

SUMMARY
To determine the feasibility of liquid biopsy for monitoring of advanced melanoma patients, cell-free DNA was extracted from plasma for 25 Stage III/IV patients, most (84.0%) having received previous therapy. DNA concentrations ranged from 0.6 to 390.0 ng/mL (median=7.8 ng/mL), and were positively correlated with tumor burden as measured by imaging (Spearman rho=0.5435, p=0.0363). Using ultra-deep sequencing for a 61-gene panel, one or more mutations were detected in 12 of 25 samples (48.0%), and this proportion did not vary significantly for patients on or off therapy at time of blood draw (52.9% and 37.5% respectively; p=0.673). Sixteen mutations were detected in 8 different genes, with the most frequent mutations detected in BRAF, NRAS, and KIT. Allele fractions ranged from 1.1% to 63.2% (median=29.1%). Among patients with tissue next-generation sequencing, 9 of 11 plasma mutations were also detected in matched tissue, for a concordance of 81.8%.
Keywords: Liquid biopsy, cell-free circulating tumor DNA, melanoma, precision medicine
INTRODUCTION
Over 70,000 people in the United States are diagnosed with melanoma each year (Siegel et al., 2017). Precision medicine and individualization of therapy have become integral to melanoma patient management as BRAF and MEK targeted therapies have led to improved survival, and been approved as standard of care for treatment of advanced V600-mutant melanoma (Larkin et al., 2014; Long et al., 2015; Robert et al., 2015a). Immune therapy has also become an essential part of the treatment of this disease with CTLA-4 and PD1 inhibitors leading to improvements in patient survival (Larkin et al., 2015; Robert et al., 2015b; Robert et al., 2011). Individualized treatment plans using these drugs require monitoring of the patient’s disease status with repeat response assessments, often over a lengthy treatment regimen. However, repeat biopsy of multiple melanoma tumor sites for patients with advanced metastatic disease is impractical, and the addition of peripheral blood assays, so-called liquid biopsies, of tumor activity and molecular alterations may more accurately represent the disease status and full molecular spectrum of the patient’s disease.
Liquid biopsy is increasingly utilized for non-invasive patient monitoring through blood samples which can contain circulating tumor cells (CTCs), cell-free circulating tumor DNA (cfDNA), and other material shed from primary and metastatic tumors. Enumeration of CTCs and monitoring of changes in CTC counts have been shown to be prognostic in multiple solid tumors (Cohen et al., 2009; Krebs et al., 2011; Riethdorf et al., 2007), however, commercially available approaches for melanoma CTC detection have low sensitivity, even among therapy-naïve patients (Khoja et al., 2013; Rao et al., 2011). A more recently developed microfluidic approach showed improved sensitivity of CTC detection in melanoma patient samples, but the device is not commercially available (Luo et al., 2014).
cfDNA has traditionally been utilized for the detection of clinically actionable driver and resistance mutations, and is readily detectable in the blood of melanoma patients prior to receiving therapy (Gonzalez-Cao et al., 2015; Kaisaki et al., 2016; Knol et al., 2016; Lipson et al., 2014; Pinzani et al., 2011). Indeed, detection of a BRAF V600 mutation is significantly associated with lower overall survival (Gonzalez-Cao et al., 2015; Knol et al., 2016), and PCR-based approaches have been used to detect a limited number of variants in BRAF and NRAS known to be associated with resistance to BRAF and MEK inhibitors, but with very limited application to melanoma patients after the advent of therapy (Girotti et al., 2016; Gray et al., 2015; Schreuer et al., 2016; Tsao et al., 2015; Xi et al., 2016). Moreover, monitoring of resistance mutations to targeted therapies is becoming increasingly complex, with multiple mutations in the MAPK and PI3K-PTEN-AKT pathways having been identified (Shi et al., 2014). Transcriptional alterations associated with therapy resistance have also been reported (Hugo et al., 2015), however, circulating RNA cannot yet be as reliably detected as cfDNA. Nevertheless, the mutational complexity of therapy resistance suggests that PCR-based interrogation of a limited number of mutations may fail to detect molecular resistance to targeted therapy. To our knowledge, no study has yet prospectively evaluated whether broad coverage ultra-deep sequencing of plasma DNA can be used to comprehensively monitor molecular alterations in advanced melanoma patients who are currently receiving or have received multiple therapies.
Cell-free DNA concentration, independent of the identification of specific mutations, has also been shown to have prognostic value (Cargnin et al., 2017; Thompson et al., 2016), although this has not yet been measured in melanoma. Plasma cfDNA yields for patients with lung and breast cancer have been associated with a worse prognosis (Couraud et al., 2014; Dawson et al., 2013; Thompson et al., 2016), and changes in cfDNA levels can precede clinical and radiographic tumor regression or progression, thus allowing for real-time, non-invasive surveillance of tumor burden (Lipson et al., 2014). Pretreatment melanoma solid tumor burden, as measured by imaging, was recently shown to be correlated with clinical response to pembrolizumab (Huang et al., 2017). This underscores the need to determine whether cfDNA concentration correlates with solid tumor burden, especially in melanoma and other cancers for which checkpoint inhibitors are routinely used.
Here we apply a minimally adapted, commercially available workflow for measuring cell-free DNA concentration, and ultra-deep sequencing of a panel of 61 genes in the cfDNA captured from patient plasma. We focus here on a cohort of melanoma patients with advanced disease, the majority of whom have received one or more therapies or are receiving therapy at the time of their blood draw.
RESULTS
Overview of patient population
In this study, we sought to explore the feasibility of using a liquid biopsy to measure cfDNA from the plasma of patients with advanced melanoma, with a focus on those who were undergoing or had previously undergone therapy. Blood was obtained for 28 melanoma patients, cfDNA extracted, concentration measured, libraries prepared, and sequencing conducted using a 61-gene panel (Supplemental Table 1). Results for 3 patients were excluded from further analysis because identified mutations did not meet our previously validated variant calling criteria (see Methods) (Janku et al., 2017). As summarized in Table 1, all patients had advanced stage melanoma and were heavily pre-treated, with 21 of 25 patients (84.0%) having received one or more previous therapies of any kind and 7 patients receiving ≥3 therapeutic regimens, including chemotherapy, a tyrosine kinase inhibitor (TKI), checkpoint inhibitor, or radiation. The majority of patients (16 of 25; 64.0%) had received a checkpoint inhibitor, such as pembrolizumab, ipilimumab, or nivolumab, at some point in their course of treatment. Six patients received a BRAF inhibitor and/or a MEK inhibitor prior to blood draw.
Table 1.
Patient characteristics
| All Patients (n = 25) |
Patients with Mutation Detected in ctDNA (n = 12) |
||||
|---|---|---|---|---|---|
| Number | %1 | Number | %2 | P-value | |
| Mean age (+/− SD) | 57.6 (+/− 16.3) | – | 57.0 (+/− 13.7) | – | 0.873 |
| Sex | |||||
| Male | 18 | 72 | 8 | 44 | 0.673 |
| Female | 7 | 28 | 4 | 57 | |
| Disease state at blood draw | |||||
| Active | 22 | 88 | 12 | 55 | 0.220 |
| NED/Inactive | 3 | 12 | 0 | 0 | |
| Disease stage at blood draw | |||||
| IIIa | 1 | 4 | 0 | 0 | 0.480 |
| IIIb | 2 | 8 | 0 | 0 | |
| IIIc | 1 | 4 | 1 | 100 | |
| IV | 21 | 84 | 11 | 52 | |
| Received any prior therapy | |||||
| Yes | 21 | 84 | 11 | 52 | 0.593 |
| No | 4 | 16 | 1 | 25 | |
| Receiving therapy at time of blood draw | |||||
| Yes | 17 | 68 | 9 | 53 | 0.673 |
| No | 8 | 32 | 3 | 38 | |
| Total number of previous therapeutic regimens | |||||
| 0 | 4 | 16 | 1 | 25 | 0.304 |
| 1–2 | 14 | 56 | 6 | 43 | |
| ≥ 3 | 7 | 28 | 5 | 71 | |
| Received chemotherapy prior to blood draw | |||||
| Yes | 2 | 8 | 2 | 100 | 0.220 |
| No | 23 | 92 | 10 | 44 | |
| Received TKI prior to blood draw | |||||
| Yes | 6 | 24 | 3 | 50 | 1.000 |
| No | 19 | 76 | 9 | 47 | |
| Received checkpoint inhibitor prior to blood draw | |||||
| Yes | 16 | 64 | 9 | 56 | 0.411 |
| No | 9 | 36 | 3 | 33 | |
| Received radiation prior to blood draw | |||||
| Yes | 10 | 40 | 7 | 70 | 0.111 |
| No | 15 | 60 | 5 | 33 | |
The denominator for this % calculation is the n=25 total patients included in this table.
The denominator for this % calculation varies row by row. It is the number of total patients shown in the left-most column for that row’s patient category.
Cell-free DNA concentration correlates with solid tumor volume
We first sought to assess whether measurement of the concentration of DNA extracted from patient plasma samples was correlated with solid tumor volume. Measurement of cfDNA was achieved for all 25 patients, with the concentration ranging from 0.6 to 390.0 ng/mL (median=7.8 ng/mL; Figure 1A). To measure solid tumor burden, we conducted a retrospective chart review of CT or PET imaging, and included results for the 15 patients who had imaging conducted within one month of blood draw. We then calculated tumor burden using a modification of RECIST 1.1 (Eisenhauer et al., 2009) in which the long diameters of all reported measurable lesions were added together, rather than limiting the number of lesions included per organ (Huang et al., 2017). Using this approach, the number of lesions measured per patient ranged from 1 to 12, the size of individual lesions ranged from 1.0 cm to 18.1 cm, and when all measurable lesions were summed for each patient, the total tumor burden per patient ranged from 0.6 cm to 48.5 cm (detailed measurements shown in Supplemental Table 2). We then compared cfDNA concentration and solid tumor volume, using Spearman rank calculation rather than Pearson’s correlation, since the cfDNA yield data was determined by Shapiro-Wilk test to be non-normally distributed. As shown in Figure 1B, for the 15 patients for whom tumor burden could be calculated, the Spearman rank calculation was significant (Spearman rho=0.5435, p=0.0363), suggesting a direct correlation between liquid biopsy- and imaging-based measurement of tumor burden. A similar comparison of cfDNA yield and serum LDH, a standard of care blood test that is prognostic in melanoma (Balch et al., 2009), was conducted but this was not significant (Spearman rho=0.3433, p=0.0929; Supplemental Figure 1). However, this analysis may have been confounded by the heterogeneity of therapies received by patients at time of blood draw (detail in Supplemental Table 2).
Figure 1. Measurement of cfDNA yield and correlation with tumor burden.
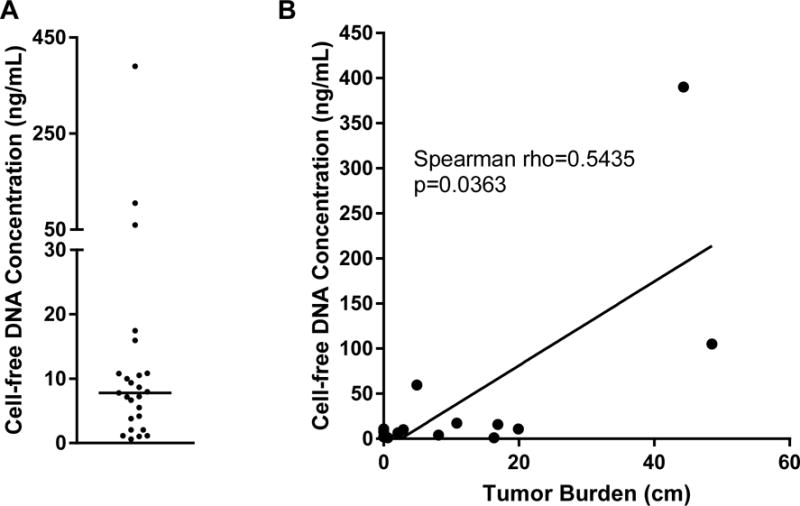
Blood was obtained in Streck DNA BCT tubes for 25 advanced melanoma patients, plasma processed within 36 hours of blood draw, and cfDNA extracted. Shown are the A. cfDNA yield per mL of plasma (horizontal line indicates median=7.8 ng/mL, range 0.6 to 390.0 ng/mL), and the B. Relationship between cfDNA concentration and tumor burden, calculated as the sum of the long diameter of all measurable lesions as detected by imaging at the timepoint closest to blood draw, with not more than one month elapsing between imaging and blood draw. Shown are the results for the 15 patients for whom tumor burden could be measured.
Mutation detection by liquid biopsy
We next sought to determine whether mutational tumor profiling could be achieved using ultra-deep sequencing of plasma cfDNA. Although 17 of 25 patients (68.0%) were receiving one or more therapies at the time of blood draw, sufficient input DNA (>2 ng) for next-generation sequencing (NGS) was extracted from 1 to 4 mL plasma for all 25 patients (median=28.4 ng, range 2.4 to 1560.0 ng; Supplemental Table 3). Sixteen total mutations were detected in the cfDNA of 12 out of 25 patients (48.0%), and this proportion did not vary significantly based on differences in disease state at time of blood draw or the number or type of prior therapies (Table 1). While others have reported low ctDNA levels for melanoma patients with subcutaneous and brain metastases (Wong et al., 2017), (metastatic sites for our patients listed in Supplemental Table 2), our study was not designed to assess the link between metastatic site and ctDNA level. Just as has been reported in other studies using NGS to detect variants in cfDNA (Chabon et al., 2016; Thompson et al., 2016), there was a wide range of allele fractions (AF; median=29.1%, range 1.1% to 63.2%) above the assay threshold of 1.0% (Figure 2A). Consistent with NGS analysis of melanoma patient tissue (Cancer Genome Atlas, 2015), BRAF V600E, NRAS Q61, and KIT mutations were detected most frequently in patient plasma samples, and BRAF and NRAS mutations were never found in the same patient plasma sample (Figure 2B).
Figure 2. Characteristics of mutations detected in plasma.
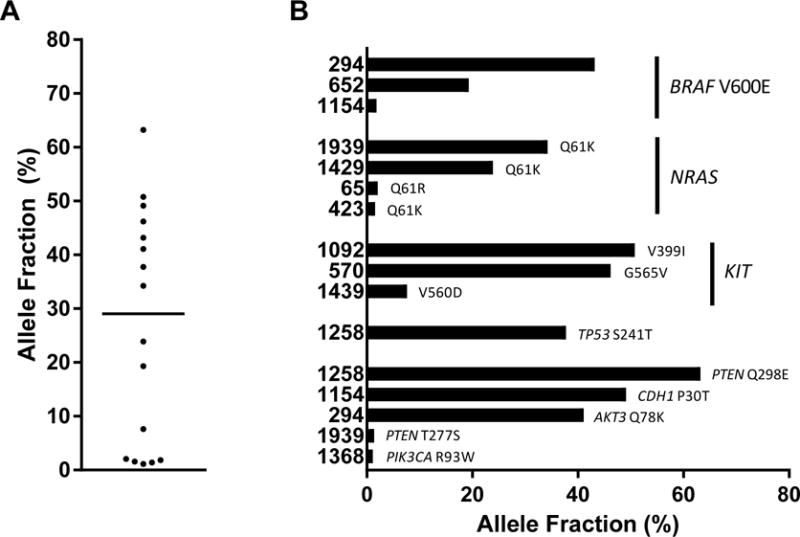
Libraries were prepared and sequencing conducted using a 61-gene panel to detect cancer-associated mutations. Shown are A. allele fractions of 16 variants detected in the plasma of 12 patients (horizontal line indicates median=29.1%, range 1.1 to 63.2%), and B. the allele fraction for each mutation detected, with mutations grouped by gene for most frequently detected mutations. Patient numbers are listed along the y-axis.
Comparison of plasma and tissue NGS
Although our study was not designed to compare plasma and tissue NGS, it was determined during chart review that 22 of 25 patients had clinical tissue NGS conducted from 1.8 to 53.5 months prior to the liquid biopsy (detailed results shown in Supplemental Table 4). Despite the gap in time between tissue and plasma collection, we reasoned that mutations detected in plasma DNA would likely reflect the mutational profile originally detected in tissue. As shown in Figure 3A, there were 10 patients with tissue NGS results for whom one or more mutations were detected in plasma. All 10 patients received one or more therapies in between tissue and plasma testing (detailed treatment history shown in Supplemental Table 4). For this group, 13 mutations were detected in the plasma, 11 of which were covered by the tissue NGS panel. Of these 11 mutations, 9 were detected in matched tissue, for a concordance of 81.8%. When the concordance calculation for these 10 patients includes 4 mutations that were detected in tissue but not plasma, the concordance drops to 9 out of 15 mutations, or 60.0%. For the 2 patients for whom a mutation was detected in plasma but not tissue (NRAS Q61K for patient 423, and PIK3CA R93W for patient 1368), we examined the tissue sequencing raw data and detected the mutation in a proportion of the total reads that was below the validated 4.0% level of detection (LOD) of the tissue NGS panel. For the 12 patients with tissue NGS results for whom no mutations were detected in plasma, 17 mutations were found in the tissue. 10 of these patients received one or more therapies in between tissue and plasma testing, and 3 had no evidence of disease by imaging at the time of their blood draw.
Figure 3. Comparison of tumor burden as measured by imaging to mutations detected in cfDNA.
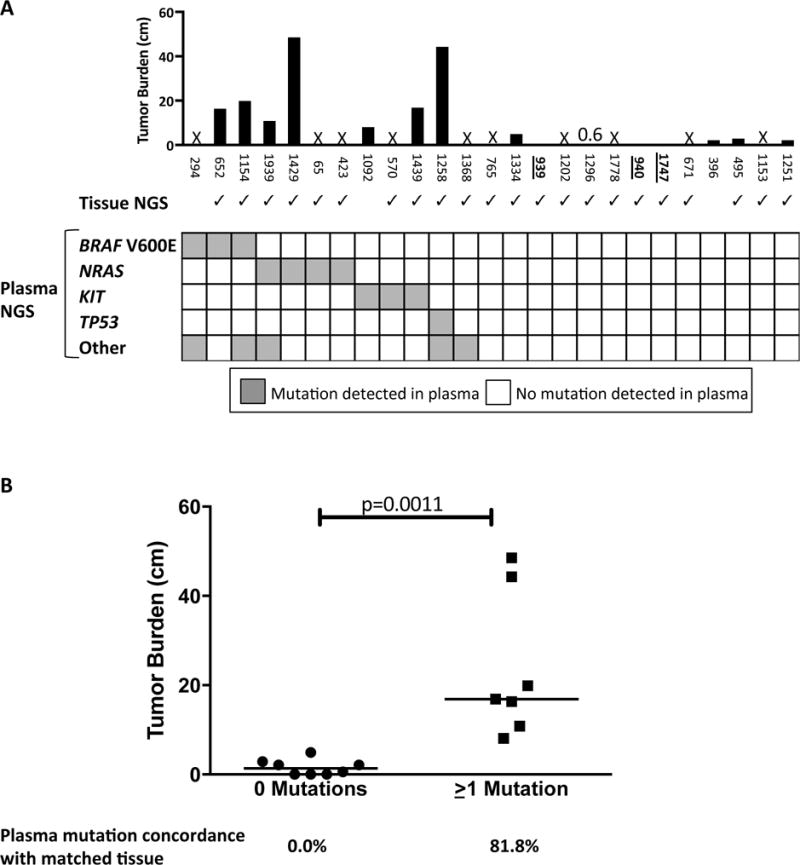
A. The black bars depict the total tumor burden (in cm) for each patient as measured by imaging conducted within one month of blood draw (see detailed measurements in Supplemental Table 2). The three patients whose numbers are bolded and underlined, 939, 940, and 1747, were determined by imaging to have no evidence of disease. An “X” indicates that tumor burden for that patient was not assessable within one month of blood draw. This was the case for 10 patients. Immediately below the bar chart, checkmarks indicate the 22 patients for whom tissue NGS was conducted. Below the check marks is a grid detailing all mutations detected for each of the 25 patients with plasma testing. Three of the NRAS mutations were Q61K, and the fourth was Q61R. The number of mutations detected per patient ranged from zero (depicted by all white squares) to two, with detected mutations depicted in grey. B. For the 15 patients with imaging-assessable tumor burden, shown is a comparison of the tumor burden for the 8 patients with no detected mutations compared to the 7 patients with one or more mutations detected in plasma. The concordance rates for any mutations detected in plasma are shown immediately below the dotplot. Horizontal line indicates the median values.
Comparison of tumor burden and sensitivity of mutation detection in plasma
Given the wide range in imaging-determined total tumor volume for our patients, we reasoned that variation in tumor burden might affect our ability to detect somatic mutations in DNA shed into plasma. To address this, we next compared tumor volume with mutations detected in plasma DNA for each of the 15 patients for whom tumor burden could be measured within one month of blood draw (Figure 3A, Supplemental Table 2). As shown in Figure 3B, the median tumor burden for patients with no detectable mutations was 1.4 cm (range 0 to 4.9 cm) as compared to 16.9 cm (range 8.1 to 48.5 cm) for patients with one or more detectable mutations (p=0.0011). No mutations were found in the plasma of any patients with a total tumor burden (the sum of all measurable lesions) of ≤ 4.9 cm. Among the 15 patients for whom tumor burden could be measured, 13 patients had tissue NGS performed, and the concordance calculations are shown in Figure 3B. Taken together, these results suggest that sensitivity and concordance of liquid biopsy-based variant detection in pre-treated patients varies with tumor burden at the time of blood draw, and that patients whose disease is well-controlled by treatment may have fewer or no detectable variants in plasma at a LOD of 1.0%. Further studies of concurrently collected, matched tissue will be necessary to more accurately assess plasma/tissue concordance.
DISCUSSION
Liquid biopsies play an increasingly important role in the application of precision medicine principles to cancer patient management, especially those cancers for which targeted therapies have become part of standard of care. However, before liquid biopsies can be more widely deployed to change clinical practice, studies to determine the clinical context in which they would be most useful are urgently needed. Others have demonstrated the ability to detect a limited number of variants in the plasma of patients at diagnosis, and reports of cfDNA for serial monitoring of melanoma patients have tended to include small numbers of patients (Girotti et al., 2016; Gray et al., 2015; Tsao et al., 2015). While these studies speak to the feasibility of liquid biopsy at diagnosis, a timepoint at which tissue biopsy is almost always simultaneously obtained, the question remains whether plasma NGS offers clinical utility once therapy has commenced. Here we describe the results of a pilot study utilizing a minimally adapted, commercially available ultra-deep sequencing platform to detect clinically relevant variants and monitor disease burden for advanced melanoma patients.
While cfDNA is more frequently thought of as a means for identifying therapeutically targetable driver and resistance mutations, we and others have also shown that measuring the amount of cfDNA in plasma can be prognostic (Cargnin et al., 2017; Thompson et al., 2016). This may be especially relevant for our cohort of heavily pre-treated patients, for whom mutations were detected in 12 of 25 plasma samples, but cfDNA yield was quantifiable for all 25 patients. Cell-free DNA yields were significantly associated with solid tumor burden as determined by imaging, suggesting that cell-free DNA may provide a blood-based means to monitor tumor burden more frequently than imaging. While our study was not designed to serially compare imaging- with blood-based measurement of tumor volume, a larger study with concurrent imaging and blood collection could be utilized to assess whether cfDNA might better discern pseudoprogression or atypical response patterns from actual progression for patients receiving checkpoint inhibitors. Further studies could also assess whether cfDNA yield can accurately predict progression prior to imaging. Although additional studies would be required to validate the utility of cfDNA yield as a prognostic marker for advanced melanoma patients, these pilot results suggest possible utility beyond detection of driver and resistance mutations.
For our study, we selected a plasma NGS panel encompassing full exon coverage for 61 genes. To be broadly clinically actionable, molecular monitoring of patients receiving targeted agents must go beyond the targeted loci to comprehensively detect associated resistance mutations. For instance, acquired resistance to the BRAF inhibitors vemurafenib and dabrafenib has been associated with multiple genetic alterations including mutations in NRAS, KRAS, MAP2K1, and CDKN2A (Shi et al., 2014). In our study, 16 mutations were detected in 8 different genes for 12 patients’ plasma samples, including 3 patients with a detected BRAF V600E mutation. Two of these 3 patients received a BRAF inhibitor prior to blood draw, and the persistent detection of the targeted mutation suggests the targeted therapy may not be adequately controlling disease. Although no known resistance mutations were detected in the plasma of any of the patients who received a TKI, repeat liquid biopsies could be utilized for detection of such therapy resistance, just as we have previously demonstrated for lung cancer (Thompson et al., 2016). This pilot study suggests that liquid biopsy detection of therapeutically targetable mutations is feasible for a pre-treated population, although larger studies will be necessary to better define the clinical utility of this data.
To our knowledge, we describe here the first prospective study using an NGS panel of a broad set of cancer-relevant genes for analysis of advanced melanoma patient plasma who have received multiple prior therapies or are currently on therapy. Given the increasing number of targeted therapies being developed with relevance for melanoma patients, and the approval of checkpoint inhibitors as standard of care, our gene panel will likely require expansion for the future management of melanoma patients. While a baseline measurement of plasma-based BRAF V600 status alone has been shown to have prognostic value (Knol et al., 2016), recent discoveries articulating the genomic heterogeneity underlying acquired resistance to BRAF inhibition speak to the need for monitoring of multiple loci over the course of a patient’s therapy (Krepler et al., 2016; Shi et al., 2014). Whole exome sequencing of paired baseline and relapse tissue samples for 4 melanoma patients who had received pembrolizumab revealed resistance-associated mutations in JAK1, JAK2, and B2M (Zaretsky et al., 2016). While JAK2 hotspots are covered on the tissue NGS panel utilized in our clinical lab, no mutations were detected in tissue for that gene, and these 3 genes are not covered by our current plasma panel. Tumor molecular burden, as assessed by whole exome sequencing of melanoma and other tumors (Rizvi et al., 2015; Van Allen et al., 2015) has been associated with response to immune checkpoint inhibitors, suggesting that expanding the coverage of our 61-gene panel would enhance the clinical actionability of the ctDNA test for patient monitoring. It may also be desirable to improve the level of detection of this assay to detect mutations with an allele fraction in plasma of less than 1.0%. Tie and colleagues recently showed that detection of therapy resistance for colorectal cancer patients by cfDNA can precede imaging by two or more months when using a sequencing based approach with a sensitivity to detect variants at or below 0.01% allele fraction (Tie et al., 2016). A similar report for breast cancer patients utilized a highly sensitive digital PCR approach (Garcia-Murillas et al., 2015). While the tumor burden was either low or undetectable for our 8 patients with available imaging and no detectable variants in plasma, our sensitivity of variant detection, especially in the context of minimal residual disease, may have been improved with a lower LOD. In summary, this proof of concept work demonstrates the feasibility of utilizing non-invasive, cfDNA-based molecular monitoring of melanoma patients. Further study with larger prospective patient cohorts, and encompassing a range of timepoints over the course of the patients’ disease, will be necessary to validate and firmly establish the clinical utility of this approach.
METHODS
Patients
This was a single-center, pilot study conducted at the Abramson Cancer Center at the University of Pennsylvania from October 2014 to February 2016. The study was approved by the Institutional Review Board of the University of Pennsylvania (IRB #703001). A single blood draw was obtained from each of 28 patients with a confirmed diagnosis of metastatic melanoma after written informed consent. Clinical variables, prior and current therapy, and results of tumor tissue sequencing were extracted via chart review. Tumor burden was calculated as the sum of the longest diameter of all reported measurable lesions as detected by CT or PET imaging performed within one month of research blood draw.
Blood sample collection and processing
Blood was drawn by conventional venipuncture into Streck DNA BCT tubes (Streck, Omaha, NE, USA), and processed for plasma collection within 36 hours of blood draw. In brief, the tubes were first spun at 1668g for 10 minutes at room temperature. The supernatant was transferred to fresh 2mL Eppendorf tubes without disturbing the cellular layer, and this plasma fraction was then centrifuged one or two additional times at 21000g for 10 minutes. Plasma samples were immediately transferred for storage at −80C.
Plasma DNA extraction and next-generation sequencing
Plasma samples were shipped overnight on dry ice to Illumina, Inc (San Diego, CA, USA). Cell-free DNA was extracted from one to four mL plasma per sample (see details in Supplemental Table 3) using the QIAamp Circulating Nucleic Acid Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. The final 200uL elution volume was either stored at −20C or immediately processed further. To determine the yield of cfDNA, a 2100 Bioanalyzer instrument (Agilent Technologies, Santa Clara, CA, USA) was used with the High Sensitivity DNA Kit (Agilent Technologies) to determine DNA yield in the 100 – 400 base pair range. Libraries were prepared and a panel of 61 genes (Supplemental Table 1) was evaluated by ultra-deep sequencing on an Illumina Next-Seq® (Illumina, Inc), using protocols adapted from TruSeq Nano (Illumina, Inc) library preparation and Nextera Rapid Capture (Illumina, Inc) target enrichment (Janku et al., 2017). Reads were mapped using hg19, and the LOD for detection of single nucleotide variants for sequencing of plasma-derived DNA was 1.0%. All cfDNA testing as described in this manuscript is for research use only, and not for use in diagnostic procedures.
Tissue sample next-generation sequencing
Chart review was used to identify 22 patients who also had tissue sequenced at a previous timepoint during the course of their therapy. The elapsed time between tissue and plasma cfDNA sequencing ranged from 49 to 1,605 days (see Supplemental Table 4). All tissue sequencing was conducted at the University of Pennsylvania Center for Personalized Diagnostics (CPD) CAP/CLIA-Certified laboratory as previously described (Hiemenz et al., 2016; Thompson et al., 2016; Yee et al., 2016). For all but one patient, the Illumina TruSeq® Amplicon – Cancer Panel (TSACP, FC-130-1008, Illumina, Inc) was used to sequence targeted hotspots or larger exonic regions for 47 genes (Supplemental Table 5). One patient (1368) had insufficient tissue DNA for the TSACP, so the sample was analyzed using the Penn Precision Panel for mutations in a smaller panel of 20 genes (Thompson et al., 2016). Analysis of results for both panels was performed using a clinically validated informatics pipeline, using hg19 genome build to map reads (Daber et al., 2013). The level of detection for sequencing of tissue was 4.0%.
Bioinformatics and statistical analysis
Nonsynonymous mutations were called from plasma-derived cfDNA if they met the following criteria: AF ≥ 1.0%, mean target coverage ≥ 300, and ≥ 2 mutant reads. These criteria have been previously validated as described (Janku et al., 2017). In brief, DNA spiking experiments were used to assess the sensitivity, specificity, and negative and positive predictive value of the test at various sequence read thresholds, with optimal sensitivity for clinical samples achieved at a threshold of ≥2 unique variant sequence reads. Allele fraction is defined as the number of mutant reads divided by the number of total reads at a given locus.
Descriptive statistics were computed for study variables. Normality was evaluated by Shapiro-Wilk test. Comparison between patients with and without mutations detected in cfDNA was done using Fisher’s exact test for categorical variables (e.g., sex, disease status, etc.), t-test for age, and Mann-Whitney test for tumor burden. Correlations between cfDNA yield and serum LDH, as well as correlations between cfDNA yield and solid tumor burden were examined by the nonparametric Spearman’s rank correlation, because the cfDNA data is not normally distributed. Significance of the correlations was evaluated based on a Fisher’s z transformation. The distribution of the tumor burden was compared between patients whose plasma and tissue sequencing results agreed and disagreed using Mann-Whitney test. Analyses were performed with GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA, USA) or Stata 14.0 (StataCorp LLC, College Station, TX, USA). All tests were two-sided. A P value < 0.05 was considered statistically significant.
Supplementary Material
SIGNIFICANCE.
Non-invasive molecular monitoring is especially promising for cancers such as melanoma where targeted and immune therapies are routinely used as part of standard of care, and yet tissue biopsy of a single tumor site may not represent the full genetic spectrum of the patient’s disease. While the utility of liquid biopsy at diagnosis has been established, its application to patients with pre-treated, advanced disease remains unknown. This study demonstrates the feasibility of non-invasive molecular monitoring of melanoma patients, even those who have received multiple prior therapies or are currently on therapy.
Acknowledgments
We gratefully acknowledge all the patients who participated in this study. This study was supported by the NCI SPORE in Skin Cancer (1 P50 CA174523) and Tara Miller Foundation.
References
- Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, Buzaid AC, Cochran AJ, Coit DG, Ding S, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27:6199–206. doi: 10.1200/JCO.2009.23.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas, N. Genomic Classification of Cutaneous Melanoma. Cell. 2015;161:1681–96. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cargnin S, Canonico PL, Genazzani AA, Terrazzino S. Quantitative Analysis of Circulating Cell-Free DNA for Correlation with Lung Cancer Survival: A Systematic Review and Meta-Analysis. J Thorac Oncol. 2017;12:43–53. doi: 10.1016/j.jtho.2016.08.002. [DOI] [PubMed] [Google Scholar]
- Chabon JJ, Simmons AD, Lovejoy AF, Esfahani MS, Newman AM, Haringsma HJ, Kurtz DM, Stehr H, Scherer F, Karlovich CA, et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun. 2016;7:11815. doi: 10.1038/ncomms11815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SJ, Punt CJ, Iannotti N, Saidman BH, Sabbath KD, Gabrail NY, Picus J, Morse MA, Mitchell E, Miller MC, et al. Prognostic significance of circulating tumor cells in patients with metastatic colorectal cancer. Ann Oncol. 2009;20:1223–9. doi: 10.1093/annonc/mdn786. [DOI] [PubMed] [Google Scholar]
- Couraud S, Vaca-Paniagua F, Villar S, Oliver J, Schuster T, Blanche H, Girard N, Tredaniel J, Guilleminault L, Gervais R, et al. Noninvasive diagnosis of actionable mutations by deep sequencing of circulating free DNA in lung cancer from never-smokers: a proof-of-concept study from BioCAST/IFCT-1002. Clin Cancer Res. 2014;20:4613–24. doi: 10.1158/1078-0432.CCR-13-3063. [DOI] [PubMed] [Google Scholar]
- Daber R, Sukhadia S, Morrissette JJ. Understanding the limitations of next generation sequencing informatics, an approach to clinical pipeline validation using artificial data sets. Cancer Genet. 2013;206:441–8. doi: 10.1016/j.cancergen.2013.11.005. [DOI] [PubMed] [Google Scholar]
- Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, Dunning MJ, Gale D, Forshew T, Mahler-Araujo B, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. The New England journal of medicine. 2013;368:1199–209. doi: 10.1056/NEJMoa1213261. [DOI] [PubMed] [Google Scholar]
- Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–47. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- Garcia-Murillas I, Schiavon G, Weigelt B, Ng C, Hrebien S, Cutts RJ, Cheang M, Osin P, Nerurkar A, Kozarewa I, et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Science translational medicine. 2015;7:302ra133. doi: 10.1126/scitranslmed.aab0021. [DOI] [PubMed] [Google Scholar]
- Girotti MR, Gremel G, Lee R, Galvani E, Rothwell D, Viros A, Mandal AK, Lim KH, Saturno G, Furney SJ, et al. Application of Sequencing, Liquid Biopsies, and Patient-Derived Xenografts for Personalized Medicine in Melanoma. Cancer Discov. 2016;6:286–99. doi: 10.1158/2159-8290.CD-15-1336. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Cao M, Mayo-De-Las-Casas C, Molina-Vila MA, De Mattos-Arruda L, Munoz-Couselo E, Manzano JL, Cortes J, Berros JP, Drozdowskyj A, Sanmamed M, et al. BRAF mutation analysis in circulating free tumor DNA of melanoma patients treated with BRAF inhibitors. Melanoma Res. 2015;25:486–95. doi: 10.1097/CMR.0000000000000187. [DOI] [PubMed] [Google Scholar]
- Gray ES, Rizos H, Reid AL, Boyd SC, Pereira MR, Lo J, Tembe V, Freeman J, Lee JH, Scolyer RA, et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget. 2015;6:42008–18. doi: 10.18632/oncotarget.5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiemenz MC, Kadauke S, Lieberman DB, Roth DB, Zhao J, Watt CD, Daber RD, Morrissette JJ. Building a Robust Tumor Profiling Program: Synergy between Next-Generation Sequencing and Targeted Single-Gene Testing. PloS one. 2016;11:e0152851. doi: 10.1371/journal.pone.0152851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, Xu W, Harmon S, Giles JR, Wenz B, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. 2017 doi: 10.1038/nature22079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W, Shi H, Sun L, Piva M, Song C, Kong X, Moriceau G, Hong A, Dahlman KB, Johnson DB, et al. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell. 2015;162:1271–85. doi: 10.1016/j.cell.2015.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janku F, Zhang S, Waters J, Liu L, Huang H, Subbiah V, Hong DS, Karp D, Fu S, Cai X, et al. Development and validation of an ultra-deep next-generation sequencing assay for testing of plasma cell-free DNA from patients with advanced cancer. Clin Cancer Res. 2017 doi: 10.1158/1078-0432.CCR-17-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaisaki PJ, Cutts A, Popitsch N, Camps C, Pentony MM, Wilson G, Page S, Kaur K, Vavoulis D, Henderson S, et al. Targeted Next-Generation Sequencing of Plasma DNA from Cancer Patients: Factors Influencing Consistency with Tumour DNA and Prospective Investigation of Its Utility for Diagnosis. PloS one. 2016;11:e0162809. doi: 10.1371/journal.pone.0162809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoja L, Lorigan P, Zhou C, Lancashire M, Booth J, Cummings J, Califano R, Clack G, Hughes A, Dive C. Biomarker utility of circulating tumor cells in metastatic cutaneous melanoma. J Invest Dermatol. 2013;133:1582–90. doi: 10.1038/jid.2012.468. [DOI] [PubMed] [Google Scholar]
- Knol AC, Vallee A, Herbreteau G, Nguyen JM, Varey E, Gaultier A, Theoleyre S, Saint-Jean M, Peuvrel L, Brocard A, et al. Clinical significance of BRAF mutation status in circulating tumor DNA of metastatic melanoma patients at baseline. Exp Dermatol. 2016;25:783–8. doi: 10.1111/exd.13065. [DOI] [PubMed] [Google Scholar]
- Krebs MG, Sloane R, Priest L, Lancashire L, Hou JM, Greystoke A, Ward TH, Ferraldeschi R, Hughes A, Clack G, et al. Evaluation and prognostic significance of circulating tumor cells in patients with non-small-cell lung cancer. J Clin Oncol. 2011;29:1556–63. doi: 10.1200/JCO.2010.28.7045. [DOI] [PubMed] [Google Scholar]
- Krepler C, Xiao M, Sproesser K, Brafford PA, Shannan B, Beqiri M, Liu Q, Xu W, Garman B, Nathanson KL, et al. Personalized Preclinical Trials in BRAF Inhibitor-Resistant Patient-Derived Xenograft Models Identify Second-Line Combination Therapies. Clin Cancer Res. 2016;22:1592–602. doi: 10.1158/1078-0432.CCR-15-1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, Mandala M, Demidov L, Stroyakovskiy D, Thomas L, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. The New England journal of medicine. 2014;371:1867–76. doi: 10.1056/NEJMoa1408868. [DOI] [PubMed] [Google Scholar]
- Larkin J, Hodi FS, Wolchok JD. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. The New England journal of medicine. 2015;373:1270–1. doi: 10.1056/NEJMc1509660. [DOI] [PubMed] [Google Scholar]
- Lipson EJ, Velculescu VE, Pritchard TS, Sausen M, Pardoll DM, Topalian SL, Diaz LA., Jr Circulating tumor DNA analysis as a real-time method for monitoring tumor burden in melanoma patients undergoing treatment with immune checkpoint blockade. Journal for immunotherapy of cancer. 2014;2:42. doi: 10.1186/s40425-014-0042-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long GV, Stroyakovskiy D, Gogas H, Levchenko E, De Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Grob JJ, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet. 2015;386:444–51. doi: 10.1016/S0140-6736(15)60898-4. [DOI] [PubMed] [Google Scholar]
- Luo X, Mitra D, Sullivan RJ, Wittner BS, Kimura AM, Pan S, Hoang MP, Brannigan BW, Lawrence DP, Flaherty KT, et al. Isolation and molecular characterization of circulating melanoma cells. Cell Rep. 2014;7:645–53. doi: 10.1016/j.celrep.2014.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinzani P, Salvianti F, Zaccara S, Massi D, De Giorgi V, Pazzagli M, Orlando C. Circulating cell-free DNA in plasma of melanoma patients: qualitative and quantitative considerations. Clinica chimica acta; international journal of clinical chemistry. 2011;412:2141–5. doi: 10.1016/j.cca.2011.07.027. [DOI] [PubMed] [Google Scholar]
- Rao C, Bui T, Connelly M, Doyle G, Karydis I, Middleton MR, Clack G, Malone M, Coumans FA, Terstappen LW. Circulating melanoma cells and survival in metastatic melanoma. Int J Oncol. 2011;38:755–60. doi: 10.3892/ijo.2011.896. [DOI] [PubMed] [Google Scholar]
- Riethdorf S, Fritsche H, Muller V, Rau T, Schindlbeck C, Rack B, Janni W, Coith C, Beck K, Janicke F, et al. Detection of circulating tumor cells in peripheral blood of patients with metastatic breast cancer: a validation study of the CellSearch system. Clin Cancer Res. 2007;13:920–8. doi: 10.1158/1078-0432.CCR-06-1695. [DOI] [PubMed] [Google Scholar]
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. The New England journal of medicine. 2015a;372:30–9. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, Mcneil C, Lotem M, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. The New England journal of medicine. 2015b;372:2521–32. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- Robert C, Thomas L, Bondarenko I, O’day S, Weber J, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. The New England journal of medicine. 2011;364:2517–26. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- Schreuer M, Meersseman G, Van Den Herrewegen S, Jansen Y, Seremet T, Bott A, Chevolet I, Wilgenhof S, Maertens G, Neyns B. Applications for quantitative measurement of BRAF V600 mutant cell-free tumor DNA in the plasma of patients with metastatic melanoma. Melanoma Res. 2016;26:157–63. doi: 10.1097/CMR.0000000000000224. [DOI] [PubMed] [Google Scholar]
- Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4:80–93. doi: 10.1158/2159-8290.CD-13-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA: a cancer journal for clinicians. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- Thompson JC, Yee SS, Troxel AB, Savitch SL, Fan R, Balli D, Lieberman DB, Morrissette JD, Evans TL, Bauml J, et al. Detection of Therapeutically Targetable Driver and Resistance Mutations in Lung Cancer Patients by Next-Generation Sequencing of Cell-Free Circulating Tumor DNA. Clin Cancer Res. 2016;22:5772–5782. doi: 10.1158/1078-0432.CCR-16-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tie J, Wang Y, Tomasetti C, Li L, Springer S, Kinde I, Silliman N, Tacey M, Wong HL, Christie M, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Science translational medicine. 2016;8:346ra92. doi: 10.1126/scitranslmed.aaf6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao SC, Weiss J, Hudson C, Christophi C, Cebon J, Behren A, Dobrovic A. Monitoring response to therapy in melanoma by quantifying circulating tumour DNA with droplet digital PCR for BRAF and NRAS mutations. Sci Rep. 2015;5:11198. doi: 10.1038/srep11198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Geukes Foppen MH, Goldinger SM, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–11. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SQ, Raleigh JM, Callahan J, Vergara IA, Ftouni S, Hatzimihalis A, Colebatch AJ, Li J, Semple T, Doig K, et al. Circulating Tumor DNA Analysis and Functional Imaging Provide Complementary Approaches for Comprehensive Disease Monitoring in Metastatic Melanoma. JCO Precis Oncol. 2017;1:14. doi: 10.1200/PO.16.00009. [DOI] [PubMed] [Google Scholar]
- Xi L, Pham TH, Payabyab EC, Sherry RM, Rosenberg SA, Raffeld M. Circulating Tumor DNA as an Early Indicator of Response to T-cell Transfer Immunotherapy in Metastatic Melanoma. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-16-0613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee SS, Lieberman DB, Blanchard T, Rader J, Zhao J, Troxel AB, Desloover D, Fox AJ, Daber RD, Kakrecha B, et al. A novel approach for next-generation sequencing of circulating tumor cells. Mol Genet Genomic Med. 2016;4:395–406. doi: 10.1002/mgg3.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. The New England journal of medicine. 2016;375:819–29. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.