

Abstract
Pirfenidone is an anti-inflammatory and anti-fibrotic drug that has shown efficacy in lung and kidney fibrosis. Because inflammation and fibrosis have been linked to the progression of osteoarthritis, we investigated the effects of oral Pirfenidone in a mouse model of cartilage injury, which results in chronic inflammation and joint-wide fibrosis in mice that lack hyaluronan synthase 1 (Has1−/−) in comparison to wild-type. Femoral cartilage was surgically injured in wild-type and Has1−/− mice, and Pirfenidone was administered in food starting after 3 days. At 4 weeks, Pirfenidone reduced the appearance, on micro-computed tomography, of pitting in subchondral bone at, and cortical bone surrounding, the site of cartilage injury. This corresponded with a reduction in fibrotic tissue deposits as observed with gross joint surface photography. Pirfenidone resulted in significant recovery of trabecular bone parameters affected by joint injury in Has1−/− mice, although the effect in wild-type was less pronounced. Pirfenidone also increased Safranin-O staining of growth plate cartilage after cartilage injury and sham operation in both genotypes. Taken together with the expression of selected extracellular matrix, inflammation, and fibrosis genes, these results indicate that Pirfenidone may confer chondrogenic and bone-protective effects, although the well-known anti-fibrotic effects of Pirfenidone may occur earlier in the wound-healing response than the time point examined in this study. Further investigations to identify the specific cell populations in the joint and signaling pathways that are responsive to Pirfenidone are warranted, as Pirfenidone and other anti-fibrotic drugs may encourage tissue repair and prevent progression of post-traumatic osteoarthritis.
Keywords: Cartilage injury, subchondral bone, joint fibrosis, hyaluronan synthase 1, Pirfenidone
Graphical abstract
Pirfenidone (PFD) – an anti-inflammatory, anti-fibrotic drug – has shown efficacy in lung and kidney fibrosis, while synovial inflammation and joint fibrosis are hallmarks of osteoarthritis progression. In a cartilage injury model in wild-type and hyaluronan synthase 1 null mice, orally administered Pirfenidone, started at 3 days after injury, conferred joint-protective effects. Pirfenidone reduced appearance of fibrotic tissue deposition on the joint surface after 4 weeks post-injury. Micro-computed tomography showed that Pirfenidone also reduced the loss of cortical, subchondral, and trabecular bone.
INTRODUCTION
The cellular response mechanisms that underlie intrinsic cartilage and subchondral bone repair after joint injury are poorly understood, although it is now clear that they cannot be delineated under the simplistic paradigm of a balance of matrix anabolism vs catabolism 1–4. Therefore, the discovery of new targetable disease mechanisms is essential to the design of effective biological therapies to complement existing strategies for osteoarthritis (OA) prevention in the clinic. With respect to post-traumatic OA, the cellular reaction to joint tissue injury can be viewed as a classical “wound-healing” response, involving a balance between “inflammatory/fibrotic” and “chondrogenic/osteogenic” pathways in the injured joint environment.
Within the classic wound healing response, tissue injury results in cell proliferation, differentiation, extracellular matrix production and remodeling; alterations in this process can lead to uncontrolled deposition of granulation tissue, fibrosis, and scar formation. These phenomena are well described in dermal wounds 5 and internal organs, like the lung and liver 6–10, where recurrent inflammation may lead to chronic fibrosis and deterioration of function. In organ fibrosis, various anti-fibrotic drugs have shown clinical promise in reducing the formation of scars and halting, or at least slowing the progression of, functional loss 11.
Strategies used in organ fibrosis may therefore hold promise for applications in the injured joint, where the processes of inflammation and fibrosis are also hallmarks of OA development. Substantial evidence for the activation of collagen synthesis and fibrotic remodeling in human OA cartilage has recently been described in at least eight independent genome-wide analysis studies 12. There is also accumulating evidence from human OA studies for scarring responses in bone 13; 14, meniscus and cartilage 15, as well as joint capsule and synovium 16, with similar findings in animal models of OA 17–19. These suggested that fibrotic scarring of joint tissues represents a potentially important drug target for OA therapy. Anti-fibrotics that are commonly used for lung and kidney fibrosis have shown both safety and efficacy with extended use 11; 20. Prominent among these is 5-methyl-1-phenyl-2-(1H)-pyridone or pirfenidone (PFD), which has been evaluated in animal models and used clinically in a wide range of inflammatory or fibrotic conditions – including idiopathic pulmonary fibrosis 21, primary sclerosing cholangitis 22, chronic hepatitis C 23, myelofibrosis 24, neurofibromatosis 25, fibrotic disorders of cardiac 26 and renal 27 origin, and corneal scarring 28 – and is currently in clinical trials for scleroderma 29.
It has been suggested that the anti-fibrotic action of PFD stems from suppression of TGF-β1 driven collagen type I expression, and the underlying mechanism is under active study 30; 31. For example, PFD inhibits the non-canonical hedgehog (Hh) pathway by shortening the half-life of endogenous full-length glioma-associated oncogene homolog 2 (GLI2), a transcription factor, by more than 3-fold, and thereby reduces Hh/TGF-β signaling and expression of collagen type I 32. PFD has also been shown to reduce IL-1β-induced hyaluronan production in orbital fibroblasts 33. PFD has shown pain relief in RA 34 and may also be able to block nitric oxide release in articular chondrocytes 35, but PFD has not yet been tested in vivo against post-traumatic OA.
Similarities between the chronic inflammation and wide-spread joint fibrosis in this cartilage injury model and the inflammatory events that lead to fibrosis in other organs 36; 37 have therefore led us to consider PFD as a possible disease-modifying drug. Accordingly, in this study we use gene expression, histology, gross pathology, and micro-computed tomography (μCT) to evaluate the effects of PFD after cartilage injury in the murine joint. Additionally, among the numerous genes linked to the tissue injury response, hyaluronan synthase 1 (Has1) has been linked to abnormal wound healing and hypertrophy in the skin, muscle, and lungs 8; 38–40, as well as the synovial membranes of OA patients 41 and murine joints after cartilage injury 42. Therefore, we also use homozygous Has1 knockout (Has1−/−) mice, which show abnormal wound healing 38 and chronic inflammation and fibrosis with joint injury 42, to examine whether PFD can alter the outcome in the absence of Has1.
METHODS
Cartilage injury model
Wild-type and Has1−/− male C57Bl/6 mice (skeletally mature, 11–12 weeks old, 28.3 ± 2.5 g and 23.9 ± 1.4 g, respectively) from in-house colonies were used under approval of the Rush University Institutional Animal Care and Use Committee. Mice were housed in groups of 4–5 males, which were treated with no surgery (naïve), sham operation, or cartilage injury, without or with PFD; individual mice were then assigned towards experimental outcomes as needed. With a mouse under anesthesia, cartilage injury to the patellar groove of the right knee was performed as previously described 42. Briefly, after medial parapatellar arthrotomy, femoral groove cartilage was debrided from a rectangular surface (~2-mm wide × 4-mm long) without penetrating the subchondral bone. Sham surgery included all steps, except cartilage debridement, and naïve mice did not receive anesthetic or surgery. All mice were maintained at cage activity with food and water provided ad libitum until sacrifice after 4 weeks.
Oral dosing with PFD
All PFD groups were switched from the standard chow to PFD chow (5 mg PFD per g chow, Envigo, formerly Harlan) at 12 weeks for naïve mice and 3 days after surgery for sham and injury groups. PFD chow was weighed at least twice weekly to monitor intake of the drug. Using an estimated absorption efficiency of ~20% 43 and average weights for each genotype, the daily dosage of PFD absorbed into the bloodstream upon ingestion of PFD chow was estimated by cage. For each group (naïve control with normal chow (NN), naïve with PFD chow (NP), sham (SN), sham with PFD (SP), injured (IN), injured with PFD (IP)), the minimal number of mice needed for each experimental outcome was determined based on previous studies 42; 44, and mouse numbers in experimental groups are given (Table S-1) with experiment timelines (Figure S-1). Treatments were the same within a cage, and mice within each cage were randomly assigned to experimental outcomes as described below.
Macroscopic joint imaging and histology
Joint wide pathology (n = 3) was assessed in naïve and operated joints, without and with PFD treatment, as previously described 42; 44 for all experimental groups. These joints were then reserved for subsequent μCT, as described further in the Methods below.
For histology (n = 3), skin and muscle were removed, joints decalcified, processed and embedded as described 42. Specimens were sectioned (6-μm thick) with sagittal sections 1–60, 61–120, and 121–190 spanning the medial, central, and lateral regions respectively 44. Sections from medial compartment, joint midline, and lateral compartment were stained with Safranin-O or biotinylated TNF-stimulated gene 6 protein (bTSG6) to localize sulfated glycosaminoglycans and hyaluronan, respectively 42. Histology was used in combination with macro-images to evaluate structural alterations in multiple tissue types adjacent to the injury and throughout the joint.
Quantitative PCR (qPCR) for selected genes and gene arrays
For gene expression in whole joints, hind legs were harvested immediately after sacrifice and whole knee joints isolated as previously described 42 before storage at −20°C in RNAlater® (Life Technologies), and RNA purification, cDNA synthesis, qPCR (3 technical replicates) with Taqman® (Life Technologies) inventoried primers (Table S-2) performed as described 44. Transcript abundance of collagens (Col1a1, Col2a1, Col3a1), hyalectans (Acan, Vcan (V2 isoform)), hyaluronan-metabolism proteins (Has1, Has2, Tnfaip6, Hyal1, Hyal2, Cemip), and Gapdh (housekeeping gene) was calculated as 1000×2−ΔCt, with ΔCt = [Ct (gene of interest) − Ct (Gapdh)] and Ct > 37 considered “not detectable”. RT2 Profiler PCR Arrays (Qiagen) were used for fibrosis (PAMM-120ZA) and NF-κB signaling targets (PAMM-225ZA) genes, again with Gapdh as the housekeeping gene. For the 15 genes present on both arrays, an average ΔCt value was taken. Fold-change in expression was calculated as 2−ΔΔCt, where ΔΔCt is the difference in average ΔCt values between two experimental groups.
μCT for bone morphology and characterization
After photography of joint surface changes, distal femurs (n = 3) from all experimental groups (naïve, sham, injured) were imaged with a SCANCO-35 μCT desktop scanner at an isotropic spatial resolution of 12μm. Trabecular bone volumes of interest were semi-automatically segmented at the endocortical boundary in the metaphysis and diaphysis (Figure S-2) with on-board software (SCANCO Medical), which was then used to estimate tissue mineral density (TMD), bone volume fraction (BV/TV), and trabecular bone parameters of number (Tb.N), thickness (Tb.Th), and spacing (Tb.Sp). Cortical bone volumes of interest were similarly segmented for estimation of cortical bone parameters.
Statistical analyses
Analysis of μCT and gene expression data was performed with software (Matlab R2016a), and all data are presented as mean ± standard deviation, unless otherwise indicated. Normality of the data sets were tested with Kolmogorov-Smirnov test prior to election of a parametric or non-parametric test. Nonparametric analyses were therefore performed in cases where data sets were found to be not normally distributed. The experiment-wide significance level was α = 0.05.
Two-tailed Student’s t test were used to compare mouse weights and chow intake. For gene expression, one-way analysis of variance (ANOVA) was used to compare Gapdh Ct values across experimental groups to confirm selection of a housekeeping gene that did not vary with treatment. Because most sets of ΔCt values were found to not fall under the normality assumption, nonparametric tests were used to compare between experimental groups. Therefore, Kruskal-Wallis Test was used first to determine the effect of surgery among the naïve, sham, and injury groups, separately for wild-type and Has1−/− groups, and then to test among SN, SP, IN, and IP groups, to determine the effects of cartilage injury and PFD dosing. Post hoc comparisons using a Dunn-Sidak correction were used to identify significantly different experimental groups. For the normally-distributed bone parameters from μCT, two-way ANOVA was used to evaluate the effects of surgery (naïve, sham, cartilage injury) and drug administration (none, PFD). For statistically significant effects, post hoc pair-wise comparisons were performed with Tukey’s honest significant difference test.
RESULTS
Wild-type and Has1−/− mice were operated on as described previously 42 and evaluated 4 weeks after injury with joint surface pathology, distal femur μCT imaging, gene expression, and histology (Figure S-1). No adverse events occurred in any experimental group. On average, wild-type and Has1−/− adult male mice ate 3.8 ± 1.2 g and 4.3 ± 0.8 g, respectively, of chow daily, equivalent to a daily PFD intake of 19.0 ± 5.8 mg and 21.6 ± 4.2 mg. Although wild-type and Has1−/− mice were significantly different in weight at the start of the experiments (p < 0.001), PFD-dosed chow intake was not significantly different between wild-type and Has1−/− (p = 0.37) nor among naïve, sham, or injured groups (p > 0.15). However, the difference in average weights between genotypes led to a higher, but statistically insignificant (p = 0.06), estimated daily dosage of PFD in plasma upon ingestion of PFD chow in Has1−/− (180 ± 35 mg/kg) compared to wild-type (135 ± 41 mg/kg).
Joint pathology 28 days after intra-articular cartilage injury in wild-type and Has1−/− mice
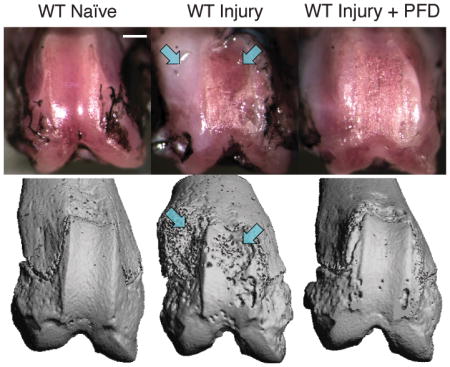
As previously described in this model 42, a fibrotic response after debridement of patellar groove cartilage was observed, specifically as an excessive deposition of fibrous extracellular matrix, at both trochlear ridges near the injury site. This fibrotic tissue deposition was generally exacerbated in the absence of Has1 (Figure 1A). μCT of these same specimens (Figure 1B) showed bony erosion and pitting co-localized with the fibrotic deposits observed with joint surface photography. No fibrosis or bony erosion was detected in age-matched naïve joints.
Figure 1. Macroscopic and μCT imaging of injury site in naïve, injured and sham-injured joints of wild-type (WT) and hyaluronan synthase 1 knockout (Has1−/−) mice with and without pirfenidone (PFD) dosing.

Joint-matched macroscopy (rows A and C) and μCT images (rows B and D) are shown with the lateral aspect to the left of each image. Representative images are shown with arrows that indicate the co-localization of fibrosis and bone erosion after cartilage injury in WT and Has1−/− mice. Images were obtained at 28 days after injury and illustrate the outcomes of no surgery (naïve), cartilage injury, and sham operation with no drug (rows A and B) and with PFD (rows C and D) dosing starting at 3 days after injury. μCT images were scaled to approximate the sizing of joint surface photographs, whose scale (bar = ~2mm) may vary with perspective and plane of focus. Has1−/− injury panel in row A is recreated42 to maintain matched images between joint pathology and μCT.
Effect of pirfenidone on joint fibrosis and bone pitting after cartilage injury
The same imaging methods showed that PFD dosing essentially eliminated the fibrotic reaction in both genotypes (Figure 1C) and also blocked the bone pitting near the cartilage injury site and the bone erosion at the periarticular margins (Figure 1D). Despite the qualitatively protective effects of PFD against fibrosis and bone pitting in both genotypes, PFD dosing of injured wild-type mice had no detectable effect on any trabecular bone parameters within the metaphysis or epiphysis (Figure S-2) because surgery effects dominated (p < 0.05, Figure 2 and Table 1). For example, cartilage injury resulted in a 41% decrease in BV/TV in wild-type metaphysis in both dosed (p < 0.01) and non-dosed (p < 0.05) mice.
Figure 2. Distal femoral trabecular bone parameters obtained from μCT of naïve, injured, and sham-operated joints in wild-type (WT) and hyaluronan synthase 1 knockout (Has1−/−) mice.

Metaphyseal and epiphyseal trabecular bone was analyzed for bone volume fraction (bone volume (BV) divided by total volume (TV)) and trabecular number (Tb.N), thickness (Tb.Th), and spacing (Tb.Sp) in naïve WT and Has1−/− mice at 28 days after cartilage injury (triangles) or sham (squares) operation, as compared to naïve (circles). WT and Has1−/− mice were either left without drug (white and light gray shapes, respectively) or treated with pirfenidone (PFD) starting 3 days after operation (medium and dark gray shapes, respectively). Mean (black bars) ± standard deviation (black lines) are indicated for each group. Two-way analysis of variance was performed, taking surgery and PFD treatment as main effects. Significant differences were found between surgery types with no dosing (solid lines above data points), between surgery types with PFD dosing (solid lines below data points), and within a surgery group without and with drug (dashed lines above data points).
Table 1.
Significance of surgery and pirfenidone effects on metaphyseal and epiphyseal trabecular bone parameters. Two-way analysis of variance was performed on, and p values (bolded if p < 0.05) reported for, trabecular bone parameters to determine the effect of surgery (naïve, injury, or sham) and drug treatment (no dosing, pirfenidone) on bone volume fraction (BV/TV) and trabecular number, thickness, and spacing (Tb.N [1/mm], Tb.Th [mm], Tb.Sp [mm], respectively). Post hoc comparisons were performed for significant effects with Tukey’s honest significant difference, with significant pair-wise comparisons indicated on Figure 2.
| WT | Has1−/− | |||||
|---|---|---|---|---|---|---|
| Metaphyseal Trabecular Bone | Surgery (p) | Drug (p) | Interaction (p) | Surgery (p) | Drug (p) | Interaction (p) |
| BV/TV | 0.001 | 0.690 | 0.400 | 0.095 | 0.0001 | 0.710 |
| Tb.N | 0.0001 | 0.380 | 0.245 | 0.063 | 0.006 | 0.110 |
| Tb.Th | 0.518 | 0.622 | 0.218 | 0.087 | 0.025 | 0.228 |
| Tb.Sp | 0.0004 | 0.517 | 0.472 | 0.041 | 0.001 | 0.059 |
| Epiphyseal Trabecular Bone | Surgery (p) | Drug (p) | Interaction (p) | Surgery (p) | Drug (p) | Interaction (p) |
| BV/TV | 0.055 | 0.265 | 0.585 | 0.002 | 0.0002 | 0.119 |
| Tb.N | 0.381 | 0.406 | 0.366 | 0.012 | 0.164 | 0.635 |
| Tb.Th | 0.073 | 0.642 | 0.426 | 0.027 | 0.0004 | 0.003 |
| Tb.Sp | 0.284 | 0.268 | 0.708 | 0.101 | 0.936 | 0.721 |
In contrast, PFD dosing was the dominant effect on Has1−/− bone parameters, while the injury effect on BV/TV or any other trabecular bone parameter was not significant (Table 1). In the metaphysis of injured Has1−/− mice, PFD dosing slightly increased BV/TV (p < 0.05) and Tb.N (p < 0.05) and reduced Tb.Sp (p < 0.01) relative to non-dosed injured mice (Table 1). Notably, naïve Has1−/− mice showed reduced trabecular bone density and bone parameters that were significantly different from naïve wild-type, including metaphyseal (p < 0.001) and epiphyseal (p < 0.01) BV/TV, metaphyseal (p < 0.01) and epiphyseal (p < 0.001) Tb.Th, metaphyseal Tb.Sp (p < 0.001), and metaphyseal Tb.N (p < 0.001). Trabecular bone tissue mineral densities (Table S-3) and cortical bone parameters (Table S-4) showed no significant differences between groups (Table S-5). It therefore appears that PFD treatment for 25 days does not alter bone parameters in any treatment group of wild-type mice but leads to a partial normalization of the low values for BV/TV and Tb.N in the injured Has1−/− group.
Effects of PFD dosing on injury-induced changes in gene expression
Given the apparently protective effects of PFD dosing against fibrosis and bone pitting at the injury site in both genotypes (Figure 1), we examined the expression of fibrosis and NF-κB pathway genes, as well as selected matrix genes, in the combined joint tissues harvested between the tibial and femoral growth plates. PFD caused a significant activation (p < 0.05) of 30 genes in wild-type (Tables 2, S-6, and S-7) whereas the same comparison for Has1−/− mice (Tables 3, S-8, and S-9) showed activation (p < 0.05) of only seven genes. Two genes (Il4, Itgb8) were significantly upregulated by PFD in both phenotypes, but to a much greater extent in wild-type than in Has1−/− mice. In wild-type mice, the activations ranged from 3.9-fold (Snai1) to 2876-fold (Timp1), and in Has1−/− mice from 2.3-fold (Vcam1) to 18.9-fold (Fasl). Assays for matrix-related genes (Table S-10) showed high variability and no injury or PFD effects that reached statistical significance.
Table 2.
Significant Gene Expression Changes with Pirfenidone Dosing After Cartilage Injury in Wild-Type Mice. Arrays for fibrosis and NF-κB pathway genes were used to determine gene expression in the joints of injured wild-type (WT) mice dosed with Pirfenidone (IP) with respect to non-dosed counterparts (IN). Transcript abundance was used to calculate effective fold change (IP:IN) and to compare between groups, with significant (p < 0.05) comparisons shown here. The full array data are presented in Supplemental Data Tables S-6 and S-7.
| Fibrosis | WT IP:IN | IP vs IN (p) | NF-κB Signaling Targets | WT IP:IN | IP vs IN (p) |
|---|---|---|---|---|---|
| Itgb6 | 56.41 | 0.015 | Cd80 | 9.11 | 0.036 |
| Itgb8 | 7.81 | 0.046 | Csf2rb | 12.08 | 0.046 |
| Mmp1a | 884.06 | 0.029 | Icam1 | 12.56 | 0.006 |
| Mmp3 | 7.62 | 0.014 | Il1r2 | 4.42 | 0.044 |
| Serpine1 | 25.53 | 0.004 | Il4 | 134.74 | 0.029 |
| Smad3 | 11.44 | 0.045 | Irf1 | 68.48 | 0.027 |
| Smad6 | 20.95 | 0.013 | Ltb | 13.09 | 0.044 |
| Snai1 | 3.95 | 0.032 | Nfkb2 | 14.26 | 0.008 |
| Tgfb2 | 8.19 | 0.044 | Nfkbia | 6.28 | 0.009 |
| Tgfbr2 | 17.78 | 0.032 | Rel | 5.77 | 0.016 |
| Timp1 | 2876.30 | 0.003 | Sele | 9.24 | 0.014 |
| Timp3 | 7.21 | 0.043 | Stat1 | 5.70 | 0.029 |
| Vegfa | 14.54 | 0.002 | Stat3 | 53.82 | 0.035 |
| Stat5b | 3.34 | 0.003 | |||
| Tnf | 14.24 | 0.028 | |||
| Tnfsf10 | 8.84 | 0.025 | |||
| Xiap | 4.94 | 0.006 |
Table 3.
Significant Gene Expression Changes with Pirfenidone Dosing After Cartilage Injury in Hyaluronan Synthase 1 Knockout Mice. Arrays for fibrosis and NF-κB pathway genes were used to determine gene expression in the joints of injured hyaluronan synthase 1 knockout (Has1−/−) mice dosed with Pirfenidone (IP) with respect to non-dosed counterparts (IN). Transcript abundance was used to calculate effective fold change (IP:IN) and to compare between groups, with significant (p < 0.05) comparisons shown here. The full array data are presented in Supplemental Data Tables S-8 and S-9.
| Fibrosis | Has1−/− IP:IN | IP vs IN (p) | NF-κB Signaling Targets | Has1−/− IP:IN | IP vs IN (p) |
|---|---|---|---|---|---|
| Col1a2 | 7.08 | 0.009 | Fasl | 18.87 | 0.011 |
| Il13ra2 | 3.13 | 0.013 | Il4 | 4.84 | 0.046 |
| Itgb8 | 2.73 | 0.021 | Vcam1 | 2.32 | 0.013 |
| Pdgfa | 4.61 | 0.015 |
The genotype-specific effects of PFD at 28 days (Tables 2 vs 3) result primarily from a marked genotypic difference in the effect of injury itself on expression levels. For wild-type mice, the expression levels (relative to age-matched naïve) after injury were either markedly (e.g. Timp1, Mmp1a, Tgfbr2, Irf1 etc.) or modestly (e.g. Itgb6, Serpine1, Itgb8, etc.) reduced at 28 days, (except for Mmp3 which was moderately activated), confirming earlier data with this model 42. In contrast, for Has1−/− mice, expression of most of those genes was not reduced but similar to naïve levels at 28 days after injury, except for Timp1 and Mmp3, which were markedly activated even in Has1−/−. Therefore for wild-type mice, the response to injury was a broad reduction in gene expression at 28 days, and this effect of injury was prevented by PFD dosing. In contrast, for Has1−/− mice, the injury did not broadly inhibit expression at 28 days and the PFD was largely without effect.
Effect of PFD dosing in sham-operated joints
To determine the extent to which the bone pitting in both genotypes (Figure 1), loss of BV/TV in wild-type mice (Figure 2) and genotype-specific changes in gene expression with and without PFD at 28 days (Tables 2 and 3) require intra-articular injury (such as cartilage debridement), we examined the effects of sham injury on these parameters. Firstly, unlike after cartilage injury, there was no evidence for an effect of sham on bone pitting in either genotype, and there was no loss of BV/TV in wild-type mice. Secondly, fibrosis and NF-κB pathway gene expression analysis (Tables S-6 to S-9) showed that for many genes (27 for wild-type and 35 for Has1−/−) the transcript abundance at 28 days after sham-injury was higher than after intra-articular injury (p < 0.05), consistent with a responsiveness of these genes to both types of injury. Nonetheless, it appears that the process that markedly reduces expression of 36 genes between day 12 and day 28 in injured wild-type mice 42 is not active in sham-operated wild-type joints over this period. However, none of the genes that were activated by PFD dosing at day 28 after intra-articular injury (30 for wild-type, Table 2, and seven for Has1−/−, Table 3) were affected by PFD dosing after sham injury (Table S-11), although such a difference might be masked by the gene-activating effect of the sham injury itself. Lastly, in Has1−/− mice, BV/TV (p < 0.05, p < 0.05) and Tb.Th (p < 0.01, p < 0.001) in the metaphysis and epiphysis, respectively, were enhanced by PFD dosing after both cartilage and sham injury, in general agreement with its effects on the intrinsically low values in Has1−/− mice (Figure 2).
Altered histological staining of sGAGs and hyaluronan after joint injury and PFD dosing
Multiple Safranin-O stained sections from naïve and injured joints of non-dosed wild-type mice (Figure 3), showed a marked injury-induced enhancement of staining in the femoral growth plate. With PFD dosing in injured mice, the enhanced growth plate staining was even greater and was accompanied by periosteal chondrogenesis on the lateral aspect. Enhanced growth plate staining in dosed mice was consistent with the loss of hyaluronan staining observed, indicative of aggrecan deposition and masking of hyaluronan binding sites. A similar response to PFD dosing was seen in injured Has1−/− joints (Figure 4), with periosteal staining on the medial aspect, indicating that the excessive fibrotic scarring response in non-dosed Has1−/− joints (Figures 2 and 4) that was also previously observed 42 was modified to a chondrogenic response with PFD dosing.
Figure 3. Effects of pirfenidone (PFD) dosing on Safranin-O (aggrecan) and biotinylated TSG6 (hyaluronan) staining after joint injury in wild-type mice.

Histological sections selected from matched locations in the lateral compartment, joint midline, and medial compartment of wild-type mice were stained with Safranin-O (for aggrecan) and Fast Green counter-stain (A). Adjacent sections were stained with biotinylated TSG6, and regions of the femoral growth plate stained with Safranin-O are shown for comparison (B). Joint injury appeared to increase aggrecan staining of the growth plate cartilage, and this effect was further enhanced by PFD dosing (arrows). This increase in aggrecan density was accompanied by a decrease in accessibility (staining) of hyaluronan for the biotinylated TSG6 probe. [Scale bars: (A) 50 μm; (B) 10 μm.]
Figure 4. Effects of pirfenidone (PFD) dosing on Safranin-O (aggrecan) and biotinylated TSG6 (hyaluronan) staining after joint injury in hyaluronan synthase 1 knockout (Has1−/−) mice.

Histological sections from the lateral compartment, joint midline, and medial compartment of Has1−/− mice were stained with Safranin-O (A). Joint injury showed increased deposition of fibrotic tissues (circles), compared to wild-type, as well as a reduced Safranin-O staining of the femoral growth plate, an effect that was then reversed by PFD dosing (arrows). There was an inverse relationship between aggrecan staining and hyaluronan staining (B), as also observed in wild-type tissues (Figure 3). [Scale bars: (A) 50 μm; (B) 10 μm.]
Safranin-O staining of sham-injured joints (Figure 5) from non-dosed wild-type mice showed weak staining in the growth plate, which was markedly enhanced by PFD dosing, but no periosteal responses were observed. This observation was also supported by the loss of hyaluronan staining. However, non-dosed Has1−/− mice showed an enhanced fibrotic scarring response in the areas of soft tissue damage made during sham injury (Figure 5), and this response was essentially absent in PFD-dosed mice, consistent with an anti-fibrotic effect of PFD on the soft tissue wounds of the sham surgery.
Figure 5. Effects of pirfenidone (PFD) dosing on Safranin-O (aggrecan) and biotinylated TSG6 (hyaluronan) staining after sham operation.

Histological sections from the lateral compartment, joint midline, and medial compartment of sham-operated wild-type (WT) and hyaluronan synthase 1 knockout (Has1−/−) mice were stained with Safranin-O (A). Sham surgery reduced Safranin-O staining of the femoral growth plate, and, in Has1−/− mice, was accompanied by a fibrotic scarring response (circles) to disruption of the joint capsule. Safranin-O staining appeared to be recovered with PFD dosing (arrows) in both genotypes. There was an inverse relationship between aggrecan staining and hyaluronan staining with bTSG6 (B), as observed after PFD treatment in both genotypes (Figures 3 and 4). [Scale bars: (A) 50 μm; (B) 10 μm.]
DISCUSSION
Post-traumatic OA is widely studied in clinical and animal model research to describe pathological remodeling in joint tissues after different types of injuries 45; 46. The severe murine cartilage injury used here had shown fibrotic, chondrogenic, and bone remodeling responses 42. Towards efforts to identify novel therapeutic targets for different cellular response networks, we have here examined efficacy of the anti-fibrotic and anti-inflammatory drug PFD during the post-injury joint tissue regeneration response.
The relationship between Has1, fibrosis, and bone metabolism
Several targetable pathways were suggested by the finding that Has1−/− mice showed a markedly exacerbated fibrotic pathology and this was accompanied by prolonged activation (relative to wild-type) of hyaluronan network genes (Has1, Tnfaip6), matrix genes (Acan, Col1a1, Col3a1) and genes related to the IL-17/IL-6 axis, extracellular matrix remodeling, and apoptosis 42. Further evaluation of this data has shown that, early in the response (day 12), the absolute expression of nine genes (Irf1, Stat3, Stat5b, Nfkb2, Nfkbia, Icam1, Csfr2b, Plau, Xiap) was inhibited greater than 5-fold in Has1−/− mice 42, raising the possibility that their low expression on day 12 might also be related to the exacerbated pathology at day 28. Notably, these nine genes are associated with NF-κB signaling rather than fibrosis, suggesting that Has1−/− joint tissues are less active in inflammation-related signaling thru these factors at day 12 42.
It appears relevant that Irf1, in addition to its role in Toll-like receptor-mediated Stat activation 47, has been implicated in the maturation and activity of osteoclasts and osteoblasts 48. Indeed, an interplay between Has1, Irf1 expression, and bone metabolism might explain the low BV/TV, Tb.Th and Tb.N values found in naïve Has1−/− mice (Figure 2) and also the apparent role of Has1 in osteophyte formation 49.
The finding that naïve Has1−/− mice had reduced trabecular bone quality, which was similar to injured wild-type values but not further diminished by injury, is consistent with the presence of injury-labile bone fraction which requires Has1 for its maintenance. Such putative Has1-dependent bone metabolism might be a consequence of the apparent role of Has1 in bone marrow mesenchymal progenitor activity 50. Further, the widely variable expression profile of Nfkb2 in post-injury Has1−/− mice (78-fold higher, 49-fold lower, and 34-fold higher than wild-type at 3, 12, and 28 days respectively 42) would also appear to link Has1 to control of post-injury bone turnover thru modulation of osteoclastic activity 51; 52.
Prochondrogenic, anti-fibrotic, and bone-protective effects of PFD after injury
PFD dosing for 25 days enhanced post-injury aggrecan deposition in the growth plate and perichondrial regions adjacent to the cartilage damage in both genotypes, thus appearing to convert the collagen-rich and proteoglycan-poor repair tissues in Has1−/− joints (Figure 4) into the cartilaginous repair tissues seen in wild-type joints (Figure 3). These results are supported by the known role of PFD in reducing collagen type 1 synthesis 30; 31. However, the expression of classic chondrogenic genes (Acan and Col2a1) at day 28 was either unaffected or moderately inhibited by PFD dosing (Table S-10). This apparent anomaly would be explained if the PFD acted indirectly by blocking catabolism of aggrecan by aggrecanases (such as Adamts9) and collagen by collagenases (such as Mmp1a). Such an effect on metalloproteinases has been previously observed with PFD-induced suppression of MMP-2 in fibrotic liver 31. Indeed, the expression of MMP inhibitors Timp3 and Timp1 was markedly higher in PFD-dosed mice of both genotypes (Table S-9), further supporting the explanation that PFD acts in the joint to block catabolism of cartilaginous tissues. Additionally, the suppression of the GLI2/Hh axis by PFD may also affect the injury response of the epiphyseal growth plate ECM, as evidenced by the increase in staining after PFD dosing being greater with cartilage injury than with sham operation. Although PFD consistently increases the Safranin-O staining of growth plate cartilage, it is of note that there remain differences in staining between individual mice without PFD treatment (Figures 3 to 5), most likely due to variations between animals in the completion of epiphyseal bone calcification processes 53.
It may also be relevant that the fibrotic tissue deposits around the cartilage injury site at 7 days stain positively with Safranin-O by 28 days, typical of a chondrophyte-like outgrowth 42. These chondrophytic deposits are associated with the pitting seen in the underlying cortical bone (Figure 1). Therefore, if PFD reduces the deposition of fibrotic tissues during the proliferative phase, remodeling of that temporary matrix into chondrophytic tissue is likewise reduced, a reduction that would be reflected in both the lower Col2a1 expression observed and the prevention of cortical bone loss at the site of injury. The corresponding PFD-driven depression of pro-fibrotic genes in the proliferative phase is consistent with reports that PFD reduces collagen expression 30; 31 and blocks fibroblast proliferation and migration by destabilizing GLI2 and suppressing both TGF-β and Hh signaling pathways 32. Moreover, inhibition of the GLI2/Hh axis by PFD 54 might underlie the pro-chondrogenic effect in both genotypes (Figures 3–5) on the growth plate ECM 55; 56 and the anabolic effect on the subchondral bone 57. However, these direct effects of PFD during early post-injury time points could not be measured with the current study design and remain to be examined in further studies.
Has1−/− mice also showed an enhanced fibrotic scarring response, relative to wild-type, in the areas of soft tissue damage made during sham injury (Figure 5). This response was essentially absent in PFD-dosed mice after sham surgery, consistent with an anti-fibrotic effect of PFD on the soft tissue wounds58; 59. PFD blocking of bone pitting and erosion near the injury site in both genotypes (Figure 1B vs 1D) might be explained by the PFD-stimulated expression of genes which have been implicated in bone turnover, such as Irf1 48 and Nfkb2 51;52. Such a mechanism for PFD-mediated bone protection appears reasonable since both Irf1 and Nfkb2 have been shown to prevent excessive osteoclastic activity in mutant mice 48; 51; 52.
Enhanced expression of Timp1, Timp3, Irf1, and Nfkb2 might also explain the pro-chondrogenic and bone-protective effects of PFD in this model. However, the anti-fibrotic effects observed cannot be explained by inhibition of the classical markers (Col1a1, Timp1, Mmp2), since they were increased with PFD rather than decreased, as shown previously in animal models of lung, liver, kidney, and heart fibrosis 60. In this regard, it is important to note that in the current work we have evaluated PFD on gene expression in both a different organ system and within a mixed tissue pool including cartilage/bone, menisci, ligaments and synovium, meaning that the effects cannot be attributed to the response of any single tissue or cell type. Overall, the effects were associated with an apparent increase in expression (relative to no PFD dosing) at 28 days after injury of almost all 164 genes (inclusive of NF-κB and fibrosis arrays and extracellular matrix genes) examined in both genotypes (154/164 in wild-type, 135/164 in Has1−/−), with the effects reaching statistical significance in 36 genes in wild-type and 7 genes in Has1−/−. Examination of the genes that were significantly upregulated in injured wild-type joints with PFD indicates that the anti-fibrotic effect might be exerted thru activation of genes involved in many processes including the TGF-β signaling genes Smad6, Tgfb2, and Tgfbr2. Notably, the 21-fold activated expression of Smad6 (Table 2) would be expected to exert a significant anti-fibrotic effect in fibroblastic tissues of the joint 61.
Therapeutic potential of PFD in post-injury musculoskeletal medicine
This study has shown that the anti-fibrotic drug PFD reduced bone loss and fibrotic tissue deposition after cartilage injury in young male mice of both wild-type and Has1-null genotypes. Female mice were not utilized in this study because the estrous cycle could have a significant effect on the acute bone and innate immune response to injury and PFD treatment; however, the potential for PFD in reducing bone loss after joint injury warrants additional study in how the effects of PFD may be altered by hormonal variation (e.g., bone turnover rates 62). Additionally, younger mice were utilized in this study of cartilage injury because of the prevalence of joint injury in younger adults in both civilian and military populations. The cascade of events that lead to the development of post-traumatic OA after a known injury event is also a readily targetable disease state for pharmaceutical intervention.
Future work is also needed to include a time course of PFD effects at the tissue and cell specific level (cartilage, subchondral bone, meniscus, synovium), as well as to identify an optimal time frame for the initiation and duration of drug administration. Such information may contribute to development of adjunct pharmacological therapies to enhance cell-based or surgical joint tissue repair strategies. Since, at the morphological level, it appears that PFD influences the pathway normally regulated by Has1−/− to avoid excessive scarring, more detailed information on the mechanism of action of Has1−/− in the post-natal tissue regeneration response may be obtained by studying in vitro effects of PFD on regulatory pathways in relevant cell populations such as osteoblast, osteoclasts, chondroprogenitors, and synoviocytes.
PFD and other anti-fibrotic drugs 18 may also be able to supplement surgical and pharmaceutical interventions 63, which individually have thus far been ineffective in preventing OA. Success rates of surgical interventions have shown arthroscopy to be ineffective in preventing OA 64. Furthermore, anti-inflammatory disease-modifying OA drugs, although often effective against inflammation and pain in the short term 65, have shown some adverse effects 66 and little to no successful long-term clinical results 67. Among those, dexamethasone has shown reduced inflammation in a post-traumatic OA model 68 as well as effectiveness against the inflammation that precipitates pulmonary fibrosis 69; 70. Likewise, some bisphosphonates have shown some promise for chondroprotection in animal models 71–73. Naproxen, while effective in short-term pain treatment, may interfere with osteochondral repair in a mechanism involving enhanced expression of GLI2 57, which is suppressed by the anti-fibrotic actions of PFD. In addition to suppression of GLI2 by PFD, other anti-fibrotic drugs could target lysophosphatidic acid, which has also been shown to block collagen type 1 production by chondrocytes and stromal cells in vitro 18.
In conclusion, the chondroprotective potential of PFD shown in this cartilage injury model in young mice indicates that further studies of PFD’s efficacy in older animals and with other models of OA are warranted. Cell and tissue specific effects of PFD should also be examined, in addition to investigation of the role that Has1 and hyaluronan metabolism may play in the joint injury response. Effective therapies for preventing or reversing OA progression after injury may well depend on a combination of various pharmaceutical, physical, and other therapies, applied in coordination with the progression of wound healing processes within the joint.
Supplementary Material
Acknowledgments
The authors acknowledge Drs. Katie Trella and Shuguang Gao for their assistance. Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number R01-AR057066. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. DDC was also supported by an Arthritis Foundation Postdoctoral Fellowship. Funding sources were not involved in the study design; the collection, analysis, and interpretation of data; the writing of the report; nor the decision to submit this work for publication.
Footnotes
Author Contributions Statements
DDC conceived of the study, performed surgeries and treatments, acquired and analyzed data, and wrote and revised the manuscript.
JL performed surgeries and treatments and acquired and analyzed data.
WL acquired data.
DNP conceived of the study and assisted in pharmaceutical preparation and administration.
BC provided discussion regarding translational relevance of the study.
AP conceived of the study and wrote and revised the manuscript.
All authors have read and approved of the final submitted manuscript.
Declaration of Interests
The authors have no conflicts of interest, perceived or actual, to declare.
References
- 1.Mobasheri A, Rayman MP, Gualillo O, et al. The role of metabolism in the pathogenesis of osteoarthritis. Nat Rev Rheumatol. 2017;13:302–311. doi: 10.1038/nrrheum.2017.50. [DOI] [PubMed] [Google Scholar]
- 2.June RK, Liu-Bryan R, Long F, et al. Emerging role of metabolic signaling in synovial joint remodeling and osteoarthritis. J Orthop Res. 2016;34:2048–2058. doi: 10.1002/jor.23420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hughes A, Oxford AE, Tawara K, et al. Endoplasmic Reticulum Stress and Unfolded Protein Response in Cartilage Pathophysiology; Contributing Factors to Apoptosis and Osteoarthritis. International journal of molecular sciences. 2017:18. doi: 10.3390/ijms18030665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang CY, Chanalaris A, Troeberg L. ADAMTS and ADAM metalloproteinases in osteoarthritis - looking beyond the ‘usual suspects’. Osteoarthritis Cartilage. 2017 doi: 10.1016/j.joca.2017.02.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu Z, Ding J, Tredget EE. The molecular basis of hypertrophic scars. Burns Trauma. 2016;4:2. doi: 10.1186/s41038-015-0026-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang C, Liu L, You Z, et al. Endothelial Dysfunction and Mechanobiology in Pathological Cutaneous Scarring: Lessons Learned from Soft Tissue Fibrosis. Br J Dermatol. 2017 doi: 10.1111/bjd.15576. [DOI] [PubMed] [Google Scholar]
- 7.Clarke DL, Carruthers AM, Mustelin T, et al. Matrix regulation of idiopathic pulmonary fibrosis: the role of enzymes. Fibrogenesis Tissue Repair. 2013;6:20. doi: 10.1186/1755-1536-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng G, Swaidani S, Sharma M, et al. Correlation of hyaluronan deposition with infiltration of eosinophils and lymphocytes in a cockroach-induced murine model of asthma. Glycobiology. 2013;23:43–58. doi: 10.1093/glycob/cws122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Y, Jiang D, Liang J, et al. Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. The Journal of experimental medicine. 2011;208:1459–1471. doi: 10.1084/jem.20102510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Su TH, Kao JH, Liu CJ. Molecular mechanism and treatment of viral hepatitis-related liver fibrosis. International journal of molecular sciences. 2014;15:10578–10604. doi: 10.3390/ijms150610578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hughes G, Toellner H, Morris H, et al. Real World Experiences: Pirfenidone and Nintedanib are Effective and Well Tolerated Treatments for Idiopathic Pulmonary Fibrosis. J Clin Med. 2016:5. doi: 10.3390/jcm5090078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Y, Barter MJ, Swan DC, et al. Identification of the pathogenic pathways in osteoarthritic hip cartilage: commonality and discord between hip and knee OA. Osteoarthritis Cartilage. 2012;20:1029–1038. doi: 10.1016/j.joca.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 13.Link TM, Li X. Bone marrow changes in osteoarthritis. Semin Musculoskelet Radiol. 2011;15:238–246. doi: 10.1055/s-0031-1278423. [DOI] [PubMed] [Google Scholar]
- 14.Nakamae A, Engebretsen L, Bahr R, et al. Natural history of bone bruises after acute knee injury: clinical outcome and histopathological findings. Knee Surg Sports Traumatol Arthrosc. 2006;14:1252–1258. doi: 10.1007/s00167-006-0087-9. [DOI] [PubMed] [Google Scholar]
- 15.Harris JD, Hussey K, Wilson H, et al. Biological knee reconstruction for combined malalignment, meniscal deficiency, and articular cartilage disease. Arthroscopy. 2015;31:275–282. doi: 10.1016/j.arthro.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 16.Campbell TM, Trudel G, Wong KK, et al. Genome wide gene expression analysis of the posterior capsule in patients with osteoarthritis and knee flexion contracture. J Rheumatol. 2014;41:2232–2239. doi: 10.3899/jrheum.140079. [DOI] [PubMed] [Google Scholar]
- 17.Remst DF, Blom AB, Vitters EL, et al. Gene expression analysis of murine and human osteoarthritis synovium reveals elevation of transforming growth factor beta-responsive genes in osteoarthritis-related fibrosis. Arthritis Rheumatol. 2014;66:647–656. doi: 10.1002/art.38266. [DOI] [PubMed] [Google Scholar]
- 18.Wu L, Petrigliano FA, Ba K, et al. Lysophosphatidic acid mediates fibrosis in injured joints by regulating collagen type I biosynthesis. Osteoarthritis Cartilage. 2014 doi: 10.1016/j.joca.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matthews JL, Chung M, Matyas JR. Indirect injury stimulates scar formation-adaptation or pathology? Connect Tissue Res. 2004;45:94–100. doi: 10.1080/03008200490464794. [DOI] [PubMed] [Google Scholar]
- 20.Valeyre D, Albera C, Bradford WZ, et al. Comprehensive assessment of the long-term safety of pirfenidone in patients with idiopathic pulmonary fibrosis. Respirology. 2014;19:740–747. doi: 10.1111/resp.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poletti V, Ravaglia C, Tomassetti S. Pirfenidone for the treatment of idiopathic pulmonary fibrosis. Expert Rev Respir Med. 2014;8:539–545. doi: 10.1586/17476348.2014.915750. [DOI] [PubMed] [Google Scholar]
- 22.Angulo P, MacCarty RL, Sylvestre PB, et al. Pirfenidone in the treatment of primary sclerosing cholangitis. Dig Dis Sci. 2002;47:157–161. doi: 10.1023/a:1013240225965. [DOI] [PubMed] [Google Scholar]
- 23.Flores-Contreras L, Sandoval-Rodriguez AS, Mena-Enriquez MG, et al. Treatment with pirfenidone for two years decreases fibrosis, cytokine levels and enhances CB2 gene expression in patients with chronic hepatitis C. BMC Gastroenterol. 2014;14:131. doi: 10.1186/1471-230X-14-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mesa RA, Tefferi A, Elliott MA, et al. A phase II trial of pirfenidone (5-methyl-1-phenyl-2-[1H]-pyridone), a novel anti-fibrosing agent, in myelofibrosis with myeloid metaplasia. Br J Haematol. 2001;114:111–113. doi: 10.1046/j.1365-2141.2001.02883.x. [DOI] [PubMed] [Google Scholar]
- 25.Widemann BC, Babovic-Vuksanovic D, Dombi E, et al. Phase II trial of pirfenidone in children and young adults with neurofibromatosis type 1 and progressive plexiform neurofibromas. Pediatr Blood Cancer. 2014;61:1598–1602. doi: 10.1002/pbc.25041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamagami K, Oka T, Wang Q, et al. Pirfenidone exhibits cardioprotective effects by regulating myocardial fibrosis and vascular permeability in pressure-overloaded hearts. Am J Physiol Heart Circ Physiol. 2015;309:H512–522. doi: 10.1152/ajpheart.00137.2015. [DOI] [PubMed] [Google Scholar]
- 27.Ji X, Naito Y, Weng H, et al. Renoprotective mechanisms of pirfenidone in hypertension-induced renal injury: through anti-fibrotic and anti-oxidative stress pathways. Biomed Res. 2013;34:309–319. doi: 10.2220/biomedres.34.309. [DOI] [PubMed] [Google Scholar]
- 28.Fink MK, Giuliano EA, Tandon A, et al. Therapeutic potential of Pirfenidone for treating equine corneal scarring. Vet Ophthalmol. 2015;18:242–250. doi: 10.1111/vop.12194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodriguez-Castellanos M, Tlacuilo-Parra A, Sanchez-Enriquez S, et al. Pirfenidone gel in patients with localized scleroderma: a phase II study. Arthritis Res Ther. 2014;16:510. doi: 10.1186/s13075-014-0510-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hisatomi K, Mukae H, Sakamoto N, et al. Pirfenidone inhibits TGF-beta1-induced over-expression of collagen type I and heat shock protein 47 in A549 cells. BMC Pulm Med. 2012;12:24. doi: 10.1186/1471-2466-12-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Di Sario A, Bendia E, Macarri G, et al. The anti-fibrotic effect of pirfenidone in rat liver fibrosis is mediated by downregulation of procollagen alpha1(I), TIMP-1 and MMP-2. Dig Liver Dis. 2004;36:744–751. doi: 10.1016/j.dld.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 32.Didiasova M, Singh R, Wilhelm J, et al. Pirfenidone exerts antifibrotic effects through inhibition of GLI transcription factors. FASEB J. 2017 doi: 10.1096/fj.201600892RR. [DOI] [PubMed] [Google Scholar]
- 33.Chung SA, Jeon BK, Choi YH, et al. Pirfenidone attenuates the IL-1beta-induced hyaluronic acid increase in orbital fibroblasts from patients with thyroid-associated ophthalmopathy. Invest Ophthalmol Vis Sci. 2014;55:2276–2283. doi: 10.1167/iovs.13-13759. [DOI] [PubMed] [Google Scholar]
- 34.Pesce E, Struma E, Giri SN, et al. Antirheumatic effect of pirfenidone in a double blind clinical pilot trial in humans. Res Commun Mol Pathol Pharmacol. 2004;115–116:39–48. [PubMed] [Google Scholar]
- 35.Benton HP, Esquivel AV, Rice AD, et al. Modulation of articular chondrocyte activity by pirfenidone. Res Commun Mol Pathol Pharmacol. 2003;113–114:275–288. [PubMed] [Google Scholar]
- 36.de la Motte CA. Hyaluronan in intestinal homeostasis and inflammation: implications for fibrosis. Am J Physiol Gastrointest Liver Physiol. 2011;301:G945–949. doi: 10.1152/ajpgi.00063.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoshino T, Okamoto M, Sakazaki Y, et al. Role of proinflammatory cytokines IL-18 and IL-1beta in bleomycin-induced lung injury in humans and mice. Am J Respir Cell Mol Biol. 2009;41:661–670. doi: 10.1165/rcmb.2008-0182OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mack JA, Feldman RJ, Itano N, et al. Enhanced inflammation and accelerated wound closure following tetraphorbol ester application or full-thickness wounding in mice lacking hyaluronan synthases Has1 and Has3. J Invest Dermatol. 2012;132:198–207. doi: 10.1038/jid.2011.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Calve S, Isaac J, Gumucio JP, et al. Hyaluronic acid, HAS1, and HAS2 are significantly upregulated during muscle hypertrophy. Am J Physiol Cell Physiol. 2012;303:C577–588. doi: 10.1152/ajpcell.00057.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Siiskonen H, Oikari S, Pasonen-Seppanen S, et al. Hyaluronan synthase 1: a mysterious enzyme with unexpected functions. Front Immunol. 2015;6:43. doi: 10.3389/fimmu.2015.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lambert C, Dubuc JE, Montell E, et al. Gene expression pattern of cells from inflamed and normal areas of osteoarthritis synovial membrane. Arthritis Rheumatol. 2014;66:960–968. doi: 10.1002/art.38315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chan DD, Xiao WF, Li J, et al. Deficiency of hyaluronan synthase 1 (Has1) results in chronic joint inflammation and widespread intra-articular fibrosis in a murine model of knee joint cartilage damage. Osteoarthritis Cartilage. 2015;23:1879–1889. doi: 10.1016/j.joca.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi S, Wu J, Chen H, et al. Single- and multiple-dose pharmacokinetics of pirfenidone, an antifibrotic agent, in healthy Chinese volunteers. J Clin Pharmacol. 2007;47:1268–1276. doi: 10.1177/0091270007304104. [DOI] [PubMed] [Google Scholar]
- 44.Li J, Gorski DJ, Anemaet W, et al. Hyaluronan injection in murine osteoarthritis prevents TGFbeta 1-induced synovial neovascularization and fibrosis and maintains articular cartilage integrity by a CD44-dependent mechanism. Arthritis Res Ther. 2012;14:R151. doi: 10.1186/ar3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blaker CL, Clarke EC, Little CB. Using mouse models to investigate the pathophysiology, treatment, and prevention of post-traumatic osteoarthritis. J Orthop Res. 2017;35:424–439. doi: 10.1002/jor.23343. [DOI] [PubMed] [Google Scholar]
- 46.Carbone A, Rodeo S. Review of current understanding of post-traumatic osteoarthritis resulting from sports injuries. J Orthop Res. 2017;35:397–405. doi: 10.1002/jor.23341. [DOI] [PubMed] [Google Scholar]
- 47.Liljeroos M, Vuolteenaho R, Rounioja S, et al. Bacterial ligand of TLR2 signals Stat activation via induction of IRF1/2 and interferon-alpha production. Cell Signal. 2008;20:1873–1881. doi: 10.1016/j.cellsig.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 48.Salem S, Gao C, Li A, et al. A novel role for interferon regulatory factor 1 (IRF1) in regulation of bone metabolism. J Cell Mol Med. 2014;18:1588–1598. doi: 10.1111/jcmm.12327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gelse K, Ekici AB, Cipa F, et al. Molecular differentiation between osteophytic and articular cartilage--clues for a transient and permanent chondrocyte phenotype. Osteoarthritis Cartilage. 2012;20:162–171. doi: 10.1016/j.joca.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 50.Calabro A, Oken MM, Hascall VC, et al. Characterization of hyaluronan synthase expression and hyaluronan synthesis in bone marrow mesenchymal progenitor cells: predominant expression of HAS1 mRNA and up-regulated hyaluronan synthesis in bone marrow cells derived from multiple myeloma patients. Blood. 2002;100:2578–2585. doi: 10.1182/blood-2002-01-0030. [DOI] [PubMed] [Google Scholar]
- 51.Seo Y, Fukushima H, Maruyama T, et al. Accumulation of p100, a precursor of NF-kappaB2, enhances osteoblastic differentiation in vitro and bone formation in vivo in aly/aly mice. Mol Endocrinol. 2012;26:414–422. doi: 10.1210/me.2011-1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soysa NS, Alles N, Weih D, et al. The pivotal role of the alternative NF-kappaB pathway in maintenance of basal bone homeostasis and osteoclastogenesis. J Bone Miner Res. 2010;25:809–818. doi: 10.1359/jbmr.091030. [DOI] [PubMed] [Google Scholar]
- 53.Larsson SE. The metabolic heterogeneity of glycosaminoglycans of the different zones of the epiphyseal growth plate and the effect of ethane-1-hydroxy-1, 1-diphosphonate (EHDP) upon glycosaminoglycan synthesis in vivo. Calcif Tissue Res. 1976;21:67–82. doi: 10.1007/BF02547384. [DOI] [PubMed] [Google Scholar]
- 54.Didiasova M, Singh R, Wilhelm J, et al. Pirfenidone exerts antifibrotic effects through inhibition of GLI transcription factors. FASEB J. 2017;31:1916–1928. doi: 10.1096/fj.201600892RR. [DOI] [PubMed] [Google Scholar]
- 55.Ho L, Stojanovski A, Whetstone H, et al. Gli2 and p53 cooperate to regulate IGFBP-3-mediated chondrocyte apoptosis in the progression from benign to malignant cartilage tumors. Cancer Cell. 2009;16:126–136. doi: 10.1016/j.ccr.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 56.Iwasaki M, Le AX, Helms JA. Expression of indian hedgehog, bone morphogenetic protein 6 and gli during skeletal morphogenesis. Mech Dev. 1997;69:197–202. doi: 10.1016/s0925-4773(97)00145-7. [DOI] [PubMed] [Google Scholar]
- 57.Salem O, Wang HT, Alaseem AM, et al. Naproxen affects osteogenesis of human mesenchymal stem cells via regulation of Indian hedgehog signaling molecules. Arthritis Res Ther. 2014;16:R152. doi: 10.1186/ar4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saito M, Yamazaki M, Maeda T, et al. Pirfenidone suppresses keloid fibroblast-embedded collagen gel contraction. Arch Dermatol Res. 2012;304:217–222. doi: 10.1007/s00403-011-1184-2. [DOI] [PubMed] [Google Scholar]
- 59.Orozco-Perez J, Aguirre-Jauregui O, Salazar-Montes AM, et al. Pirfenidone prevents rat esophageal stricture formation. J Surg Res. 2014 doi: 10.1016/j.jss.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 60.Schaefer CJ, Ruhrmund DW, Pan L, et al. Antifibrotic activities of pirfenidone in animal models. Eur Respir Rev. 2011;20:85–97. doi: 10.1183/09059180.00001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shen ZJ, Braun RK, Hu J, et al. Pin1 protein regulates Smad protein signaling and pulmonary fibrosis. J Biol Chem. 2012;287:23294–23305. doi: 10.1074/jbc.M111.313684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sengupta S, Arshad M, Sharma S, et al. Attainment of peak bone mass and bone turnover rate in relation to estrous cycle, pregnancy and lactation in colony-bred Sprague-Dawley rats: suitability for studies on pathophysiology of bone and therapeutic measures for its management. J Steroid Biochem Mol Biol. 2005;94:421–429. doi: 10.1016/j.jsbmb.2004.12.039. [DOI] [PubMed] [Google Scholar]
- 63.Yu SP, Hunter DJ. Emerging drugs for the treatment of knee osteoarthritis. Expert Opin Emerg Drugs. 2015;20:361–378. doi: 10.1517/14728214.2015.1037275. [DOI] [PubMed] [Google Scholar]
- 64.Adelani MA, Harris AH, Bowe TR, et al. Arthroscopy for Knee Osteoarthritis Has Not Decreased After a Clinical Trial. Clin Orthop Relat Res. 2016;474:489–494. doi: 10.1007/s11999-015-4514-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Towheed TE, Maxwell L, Judd MG, et al. Acetaminophen for osteoarthritis. Cochrane Database Syst Rev. 2006:CD004257. doi: 10.1002/14651858.CD004257.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Makris UE, Kohler MJ, Fraenkel L. Adverse effects of topical nonsteroidal antiinflammatory drugs in older adults with osteoarthritis: a systematic literature review. J Rheumatol. 2010;37:1236–1243. doi: 10.3899/jrheum.090935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lapane KL, Yang S, Driban JB, et al. Effects of prescription nonsteroidal antiinflammatory drugs on symptoms and disease progression among patients with knee osteoarthritis. Arthritis Rheumatol. 2015;67:724–732. doi: 10.1002/art.38933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huebner KD, Shrive NG, Frank CB. Dexamethasone inhibits inflammation and cartilage damage in a new model of post-traumatic osteoarthritis. J Orthop Res. 2014;32:566–572. doi: 10.1002/jor.22568. [DOI] [PubMed] [Google Scholar]
- 69.Shi K, Jiang J, Ma T, et al. Dexamethasone attenuates bleomycin-induced lung fibrosis in mice through TGF-beta, Smad3 and JAK-STAT pathway. Int J Clin Exp Med. 2014;7:2645–2650. [PMC free article] [PubMed] [Google Scholar]
- 70.Porte J, Jenkins G. Assessment of the effect of potential antifibrotic compounds on total and alphaVbeta6 integrin-mediated TGF-beta activation. Pharmacol Res Perspect. 2014;2:e00030. doi: 10.1002/prp2.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lampropoulou-Adamidou K, Dontas I, Stathopoulos IP, et al. Chondroprotective effect of high-dose zoledronic acid: An experimental study in a rabbit model of osteoarthritis. J Orthop Res. 2014;32:1646–1651. doi: 10.1002/jor.22712. [DOI] [PubMed] [Google Scholar]
- 72.Dearmin MG, Trumble TN, Garcia A, et al. Chondroprotective effects of zoledronic acid on articular cartilage in dogs with experimentally induced osteoarthritis. Am J Vet Res. 2014;75:329–337. doi: 10.2460/ajvr.75.4.329. [DOI] [PubMed] [Google Scholar]
- 73.Cinar BM, Ozkoc G, Bolat F, et al. Intra-articular zoledronic acid in a rat osteoarthritis model: significant reduced synovitis may indicate chondroprotective effect. Knee Surg Sports Traumatol Arthrosc. 2015;23:1410–1418. doi: 10.1007/s00167-014-2955-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.