SR, Roop RM. Identification and characterization of a 14-kilodalton Brucella abortus protein reactive with antibodies from naturally and experimentally infected hosts and T lymphocytes from experimentally infected BALB/c mice. Infect Immun. 1998;66:4000–4003. doi: 10.1128/iai.66.8.4000-4003.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crasta O, Folkerts O, Fei Z, Mane S, Evans C, Martino-Catt S, Bricker B, Yu G, Du L, Sobral B. Genome sequence of Brucella abortus vaccine strain S19 compared to virulent strains yields candidate virulence genes. PLoS One. 2008;3:e2193. doi: 10.1371/journal.pone.0002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lowry J, Isaak D, Leonhardt J, Vernati G, Pate J, Andrews G. Vaccination with Brucella abortus recombinant in vivo-induced antigens reduces bacterial load and promotes clearance in a mouse model for infection. PLoS One. 2011;6:e17425. doi: 10.1371/journal.pone.0017425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garrity GM, Bell JA, Lilburn T. Phylum XIV. Proteobacteria phyl. Nov. in: Garrity GM, Brenner D, Krieg N, Staley JA, editors. Bergey’s manual of systematic bacteriology. New York: Springer; 2005.
- 26.Euzéby J. Validation list no. 107. List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol. 2006;56:1–6. doi: 10.1099/ijs.0.64188-0. [DOI] [PubMed] [Google Scholar]
- 27.Garrity GM, Bell JA, Lilburn T. Class I. Alphaproteobacteria class. Nov. in: Garrity GM, Brenner D, Krieg N, Staley JA, editors. Bergey’s manual of systematic bacteriology. New York: Springer; 2005. [Google Scholar]
- 28.Kuykendall LD. Order VI. Rhizobiales ord. nov. In: Garrity GM, Brenner DJ, Kreig NR, Staley JT. Bergey’s Manual of Systematic Bacteriology. 2nd ed. New York: Springer - Verlag; 2005: 324.
- 29.Breed RS, Murray EGD, Smith NR. Family V Brucellaceae, nom. nov. Bergey's Manual of Determinative Bacteriology. 1957;394–423.
- 30.Skerman VBD, McGowan V, Sneath PHA. Approved lists of bacterial names. Int J Syst Bacteriol. 1980;30:225–420. doi: 10.1099/00207713-30-1-225. [DOI] [PubMed] [Google Scholar]
- 31.Meyer KF, Shaw EBA. Comparison of the morphologic, cultural and biochemical characteristics of B. Abortus and B. Melitensis from cattle. Studies on the genus Brucella nov. gen. Int J Infect Dis. 1920;27:173–184. doi: 10.1093/infdis/27.3.173. [DOI] [Google Scholar]
- 32.López-Merino A, Monnet DL, Hernández I, Sánchez NL, Boeufgras JM, Sandoval H, Freney J. Identification of Brucella abortus, B. canis, B. melitensis, and B. suis by carbon substrate assimilation tests. Vet Microbiol. 2001;80:359. doi: 10.1016/S0378-1135(01)00312-1. [DOI] [PubMed] [Google Scholar]
- 33.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 34.Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- 35.Tamura K, Nei M, Kumar S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci U S A. 2004;101:11030–11035. doi: 10.1073/pnas.0404206101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol Biol Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genes that differentiate Brucella abortus strain Ba Col-B012 from the type strain. (DOCX 56 kb)
Alignment of polymorphic genes, sets 0–41. (ZIP 58 kb)