

Abstract
Melanomas are metabolically heterogeneous, and they are able to adapt in order to utilize a variety of fuels that facilitate tumor progression and metastasis. The significance of metabolism in melanoma is supported by growing evidence of impact on the efficacy of contemporary therapies for this disease. There is also data to support that the metabolic phenotypes of melanoma cells depend upon contributions from both intrinsic oncogenic pathways and extrinsic factors in the tumor microenvironment. This review summarizes current understanding of the metabolic processes that promote cutaneous melanoma tumorigenesis and progression, the regulation of cancer cell metabolism by the tumor microenvironment, and the impact of metabolic pathways on targeted and immune therapies.
Keywords: melanoma, metabolism, oxidative phosphorylation, glycolysis, tumor microenvironment
INTRODUCTION
Cutaneous melanoma is the most aggressive form of skin cancer, accounting for >80% of skin-cancer related deaths (Siegel et al., 2017). Characteristically resistant to conventional chemotherapy and radiation, significant advances in metastatic melanoma treatments and outcomes have been made in the last several years due to improved understanding of the features and drivers of this disease. The Melanoma Cancer Genome Atlas (TCGA) effort and other studies have shown that the mitogen-activated protein kinase (MAPK) pathway is ubiquitously activated in cutaneous melanoma as a result somatic mutations in multiple genes, including point mutations in the v-Raf murine sarcoma viral oncogene homolog (BRAF) (35%–50% of melanomas) and Neuroblastoma RAS viral oncogene homolog (NRAS) (10%–25%), and loss of function mutations affecting Neurofibromin 1 (NF1) (~15%). Efforts to target this pathway therapeutically culminated in the Food and Drug Administration (FDA) approval of the BRAF inhibitors vemurafenib and dabrafenib; the mitogen-activated protein kinase (MEK) inhibitor trametinib; and the combination regimens of dabrafenib + trametinib and vemurafenib + cobimetinib (Luke et al., 2017). All of these agents were approved specifically for melanoma patients with mutations that cause substitution of the V600 residue of the BRAF protein (Luke et al., 2017). While the BRAF inhibitors, alone and in combination with MEK inhibitors, reduce tumor burden in almost all patients with a BRAFV600 mutation, most patients will eventually develop resistance (Gopal et al., 2014). In addition to these targeted therapies, several immune therapies have also been approved for patients with metastatic melanoma in recent years, including the checkpoint inhibitors ipilimumab, nivolumab, and pembrolizumab, and the combination of ipilimumab with nivolumab (Luke et al., 2017). Clinical responses and benefit with these immunotherapies can last for several years, and may even be curative. However, the response rates with these immune therapies are lower than for the targeted therapies (i.e. 10–15% for ipilimumab; 35–45% for nivolumab and pembrolizumab; ~55% for ipilimumab + nivolumab) (Luke et al., 2017). Thus, while there has been tremendous progress, there remains a need for additional effective therapeutic strategies for this disease.
There is a growing appreciation and data about the frequency and significance of metabolic reprogramming in cancer initiation, maintenance, and progression (Hanahan and Weinberg, 2011). However, tumor metabolism is complex, and metabolic phenotypes may reflect both intrinsic properties of tumor cells and interactions between tumor cells and the tumor microenvironment (TME). Efforts to improve our understanding of this hallmark of cancer have produced encouraging results that deserve further investigation. Thus, this review will describe several of the central metabolic pathways that have been implicated in the pathogenesis of cutaneous melanoma. We will review the regulation of metabolic pathways by both oncogenic signaling in tumor cells and by the TME, and the therapeutic relevance and implications of the metabolic reprogramming of melanoma cells. Together this information demonstrates that metabolic reprogramming contributes to the pathogenesis and heterogeneity of melanoma, and that further study of cellular metabolism should be embraced in order to improve our understanding of, and treatments for, this highly aggressive disease.
AN OVERVIEW OF CENTRAL CARBON METABOLIC PATHWAYS
Central carbon metabolism consists of glycolysis, the pentose phosphate pathway (PPP), and tricarboxylic acid (TCA) cycle (Pavlova and Thompson, 2016; Sudarsan et al., 2014) (Figure 1). In normoxic, nonmalignant cells, a molecule of glucose undergoes the process of glycolysis in the cytosol to produce pyruvate, adenosine triphosphate (ATP), and reduced nicotinamide adenine dinucleotide (NADH) (Lehninger et al., 2013). Decarboxylation of pyruvate by mitochondrial pyruvate dehydrogenase (PDH) initiates the highly efficient process of mitochondrial oxidative phosphorylation (OXPHOS) by feeding acetyl coenzyme A (acetyl-CoA) into the TCA cycle (Lodish, 2000). NADH and reduced flavin adenine dinucleotide (FADH2) molecules transfer electrons to the electron transport chain (ETC), which responds by transporting protons across the mitochondrial membrane (Lodish, 2000). The resulting electrochemical gradient then drives ATP synthesis, with oxygen serving as the final electron acceptor, producing water as a byproduct (Lodish, 2000) (Figure 1). As oxygen is necessary for the coupling of glycolysis and OXPHOS to occur, hypoxic cells rely more on glycolysis to meet their energy needs (Chatterjea and Shinde, 2012). In a process known as lactic acid fermentation, lactate dehydrogenase (LDH) transfers electrons from NADH to pyruvate, producing lactate and replenishing the supply of NAD+ in the cytosol to allow further glycolysis to continue (Chatterjea and Shinde, 2012). Melanoma cells take advantage of these available metabolic pathways, and others, by using them to meet their needs in a wide variety of contexts.
Figure 1. Signaling pathways promote melanoma progression by regulating critical metabolic reactions.

Central carbon metabolism and one-carbon metabolism play critical roles in melanoma biology. Glycolysis produces ATP and intermediates that fuel the pentose-phosphate pathway (PPP) and serine-glycine/one-carbon metabolism pathways, which produce NADPH vital for maintaining the redox balance of the cell and for anabolic reactions. The TCA cycle uses acetyl-CoA derived from pyruvate or β-oxidation of fatty acids to produce NADH, which powers the ATP-generating electron-transport chain (ETC). The TCA cycle can also generate NADPH via isocitrate dehydrogenase 2 (IDH2). Like other tumor cells, the TCA cycle of melanoma cells can alternatively oxidize glutamine, which enters the TCA cycle as α-ketoglutarate (α-KG). In addition to its bioenergetic function, the TCA cycle produces critical biosynthetic precursors. Oxaloacetate (OAA) fuels reactions that generate NADPH via malic enzyme (ME) and also promotes nucleotide synthesis by supplying the cell with aspartate. Citrate provides the cell with acetyl-CoA necessary for fatty acid synthesis and acetylation reactions. Citrate also promotes NADPH production via isocitrate dehydrogenase 1 (IDH1). Signaling pathways play a critical role in determining which metabolic pathways are favored by melanoma cells. The MAPK and PI3K pathways promote glycolysis and the decoupling of glycolysis from the TCA cycle via HIF1α and MYC. However, through mechanisms currently unknown, a subset of melanomas highly expresses PGC1α, which promotes OXPHOS and makes the cells highly dependent on the oxidation of glutamine. GLUT1, glucose transporter 1; HK2, hexokinase 2; G6PD, glucose-6-phosphate dehydrogenase; PHGDH, phosphoglycerate dehydrogenase; ENO1, enolase 1; LDH, lactate dehydrogenase; SHMT1, serine hydroxymethyltransferase 1; MTHFD1, methylenetetrahydrofolate dehydrogenase 1; THF, tetrahydrofolate; DHF, dihydrofolate; MDH, malate dehydrogenase; FASN, fatty acid synthase; ACLY, ATP citrate lyase; PDH, pyruvate dehydrogenase; GLS, glutaminase; ALT, alanine aminotransferase; GDH, glutamate dehydrogenase. Red arrows indicate NADPH-producing reactions, blue bubbles indicate metabolites, orange bubbles indicate gene products known to regulate metabolic processes in melanomas, and green boxes indicate metabolic pathways.
UTILIZATION AND REGULATION OF METABOLIC PATHWAYS IN MELANOMA PROLIFERATION, INVASION, AND METASTASIS
Glycolysis – An Essential Source of ATP and Molecular Precursors
Like many other cancer types, the majority of rapidly proliferating melanoma cells metabolize glucose into lactate regardless of oxygen levels, a process known as aerobic glycolysis, or the “Warburg effect,” in honor of Otto Warburg, who first described the propensity of tumor cells to utilize glycolysis (Scott et al., 2011). When glycolysis is utilized at high rates in proliferating tumor cells, it can supply the ATP necessary for survival while also supplying materials required for proliferation. (Jose et al., 2011). Scott et al. found that in normoxic conditions, only 25% of pyruvate enters the mitochondria of melanoma cells exhibiting the Warburg phenotype, where it is converted to acetyl-CoA by PDH (Scott et al., 2011). Instead of fully circulating in the TCA cycle, the citrate is transported to the cytosol, where ATP citrate lyase (ACL) cleaves it into acetyl-CoA and oxaloacetate. Acetyl-CoA either enters the fatty acid synthesis pathway, which is critical for the proliferation of melanoma cells, or fuels acetylation of histones in epigenetic regulation reactions while oxaloacetate is converted to malate, which produces pyruvate via malic enzyme and subsequently lactate via LDH (DeBerardinis and Chandel, 2016; Ratnikov et al., 2017). The malic enzyme serves as a source of reduced nicotinamide adenine dinucleotide phosphate (NADPH), necessary for detoxifying melanoma cells of reactive oxygen species (ROS), and LDH mitigates the surplus of NADH present from high glycolytic rates. Consistent with the role of glycolysis as a source of citrate, treatment of glycolytic B16–F10 melanoma cells with the PDH cofactor α-lipoic acid and the ACL competitive inhibitor hydroxycitrate results in significant growth inhibition (Porporato et al., 2014; Schwartz et al., 2010).
Glycolysis also supports the PPP and serine/glycine synthetic pathway in melanoma cells. Glucose-6-phosphate dehydrogenase (G6PD) oxidizes glucose-6-phosphate (G6P), a glycolytic intermediate, in the rate-limiting step of the PPP (Lehninger et al., 2013). After numerous additional reactions, the PPP produces a net gain of the reducing equivalent NADPH and 5-carbon sugars used in nucleotide synthesis. G6PD is highly expressed in melanomas, and inhibition of G6PD in A375 cells results in G1/S phase cell cycle arrest due to ROS-mediated inhibition of signal transducer and activator of transcription 3 (STAT3) (Cai et al., 2015). Phosphoglycerate dehydrogenase (PHGDH) catalyzes the initial step of the serine/glycine synthesis pathway, oxidizing the glycolytic intermediate 3-phosphoglycerate (3-PG). Serine plays a critical role in the biosynthesis of purines and pyrimidines, and it is the precursor of glycine. A bioinformatics analysis of 3131 human cancer samples found recurring copy number gains in PHGDH at a frequency of 40% in melanomas, and melanomas with PHGDH copy number gains were particularly sensitive to PHGDH inhibition (Beroukhim et al., 2010; Locasale et al., 2011).
The utilization of the Warburg phenotype by melanomas is driven in part by activation of intrinsic signaling pathways, particularly the MAPK pathway (Figure 1 and Table 1). Activation of the MAPK pathway increases the transcription of the hypoxia inducible factor 1α (HIF1α) and v-MYC avian myelocytomatosis viral oncogene homolog (MYC) (Kumar et al., 2007; Parmenter et al., 2014). Additionally, the MAPK pathway stabilizes HIF1α, allowing it to partner with HIF1β to become active and promote glycolysis via transcription of LDH, aldolase, and enolase 1 (ENO1) (Semenza et al., 1996). Furthermore, HIF1α activates pyruvate dehydrogenase kinase (PDK), which inhibits PDH, preventing pyruvate from entering the TCA cycle for use in OXPHOS (Kim et al., 2006). Besides HIF1α, MYC also promotes increased glycolytic activity, either as a downstream effector of a constitutively active MAPK pathway or due to copy number gains, which Kraehn et al. identified in 4/8 primary melanomas and 11/33 metastatic melanomas (Kraehn et al., 2001). The MYC transcription factor activates LDH, glucose transporter 1 (GLUT1), and hexokinase 2 (HK2), thereby promoting glucose uptake and glycolytic activity (Stine et al., 2015; Zeller et al., 2003). The MAPK pathway also promotes the Warburg phenotype through its inhibition of the microphthalmia-associated transcription factor (MITF), a critical regulator and promoter of OXPHOS in tumor cells (Haq et al., 2013).
Table 1.
Impact of Oncogenic Signaling Pathways on Melanoma Metabolism
| Signaling Pathway | Alterations in Melanoma | Metabolic Effects |
|---|---|---|
| MAPK |
|
|
| PI3K-AKT |
|
|
| mTOR |
|
|
| HIF1α |
|
|
| MYC |
|
|
| MITF |
|
|
| LKB1-AMPK |
|
|
The phosphoinositide 3-kinase/protein kinase B/mechanistic target of rapamycin (PI3K/AKT/mTOR) signaling pathway is activated multiple ways in melanoma, including by loss of function of the PTEN tumor suppressor, activating mutations in AKT and PIK3CA, and compensatory signaling through growth factor receptors (Davies et al., 2008; Guldberg et al., 1997; Kwong and Davies, 2013; Omholt et al., 2006; Tsao et al., 1998). Activation of this pathway promotes glycolytic metabolism in melanoma (Figure 1 and Table 1). Phosphorylation and activation of AKT drives mTOR signaling, which can promote HIF1α transcription and activity, thereby driving the synthesis of glycolytic machinery (Hudson et al., 2002; Land and Tee, 2007). Additionally, Zundel et al. demonstrated that restoring wild-type PTEN expression in glioblastoma cells lacking functional PTEN corrected the deregulation of cellular AKT and suppressed the transcription of numerous HIF1α-regulated genes, highlighting the interplay between the PI3K/AKT/mTOR pathway and HIF1α signaling (Zundel et al., 2000).
Lactate/H+ Metabolism - A Major Contributor to Melanoma Metastasis
Produced at the end of glycolysis in order to maintain NAD+, lactate is subsequently secreted into the TME via monocarboxylate transporters (MCTs), with MCT4 as the most significant transporter (Dimmer et al., 2000). Pinheiro et al. demonstrated that GLUT1 and MCT4 overexpression significantly correlated with progression from primary tumor to lymph node metastasis in a cohort of patient-derived melanoma samples, indicating that the Warburg phenotype and lactate secretion cooperate to promote melanoma metastasis (Pinheiro et al., 2016). In glycolytic tumor cells, HIF1α and MYC upregulate MCT4 to promote the secretion of lactate into the TME (Payen et al., 2016; Ullah et al., 2006). Lactate drastically alters the TME, facilitating angiogenesis, promoting metastasis, and suppressing the immune system (Romero-Garcia et al., 2016). Once secreted, lactate is taken up by surrounding endothelial cells via MCT1, where it facilitates nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB), HIF1α, and interleukin-8 (IL-8) signaling, resulting in upregulation of vascular endothelial growth factor receptor 2 (VEGFR2) and basic fibroblast growth factor (bFGF) signaling (Payen et al., 2016; Sonveaux et al., 2012; Vegran et al., 2011) (Figure 2). Intracellular protons produced via dissociation of lactic acid are secreted into the melanoma TME via sodium-proton exchanger 1 (NHE1), lowering the pH of extracellular space (Payen et al., 2016; Stuwe et al., 2007; Vahle et al., 2014). Additional membrane protein complexes, including carbonic anhydrase IX (CAIX), sodium-bicarbonate transporter 1, and anion exchanger 2 (AE2) also contribute to extracellular acidosis (Alper et al., 2002; Benej et al., 2014; Donowitz et al., 2013; Payen et al., 2016; Pinheiro et al., 2016) (Figure 2).
Figure 2. Melanoma cell metabolism promotes disease progression by shaping the tumor microenvironment.

The secretion of lactate into the tumor microenvironment (TME) by monocarboxylate transporter 4 (MCT4) results in immunosuppression and angiogenesis, facilitating tumor progression. Lactate inhibits CD8+ T cell proliferation and function, prevents the maturation of dendritic cells, and converts M1 macrophages to M2 macrophages. It also facilitates angiogenesis by increasing IL-8 and VEGF signaling in endothelial cells. Tryptophan metabolism further promotes immunosuppression via depletion of tryptophan and secretion of kynurenine into the TME. Acidification of the TME via proton transport activates cathepsins and matrix metalloproteases (MMPs), which promote migration and metastasis through degradation of the extracellular matrix. IFN-γ, interferon-gamma; IFN-γ R, interferon-gamma receptor; GLUT1, glucose transporter 1; NHE-1, sodium-hydrogen antiporter 1; SLC5A1, solute carrier family 5 member 1; NBC, sodium-coupled bicarbonate transporter; CA-IX, carbonic anhydrase 9; IDO, indoleamine 2,3-dioxygenase; OAA, oxaloacetate; α-KG, α-ketoglutarate.
Acidification of the TME facilitates melanoma metastasis in several ways. First, a low extracellular pH promotes the formation of collagen–integrin interactions in the lamellipodia of melanoma cells, allowing focal adhesion complexes to form and increasing migration of melanoma cells (Payen et al., 2016; Stock et al., 2005). In addition, extracellular acidosis disrupts cellular junctions through the induction of matrix metalloproteases (MMPs) 2 and 9 and cathepsins B and L, which are secreted into the TME as inactive zymogens and require a low pH to be activated (Rofstad et al., 2006) (Figure 2). Rofstad et al. demonstrated that melanoma cells grown in an acidic pH are significantly more metastatic than those grown in normal pH and that treatment with MMP and cathepsin inhibitors prevented the metastasis of the acid-treated cells following tail vein injection in immunodeficient mice (Rofstad et al., 2006). As further evidence, inhibition of NHE1 in vitro prevented the invasive behaviors of melanoma cells to a significantly greater extent than vehicle controls (Stuwe et al., 2007; Vahle et al., 2014). Additionally, inhibitors of AE2 and CAIX block invasiveness of a variety of other tumor cell types (Klein et al., 2000; Lagana et al., 2000; Payen et al., 2016; Svastova et al., 2012). The direct effects of lactate metabolism on the immune system will be covered more extensively later in this review.
Oxidative Phosphorylation (OXPHOS) – A Driver of Growth and Progression in Subsets of Human Melanomas
While many melanomas are characterized by the Warburg phenotype, OXPHOS also plays a critical role in melanoma. OXPHOS is significantly more efficient at generating ATP than glycolysis. While only 7% of pyruvate accesses the TCA cycle in hypoxic cells, OXPHOS still contributes a significant portion of ATP to these cells as large quantities of ATP are produced from a few molecules of pyruvate (Scott et al., 2011). Furthermore, a subset of melanomas rely extensively on OXPHOS to meet their bioenergetic needs. Our lab and others have identified that between 35–50% of BRAF-mutant and wild-type cell lines and patient samples can be characterized as “High-OXPHOS” (Gopal et al., 2014; Haq et al., 2013; Zhang et al., 2016). This phenotype is predominantly driven by peroxisome proliferator-activated receptor γ 1-α (PPARGC1A, named hereafter PGC1α), a transcriptional co-factor that regulates multiple mitochondrial genes. Examination of PGC1α gene expression in the Broad Cancer Cell Line Encyclopedia (CCLE) database revealed that melanomas have the highest expression of PGC1α among >900 cell lines representing 23 cancer types (Gopal et al., 2014). Melanomas highly expressing PGC1α also express numerous nuclear respiratory factors, mitochondrial transcriptional factors, mitochondrial DNA replication factors, mitochondrial fission mediators, and mitochondrial fusion mediators (Zhang et al., 2016). Elevated PGC1α significantly correlates with decreased overall survival in patients with stage III melanoma (Vazquez et al., 2013). As detailed later in this review, basal and compensatory upregulation of PGC1α expression also correlates with de novo and acquired resistance to MAPK pathway inhibitors (Gopal et al., 2014; Zhang et al., 2016). Also, PGC1α-high melanoma cells tolerate oxidative stress to a significantly greater extent than PGC1α-low cells (Vazquez et al., 2013). As tumor cells experience a great deal of oxidative stress, particularly during metastasis, decreased sensitivity to ROS is beneficial for melanoma cells. Consistent with this notion, shRNA knockdown of PGC1α in PGC1α-high melanoma cells inhibited expression of numerous ROS-scavenging genes and sensitized these cells to ROS (Vazquez et al., 2013). Furthermore, LeBleu et al. demonstrated that inhibition of PGC1α-mediated OXPHOS prevented metastatic spread of B16–F10 melanoma cells in a murine model of melanoma metastasis (LeBleu et al., 2014).
PGC1α expression is regulated by MITF in melanoma cells (Haq et al., 2013). HIF1α prevents the transcription of MITF in glycolytic melanomas through deleted in esophageal cancer 1 (DEC1) (Abildgaard and Guldberg, 2015; Feige et al., 2011). In High-OXPHOS melanomas, however, this regulation fails to occur (Figure 1). Instead, mTOR promotes the nuclear translocation of MITF, which drives the transcription of PGC1α and, subsequently, OXPHOS genes (Table 1 and Figure 3) (Gopal et al., 2014; McQuade and Vashisht Gopal, 2015). mTOR is the core component of two distinct complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) (Shimobayashi and Hall, 2014). The PI3K-AKT pathway activates both complexes by suppressing inhibitory inputs from tuberous sclerosis complexes 1/2 (TSC1/2) (Shimobayashi and Hall, 2014). mTORC1 also becomes active in the presence of amino acids. Once active, mTORC1 phosphorylates ribosomal protein S6 kinase (p70S6K) and eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1), promoting protein synthesis, including the translation of MYC and HIF1α (Shimobayashi and Hall, 2014). Active mTORC2 primarily functions to phosphorylate AKT at serine 473, resulting in further activation of AKT (Shimobayashi and Hall, 2014). These interactions have been shown to promote protein synthesis, lipid synthesis, nucleotide synthesis, and glycolysis (Shimobayashi and Hall, 2014). mTOR is also known to increase mitochondrial activity and OXPHOS via activation of YYI-PGC1α transcriptional complex (Cunningham et al, 2007). How mTORC1/2 signaling selects between promoting glycolysis or OXPHOS remains unclear.
Figure 3. mTOR regulates the subcellular localization of MITF.
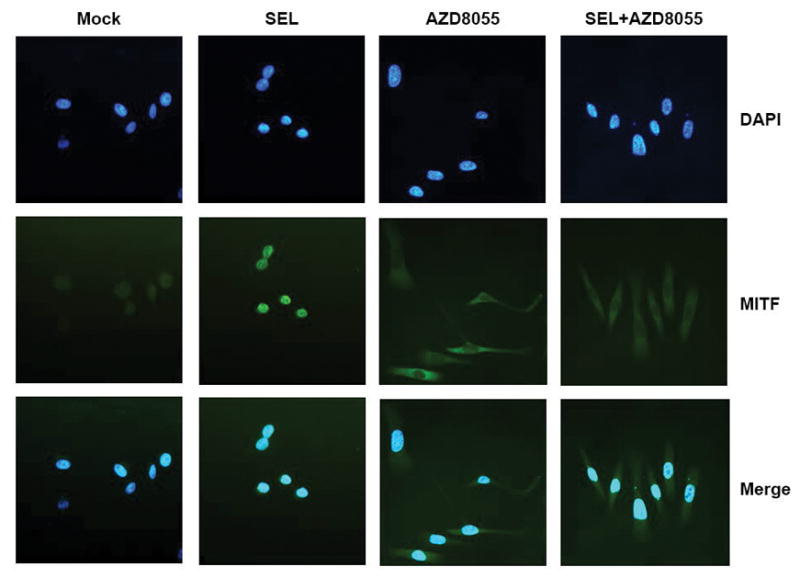
In our prior studies, High-OXPHOS MEL624 cells underwent 24-hour treatments with DMSO (Mock), the MEKi selumetinib (SEL), the mTORC1/2 inhibitor AZD8055, and selumetinib + AZD8055 (SEL + AZD8055). To identify the subcellular localization of MITF following each treatment, cells were fixed and probed with MITF antibody and stained with a FITC-labeled secondary antibody. Nuclei were stained with DAPI. Images were evaluated via immunofluorescent microscopy. AZD8055 caused a significant increase in cytoplasmic MITF staining and significant decrease in nuclear MITF staining, compared to mock treatments. In contrast, nuclear staining of MITF increased in MEL624 cells treated with selumetinib, relative to mock treatments. Gene expression profiling experiments demonstrated that 24-hour selumetinib treatments markedly increased MITF and PGC1α transcript levels in MEL624 cells, while AZD8055 inhibited basal and selumetinib-induced PGC1α expression. Image reused with permission (Gopal et al., 2014). An illustration of the mTOR-MITF-PGC1α signaling axis can also be viewed in one of our prior reviews (McQuade and Vashisht Gopal, 2015).
AMP-activated protein kinase (AMPK) expression also positively correlates with PGC1α expression (Shackelford and Shaw, 2009). In low cellular energy states, designated by a high ratio of AMP/ATP, liver kinase B1 (LKB1) activates AMPK, which inhibits anabolic reactions by suppressing mTORC1 and promotes mitochondrial gene expression, likely through PGC1α (Canto et al., 2009; Shackelford and Shaw, 2009). While the mechanism by which AMPK acts through PGC1α remains unclear, one possibility is that AMPK directly drives PGC1α expression (Canto et al., 2009). Alternatively, AMPK could promote PGC1α activation by either direct phosphorylation or indirectly through sirtuin-1 (SIRT1)-mediated deacetylation (Canto et al., 2009; Jager et al., 2007). In glycolytic tumors, phosphorylated ERK (pERK) suppresses the activation of LKB1, inhibiting the typical response to energy deficiency (Zheng et al., 2009). These cells are less responsive to high AMP/ATP ratios and less dependent on OXPHOS to meet their bioenergetic needs. It is likely that suppression of LKB1 also inhibits PGC1α expression, though this has not been tested.
TCA Cycle Metabolism – A Supplier of Cellular Building Blocks
While mitochondria serve as the primary bioenergetic source for a subset of melanomas, functional mitochondria are critical for all melanoma cells by providing intermediates utilized by biosynthetic and redox reactions (Wallace, 2012). In addition to producing the fatty acid precursor citrate via condensation with acetyl-CoA, oxaloacetate produced from the TCA cycle is converted to aspartate via transamination reactions involving glutamate. This aspartate cooperates with malate, another TCA intermediate, to power the malate-aspartate shuttle, which moves NADH from glycolysis to the mitochondria (Lehninger et al., 2013). Furthermore, aspartate transported into the cytosol by the shuttle contributes to the synthesis of nucleic acids (DeBerardinis and Chandel, 2016) (Figure 1). Rabinovich et al. demonstrated the role of cytoplasmic aspartate in promoting melanoma progression by suppressing arginosuccinate synthetase 1 (ASS1), which utilizes aspartate to produce arginosuccinate in the urea cycle. As a result, cytoplasmic levels of aspartate increased, activating carbamoyl phosphate synthetase II (CAD), the rate-limiting enzyme in pyrimidine synthesis, thereby facilitating the synthesis of nucleotides and promoting melanoma cell proliferation (Rabinovich et al., 2015).
To continue utilizing citrate and oxaloacetate as biosynthetic precursors, melanoma cells must replenish TCA intermediates in a process called anaplerosis (Owen et al., 2002). While some cancers replenish oxaloacetate from pyruvate via pyruvate carboxylase, the contribution of pyruvate carboxylase to the TCA cycle is minimal in cultured melanoma cells characterized by the Warburg phenotype (Phannasil et al., 2015; Scott et al., 2011; Sellers et al., 2015). Instead, these cells utilize glutamine as the primary source of carbon entering the TCA cycle (Scott et al., 2011). Upon transport into the cell via the glutamine receptor SLC1A5, glutamine is converted to glutamate via cytosolic glutaminase (Ratnikov et al., 2017). Glutamate enters the TCA cycle as α-ketoglutarate via reactions catalyzed by either glutamate dehydrogenase 1 (GDH) or mitochondrial alanine and aspartate aminotransferase (GOT2 and GPT2, respectively) (Ratnikov et al., 2015). Reactions catalyzed by GDH generate free ammonia, which can promote autophagy, while the aminotransferases transfer nitrogen to α-keto acids to form α-ketoglutarate and amino acids. Ratkinov et al. labeled Lu1205 melanoma cells with 13C-glutamine to demonstrate that GDH and aminotransferases interchangeably catalyze this anaplerotic reaction and can maintain anaplerosis upon suppression of the alternative route (Ratnikov et al., 2015). However, in vitro studies of SF188 glioblastoma cells demonstrated induction of GDH upon deprivation of glucose and that GDH mediated survival in glucose-deprived conditions (Yang et al., 2009). In glioblastoma cells cultured in glucose-rich conditions, transamination reactions play a greater role in converting the glutamate to α-ketoglutarate (Hensley et al., 2013; Yang et al., 2009). Additional studies are required to further determine the contexts in which GDH and aminotransferase-catalyzed reactions predominate in melanoma cells and whether or not these findings can be verified in vivo.
Like other cancers, melanoma cells can reverse the flow of the TCA cycle via reductive carboxylation of α-ketoglutarate. This process increases the mitochondrial supply of citrate for use in fatty acid synthesis. For hypoxic melanoma cells, reductive carboxylation of glutamine-derived α-ketoglutarate provides over one-third of citrate necessary for fatty acid synthesis (Scott et al., 2011). In hypoxic WM35 and LU1205 melanoma cells, cytosolic isocitrate dehydrogenase 1 (IDH1) and mitochondrial isocitrate 2 (IDH2), 2 of 3 isoforms of IDH, are necessary and sufficient for the production of reductive carboxylation in hypoxic conditions (Filipp et al., 2012b) (Figure 1). Likewise, Mullen et al. demonstrated that IDH1 and IDH2 mediate reductive carboxylation in 143B human osteosarcoma cells with mitochondrial defects (Mullen et al., 2011). In contrast, Metallo et al. utilized cells from multiple cancer types to demonstrate that IDH1 predominates in hypoxic conditions (Metallo et al., 2011). Furthermore, IDH1 also predominates in monolayer detachment of multiple lung adenocarcinoma cell lines (Jiang et al., 2016). With melanoma-specific findings limited to work performed on only two cell lines, additional work remains to fully understand the relative contributions of IDH1 and IDH2 to reductive carboxylation in melanoma and if these findings can be replicated in in vivo experiments.
A great deal of attention has been placed on IDH1 and IDH2 in recent years as heterozygous mutations have been identified at R132 in the cytosolic IDH1 and at R172 in mitochondrial IDH2 in glioblastoma and acute myeloid leukemia (Abbas et al., 2010; Filipp et al., 2012a; Mardis et al., 2009; Parsons et al., 2008). These mutated enzymes produce 2-hydroxyglutarate (2-HG) from alpha-ketoglutarate. 2-HG acts as an oncometabolite by inducing a variety of epigenetic changes in the tumor cells (Filipp et al., 2012a; Rakheja et al., 2013). Mutations in IDH1/2 are rare in melanoma and typically affect IDH1 (Filipp et al., 2012a; Lopez et al., 2010). These mutations have shown to provide a growth advantage to melanoma cell lines with concurrent BRAF mutations (Shibata et al., 2011). While succinate dehydrogenase and fumarate hydratase mutations have also been identified in a variety of cancers, they have not been identified in melanomas (DeBerardinis and Chandel, 2016).
Protein/Amino Acid Metabolism – Arginine, Proline, and Proteasomal Degradation in Human Melanomas
In addition to glutamine and aspartate, a considerable amount of amino acid research in melanoma has focused on arginine and proline. Interestingly, melanomas cannot synthesize their own arginine, as they lack expression of arginine synthetase (Dillon et al., 2004) Thus, they are solely dependent on external sources of arginine in the TME for protein synthesis (Delage et al., 2010). While efforts have been made to starve melanoma cells by depleting them of arginine, this strategy has been complicated by resistance mechanisms in which arginine synthetase is stimulated following arginine depletion (Manca et al., 2011).
The limited supply of arginine directly impacts the synthesis of proline in melanoma cells, as it makes the cells largely dependent on glutamine for the synthesis of proline as opposed to using both arginine and glutamine to synthesize proline (Filipp et al., 2012a). Melanoma cells synthesize proline to a significantly greater extent than melanocytes (De Ingeniis et al., 2012; Scott et al., 2011) Three isoforms of pyrroline-5-carboxylate reductase (PYCR) catalyze the rate-limiting reaction of proline synthesis (De Ingeniis et al., 2012). PYCR1 and 2 are localized to the mitochondria and utilize glutamate and NADH to produce proline, while PYCRL is localized to the cytoplasm and utilizes NADPH and arginine-derived ornithine to produce proline (De Ingeniis et al., 2012). Only PYCR1 and 2 are overexpressed in melanoma cells compared to melanocytes, suggesting that only glutamate-derived proline synthesis provides melanoma cells with a survival advantage (De Ingeniis et al., 2012). Given NADPH’s critical role in maintaining redox balance in melanoma cells, it has been proposed that cells select against PYCRL expression in order to preserve NADPH levels, though this explanation remains to be explored further (Filipp et al., 2012a; Ratnikov et al., 2017). The potential role of proline in redox balance is further supported by the fact that melanoma cells synthesize proline regardless of the concentration of proline in the surrounding environment, suggesting that the act of synthesizing proline is significantly more important than proline itself (Ratnikov et al., 2017). It is possible that melanoma cells synthesize proline via PYCR1 and PYCR2 to replenish the cellular supply of NAD+ for use in glycolysis while preserving NADPH necessary for anabolic reactions and for managing oxidative stress.
While physiological proteasomal degradation of proteins is essential for the health of normal cells, melanoma cells take advantage of this existing machinery to rid themselves of various tumor suppressor proteins. For example, Healey et al. utilized Tyr/Tet-Ras INK4a−/− transgenic mice, and R545 melanoma cells isolated from their tumors, to demonstrate that Ras-mediated melanomas target the Inducible cAMP early repressor (ICER) for proteasomal degradation (Healey et al., 2013). Without functional ICER protein, the tumor cells upregulated the transcription of cyclin D1, an essential mediator of cell cycle progression (Healey et al., 2013). Likewise, p53 mutations are relatively uncommon (~20%) in melanoma (Albino et al., 1994; Cancer Genome Atlas Network, 2015; Hodis et al., 2012; Volkenandt et al., 1991; Weiss et al., 1993). However, Mouse double minute 2 homolog (MDM2) can still freely suppress the transcriptional activity of p53 and target p53 for proteasomal degradation (Michael and Oren, 2003). Anwar et al. derived 8B20 cells from a genetically engineered mouse model (GEMM) of melanoma to demonstrate that low levels of p53 protein in the cells resulted from increased ubiquitin-mediated proteasomal degradation (Anwar et al., 2011). Their attempts at disrupting this uncontrolled degradation of p53 protein triggered G1 cell cycle arrest in the 8B20 cells (Anwar et al., 2011). Similarly, nutlins are a class of compounds that disrupt the interactions between MDM2 and p53. By preventing the unregulated degradation of p53, nutlin-3 restores p53 levels in human melanoma cell lines, regardless of p53 mutational status (Ji et al., 2012; Worrall et al., 2017). However, nutlin-3 selectively decreases the viability of cells with wild-type p53 (Ji et al., 2012; Worrall et al., 2017). Interestingly, nutlin-3 treatment increases cellular levels of MDM2, resulting in ubiquination and degradation of Insulin-like growth factor type 1 receptor (IGF-1R) (Worrall et al., 2017). The disruption of the IGF-1R signaling axis causes an initial increase in melanoma cell proliferation but ultimately suppresses proliferation and inhibits migration (Worrall et al., 2017). These examples demonstrate the tendency of melanoma cells to hijack common cellular machinery to promote their own survival and how doing so makes them vulnerable to novel classes of therapeutics.
Lipid Metabolism – Synthesis and Degradation of Fatty Acids Promotes Melanoma Progression
Fatty acid synthase (FASN) catalyzes the rate-limiting step of the endogenous synthesis of fatty acids (Menendez and Lupu, 2007). FASN is positively regulated in normal tissue by insulin and hormones such as estrogen and progesterone and negatively regulated by leptin, which in turn is stimulated by free fatty acids (FFAs) (Kersten, 2001; Kim et al., 1998; Menendez and Lupu, 2007). Insulin, estrogen, and progesterone activate the transcription factor sterol regulatory element-binding protein 1c (SREBPC1c), which is responsible for driving FASN transcription (Kersten, 2001; Kim et al., 1998; Menendez and Lupu, 2007). In many tumor types, including melanoma, SREBPC1c is constitutively driven by the MAPK and PI3K/AKT pathways, which causes significant upregulation of FASN (Kersten, 2001; Kim et al., 1998; Menendez and Lupu, 2007) (Figure 1 and Table 1). These signaling pathways promote the maturation of SREBP1c from its precursors and its nuclear localization from the Golgi apparatus following its maturation (Menendez and Lupu, 2007). The increased FASN is believed to provide a survival benefit to tumor cells by promoting a “lipogenic phenotype.” Fatty acids form the structural foundation of cellular and organelle membranes. Proliferating cells require a large supply of fatty acids to form these structures. Constitutively active FASN ensures an adequate concentration of phospholipids necessary for this rapid proliferation (Menendez and Lupu, 2007). Saab et al. demonstrated that patient-derived metastatic melanomas test positive for FASN staining, as opposed to benign intracapsular nodal nevi (Saab et al., 2017). In addition, FASN expression levels correlate with tumor invasion and poor prognosis in cutaneous melanoma (Innocenzi et al., 2003; Zecchin et al., 2011). Targeting FASN prevents the proliferation of B16–F10 melanoma cells in vitro and activates the intrinsic apoptotic pathway in melanoma cells (Zecchin et al., 2011). Furthermore, inhibiting the thioesterase domain of FASN with orlistat, a drug originally approved by the FDA for the treatment of obesity, prevented the metastasis of B16–F10 mouse melanoma cells following implantation in the peritoneal cavity of C57BL/6 mice (Carvalho et al., 2008).
Additionally, melanomas overexpress numerous genes involved in synthesis of sialylated glycosphingolipids known as gangliosides. For example, microarray analysis performed by Sumantran et al. determined that UDP-glucose ceramide glucosyltransferase (UGCG), hexosaminidase A (HEXA), ST3 β-galactoside α-2,3-sialyltransferase 5 (ST3GAL5), and ST8 α-N-acetyl-neuramide-α-2,8-sialyltransferaase 1 (ST8SIA1), all of which are involved in ganglioside synthesis, are upregulated in melanomas between 3 and 9-fold, relative to benign nevi (Sumantran et al., 2015). Subclones of M4BE melanoma cells enriched in gangliosides survive radiation treatment significantly better than parental cells (Thomas et al., 1996). Furthermore, ganglioside GD3− SK-MEL-28-N1 melanoma cells became significantly more proliferative and invasive following transfection with GD3 via stimulation of p130Cas and paxillin signaling (Hamamura et al., 2005). siRNA-mediated knockdown of p130Cas and paxillin subsequently inhibited ganglioside GD3 signaling in these cells, though the effects remain to be validated in vivo (Hamamura et al., 2005).
In addition to the synthesis of fatty acids, fatty acid oxidation (FAO) appears to play an important role in promoting melanoma progression. For example, carnitine palmitoyltransferase 2 (CPT2), the enzyme critical for translocation of long-chain fatty acids (LCFAs) in preparation for β-oxidation, is one of the most significantly upregulated genes in melanomas, relative to benign nevi (Sumantran et al., 2015). Furthermore, Rodrigues et al. derived metastatic 4C11+ cells from melan-a melanocytes after sequential detachment-re-adhesion cycles, and FAO contributed significantly to the energy reserves of these 4C11+ cells, relative to nonmetastatic controls (Rodrigues et al., 2016). How FAO promotes melanoma progression remains unclear. Studies in other tumor types have demonstrated that fatty acids can provide an ATP boost for tumor cells when necessary under nutrient-depleted conditions (Carracedo et al., 2012; Carracedo et al., 2013; Zaugg et al., 2011). Indeed, the process of metastasis is a highly demanding process. Highly efficient β-oxidation could provide a survival advantage for cells away from the primary tumor site. Furthermore, fatty acids can serve as a valuable source of acetyl-CoA that contributes to citrate formation after entering the TCA cycle (Carracedo et al., 2013). As detailed earlier, this citrate can enter metabolic reactions that produce NADPH via IDH1, thereby contributing to redox balance in the tumor cells.
Numerous lipid-derived second messengers such as phospholipase D3 (PLD3), inositol triphosphate protein kinase B (ITPKB), and inositol triphosphate receptor 3 (ITPR3) are significantly upregulated in melanomas, relative to benign nevi (Sumantran et al., 2015). Furthermore, ITPKB is significantly upregulated in MALME_3M and UACC_257 melanoma cell lines of the National Cancer Institute (NCI) Cell Miner database (Sumantran et al., 2015). Additionally, fatty acid binding proteins, which are regulators of fatty acid uptake and lipid trafficking, appear to play important roles in melanoma progression. Fatty acid binding protein 3 (FABP3) expression is 3-fold higher in melanomas than benign nevi (Sumantran et al., 2015). Furthermore, Slipicevic et al. demonstrated that fatty acid binding protein 7 (FABP7) is involved in proliferation and invasion of melanoma cells in vitro (Slipicevic et al., 2008). siRNA-mediated suppression of FABP7 inhibited invasion and proliferation of melanoma cells without affecting apoptosis (Slipicevic et al., 2008). However, the relevance of FABP7 in in vivo models of metastasis remains to be validated.
Finally, Nath and Chan identified a crucial role for fatty acid metabolism in melanoma progression while deriving a gene signature to detect activation of epithelial-mesenchymal transition (EMT) programming across multiple cancers (Nath and Chan, 2016). TCGA melanoma patients enriched in the authors’ 5-gene signature, which included the fatty acid uptake genes caveolin-1 (CAV1) and cluster of differentiation 36 (CD36) and the fatty acid oxidation gene carnitine palmitoyltransferase 1C (CPT1C), have significantly worse overall survival than those whose melanomas are not enriched for the signature (Nath and Chan, 2016). Additionally, CD36 amplifications significantly correlate with invasiveness in 501mel melanoma cells (Nath and Chan, 2016; Pascual et al., 2017). In contrast, melanoma cells lacking CD36 fail to metastasize in NOD scid gamma (NSG) mice following tail vein inoculation (Pascual et al., 2017). These studies further implicate lipid metabolism in the progression of human melanoma.
One-Carbon Metabolism – A Critical Deterrent of Oxidative Stress
Melanoma cells are subjected to oxidative stress, with ROS formed secondary to oncogenic mutations, aberrant signaling pathways, unregulated proliferation, and hypoxia (DeBerardinis and Chandel, 2016). While minimal levels of ROS are actually beneficial to these cells by stimulating proliferation and inducing mutations, excessive ROS can induce damage and eventually apoptosis (DeBerardinis and Chandel, 2016). As oxidative stress increases during tumorigenesis progression and metastasis, melanoma cells must adjust and strengthen their antioxidant defenses to survive (Gorrini et al., 2013). In addition to activating nuclear factor (erythroid-derived 2)–related factor-2 (NRF2) to drive the transcription of antioxidant proteins, tumor cells increase their reliance on glutathione peroxidase to manage oxidative stress (Jaramillo and Zhang, 2013). This protein utilizes reduced glutathione (GSH) to convert peroxides to water. Glutathione reductase utilizes NADPH to restore GSH to its oxidized state (GSSG), making NADPH a critical component of redox balance in melanoma cells. Early in tumorigenesis, the PPP supplies the majority of the NADPH used by this enzyme. Together with NRF2-regulated genes, the PPP is sufficient to manage the oxidative stress secondary to intense proliferation. However, the role of one-carbon metabolism, mediated by the folate cofactor, becomes increasingly important as tumorigenesis progresses, glucose becomes scarce, and hypoxia becomes a more significant source of ROS (DeBerardinis and Chandel, 2016). In this pathway, serine is converted to glycine via serine hydroxymethyltransferase (SHMT1) in a reaction that also converts tetrahydrofolate (THF) to 5,10-methylenetetrahydrofolate. Methylenetetrahydrofolate reductase dehydrogenase 1 (MTHFD1) subsequently catalyzes a series of three reactions, the first of which produces NADPH for use in redox reactions (Lehninger et al., 2013) (Figure 1). One-carbon metabolism is critical in the management of deleterious ROS during metastasis. Piskounova et al. demonstrated that melanoma cells that successfully metastasized to distant sites in NSG mice increase their dependence on one-carbon metabolism. Inhibition of one-carbon metabolism prevented the formation of distant metastases without affecting subcutaneous tumor growth (Piskounova et al., 2015).
IMPACT OF THE TUMOR MICROENVIRONMENT ON TUMOR METABOLISM
Metabolic Adaptation to Nutrient-Depleted Conditions
While it is most convenient to classify melanomas as either glycolytic or oxidative, an increasing body of evidence suggests that metabolic phenotypes of tumors are more accurately viewed as dynamic in nature (Jose et al., 2011). While key oncogenic pathways implicated in melanomas (RAS-RAF-MAPK and PI3K-AKT) typically promote glycolysis, this metabolic pathway requires an extensive supply of glucose to fuel reactions. Tumor cells are not equally supplied with oxygen and nutrients due to inadequate vascularization, and frequently they must adapt to hypoxia and to glucose deprivation (Jose et al., 2011). In the presence of adequate glucose, hypoxic melanoma cells almost completely decouple glycolysis from the TCA cycle (Scott et al., 2011). Glutamine fuels the TCA cycle and provides citrate for fatty acid synthesis while glutamine and glucose oxidation contribute over 90% of ATP to the cell (Scott et al., 2011). Alternative pathways such as glutamine and fatty acid oxidation are essential in glucose-depleted conditions. For example, adipocytes in the TME can communicate with melanoma cells via exosomes carrying proteins involved in FAO (Lazar et al., 2016). Upon uptake by melanoma cells, via an uncharacterized mechanism, these exosomes force a metabolic reprogramming in melanoma cells characterized by high levels of FAO and aggressiveness (Lazar et al., 2016). Studies in other cancer types have provided numerous insights into the role of the TME in shaping tumor cell metabolism. For example, the MDA-MB-231Br3 cell line, derived from MDA-MB-231 parental cells after 3 rounds of in vivo selection from brain metastases following injections into the internal carotid artery, adapted to the glucose deprived conditions of the brain TME by becoming dependent on gluconeogenesis and oxidation of glutamine and branched chain amino acids instead of glycolysis (Chen et al., 2015). Similarly, Smolkova et al. demonstrated that 4 days of glucose deprivation completely reprogrammed HTB-126 breast cancer cells, significantly elevating OXPHOS to compensate for aglycemia (Smolkova et al., 2011). Mitochondria are actually able to function at oxygen concentrations as low as 0.5%, allowing this flexibility to occur even under hypoxic conditions (DeBerardinis and Chandel, 2016; Rumsey et al., 1990; Weinberg and Chandel, 2015). The LKB1-AMPK-PGC1α signaling axis stimulates OXPHOS in these scenarios (Smolkova et al., 2011). Redox balance is maintained through one-carbon metabolism, cytoplasmic malic enzyme, and IDH1/2-mediated production of NADPH (DeBerardinis and Chandel, 2016) (Figure 1). Metabolic flexibility allows tumor cells to survive inhibition of a single metabolic pathway and provides a survival advantage necessary for survival in harsh TMEs (Chen et al., 2007; Simoes et al., 2015). Furthermore, this metabolic flexibility has complicated attempts to target single metabolic pathways as a therapeutic strategy. However, targeting glycolysis and OXPHOS via simultaneous treatment with the electron transport chain complex I inhibitor metformin and the LDH inhibitor oxamate has produced promising results, significantly inhibiting B16–F10 tumor growth in C57BL/6 mice (Chaube et al., 2015).
Analysis of the TCGA melanoma dataset identified three broad categories of melanomas: “Immune”, “Keratin”, and “MITF-low” subtypes (Cancer Genome Atlas Network, 2015). The MITF-low melanomas express numerous genes involved in invasion and migration (Cancer Genome Atlas Network, 2015). In vitro analyses have demonstrated that temporary suppression of MITF triggers a slow-cycling, invasive phenotype, while long-term suppression of MITF results in senescence (Cheli et al., 2011; Giuliano et al., 2010). However, MITF-low cells do not experience senescence in vivo (Goodall et al., 2008; Riesenberg et al., 2015). In studying this paradox, Falletta et al. determined that distressed melanomas increase cellular levels of Activating transcription factor 4 (ATF4), which subsequently suppresses MITF and prevents senescence (Falletta et al., 2017). Further experiments determined that glutamine deprivation activated the aforementioned response (Falletta et al., 2017). Specifically, glutamine deprivation resulted in a transient increase in MITF gene expression due to calmodulin-dependent kinase II (CAMKII)-mediated stimulation of cyclic AMP response element-binding protein (CREB) (Falletta et al., 2017). The MAPK pathway subsequently phosphorylated MITF, which promoted ATF4 transcription (Falletta et al., 2017). In parallel, eIF2B suppression triggered a global reprogramming that involved further suppression of MITF and increased expression of ATF4 and was necessary for MITF-low cells to invade (Falletta et al., 2017). Likewise, Ferguson et al. demonstrated that glucose deprivation facilitates expression of ATF4, which suppresses MITF by competing with CREB at the MITF promoter (Ferguson et al., 2017). Together, these findings demonstrate how nutrient supply in the TME induces significant changes in signaling pathways that drive metastasis.
In recent years, the role of autophagy and macropinocytosis in promoting survival of melanoma cells in hostile microenvironments has gained considerable interest. Autophagy is a highly regulated process that involves the degradation of macromolecules and organelles into useful constituent components. In this manner, large proteins can be degraded to provide nitrogen and carbon for bioenergetic maintenance. MEK/ERK signaling in melanoma upregulates phorbol-12-myristate-13-acetate-induced protein 1 (NOXA), which promotes an increase in autophagy through cAMP responsive element binding protein (CREB) (Liu et al., 2014; Ndoye and Weeraratna, 2016). NOXA-driven autophagy delays the apoptosis of human melanoma cells in nutrient-depleted conditions (Liu et al., 2014). Furthermore, autophagy provides a survival advantage to melanoma cells cultured in harsh, acidic conditions (Marino et al., 2012; Ndoye and Weeraratna, 2016). In addition to autophagy, a variety of tumor types utilize macropinocytosis to scavenge extracellular nutrients when grown in harsh conditions (DeBerardinis and Chandel, 2016). Macropinocytosis can supply tumor cells with necessary nitrogen and carbon (Commisso et al., 2013; DeBerardinis and Chandel, 2016). In melanoma, hyperactive PI3K-AKT pathway signaling drives macropinocytosis so efficiently that the cells require activation of RAB7 to clear the vesicles taken into cells before a form of cell death called methuosis occurs (Alonso-Curbelo et al., 2015). In scenarios of widespread nutrient starvation, the ratio of ATP to AMP declines, stimulating the LKB1-AMPK axis to suppress mTOR-mediated anabolic processes and to stimulate autophagy and macropinocytosis (DeBerardinis and Chandel, 2016).
Lactate Shuttling – An Untapped Avenue in Melanoma Metabolism
Directly in contrast with the Warburg effect, the reverse Warburg effect was originally proposed by Pavlides et al. in 2009. In this model, tumor cells and surrounding stromal cells engage in a symbiotic relationship where tumor cells corrupt caveolin-1-deficient cancer associated fibroblasts (CAFs) via secretion of hydrogen peroxide. Mitochondrial activity in the CAFs is inhibited, forcing the cells to depend exclusively on glycolysis. These cells secrete lactate into the TME via MCT4, which tumor cells take up via MCT1 to fuel oxidative metabolism (Gonzalez et al., 2014; Pavlides et al., 2009; Pavlides et al., 2010). While this model originally was described in breast cancer, similar tumor-stroma interactions have been demonstrated in a variety of other cancers, including oral squamous cell carcinoma and osteosarcoma (Jensen et al., 2015; Sotgia et al., 2014). Though this model could offer novel therapeutic approaches via disruption of lactate shuttling, it remains to be investigated in melanoma, as does the concept of metabolic symbiosis. Similar to the reverse Warburg effect, this version of lactate shuttling involves hypoxic glycolytic tumor cells secreting lactate into the TME via MCT4, which oxidative tumor cells uptake via MCT1 and utilize for energy. Interestingly, Ho et al. demonstrated that increased expression of MCT1 and MCT4 correlate with melanoma progression (Ho et al., 2012). Together with the evidence of dependence on OXPHOS in melanoma metastasis, these findings suggests that metabolic symbiosis could play a role in melanoma progression, and a potential therapeutic opportunity. For example, inhibition of MCT1 in a mouse model of lung carcinoma resulted in oxygenated cells switching to glycolysis for ATP production. As a result, hypoxic tumor cells died secondary to glucose starvation (Sonveaux et al., 2008).
IMMUNE EFFECTS OF METABOLIC REPROGRAMMING
As immune cells compose a critical component of the TME, there is an increasing interest in studying the metabolic interactions that occur between melanoma cells and immune cells. To fully understand the complexity of these dynamic interactions, it is necessary to review the typical metabolic statuses of the key immune cells present in the TME.
Neutrophils contain few mitochondria and therefore use glycolysis extensively to meet their bioenergetic demands. Upon activation, these cells consume large quantities of glucose to feed the PPP for production of NADPH, needed to activate NADPH oxidase, which in turn produces hydrogen peroxide, a bactericidal byproduct (Pearce and Pearce, 2013). Unlike neutrophils, dendritic cells are a highly complex group of cells responsible for connecting the innate and adaptive arms of the immune system by responding to pathogen associated molecular patterns (PAMPS) and subsequently controlling the fates of T cells. Naïve dendritic cells rely extensively on OXPHOS to produce ATP. Activation of dendritic cells stimulates the PI3K-AKT pathway and shifts the metabolic dependency to glycolysis (Romero-Garcia et al., 2016). The two categories (M1 and M2) of macrophages differ in their preferred bioenergetic pathways. M1 macrophages, which have antimicrobial and antitumor functions, exhibit a glycolytic phenotype. In contrast, M2 macrophages, which are critical regulators of wound healing with minimal antitumor activity, utilize OXPHOS as their main source of ATP (Pearce and Pearce, 2013). T cells, the most researched component of the adaptive immune system, utilize a spectrum of metabolic phenotypes depending on their activation state and the subclass of T cells to which they belong. Activated CD4+ and CD8+ effector T cells adopt a glycolytic phenotype, upregulating glucose transporters and glycolytic enzymes in the process (Gonzalez et al., 2014). Unlike cells of the innate immune system, T cells produce memory cells, which catabolize fatty acids and utilize OXPHOS to produce ATP (Romero-Garcia et al., 2016). Regulatory CD4+ T cells (Tregs) are highly oxidative cells while regulatory T helper-17 (Th17) cells depend on glycolysis (Pearce and Pearce, 2013). While much work remains to full understand the metabolic dependencies of B lymphocytes, these cells appear to utilize both OXPHOS and glycolysis (Romero-Garcia et al., 2016).
Tumor metabolism can suppress the function of nearby immune cells (Figure 2). As CD8+ T cells are glycolytic, they must compete with highly glycolytic tumor cells for access to glucose (Chang et al., 2015). Without glucose, CD8+ T cell activity is inhibited. Furthermore, high concentrations of lactate in the TME prevent adequate secretion of lactate out of T cells, again inhibiting their function (Fischer et al., 2007; Romero-Garcia et al., 2016). By inducing P38 MAPK and c-Jun N-terminal kinase (JNK)/c-JUN signaling pathways, lactate-based immunosuppression has been demonstrated in numerous cancers, including melanoma (Mendler et al., 2012; Romero-Garcia et al., 2016). Melanoma spheroids inhibit cytokine secretion from T cells by secreting lactate into the TME (Feder-Mengus et al., 2007; Romero-Garcia et al., 2016). However, T cells can recover from this inhibition upon lactate clearance (Fischer et al., 2007). Lactate can also suppress cells of the innate immune system. It converts M1 macrophages to M2 macrophages, promoting tumor progression (Romero-Garcia et al., 2016). In addition, it prevents the maturation of dendritic cells, resulting in an increase in the immunosuppressive IL-10 cytokine in the TME (Romero-Garcia et al., 2016).
Tumor cell metabolism can also profoundly impact the immune system via tryptophan metabolism. Once activated, effector T helper-1 (Th1) cells secrete interferon-γ (IFNγ), which stimulates indoleamine 2,3-dioxygenase (IDO) in tumor cells. In turn, IDO metabolizes tryptophan, producing kynurenine in the process. IDO promotes melanoma tumorigenesis and diminishes survival through two mechanisms (Brody et al., 2009; Sucher et al., 2010). First, IDO starves effector T cells of tryptophan, resulting in general control nonderepressible 2 (GCN2)-mediated suppression of proliferation and promotion of apoptosis (Lee et al., 2002; Munn et al., 2005; Munn and Mellor, 2016; Sucher et al., 2010). While a minimum level of tryptophan for T cell maintenance has not been demonstrated, low serum tryptophan levels correlate with a poor prognosis in melanoma patients (Sucher et al., 2010; Weinlich et al., 2007). In addition, kynurenine binds to the aryl hydrocarbon receptor (AhR), inducing Treg differentiation and polarizing dendritic cells and macrophages to immunosuppressive phenotypes (Mezrich et al., 2010; Munn and Mellor, 2016). Moreno et al. demonstrated that targeting IDO with the competitive inhibitor 1-methyl-tryptophan (1-MT) retards the proliferation of melanoma cells in vitro (Moreno et al., 2013). Furthermore, 1-MT treatment delays the outgrowth of Lewis lung cancer cells in syngeneic mice (Friberg et al., 2002). Though monotherapy with 1-MT has little effect on the growth of subcutaneous B16–F10 tumors, 1-MT sensitizes the tumors to chemotherapy and whole-body radiation (Hou et al., 2007). While promising, additional exploration is required to further define how IDO mediates immunosuppression in melanoma and whether or not 1-MT can be combined with currently approved therapies.
THERAPEUTIC IMPLICATIONS OF MELANOMA METABOLISM
The development of effective immune and targeted therapies has revolutionized the management of patients with metastatic melanoma. However, these treatments have metabolic implications that must be understood in order to identify potential mechanisms of resistance and novel treatment combinations.
Immunotherapy
Metabolic reprogramming of tumor cells has significant implications for immunotherapy. Ipilimumab, an antibody that inhibits signaling by cytotoxic T-lymphocyte-associated protein (CTLA-4) on T cells, was approved by the US FDA in 2011 for the treatment of patients with metastatic melanoma. Three years later, the programmed cell death protein-1 (PD-1) blocking antibodies pembrolizumab and nivolumab were also approved. These three antibodies overcome inhibitory signals encountered by T cells in their attempt to locate and destroy tumor cells. Specifically, the expression of CTLA-4, also known as CD152, increases significantly following the activation of CD4+ and CD8+ T cells (Alegre et al., 2001). This T cell surface molecule subsequently competes with CD28 for interactions with CD80 and CD86 (B7-1 and B7-2, respectively) on the surface of antigen presenting cells, such as dendritic cells and macrophages. The interactions between CTLA-4 and CD80 and CD86 are more favorable than those between CD28 and CD80 and CD86, resulting in CTLA-4 binding with these molecules in place of CD28. In response, inhibitory signals are transmitted to the effector CD8+ T cells, resulting in suppression of the immune response (Krummel and Allison, 1995; Walunas et al., 1996). Like CTLA-4, PD-1 is expressed on the surface of T cells. Its ligand, PD-L1, is expressed on the surface of dendritic cells, M2 macrophages, fibroblasts, and tumor cells (Pardoll, 2012). Activation of PD-1 following binding to PD-L1 inhibits effector CD8+ T cells, suppressing the antitumor response (Lee et al., 2016). Mechanistically, CTLA-4 and PD-1 both inhibit the PI3K-AKT pathway in distinct yet synergistic ways, with CTLA-4 signaling inhibiting AKT through protein phosphatase 2A (PP2A) and PD-1 signaling inhibiting PI3K (Parry et al., 2005). Since this pathway promotes glycolysis, CTLA-4 and PD-1 signaling promote OXPHOS at the expense of glycolysis, and PD-1 signaling augments fatty acid oxidation (Patsoukis et al., 2015). As activated effector T cells utilize glycolysis, these metabolic alterations provide a metabolic explanation for CTLA-4- and PD-1-mediated immunosuppression. Checkpoint inhibitors remove the suppression of the PI3K-AKT in effector T cells, allowing them to become active once again and adopt a glycolytic phenotype. Once this occurs, tumor cell death caused by the checkpoint inhibitors results in the release of glucose into the TME, promoting further activation of effector CD8+ T cells (Chang et al., 2015). It remains to be seen if directly inhibiting glycolysis in tumor cells will improve the efficacy of checkpoint inhibitors.
In addition to central carbon metabolism, tryptophan metabolism also appears to play a critical role in mediating response to immunotherapy. Holmgaard et al. demonstrated that treatment of IDO-knockout mice with anti-CTLA-4 and anti-PD-1 inhibited the growth of B16-F10 melanoma tumors compared to wild-type mice. Furthermore, IDO inhibitors synergized with anti-CTLA-4 treatment, promoting CD8+ T cell recruitment and inhibiting tumor growth to a significantly greater extent than anti-CTLA-4 treatment alone (Holmgaard et al., 2013). When given to 19 treatment-naïve metastatic melanoma patients participating in a phase I clinical trial (NCT02178722), the combination of pembrolizumab and the IDO inhibitor Epacadostat achieved complete responses in 4 patients, partial responses in 7 patients, and stable disease in 3 patients (Gangadhar et al., 2016). Furthermore, the combination demonstrated an acceptable safety profile (Gangadhar et al., 2016). Treatment-naive metastatic melanoma patients are currently being enrolled in phase II and III clinical trials (NCT02752074) designed to evaluate this treatment strategy.
Targeted Therapy
While the BRAF inhibitors dabrafenib and vemurafenib and the MEK inhibitors trametinib and cobimetinib are highly effective in metastatic melanoma patients with activating BRAF mutations, the depth and duration of clinical responses to these agents are variable, and the majority of patients will eventually develop resistance to them (Gopal et al., 2014). As the MAPK pathway promotes aerobic glycolysis in tumor cells (Figure 1), inhibition of the pathway results in significant reduction in glucose utilization and glycolytic flux. This suppression results from modulation of the glycolytic regulators HIF1α and MYC and is necessary to achieve clinical responses to BRAF inhibitors (Parmenter et al., 2014). Studies have shown that glycolytic flux is restored in cells resistant to BRAF inhibition, and that combined inhibition of BRAF and glycolysis could overcome targeted therapy resistance (Parmenter et al., 2014). While those observations were based largely on experiments performed after 24-hour treatments, Haq et al. demonstrated increased PGC1α-driven OXPHOS in response to longer (i.e., ≥ 72 hours) treatments with MAPK inhibitors (Haq et al., 2013). Induction of OXPHOS has been identified in 30–50% of BRAF-mutant melanomas with both de novo and acquired resistance to MAPK pathway inhibitors (Gopal et al., 2014). Enforced expression of PGC1α induces resistance in cells previously sensitive to MAPK inhibitors (Haq et al., 2013), while genetic knockdown of PGC1α results in synergistic growth inhibition and apoptosis induction with MAPK pathway therapies in PGC1α-mediated High-OXPHOS cells (Gopal et al., 2014). In addition, studies by Herlyn and colleagues previously identified a population of slow-cycling, treatment-resistant cells characterized by expression of the histone 3 K4 demethylase JARID1B. They further showed that these slow-cycling cells are characterized by High-OXPHOS. Interestingly, Transcription factor A, mitochondrial (TFAM), not PGC1α, facilitates the High-OXPHOS phenotype in these cells, indicating that some High-OXPHOS melanoma cells adopt this metabolic phenotype independent of the MITF-PGC1α signaling axis, (Roesch et al., 2010; Roesch et al., 2013; Zhang et al., 2016). Combination treatments with vemurafenib and mitochondrial inhibitors overcame the multi-drug resistance of JARID1Bhigh cells in vitro (Roesch et al., 2013). This strategy of targeting both the MAPK pathway and OXPHOS has been highly effective in other studies as well. For example, our lab determined that the High-OXPHOS phenotype predicts sensitivity to combination treatment with MAPK pathway inhibitors and mTORC1/2 inhibitors (Gopal et al., 2014). Mechanistically, mTORC1/2 inhibitors prevented the nuclear translocation of MITF, thereby inhibiting PGC1α transcription (Figure 3) (Gopal et al., 2014). Similarly, directly targeting mitochondrial biogenesis with the mitochondria-targeted, small-molecule HSP90 inhibitor gamitrinib, has also been shown to overcome MAPK pathway resistance in melanomas with the High-OXPHOS phenotype (Zhang et al., 2016).
Numerous studies have also examined the effects of the biguanide metformin as a combinatorial strategy in melanoma. Commonly used for the treatment of diabetes mellitus type 2, metformin became the subject of extensive oncological research after an initial report indicated that diabetic patients treated with metformin had lower rates of cancer (Pulito et al., 2013). As an inhibitor of complex I of the mitochondrial electron transport chain, metformin deprives cells of ATP, stimulating AMPK and resulting in mTOR inhibition, p53 activation, and apoptosis (Cerezo et al., 2015). Furthermore, metformin independently inhibits mTOR likely through a RAS-related GTPase and suppresses NFκB-STAT3 signaling (Cerezo et al., 2015). Experiments testing metformin as a single-agent therapy have produced mixed results (Cerezo et al., 2015). While several studies demonstrated the anti-melanoma effect of metformin therapy in vitro, Martin et al. found that metformin treatments promoted the growth of BRAFV600E-mutated melanomas in vivo by inducing angiogenesis (Janjetovic et al., 2011; Martin et al., 2012; Niehr et al., 2011; Tomic et al., 2011). Despite this effect, metformin treatment also sensitized melanoma cells to treatment with angiogenesis inhibitors (Martin et al., 2012). Other combination studies have been very promising, with Niehr et al. demonstrating synergy between vemurafenib and metformin in BRAFV600E-mutated melanomas (Niehr et al., 2011). Currently, two phase I/II trials are enrolling patients to test combinations of vemurafenib and metformin (NCT01638676) and dabrafenib, trametinib, and metformin (NCT02143050) in metastatic melanoma patients.
Recent work also supports that BRAF inhibitor-resistant, High-OXPHOS melanomas prefer glutamine metabolism over glucose metabolism (Baenke et al., 2016). This finding suggests a new therapeutic opportunity, and experiments demonstrated that the glutaminase inhibitor BPTES enhanced the antitumor effects of MAPK pathway inhibitors in the treatment of these High-OXPHOS melanomas (Baenke et al., 2016). While promising, additional work remains to understand optimal dosing of treatment combinations with targeted therapies and metabolic inhibitors.
CONCLUSIONS
The understanding of metabolic reprogramming in melanoma has expanded significantly in recent years. Much of this work has focused on the regulation and roles of glycolysis and OXPHOS, which appear to be critical to many processes involved in melanoma maintenance, progression, and resistance. However, numerous additional metabolic pathways have been identified that promote melanoma survival, allowing adaptation to a wide range of environments. These pathways should be evaluated further to improve our understanding of the biology of melanoma and identify novel therapeutic strategies. Furthermore, there is growing understanding of the regulation of metabolic phenotypes in this disease by both intrinsic signaling aberrations and extrinsic signaling by the TME. Finally, there is also growing evidence that the metabolic state of the tumor cells can affect the surrounding TME, with implications for both targeted and immune therapies.
Additional investigation is needed to further define the interplay between metabolic pathways and oncogenic signaling pathways characteristic of melanoma. Further defining how metabolic pathways cooperate with each other and the TME to promote melanoma progression and metastasis should also be investigated. In addition, our understanding of how melanoma signaling networks hijack the metabolic processes available in different microenvironments remains incomplete. It is possible that melanoma cells require different metabolic processes to survive in different organs, which would have significant therapeutic implications. Finally, currently there is very little known about the heterogeneity of metabolic states and dependencies between cells within individual tumors. To address these questions, efforts must be taken to study metabolic pathways in vivo. While informative, in vitro metabolic profiling experiments completely neglect intratumoral heterogeneity and the role of the TME in shaping the metabolic phenotype of melanoma cells. Metabolic profiling of samples from patients and in vivo preclinical models will likely be necessary to address these highly relevant questions.
Targeting metabolic pathways remains a difficult task due to toxicity, as well as the metabolic flexibility of melanoma cells. While some activity with single agents has been observed in preclinical models, metabolic inhibitors will likely be most useful in combination with approved targeted and/or immune therapies. Further, the likelihood of success for such strategies will be enhanced by the development of biomarkers that can be used to select patients most likely to benefit from metabolic inhibitors.
The field of melanoma metabolism has moved far beyond an exclusive focus on the Warburg effect. While aerobic glycolysis still plays a central role in melanoma metabolism, this review has highlighted additional pathways that also contribute to tumorigenesis and metastasis. Each of these pathways provides an array of therapeutic targets that could translate to novel treatments for this highly aggressive, metabolically heterogeneous disease.
Acknowledgments
G.M.F. is supported by the NIH National Center for Advancing Translational Sciences (TL1TR000369 and UL1TR000371). M.A.D. is supported by the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation and philanthropic contributions to the Melanoma Moon Shots Program of the University of Texas MD Anderson Cancer Center. M.A.D. and Y.N.V.G. are supported by the Cancer Prevention Research Institute of Texas (RP160183). M.A.D. and W.P. are supported by the NIH/NCI (R01 CA121118-06A1) and the Cancer Prevention Research Institute of Texas (RP170401). Y.N.V.G. is supported by Melanoma Research Alliance Young Investigator Award (348483). J.L.M. is supported by an ASCO/CCF Young Investigator Award, an MDACC Melanoma SPORE Developmental Research Award (P50 CA093459), and an NIH T32 Training Grant Award (CA009666). R.J.D. is supported by the NIH/NCI (R35 CA220449). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
M.A.D. has served on advisory committees for Roche/Genentech, BMS, Novartis, GSK, Sanofi-Aventis, and Vaccinex and has been the PI of funded research grants to his institution by Roche/Genentech, GSK, Sanofi-Aventis, Merck, Myriad, and Oncothyreon. R.J.D. is an adviser at Agios Pharmaceuticals. No potential conflicts of interest were disclosed by other authors.
References
- Abbas S, Lugthart S, Kavelaars FG, Schelen A, Koenders JE, Zeilemaker A, van Putten WJ, Rijneveld AW, Lowenberg B, Valk PJ. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: Prevalence and prognostic value. Blood. 2010;116:2122–2126. doi: 10.1182/blood-2009-11-250878. [DOI] [PubMed] [Google Scholar]
- Abildgaard C, Guldberg P. Molecular drivers of cellular metabolic reprogramming in melanoma. Trends Mol Med. 2015;21:164–171. doi: 10.1016/j.molmed.2014.12.007. [DOI] [PubMed] [Google Scholar]
- Albino AP, Vidal MJ, McNutt NS, Shea CR, Prieto VG, Nanus DM, Palmer JM, Hayward NK. Mutation and expression of the p53 gene in human malignant melanoma. Melanoma Res. 1994;4:35–45. doi: 10.1097/00008390-199402000-00006. [DOI] [PubMed] [Google Scholar]
- Alegre ML, Frauwirth KA, Thompson CB. T-cell regulation by CD28 and CTLA-4. Nat Rev Immunol. 2001;1:220–228. doi: 10.1038/35105024. [DOI] [PubMed] [Google Scholar]
- Alonso-Curbelo D, Osterloh L, Canon E, Calvo TG, Martinez-Herranz R, Karras P, Martinez S, Riveiro-Falkenbach E, Romero PO, Rodriguez-Peralto JL, et al. RAB7 counteracts PI3K-driven macropinocytosis activated at early stages of melanoma development. Oncotarget. 2015;6:11848–11862. doi: 10.18632/oncotarget.4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alper SL, Chernova MN, Stewart AK. How pH regulates a pH regulator: A regulatory hot spot in the N-terminal cytoplasmic domain of the AE2 anion exchanger. Cell Biochem Biophys. 2002;36:123–136. doi: 10.1385/CBB:36:2-3:123. [DOI] [PubMed] [Google Scholar]
- Anwar A, Norris DA, Fujita M. Ubiquitin proteasomal pathway mediated degradation of p53 in melanoma. Arch Biochem Biophys. 2011;508:198–203. doi: 10.1016/j.abb.2010.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baenke F, Chaneton B, Smith M, Van Den Broek N, Hogan K, Tang H, Viros A, Martin M, Galbraith L, Girotti MR, et al. Resistance to BRAF inhibitors induces glutamine dependency in melanoma cells. Mol Oncol. 2016;10:73–84. doi: 10.1016/j.molonc.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benej M, Pastorekova S, Pastorek J. Carbonic anhydrase IX: Regulation and role in cancer. Subcell Biochem. 2014;75:199–219. doi: 10.1007/978-94-007-7359-2_11. [DOI] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody JR, Costantino CL, Berger AC, Sato T, Lisanti MP, Yeo CJ, Emmons RV, Witkiewicz AK. Expression of indoleamine 2,3-dioxygenase in metastatic malignant melanoma recruits regulatory T cells to avoid immune detection and affects survival. Cell Cycle. 2009;8:1930–1934. doi: 10.4161/cc.8.12.8745. [DOI] [PubMed] [Google Scholar]
- Cai T, Kuang Y, Zhang C, Zhang Z, Chen L, Li B, Li Y, Wang Y, Yang H, Han Q, et al. Glucose-6-phosphate dehydrogenase and NADPH oxidase 4 control STAT3 activity in melanoma cells through a pathway involving reactive oxygen species, c-SRC and SHP2. Am J Cancer Res. 2015;5:1610–1620. [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: Fatty acid oxidation in the limelight. Nat Rev Cancer. 2013;13:227–232. doi: 10.1038/nrc3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carracedo A, Weiss D, Leliaert AK, Bhasin M, de Boer VC, Laurent G, Adams AC, Sundvall M, Song SJ, Ito K, et al. A metabolic prosurvival role for PML in breast cancer. J Clin Invest. 2012;122:3088–3100. doi: 10.1172/JCI62129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho MA, Zecchin KG, Seguin F, Bastos DC, Agostini M, Rangel AL, Veiga SS, Raposo HF, Oliveira HC, Loda M, et al. Fatty acid synthase inhibition with orlistat promotes apoptosis and reduces cell growth and lymph node metastasis in a mouse melanoma model. Int J Cancer. 2008;123:2557–2565. doi: 10.1002/ijc.23835. [DOI] [PubMed] [Google Scholar]
- Cerezo M, Tomic T, Ballotti R, Rocchi S. Is it time to test biguanide metformin in the treatment of melanoma? Pigment Cell Melanoma Res. 2015;28:8–20. doi: 10.1111/pcmr.12267. [DOI] [PubMed] [Google Scholar]
- Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJ, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162:1229–1241. doi: 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjea MN, Shinde R. Textbook of Medical Biochemistry. New Delhi: Jaypee Brothers Medical Publications P Ltd; 2012. p. 876. [Google Scholar]
- Chaube B, Malvi P, Singh SV, Mohammad N, Meena AS, Bhat MK. Targeting metabolic flexibility by simultaneously inhibiting respiratory complex I and lactate generation retards melanoma progression. Oncotarget. 2015;6:37281–37299. doi: 10.18632/oncotarget.6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheli Y, Giuliano S, Botton T, Rocchi S, Hofman V, Hofman P, Bahadoran P, Bertolotto C, Ballotti R. Mitf is the key molecular switch between mouse or human melanoma initiating cells and their differentiated progeny. Oncogene. 2011;30:2307–2318. doi: 10.1038/onc.2010.598. [DOI] [PubMed] [Google Scholar]
- Chen EI, Hewel J, Krueger JS, Tiraby C, Weber MR, Kralli A, Becker K, Yates JR, 3rd, Felding-Habermann B. Adaptation of energy metabolism in breast cancer brain metastases. Cancer Res. 2007;67:1472–1486. doi: 10.1158/0008-5472.CAN-06-3137. [DOI] [PubMed] [Google Scholar]
- Chen J, Lee HJ, Wu X, Huo L, Kim SJ, Xu L, Wang Y, He J, Bollu LR, Gao G, et al. Gain of glucose-independent growth upon metastasis of breast cancer cells to the brain. Cancer Res. 2015;75:554–565. doi: 10.1158/0008-5472.CAN-14-2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, et al. Macropinocytosis of protein is an amino acid supply route in ras-transformed cells. Nature. 2013;497:633–637. doi: 10.1038/nature12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies MA, Stemke-Hale K, Tellez C, Calderone TL, Deng W, Prieto VG, Lazar AJ, Gershenwald JE, Mills GB. A novel AKT3 mutation in melanoma tumours and cell lines. Br J Cancer. 2008;99:1265–1268. doi: 10.1038/sj.bjc.6604637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ingeniis J, Ratnikov B, Richardson AD, Scott DA, Aza-Blanc P, De SK, Kazanov M, Pellecchia M, Ronai Z, Osterman AL, et al. Functional specialization in proline biosynthesis of melanoma. PLoS One. 2012;7:e45190. doi: 10.1371/journal.pone.0045190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2:e1600200. doi: 10.1126/sciadv.1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delage B, Fennell DA, Nicholson L, McNeish I, Lemoine NR, Crook T, Szlosarek PW. Arginine deprivation and argininosuccinate synthetase expression in the treatment of cancer. Int J Cancer. 2010;126:2762–2772. doi: 10.1002/ijc.25202. [DOI] [PubMed] [Google Scholar]
- Dillon BJ, Prieto VG, Curley SA, Ensor CM, Holtsberg FW, Bomalaski JS, Clark MA. Incidence and distribution of argininosuccinate synthetase deficiency in human cancers: A method for identifying cancers sensitive to arginine deprivation. Cancer. 2004;100:826–833. doi: 10.1002/cncr.20057. [DOI] [PubMed] [Google Scholar]
- Dimmer KS, Friedrich B, Lang F, Deitmer JW, Broer S. The low-affinity monocarboxylate transporter MCT4 is adapted to the export of lactate in highly glycolytic cells. Biochem J. 2000;350(Pt 1):219–227. [PMC free article] [PubMed] [Google Scholar]
- Donowitz M, Ming Tse C, Fuster D. SLC9/NHE gene family, a plasma membrane and organellar family of na(+)/H(+) exchangers. Mol Aspects Med. 2013;34:236–251. doi: 10.1016/j.mam.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falletta P, Sanchez-Del-Campo L, Chauhan J, Effern M, Kenyon A, Kershaw CJ, Siddaway R, Lisle R, Freter R, Daniels MJ, et al. Translation reprogramming is an evolutionarily conserved driver of phenotypic plasticity and therapeutic resistance in melanoma. Genes Dev. 2017;31:18–33. doi: 10.1101/gad.290940.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feder-Mengus C, Ghosh S, Weber WP, Wyler S, Zajac P, Terracciano L, Oertli D, Heberer M, Martin I, Spagnoli GC, et al. Multiple mechanisms underlie defective recognition of melanoma cells cultured in three-dimensional architectures by antigen-specific cytotoxic T lymphocytes. Br J Cancer. 2007;96:1072–1082. doi: 10.1038/sj.bjc.6603664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feige E, Yokoyama S, Levy C, Khaled M, Igras V, Lin RJ, Lee S, Widlund HR, Granter SR, Kung AL, et al. Hypoxia-induced transcriptional repression of the melanoma-associated oncogene MITF. Proc Natl Acad Sci U S A. 2011;108:E924–33. doi: 10.1073/pnas.1106351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson J, Smith M, Zudaire I, Wellbrock C, Arozarena I. Glucose availability controls ATF4-mediated MITF suppression to drive melanoma cell growth. Oncotarget. 2017;8:32946–32959. doi: 10.18632/oncotarget.16514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipp FV, Ratnikov B, De Ingeniis J, Smith JW, Osterman AL, Scott DA. Glutamine-fueled mitochondrial metabolism is decoupled from glycolysis in melanoma. Pigment Cell Melanoma Res. 2012a;25:732–739. doi: 10.1111/pcmr.12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipp FV, Scott DA, Ronai ZA, Osterman AL, Smith JW. Reverse TCA cycle flux through isocitrate dehydrogenases 1 and 2 is required for lipogenesis in hypoxic melanoma cells. Pigment Cell Melanoma Res. 2012b;25:375–383. doi: 10.1111/j.1755-148X.2012.00989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G, Hoves S, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109:3812–3819. doi: 10.1182/blood-2006-07-035972. [DOI] [PubMed] [Google Scholar]
- Friberg M, Jennings R, Alsarraj M, Dessureault S, Cantor A, Extermann M, Mellor AL, Munn DH, Antonia SJ. Indoleamine 2,3-dioxygenase contributes to tumor cell evasion of T cell-mediated rejection. Int J Cancer. 2002;101:151–155. doi: 10.1002/ijc.10645. [DOI] [PubMed] [Google Scholar]
- Gangadhar TC, Hamid O, Smith DC, Bauer TM, Wasser JS, Olszanski AJ, Luke J, Balmanoukian AS, Kaufman DR, Zhao Y, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: Updated phase 1 results from ECHO-202/KEYNOTE-037. Ann Onc. 2016;27:379–400. [Google Scholar]
- Giuliano S, Cheli Y, Ohanna M, Bonet C, Beuret L, Bille K, Loubat A, Hofman V, Hofman P, Ponzio G, et al. Microphthalmia-associated transcription factor controls the DNA damage response and a lineage-specific senescence program in melanomas. Cancer Res. 2010;70:3813–3822. doi: 10.1158/0008-5472.CAN-09-2913. [DOI] [PubMed] [Google Scholar]
- Gonzalez CD, Alvarez S, Ropolo A, Rosenzvit C, Bagnes MF, Vaccaro MI. Autophagy, warburg, and warburg reverse effects in human cancer. Biomed Res Int. 2014;2014:926729. doi: 10.1155/2014/926729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodall J, Carreira S, Denat L, Kobi D, Davidson I, Nuciforo P, Sturm RA, Larue L, Goding CR. Brn-2 represses microphthalmia-associated transcription factor expression and marks a distinct subpopulation of microphthalmia-associated transcription factor-negative melanoma cells. Cancer Res. 2008;68:7788–7794. doi: 10.1158/0008-5472.CAN-08-1053. [DOI] [PubMed] [Google Scholar]
- Gopal YN, Rizos H, Chen G, Deng W, Frederick DT, Cooper ZA, Scolyer RA, Pupo G, Komurov K, Sehgal V, et al. Inhibition of mTORC1/2 overcomes resistance to MAPK pathway inhibitors mediated by PGC1alpha and oxidative phosphorylation in melanoma. Cancer Res. 2014;74:7037–7047. doi: 10.1158/0008-5472.CAN-14-1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- Guldberg P, thor Straten P, Birck A, Ahrenkiel V, Kirkin AF, Zeuthen J. Disruption of the MMAC1/PTEN gene by deletion or mutation is a frequent event in malignant melanoma. Cancer Res. 1997;57:3660–3663. [PubMed] [Google Scholar]
- Hamamura K, Furukawa K, Hayashi T, Hattori T, Nakano J, Nakashima H, Okuda T, Mizutani H, Hattori H, Ueda M, et al. Ganglioside GD3 promotes cell growth and invasion through p130Cas and paxillin in malignant melanoma cells. Proc Natl Acad Sci U S A. 2005;102:11041–11046. doi: 10.1073/pnas.0503658102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell. 2013;23:302–315. doi: 10.1016/j.ccr.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healey M, Crow MS, Molina CA. Ras-induced melanoma transformation is associated with the proteasomal degradation of the transcriptional repressor ICER. Mol Carcinog. 2013;52:692–704. doi: 10.1002/mc.21908. [DOI] [PubMed] [Google Scholar]
- Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J Clin Invest. 2013;123:3678–3684. doi: 10.1172/JCI69600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho J, de Moura MB, Lin Y, Vincent G, Thorne S, Duncan LM, Hui-Min L, Kirkwood JM, Becker D, Van Houten B, et al. Importance of glycolysis and oxidative phosphorylation in advanced melanoma. Mol Cancer. 2012;11:76-4598-11-76. doi: 10.1186/1476-4598-11-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med. 2013;210:1389–1402. doi: 10.1084/jem.20130066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou DY, Muller AJ, Sharma MD, DuHadaway J, Banerjee T, Johnson M, Mellor AL, Prendergast GC, Munn DH. Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res. 2007;67:792–801. doi: 10.1158/0008-5472.CAN-06-2925. [DOI] [PubMed] [Google Scholar]
- Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, Kaper F, Giaccia AJ, Abraham RT. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol. 2002;22:7004–7014. doi: 10.1128/MCB.22.20.7004-7014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innocenzi D, Alo PL, Balzani A, Sebastiani V, Silipo V, La Torre G, Ricciardi G, Bosman C, Calvieri S. Fatty acid synthase expression in melanoma. J Cutan Pathol. 2003;30:23–28. doi: 10.1034/j.1600-0560.2003.300104.x. [DOI] [PubMed] [Google Scholar]
- Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janjetovic K, Harhaji-Trajkovic L, Misirkic-Marjanovic M, Vucicevic L, Stevanovic D, Zogovic N, Sumarac-Dumanovic M, Micic D, Trajkovic V. In vitro and in vivo anti-melanoma action of metformin. Eur J Pharmacol. 2011;668:373–382. doi: 10.1016/j.ejphar.2011.07.004. [DOI] [PubMed] [Google Scholar]
- Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013;27:2179–2191. doi: 10.1101/gad.225680.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen DH, Therkildsen MH, Dabelsteen E. A reverse warburg metabolism in oral squamous cell carcinoma is not dependent upon myofibroblasts. J Oral Pathol Med. 2015;44:714–721. doi: 10.1111/jop.12297. [DOI] [PubMed] [Google Scholar]
- Ji Z, Njauw CN, Taylor M, Neel V, Flaherty KT, Tsao H. p53 rescue through HDM2 antagonism suppresses melanoma growth and potentiates MEK inhibition. J Invest Dermatol. 2012;132:356–364. doi: 10.1038/jid.2011.313. [DOI] [PubMed] [Google Scholar]
- Jiang L, Shestov AA, Swain P, Yang C, Parker SJ, Wang QA, Terada LS, Adams ND, McCabe MT, Pietrak B, et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature. 2016;532:255–258. doi: 10.1038/nature17393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jose C, Bellance N, Rossignol R. Choosing between glycolysis and oxidative phosphorylation: A tumor’s dilemma? Biochim Biophys Acta. 2011;1807:552–561. doi: 10.1016/j.bbabio.2010.10.012. [DOI] [PubMed] [Google Scholar]
- Kersten S. Mechanisms of nutritional and hormonal regulation of lipogenesis. EMBO Rep. 2001;2:282–286. doi: 10.1093/embo-reports/kve071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JB, Sarraf P, Wright M, Yao KM, Mueller E, Solanes G, Lowell BB, Spiegelman BM. Nutritional and insulin regulation of fatty acid synthetase and leptin gene expression through ADD1/SREBP1. J Clin Invest. 1998;101:1–9. doi: 10.1172/JCI1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Klein M, Seeger P, Schuricht B, Alper SL, Schwab A. Polarization of na(+)/H(+) and cl(−)/HCO (3)(−) exchangers in migrating renal epithelial cells. J Gen Physiol. 2000;115:599–608. doi: 10.1085/jgp.115.5.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraehn GM, Utikal J, Udart M, Greulich KM, Bezold G, Kaskel P, Leiter U, Peter RU. Extra c-myc oncogene copies in high risk cutaneous malignant melanoma and melanoma metastases. Br J Cancer. 2001;84:72–79. doi: 10.1054/bjoc.2000.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182:459–465. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar SM, Yu H, Edwards R, Chen L, Kazianis S, Brafford P, Acs G, Herlyn M, Xu X. Mutant V600E BRAF increases hypoxia inducible factor-1alpha expression in melanoma. Cancer Res. 2007;67:3177–3184. doi: 10.1158/0008-5472.CAN-06-3312. [DOI] [PubMed] [Google Scholar]
- Kwong LN, Davies MA. Navigating the therapeutic complexity of PI3K pathway inhibition in melanoma. Clin Cancer Res. 2013;19:5310–5319. doi: 10.1158/1078-0432.CCR-13-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagana A, Vadnais J, Le PU, Nguyen TN, Laprade R, Nabi IR, Noel J. Regulation of the formation of tumor cell pseudopodia by the na(+)/H(+) exchanger NHE1. J Cell Sci. 2000;113( Pt 20):3649–3662. doi: 10.1242/jcs.113.20.3649. [DOI] [PubMed] [Google Scholar]
- Land SC, Tee AR. Hypoxia-inducible factor 1alpha is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J Biol Chem. 2007;282:20534–20543. doi: 10.1074/jbc.M611782200. [DOI] [PubMed] [Google Scholar]
- Lazar I, Clement E, Dauvillier S, Milhas D, Ducoux-Petit M, LeGonidec S, Moro C, Soldan V, Dalle S, Balor S, et al. Adipocyte exosomes promote melanoma aggressiveness through fatty acid oxidation: A novel mechanism linking obesity and cancer. Cancer Res. 2016;76:4051–4057. doi: 10.1158/0008-5472.CAN-16-0651. [DOI] [PubMed] [Google Scholar]
- LeBleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, de Carvalho FM, Damascena A, Domingos Chinen LT, Rocha RM, et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol. 2014;16:992–1003. 1–15. doi: 10.1038/ncb3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee GK, Park HJ, Macleod M, Chandler P, Munn DH, Mellor AL. Tryptophan deprivation sensitizes activated T cells to apoptosis prior to cell division. Immunology. 2002;107:452–460. doi: 10.1046/j.1365-2567.2002.01526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kefford R, Carlino M. PD-1 and PD-L1 inhibitors in melanoma treatment: Past success, present application and future challenges. Immunotherapy. 2016;8:733–746. doi: 10.2217/imt-2016-0022. [DOI] [PubMed] [Google Scholar]
- Lehninger AL, Nelson DL, Cox MM. Lehninger Principles of Biochemistry. New York: W.H. Freeman; 2013. [Google Scholar]
- Liu YL, Lai F, Wilmott JS, Yan XG, Liu XY, Luan Q, Guo ST, Jiang CC, Tseng HY, Scolyer RA, et al. Noxa upregulation by oncogenic activation of MEK/ERK through CREB promotes autophagy in human melanoma cells. Oncotarget. 2014;5:11237–11251. doi: 10.18632/oncotarget.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, Heffron G, Metallo CM, Muranen T, Sharfi H, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet. 2011;43:869–874. doi: 10.1038/ng.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodish HF. Molecular Cell Biology. New York: W.H. Freeman; 2000. p. 1084.p. G-17.p. I-36. [Google Scholar]
- Lopez GY, Reitman ZJ, Solomon D, Waldman T, Bigner DD, McLendon RE, Rosenberg SA, Samuels Y, Yan H. IDH1(R132) mutation identified in one human melanoma metastasis, but not correlated with metastases to the brain. Biochem Biophys Res Commun. 2010;398:585–587. doi: 10.1016/j.bbrc.2010.06.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke JJ, Flaherty KT, Ribas A, Long GV. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat Rev Clin Oncol. 2017 doi: 10.1038/nrclinonc.2017.43. [DOI] [PubMed] [Google Scholar]
- Manca A, Sini MC, Izzo F, Ascierto PA, Tatangelo F, Botti G, Gentilcore G, Capone M, Mozzillo N, Rozzo C, et al. Induction of arginosuccinate synthetase (ASS) expression affects the antiproliferative activity of arginine deiminase (ADI) in melanoma cells. Oncol Rep. 2011;25:1495–1502. doi: 10.3892/or.2011.1220. [DOI] [PubMed] [Google Scholar]
- Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino ML, Pellegrini P, Di Lernia G, Djavaheri-Mergny M, Brnjic S, Zhang X, Hagg M, Linder S, Fais S, Codogno P, et al. Autophagy is a protective mechanism for human melanoma cells under acidic stress. J Biol Chem. 2012;287:30664–30676. doi: 10.1074/jbc.M112.339127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MJ, Hayward R, Viros A, Marais R. Metformin accelerates the growth of BRAF V600E-driven melanoma by upregulating VEGF-A. Cancer Discov. 2012;2:344–355. doi: 10.1158/2159-8290.CD-11-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuade JL, Vashisht Gopal Y. Counteracting oxidative phosphorylation-mediated resistance of melanomas to MAPK pathway inhibition. Mol Cell Oncol. 2015;2:e991610. doi: 10.4161/23723556.2014.991610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendler AN, Hu B, Prinz PU, Kreutz M, Gottfried E, Noessner E. Tumor lactic acidosis suppresses CTL function by inhibition of p38 and JNK/c-jun activation. Int J Cancer. 2012;131:633–640. doi: 10.1002/ijc.26410. [DOI] [PubMed] [Google Scholar]
- Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7:763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2011;481:380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. 2010;185:3190–3198. doi: 10.4049/jimmunol.0903670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael D, Oren M. The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol. 2003;13:49–58. doi: 10.1016/s1044-579x(02)00099-8. [DOI] [PubMed] [Google Scholar]
- Moreno AC, Clara RO, Coimbra JB, Julio AR, Albuquerque RC, Oliveira EM, Maria-Engler SS, Campa A. The expanding roles of 1-methyl-tryptophan (1-MT): In addition to inhibiting kynurenine production, 1-MT activates the synthesis of melatonin in skin cells. FEBS J. 2013;280:4782–4792. doi: 10.1111/febs.12444. [DOI] [PubMed] [Google Scholar]
- Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2011;481:385–388. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn DH, Mellor AL. IDO in the tumor microenvironment: Inflammation, counter-regulation, and tolerance. Trends Immunol. 2016;37:193–207. doi: 10.1016/j.it.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, Mellor AL. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005;22:633–642. doi: 10.1016/j.immuni.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Nath A, Chan C. Genetic alterations in fatty acid transport and metabolism genes are associated with metastatic progression and poor prognosis of human cancers. Sci Rep. 2016;6:18669. doi: 10.1038/srep18669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndoye A, Weeraratna AT. Autophagy- an emerging target for melanoma therapy. F1000Res. 2016;5:10. doi: 10.12688/f1000research.8347.1. 12688/f1000research.8347.1. eCollection 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehr F, von Euw E, Attar N, Guo D, Matsunaga D, Sazegar H, Ng C, Glaspy JA, Recio JA, Lo RS, et al. Combination therapy with vemurafenib (PLX4032/RG7204) and metformin in melanoma cell lines with distinct driver mutations. J Transl Med. 2011;9:76-5876-9-76. doi: 10.1186/1479-5876-9-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omholt K, Krockel D, Ringborg U, Hansson J. Mutations of PIK3CA are rare in cutaneous melanoma. Melanoma Res. 2006;16:197–200. doi: 10.1097/01.cmr.0000200488.77970.e3. [DOI] [PubMed] [Google Scholar]
- Owen OE, Kalhan SC, Hanson RW. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem. 2002;277:30409–30412. doi: 10.1074/jbc.R200006200. [DOI] [PubMed] [Google Scholar]
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmenter TJ, Kleinschmidt M, Kinross KM, Bond ST, Li J, Kaadige MR, Rao A, Sheppard KE, Hugo W, Pupo GM, et al. Response of BRAF-mutant melanoma to BRAF inhibition is mediated by a network of transcriptional regulators of glycolysis. Cancer Discov. 2014;4:423–433. doi: 10.1158/2159-8290.CD-13-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, Linsley PS, Thompson CB, Riley JL. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25:9543–9553. doi: 10.1128/MCB.25.21.9543-9553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS, Berenguer A, Prats N, Toll A, Hueto JA, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature. 2017;541:41–45. doi: 10.1038/nature20791. [DOI] [PubMed] [Google Scholar]
- Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, Karoly ED, Freeman GJ, Petkova V, Seth P, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun. 2015;6:6692. doi: 10.1038/ncomms7692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, Wang C, Pestell RG, Martinez-Outschoorn UE, Howell A, et al. Transcriptional evidence for the “reverse warburg effect” in human breast cancer tumor stroma and metastasis: Similarities with oxidative stress, inflammation, alzheimer’s disease, and “neuron-glia metabolic coupling”. Aging (Albany NY) 2010;2:185–199. doi: 10.18632/aging.100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S, et al. The reverse warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8:3984–4001. doi: 10.4161/cc.8.23.10238. [DOI] [PubMed] [Google Scholar]
- Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payen VL, Porporato PE, Baselet B, Sonveaux P. Metabolic changes associated with tumor metastasis, part 1: Tumor pH, glycolysis and the pentose phosphate pathway. Cell Mol Life Sci. 2016;73:1333–1348. doi: 10.1007/s00018-015-2098-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38:633–643. doi: 10.1016/j.immuni.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phannasil P, Thuwajit C, Warnnissorn M, Wallace JC, MacDonald MJ, Jitrapakdee S. Pyruvate carboxylase is up-regulated in breast cancer and essential to support growth and invasion of MDA-MB-231 cells. PLoS One. 2015;10:e0129848. doi: 10.1371/journal.pone.0129848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro C, Miranda-Goncalves V, Longatto-Filho A, Vicente AL, Berardinelli GN, Scapulatempo-Neto C, Costa RF, Viana CR, Reis RM, Baltazar F, et al. The metabolic microenvironment of melanomas: Prognostic value of MCT1 and MCT4. Cell Cycle. 2016;15:1462–1470. doi: 10.1080/15384101.2016.1175258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, DeBerardinis RJ, Morrison SJ. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;527:186–191. doi: 10.1038/nature15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porporato PE, Payen VL, Perez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, Dhup S, Tardy M, Vazeille T, Bouzin C, et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. 2014;8:754–766. doi: 10.1016/j.celrep.2014.06.043. [DOI] [PubMed] [Google Scholar]
- Pulito C, Sanli T, Rana P, Muti P, Blandino G, Strano S. Metformin: On ongoing journey across diabetes, cancer therapy and prevention. Metabolites. 2013;3:1051–1075. doi: 10.3390/metabo3041051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich S, Adler L, Yizhak K, Sarver A, Silberman A, Agron S, Stettner N, Sun Q, Brandis A, Helbling D, et al. Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature. 2015;527:379–383. doi: 10.1038/nature15529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakheja D, Medeiros LJ, Bevan S, Chen W. The emerging role of d-2-hydroxyglutarate as an oncometabolite in hematolymphoid and central nervous system neoplasms. Front Oncol. 2013;3:169. doi: 10.3389/fonc.2013.00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnikov B, Aza-Blanc P, Ronai ZA, Smith JW, Osterman AL, Scott DA. Glutamate and asparagine cataplerosis underlie glutamine addiction in melanoma. Oncotarget. 2015;6:7379–7389. doi: 10.18632/oncotarget.3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnikov BI, Scott DA, Osterman AL, Smith JW, Ronai ZA. Metabolic rewiring in melanoma. Oncogene. 2017;36:147–157. doi: 10.1038/onc.2016.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riesenberg S, Groetchen A, Siddaway R, Bald T, Reinhardt J, Smorra D, Kohlmeyer J, Renn M, Phung B, Aymans P, et al. MITF and c-jun antagonism interconnects melanoma dedifferentiation with pro-inflammatory cytokine responsiveness and myeloid cell recruitment. Nat Commun. 2015;6:8755. doi: 10.1038/ncomms9755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues MF, Obre E, de Melo FH, Santos GC, Jr, Galina A, Jasiulionis MG, Rossignol R, Rumjanek FD, Amoedo ND. Enhanced OXPHOS, glutaminolysis and beta-oxidation constitute the metastatic phenotype of melanoma cells. Biochem J. 2016;473:703–715. doi: 10.1042/BJ20150645. [DOI] [PubMed] [Google Scholar]
- Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, Basu D, Gimotty P, Vogt T, Herlyn M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141:583–594. doi: 10.1016/j.cell.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, Korbel C, Laschke MW, Gimotty PA, Philipp SE, et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell. 2013;23:811–825. doi: 10.1016/j.ccr.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rofstad EK, Mathiesen B, Kindem K, Galappathi K. Acidic extracellular pH promotes experimental metastasis of human melanoma cells in athymic nude mice. Cancer Res. 2006;66:6699–6707. doi: 10.1158/0008-5472.CAN-06-0983. [DOI] [PubMed] [Google Scholar]
- Romero-Garcia S, Moreno-Altamirano MM, Prado-Garcia H, Sanchez-Garcia FJ. Lactate contribution to the tumor microenvironment: Mechanisms, effects on immune cells and therapeutic relevance. Front Immunol. 2016;7:52. doi: 10.3389/fimmu.2016.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumsey WL, Schlosser C, Nuutinen EM, Robiolio M, Wilson DF. Cellular energetics and the oxygen dependence of respiration in cardiac myocytes isolated from adult rat. J Biol Chem. 1990;265:15392–15402. [PubMed] [Google Scholar]
- Saab J, Santos-Zabala ML, Loda M, Stack EC, Hollmann TJ. Fatty acid synthase and acetyl-CoA carboxylase are expressed in nodal metastatic melanoma but not in benign intracapsular nodal nevi. Am J Dermatopathol. 2017 doi: 10.1097/DAD.0000000000000939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz L, Abolhassani M, Guais A, Sanders E, Steyaert JM, Campion F, Israel M. A combination of alpha lipoic acid and calcium hydroxycitrate is efficient against mouse cancer models: Preliminary results. Oncol Rep. 2010;23:1407–1416. doi: 10.3892/or_00000778. [DOI] [PubMed] [Google Scholar]
- Scott DA, Richardson AD, Filipp FV, Knutzen CA, Chiang GG, Ronai ZA, Osterman AL, Smith JW. Comparative metabolic flux profiling of melanoma cell lines: Beyond the warburg effect. J Biol Chem. 2011;286:42626–42634. doi: 10.1074/jbc.M111.282046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellers K, Fox MP, Bousamra M, 2nd, Slone SP, Higashi RM, Miller DM, Wang Y, Yan J, Yuneva MO, Deshpande R, et al. Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J Clin Invest. 2015;125:687–698. doi: 10.1172/JCI72873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, Giallongo A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem. 1996;271:32529–32537. doi: 10.1074/jbc.271.51.32529. [DOI] [PubMed] [Google Scholar]
- Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata T, Kokubu A, Miyamoto M, Sasajima Y, Yamazaki N. Mutant IDH1 confers an in vivo growth in a melanoma cell line with BRAF mutation. Am J Pathol. 2011;178:1395–1402. doi: 10.1016/j.ajpath.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimobayashi M, Hall MN. Making new contacts: The mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol. 2014;15:155–162. doi: 10.1038/nrm3757. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- Simoes RV, Serganova IS, Kruchevsky N, Leftin A, Shestov AA, Thaler HT, Sukenick G, Locasale JW, Blasberg RG, Koutcher JA, et al. Metabolic plasticity of metastatic breast cancer cells: Adaptation to changes in the microenvironment. Neoplasia. 2015;17:671–684. doi: 10.1016/j.neo.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slipicevic A, Jorgensen K, Skrede M, Rosnes AK, Troen G, Davidson B, Florenes VA. The fatty acid binding protein 7 (FABP7) is involved in proliferation and invasion of melanoma cells. BMC Cancer. 2008;8:276-2407-8-276. doi: 10.1186/1471-2407-8-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolkova K, Plecita-Hlavata L, Bellance N, Benard G, Rossignol R, Jezek P. Waves of gene regulation suppress and then restore oxidative phosphorylation in cancer cells. Int J Biochem Cell Biol. 2011;43:950–968. doi: 10.1016/j.biocel.2010.05.003. [DOI] [PubMed] [Google Scholar]
- Sonveaux P, Copetti T, De Saedeleer CJ, Vegran F, Verrax J, Kennedy KM, Moon EJ, Dhup S, Danhier P, Frerart F, et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS One. 2012;7:e33418. doi: 10.1371/journal.pone.0033418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008;118:3930–3942. doi: 10.1172/JCI36843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotgia F, Martinez-Outschoorn UE, Lisanti MP. The reverse warburg effect in osteosarcoma. Oncotarget. 2014;5:7982–7983. doi: 10.18632/oncotarget.2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, metabolism, and cancer. Cancer Discov. 2015;5:1024–1039. doi: 10.1158/2159-8290.CD-15-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stock C, Gassner B, Hauck CR, Arnold H, Mally S, Eble JA, Dieterich P, Schwab A. Migration of human melanoma cells depends on extracellular pH and na+/H+ exchange. J Physiol. 2005;567:225–238. doi: 10.1113/jphysiol.2005.088344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuwe L, Muller M, Fabian A, Waning J, Mally S, Noel J, Schwab A, Stock C. pH dependence of melanoma cell migration: Protons extruded by NHE1 dominate protons of the bulk solution. J Physiol. 2007;585:351–360. doi: 10.1113/jphysiol.2007.145185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sucher R, Kurz K, Weiss G, Margreiter R, Fuchs D, Brandacher G. IDO-mediated tryptophan degradation in the pathogenesis of malignant tumor disease. Int J Tryptophan Res. 2010;3:113–120. doi: 10.4137/ijtr.s4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudarsan S, Dethlefsen S, Blank LM, Siemann-Herzberg M, Schmid A. The functional structure of central carbon metabolism in pseudomonas putida KT2440. Appl Environ Microbiol. 2014;80:5292–5303. doi: 10.1128/AEM.01643-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumantran VN, Mishra P, Sudhakar N. Microarray analysis of differentially expressed genes regulating lipid metabolism during melanoma progression. Indian J Biochem Biophys. 2015;52:125–131. [PubMed] [Google Scholar]
- Svastova E, Witarski W, Csaderova L, Kosik I, Skvarkova L, Hulikova A, Zatovicova M, Barathova M, Kopacek J, Pastorek J, et al. Carbonic anhydrase IX interacts with bicarbonate transporters in lamellipodia and increases cell migration via its catalytic domain. J Biol Chem. 2012;287:3392–3402. doi: 10.1074/jbc.M111.286062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CP, Buronfosse A, Combaret V, Pedron S, Fertil B, Portoukalian J. Gangliosides protect human melanoma cells from ionizing radiation-induced clonogenic cell death. Glycoconj J. 1996;13:377–384. doi: 10.1007/BF00731470. [DOI] [PubMed] [Google Scholar]
- Tomic T, Botton T, Cerezo M, Robert G, Luciano F, Puissant A, Gounon P, Allegra M, Bertolotto C, Bereder JM, et al. Metformin inhibits melanoma development through autophagy and apoptosis mechanisms. Cell Death Dis. 2011;2:e199. doi: 10.1038/cddis.2011.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao H, Zhang X, Benoit E, Haluska FG. Identification of PTEN/MMAC1 alterations in uncultured melanomas and melanoma cell lines. Oncogene. 1998;16:3397–3402. doi: 10.1038/sj.onc.1201881. [DOI] [PubMed] [Google Scholar]
- Ullah MS, Davies AJ, Halestrap AP. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J Biol Chem. 2006;281:9030–9037. doi: 10.1074/jbc.M511397200. [DOI] [PubMed] [Google Scholar]
- Vahle AK, Domikowsky B, Schwoppe C, Krahling H, Mally S, Schafers M, Hermann S, Shahin V, Haier J, Schwab A, et al. Extracellular matrix composition and interstitial pH modulate NHE1-mediated melanoma cell motility. Int J Oncol. 2014;44:78–90. doi: 10.3892/ijo.2013.2158. [DOI] [PubMed] [Google Scholar]
- Vazquez F, Lim JH, Chim H, Bhalla K, Girnun G, Pierce K, Clish CB, Granter SR, Widlund HR, Spiegelman BM, et al. PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell. 2013;23:287–301. doi: 10.1016/j.ccr.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vegran F, Boidot R, Michiels C, Sonveaux P, Feron O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-kappaB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011;71:2550–2560. doi: 10.1158/0008-5472.CAN-10-2828. [DOI] [PubMed] [Google Scholar]
- Volkenandt M, Schlegel U, Nanus DM, Albino AP. Mutational analysis of the human p53 gene in malignant melanoma. Pigment Cell Res. 1991;4:35–40. doi: 10.1111/j.1600-0749.1991.tb00311.x. [DOI] [PubMed] [Google Scholar]
- Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12:685–698. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walunas TL, Bakker CY, Bluestone JA. CTLA-4 ligation blocks CD28-dependent T cell activation. J Exp Med. 1996;183:2541–2550. doi: 10.1084/jem.183.6.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol. 2015;11:9–15. doi: 10.1038/nchembio.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinlich G, Murr C, Richardsen L, Winkler C, Fuchs D. Decreased serum tryptophan concentration predicts poor prognosis in malignant melanoma patients. Dermatology. 2007;214:8–14. doi: 10.1159/000096906. [DOI] [PubMed] [Google Scholar]
- Weiss J, Schwechheimer K, Cavenee WK, Herlyn M, Arden KC. Mutation and expression of the p53 gene in malignant melanoma cell lines. Int J Cancer. 1993;54:693–699. doi: 10.1002/ijc.2910540427. [DOI] [PubMed] [Google Scholar]
- Worrall C, Suleymanova N, Crudden C, Trocoli Drakensjo I, Candrea E, Nedelcu D, Takahashi SI, Girnita L, Girnita A. Unbalancing p53/Mdm2/IGF-1R axis by Mdm2 activation restrains the IGF-1-dependent invasive phenotype of skin melanoma. Oncogene. 2017;36:3274–3286. doi: 10.1038/onc.2016.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Sudderth J, Dang T, Bachoo RM, McDonald JG, DeBerardinis RJ. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or akt signaling. Cancer Res. 2009;69:7986–7993. doi: 10.1158/0008-5472.CAN-09-2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaugg K, Yao Y, Reilly PT, Kannan K, Kiarash R, Mason J, Huang P, Sawyer SK, Fuerth B, Faubert B, et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011;25:1041–1051. doi: 10.1101/gad.1987211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecchin KG, Rossato FA, Raposo HF, Melo DR, Alberici LC, Oliveira HC, Castilho RF, Coletta RD, Vercesi AE, Graner E. Inhibition of fatty acid synthase in melanoma cells activates the intrinsic pathway of apoptosis. Lab Invest. 2011;91:232–240. doi: 10.1038/labinvest.2010.157. [DOI] [PubMed] [Google Scholar]
- Zeller KI, Jegga AG, Aronow BJ, O’Donnell KA, Dang CV. An integrated database of genes responsive to the myc oncogenic transcription factor: Identification of direct genomic targets. Genome Biol. 2003;4:R69. doi: 10.1186/gb-2003-4-10-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Frederick DT, Wu L, Wei Z, Krepler C, Srinivasan S, Chae YC, Xu X, Choi H, Dimwamwa E, et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J Clin Invest. 2016;126:1834–1856. doi: 10.1172/JCI82661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Jeong JH, Asara JM, Yuan YY, Granter SR, Chin L, Cantley LC. Oncogenic B-RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Mol Cell. 2009;33:237–247. doi: 10.1016/j.molcel.2008.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zundel W, Schindler C, Haas-Kogan D, Koong A, Kaper F, Chen E, Gottschalk AR, Ryan HE, Johnson RS, Jefferson AB, et al. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 2000;14:391–396. [PMC free article] [PubMed] [Google Scholar]