

Abstract
Microglia, the resident innate immune cells in the brain, have long been understood to be crucial to maintenance in the nervous system, by clearing debris, monitoring for infiltration of infectious agents, and mediating the brain’s inflammatory and repair response to traumatic injury, stroke, or neurodegeneration. A wave of new research has shown that microglia are also active players in many basic processes in the healthy brain, including cell proliferation, synaptic connectivity, and physiology. Microglia, both in their capacity as phagocytic cells and via secretion of many neuroactive molecules, including cytokines and growth factors, play a central role in early brain development, including sexual differentiation of the brain. In this review, we present the vast roles microglia play in normal brain development and how perturbations in the normal neuroimmune environment during development may contribute to the etiology of brain-based disorders. There are notable differences between microglia and neuroimmune signaling in the male and female brain throughout the life span, and these differences may contribute to the vast differences in the incidence of neuropsychiatric and neurological disorders between males and females.
Keywords: microglia, development, sex differences, hormone, inflammation, immune, cytokine, sex
Introduction
Microglia are brain-resident macrophages, comprising 5% to 12% of total cells in the brain depending on the region examined (Lawson and others 1990). As non-neuronal cells, microglia arise from common myeloid precursors in the embryonic yolk sac early in ontogeny (Alliot and others 1999; Ginhoux and others 2010; Fig. 1A). In rodents, microglia progenitors infiltrate the developing neural tube to colonize the primitive brain at approximately embryonic days 8 to 9 (Alliot and others 1999; Ginhoux and others 2010) and differentiate into microglia once in the brain (Fig. 1A). Following this initial colonization, microglia are thought to be a long-lived, stable population of cells in the brain that proliferate rapidly during perinatal development (Alliot and others 1999) and constitutively throughout the lifespan (Ajami and others 2007) and which are distinct from peripheral monocytes that can infiltrate the brain during injury, degeneration, or chronic stress (Ajami and others 2007; Hickey and others 1992; Wohleb and others 2013). Microglia have long been known to perform a vital, if supporting role in the brain as resident “garbage collectors.” But as with astrocytes, clinicians and research scientists alike underestimated the active and versatile roles that microglia play in brain development, plasticity, and neural function in both healthy and pathological brain states. This review will focus on an additional surprising role for microglia as key contributors to the developmental sculpting of sex-specific neural architecture, which mediates adult reproductive behavior.
Figure 1.

(A) Microglial cells are derived from the embryonic yolk sac from myeloid progenitors that are generated prior to the initiation of hematopoiesis in the fetal liver. These progenitors migrate to the developing brain, colonize and differentiate into microglia, which is a long-lived self-sustaining population of the cells in the brain throughout life. (B). There is a sex bias in the number of microglia in the developing rodent brain, with males having more than females across the neonatal period. It is currently unknown whether more microglia progenitors are recruited into the male brain early in life, whether more proliferation occurs, or whether more cells survive in males than females (or some combination thereof).
Historical Overview
Microglia are the primary immunocompetent cells of the brain. They were first discovered and named in the 1920s by Pío del Río Hortega (a scientific protégé of Santiago Ramon y Cajal) who investigated the role of these cells in brain pathology (Barron 1995). In their primary immunocompetent capacity, microglia have received ample research attention as mediators of brain infection, injury, and degenerative disorders.
The brain is considered immune privileged relative to the rest of the body as a consequence of the blood-brain barrier, yet it is still possible for bacterial or viral agents to infect the brain. In the absence of immune activating stimuli, microglia are found largely in what used to be considered a quiescent form but now referred to as “surveying,” with small cell bodies and long, ramified processes. These microglia are expert sensors of even small changes in the neural environment and are considered to be the first line of defense in the central nervous system. If microglia detect an infectious agent in the brain, they respond with several classical defense mechanisms of the innate immune system, including recognizing foreign substances through toll-like receptors, phagocytosis of foreign material and subsequent antigen presentation to attract T cells into the brain and destroy infectious agents.
Microglia are also central in the nervous system response to injury. Following traumatic brain or spinal cord injury or hypoxic/ischemic damage, microglia rapidly undergo a morphological transformation and retract their highly ramified, branched processes to take on an ameboid form. This ameboid form is highly motile, allowing microglia to swarm to the site of injury within minutes. Once at the site of injury, microglia first engage in robust proinflammatory signaling and release cytokines, such as interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α to recruit additional innate immune cells to the site of injury (Kreutzberg 1996; Lenzlinger and others 2001).
Microglia then phagocytose dead and dying cells to limit the spread of damage caused by the release of cell contents into the neuropil (Hanisch and Kettenmann 2007). Secondarily, microglia engage in anti-inflammatory signaling and growth factor secretion to limit chronic inflammation and promote repair (Kreutzberg 1996). Microglia can take on both classical pro-inflammatory activation (called M1) and alternative anti-inflammatory activation (called M2). These subpopulations of M1 and M2 microglia may act in concert following injury (Kigerl and others 2009), yet it remains unclear to what extent the different microglial phenotypes are responsible for the resolution or exacerbation of injury (Aguzzi and others 2013). In fact, some are disputing the existence of clear and absolute boundaries between M1 and M2 polarization, given the vastly more complex spectrum of macrophage phenotypes characterized in recent work (Xue and others 2014). Nonetheless, in states of neurodengerative disease, such as Alzheimer’s or Parkinson’s disease, microglia become chronically activated in a proinflammatory state, and thereby intensify the physiological, cognitive, and behavioral symptoms of these disorders.
A New Synthesis
Microglia Play a Novel Role in Adult Neural Plasticity
Microglia in the healthy adult brain are distributed fairly evenly throughout the parenchyma, and appear to have a home territory that they are responsible for monitoring (Nimmerjahn and others 2005). Recent advances in imaging microglia in the live brain via 2-photon microscopy reveal that so-called “resting” microglia are actually very active in the healthy brain, keeping their cell bodies relatively stationary but rapidly extending their processes to survey the surrounding tissue (Nimmerjahn and others, 2005). Microglia make regular physical contact with other cells, including neurons and astrocytes and retract and reextend their processes to different locations within minutes (Nimmerjahn and others 2005). This basal activity has led to a shift in nomenclature, with quiescent, non-ameboid microglia no longer called “resting” but instead called “surveying” (Hanisch and Kettenmann 2007). Extrapolating the rate of microglial sampling of the parenchyma leads to the estimate that the entire brain can be surveyed and assessed within hours. But just what are microglia doing during this surveillance? Accumulating evidence points to microglial regulation of neuronal excitability, synaptic activity, connectivity, neurogenesis, and clearance of apoptotic cells in the healthy adult brain.
Synaptic Activity
Microglia and microglial-derived inflammatory cytokines regulate both long-term potentiation (LTP) and long-term depression (LTD). In the dorsal horn of the spinal cord, pharmacological inhibition of microglia prevents LTP that is normally induced at C-fiber synapses by high-frequency stimulation; in fact microglial inhibition instead leads to the induction of LTD (Zhong and others 2010). In the hippocampus, mice deficient in CD200, a glycoprotein necessary for maintaining microglial in a ramified state, have impaired LTP that is dependent on increases TNFα (Costello and others 2011). In a similar model, fracktalkine receptor knockout animals, which have increased microglial activation in the brain, have impaired LTP and hippocampal-dependent learning (Rogers and others 2011). These deficits in hippocampal LTP are accompanied by increased IL-1β, and antagonism of IL-1β function rescues LTP (Rogers and others 2011).
Synaptic Architecture and Connectivity
Microglia are intimately involved in shaping synaptic connectivity in the adult brain. Surveying microglial processes regularly and transiently contact both dendritic spines as well as presynaptic terminals of excitatory synapses under normal physiological conditions (Wake and others 2009). Thus microglia are contacting (and may thus be regulating) both presynaptic and postsynaptic sides of a neuronal circuit.
Adult Neurogenesis and Apoptosis
Microglia can regulate adult neurogenesis and apoptosis in several capacities: (a) as producers of inflammatory molecules and neurotrophins that regulate apoptosis and cell survival and (b) as phagocytic cells, “eating” both apoptotic cells as well as neural progenitors. Under non-pathological conditions, microglia phagocytose newly born cells undergoing apoptosis in the subgranular zone (Sierra and others 2010). This process does not engage pro-inflammatory signaling pathways and does not result in subsequent microglial activation (Sierra and others 2010); in fact, this phagocytosis prevents proinflammatory responses induced by release of intracellular contents of dying cells into the local microenvironment (Gregory and Pound 2011). In response to experiences that increase neurogenesis, such as exercise, microglia themselves proliferate in the hippocampus and cortex (Ehninger and Kempermann 2003; Olah and others 2009). Microglia also promote neurosphere formation and hippocampal neural proliferation following exercise, via the neuronally derived chemokine, fracktalkine, and its receptor on microglia, CX3CR1 (Vukovic and others 2012). Thus microglia can regulate neurogenesis and apoptosis in the adult brain under both basal and pathological conditions. What remains to be determined is how microglia functions interact—for example, does microglial secretion of inflammatory and neurotrophic factors affect rates of phagocytosis, or vice versa? It also remains to be seen how microglia regulate neurogenesis and apoptosis differentially in the healthy and inflamed brain, both in terms of the mechanisms controlling the induction of proliferation and phagocytosis and the signals that specify which cells should be supported and which should be eliminated.
Microglia Play a Novel Role in Normal Development
Microglia colonize the brain early in embryonic development (Ginhoux and others 2010) and a host of newly emerging data illustrate that under normal healthy conditions, microglia sculpt developing brain circuits and cell populations (Fig. 2).
Figure 2.
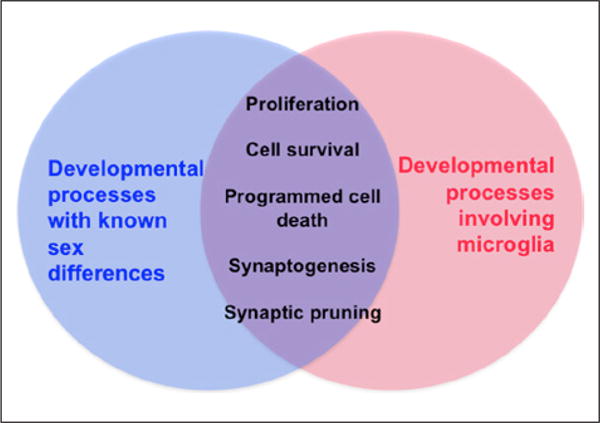
Microglia have been implicated as players in many normal developmental processes in the brain, including cell proliferation, survival, apoptosis, and synaptic patterning. Virtually all of these processes show prominent sex differences, thus the sex difference in microglia in the developing brain may contribute to each of these processes.
Cell Genesis and Stem Cell Properties
During embryonic neurogenesis, microglia are highly localized to proliferative zones in the developing brain (Cunningham and others 2013), and regulate stem cell pools and cell genesis via secretion of putative trophic factors and via phagocytosis. In culture, neural stem cells from the subventricular zone are responsive to microglia, with microglia co-culture or microglia-conditioned medium leading to decreased neuroblast numbers and highly increased numbers of newly born cells (Walton and others 2006). Without microglia present in the cultures, inducible neurogenesis does not occur (Walton and others 2006). Microglia also promote astrogliogenesis from stem cells in culture, via the release of IL-6 (Nakanishi and others 2007). Activated microglia in particular are necessary for both neurogenesis and oligodendrogenesis in the subventricular zone, with administration of the microglia inhibitor minocycline reducing postnatal cell genesis in a pro-inflammatory cytokine-dependent fashion (Shigemoto-Mogami and others 2014).
Microglia may also regulate critical periods for neural proliferation in the developing brain by limiting the size of neural precursor cell pools in proliferative zones (Cunningham and others 2013). At the end of the robust period of perinatal neurogenesis of cortical cells, microglia phagocytose not only apoptotic cells but also healthy, non-apoptotic neural precursor cells and therefore restrain overpopulation of the developing brain. If microglia are pharmacologically inhibited or ablated prenatally, precursor pool size increases, and if microglia are activated by prenatal immune challenge with lipopolysaccharide, precursor pool size decreases without any increase in cell death (Cunningham and others 2013). The possibility that microglia actively shape brain development and particularly control the timing of critical periods in brain development is completely contrary to the notion that they are programmed only to clear up debris or phagocytose only cells that have already committed to cell death, that is, the brain’s “garbage collectors.” It is currently unknown what directs microglia to phagocytose healthy progenitor cells, and whether this action is intrinsic to microglia, directed by signals from the progenitor cells themselves or originates in the broader neural environment.
Programmed Cell Death
Microglia are expert sensors of apoptotic cells, showing strong chemoattraction toward death signals such as caspase-3 (Marín-Teva and others 2004). In both the developing cerebellum and hippocampus, once cells begin the programmed cell death process and upregulate caspase-3, microglia rapidly engulf the cells and actively promote their death via a superoxide-mediated respiratory burst (Marín-Teva and others 2004). Without microglia present, cells will still express caspase-3 at similar proportions but fewer cells will undergo cell death (Marín-Teva and others 2004). Microglial processes detect apoptotic cells by surveying for phosphatidylserine, a lipid up-regulated on the surface of dying cells early in the apoptosis process (Ravichandran 2011). According to Ravichandran (2011), though the steps and molecular players in phosphatidylserine-guided phagocytosis of apoptotic cells has been most thoroughly characterized, there are several parallel signaling pathways that also tag apoptotic cells for phagocytosis. Indeed, during brain development, microglia appear to target dying cells via an immunoreceptor only expressed in the immature brain called DAP12, and along with the integrin protein, CD11b, deliver a superoxide burst to the dying cells (Wakselman and others 2008). In this way, microglia sense neurons that have begun the cell death process and then finish the cells off. There is emerging interest in the notion that microglia may also sense stressed but healthy cells and inappropriately initiate phagocytosis, thus rather than facilitating suicide, they are instead committing cellular murder.
Synaptic Development, Maturation, and Connectivity
The best studied system in which microglia regulate synaptic development is the retina and retinogeniculate circuit. During the first postnatal week of life, the critical period for synaptic pruning in the thalamus, microglia phagocytose weak inputs from the retina in an activity-dependent manner (Schafer and others 2012). This phagocytosis depends on the tagging of “weak” synapses with complement proteins, which attracts phagocytic microglia and directs lysosomal engulfment of the presynaptic terminal (Schafer and others 2012). The cytokine transforming growth factor (TGF)-β, is necessary for the neuronal expression of complement proteins in the developing thalamus (Bialas and Stevens 2013), but it remains unknown how synaptic activity and strength regulate TGF-β and how specific synapses are directed to up-regulate complement proteins while others are spared. Later in the development of the visual system, in primary visual cortex microglia sample the environment and preferentially contact synaptic elements, including presynaptic terminals and postsynaptic dendritic spines (Tremblay and others 2010). Microglia preferentially contact synaptic inputs that have been weakened by visual deprivation and appear to engulf them based on the observation that following visual deprivation microglia have increased numbers of phagocytic bodies and greater phagocytic activity (Tremblay and others 2010).
The developing hippocampus also depends on microglia to refine synapses. Using high-resolution microscopy methods, microglia have been detected phagocytosing PSD-95 containing post-synaptic dendritic spines (Paolicelli and others 2011). This synapse elimination depends on the fractalkine system that is so often implicated in microglia-neuron interactions: fractalkine receptor deficient mice have fewer microglia in the hippocampus and decreased microglial arborization, as well as increased dendritic spine density, increased presynaptic release sites, and less mature physiological properties, including enhanced LTD responses (Paolicelli and others 2011; Zhan and others 2014). Together, these data implicate surveying microglia as necessary regulators of synaptic pruning and refinement during normal brain development.
Sexual Differentiation of the Developing Brain
Sex determination in mammals, that is, whether an individual is a male or a female, is determined by the Y chromosome gene, Sry, which codes for a protein called the testis determining factor because of its obligatory role in testicular development. In the absence of Sry, such as in XX individuals, the bipotential gonad develops into an ovary (Goodfellow and Lovell-Badge 1993). The process of gonadal differentiation occurs remarkably quickly and early in development. All other phenotypic traits associated with sex occur subsequent to gonadal development. Included among these is the phenotypic sex of the brain, which like the primordial gonad is bipotential before being subject to either masculinization or feminization. Elucidating the origins, mechanisms, and impact of sexual differentiation of the brain has been the topic of investigation for more than 50 years and many fundamental principles have been established.
Gonadal Steroids in Males Masculinize the Brain during a Sensitive Perinatal Period
As noted above, the fetal gonad develops early and in the male is fully active (with the exception of spermatogenesis) by mid to late gestation. In rodents, there is a surge in fetal testis androgen production beginning the last few days of gestation and enduring until shortly after birth. In primates androgen production occurs earlier, from the end of the first trimester and well into the second with another surge at birth (Forest and others 1973; Gendrel and others 1980; Reyes and others 1973). After gaining access to the brain, testosterone (T) can be either aromatized to estradiol (E2) or 5-α reduced to dihyrdotestosterone (DHT). Both T and DHT induce some masculine endpoints but it is E2 that is the dominant masculinizing hormone in the rodent brain (for review, see McCarthy 2008).
Confirmation of the critical role of E2 in brain masculinization comes from multiple sources, including many studies in which newborn females are masculinized by treatment with exogenous hormones to mimic the levels normally experienced by males (for review, see Lenz and others 2012), or studies showing that males fail to masculinize if the estrogen receptors or aromatase enzyme have been genetically ablated (Honda and others 1998; Kudwa and others 2005; Ogawa and others 2000). Naturally occurring masculinization in males is also prevented by treatment with inhibitors of the aromatase enzyme or antagonists of the estrogen receptor (ER). Importantly, in both males and females the treatment must occur during a narrowly defined critical, or sensitive, period. We now define this sensitive period as beginning with the onset of androgen production by the fetal testis and ending with the time at which exogenous treatments are no longer effective at either inducing or preventing masculinization, which in the rodent is approximately the end of the first week of life. In primates, including humans, the sensitive period is largely prenatal (Collaer and Hines 1995; van de Beek and others 2004).
Early Life Programming Permanently Alters Brain and Behavior
The actions of gonadal steroids during the perinatal sensitive period are often referred to as being “organizational” but really are akin to early life programming. Just as nutritional status, stress or trauma can permanently influence the sensitivity of the adult to various challenges, so too does early hormone exposure influence the sensitivity of the adult brain to additional hormone exposure that occurs after puberty. However, in this instance, the intent is for adult hormone exposure to match the conditions perinatally. Thus, high testosterone in an adult male will induce male reproductive physiology (pulsatile luteinizing hormone release) and behavior (mounting females), provided the male was exposed to androgens and its metabolite estradiol early in development. Likewise, a feminized brain responds to cyclical changes in estrogens and progestins in adulthood with female reproductive physiology (ovulation inducing luteinizing hormone surges) and behavior (lordosis). If the adult hormonal profile does not match the perinatal profile, the animal is essentially blind to the inducing effects of the hormones.
The masculinization of sexual behavior in the rodent has proven a highly valuable model system for investigation of early life programming effects of gonadal steroids. We now know that the preoptic area (POA) is an essential brain region for male sexual behavior and is characterized by marked neuroanatomical sex differences. These include the much larger sexually dimorphic nucleus (SDN), more highly stellate and ramified astrocytes and a density of dendritic spine synapses that is twice as great as that observed in the female POA (Gorski and others 1980; Amateau and McCarthy 2002a, 2002b). Elucidating the mechanism(s) by which steroids regulate neuronal cell death, astrocyte differentiation and synaptogenesis is of value to more than just the sexual differentiation of reproductive behavior and it elucidates novel but fundamental principles of brain development (Fig. 2).
Induction of Prostaglandin Production by Steroids Is Necessary and Sufficient to Masculinize Both Brain and Behavior
A question essential to the phenomenon of sexual differentiation of the brain is the cellular events by which steroids exert their early life programming effect. Given the centrality of neurotransmitters to neurophysiology and behavior, a strong emphasis was placed on identifying which might be the key system subject to steroid modification. But the answer came from a surprising place. Rather than a classic neurotransmitter as the primary target of steroid hormone modification, it is the membrane-derived signaling molecule, prostaglandin E2 (PGE2) that transduces the hormonal message. The process begins with up regulation of the cyclooxygenase (COX)-1 and COX-2 enzymes leading to increased production of PGE2, which binds to and activates the GPCR-adenylyl cyclase–linked receptors EP2 and EP4 (Amateau and McCarthy 2004; Wright and others 2008). Increased cAMP activates PKA, which phosphorylates a critical subunit of the glutamate AMPA receptor, inducing it to traffic to the membranes of both neurons and astrocytes (Lenz and others 2011; Wright and McCarthy 2009). Activation of AMPA receptors leads to induction of dendritic spine synapses during the critical period (Wright and McCarthy 2009). Demonstration that the production of prostaglandins is the linchpin in this process comes from the ability of a single injection of PGE2 into the brain of neonatal female pups to fully masculinize their adult brain and behavior (Wright and McCarthy 2009). Similarly, neonatal males in which the COX enzymes were briefly inhibited failed to masculinize normally.
The observation that a single injection of PGE2, a small molecule with a short half-life, could permanently program the developing brain was both startling and puzzling. That something so ephemeral as a brief rise in prostaglandin production would have such an enduring and consequential effect seemed to defy the rules governing brain development. Pondering this turned us to consideration of a potential feed-forward mechanism in which a small amount of PGE2 stimulates production of more PGE2 and possibly other associated molecules. The COX enzymes are ubiquitously distributed throughout the brain and are expressed by neurons, astrocytes, and most importantly, microglia.
Microglia as Critical Mediators of Brain Sexual Differentiation
Microglia Are Sexually Dimorphic
Microglia colonize the rodent brain prenatally (Ginhoux and others 2010) and by birth, a majority of the microglial population in the brain has been established (Schwarz and others 2012). By parturition, there are significant sex differences in the number and morphology of microglia in almost every brain region assessed. We have found significant sex differences in the overall number of microglia in the preoptic area (Lenz and others 2013; Fig. 1B), and others have documented similar sex differences in the hippocampus, parietal cortex, and amygdala (Schwarz and others 2012). We further found that in the preoptic area males have more microglia with an “activated” morphology characterized by an increase in cell body size and a decrease in process length and branching (Lenz and others 2013). Interestingly in the immature brain, this morphological activation does not map on to a clear microglial phenotype (M1 or M2) as is so often the case in the injured or degenerating brain. Instead, all microglia in the immature brain express markers for classical pro-inflammatory (M1) activation as well as alternative (M2) activation (Lenz and others 2013). These data match that of other groups who found microglia to highly express both M1 and M2 markers in the healthy, developing brain (Crain and others 2013; Cunningham and others 2013) and underscore that microglia in the immature brain are phenotypically distinct from adult microglia. It should be no surprise, then, that microglia perform distinct functions in the developing brain versus the injured or inflamed adult brain.
Most reproductively relevant sex differences in the brain are induced by steroid hormones during development and this is also true for the microglia of the POA. Treating females on the first two days of birth with estradiol leads to the full masculinization of microglial numbers and an increase in the number of ameboid microglia within a day of the final hormone treatment (Lenz and others 2013). Microglia in the developing POA do not possess estrogen receptor alpha (Lenz and others 2013), but it is unknown whether estradiol induces an ameboid morphology by acting on microglia directly via other estrogen receptors or indirectly by stimulating production and secretion of pro-inflammatory molecules in neighboring cell types. However it is established that microglia are the necessary node in the feed-forward mechanism through which PGE2 begets more PGE2 in the POA: If microglial activation is prevented with the pharmacological inhibitor, minocycline, a dose of estradiol to a neonate is no longer effective at increasing PGE2 levels in the POA (Lenz and others 2013). Microglial activation is also necessary to induce the masculine pattern of dendritic spines in the POA: If neonatal microglial activation is blocked with the inhibitor, minocycline, in vivo or in vitro, or if microglia are removed from POA primary cell cultures, estradiol treatment is no longer sufficient to induce a masculine dendritic spine phenotype. This early life programming by microglia has lifelong implications, with minocycline treatment preventing the masculinization of adult sexual behavior that is normally induced by estradiol (Lenz et al. 2013; Fig. 3).
Figure 3.

Microglia contribute to sexual differentiation of the preoptic area (POA) via release of the proinflammatory molecule, prostaglandin E2, which leads to sexually dimorphic patterning of dendritic spine synpases in the POA and resultant male-typical copulatory behavior in adulthood.
Recent data suggest that other inflammatory players may participate in sex differentiation in the brain, particularly during development. We have found significant sex differences in the expression of several inflammatory genes and pro-inflammatory cytokine levels in the developing POA, which may contribute to masculinization of the sex behavior circuit. Sex differences have also been detected in multiple cytokines and chemokines in the developing hippocampus, amygdala, and cortex, including CCL4, CCL20, and CD206, which may direct microglial colonization of the developing brain (Schwarz and others 2012). Ex vivo, microglia derived from male and female brains have shown divergent inflammatory signaling responses to lipopolysaccharide and estradiol treatment (Loram and others 2012), which argues that sex differences in microglia in the brain may be more extensive than just a difference in number or morphology, but may also be phenotypically distinct.
We are now extending our experimental questions beyond microglia to another innate immune cell in the brain, the mast cell. Mast cells are an innate immune cell largely studied in the context of asthma, anaphylaxis, and atopic allergic responses, yet there is also a population of mast cells in the brain that is first detected during the embryonic period and persists throughout the lifespan (Lambracht-Hall and others 1990). Mast cells are detected near the blood-brain barrier, but are almost entirely on the brain side in close apposition to blood vessels (Khalil and others 2007; Silverman and others 2000). Mast cells have long been implicated in the modulation of sexual behavior in birds, rapidly releasing hormones, such as GnRH as well as neurotransmitters, proteases, and cytokines into the brain parenchyma (Silver and Curley 2013; Silverman and others 1994). Though mast cells are known to populate the healthy developing brain, their function is an almost complete mystery. We have found that, just like microglia, there is a significant sex difference in the number of mast cells in both the developing preoptic area and the developing hippocampus. Our recent data implicates mast cells as key players in developmental masculinization of microglia in the POA and ultimately synaptic patterning and sexual behavior.
Implications of Immune Mediators as Normal Agents of Brain Development
Developmental Disorders in Which Neural Inflammation or Immune Signaling Is Implicated
Epidemiological studies have persuasively shown that prenatal or neonatal inflammation or infection increases risk for developing several neuropsychiatric disorders (Fig. 4). Schizophrenia is three times more common in offspring of women who suffered from influenza during the first trimester of pregnancy (Brown and others 2010), and bipolar disorder is four times more common (Parboosing and others 2013). Autism is significantly more common in children whose mothers had severe viral infections during the first trimester or bacterial infections during the second trimester of gestation (Atladóttir and others 2010). Corroborating animal studies investigating these effects of developmental neuroinflammation are largely focused on schizophrenia and autism, likely due to the stronger developmental models and understanding of these conditions than models for bipolar disorder.
Figure 4.

There is substantial overlap between neuropsychiatric and neurological disorders that show microglial involvement, sex biases in prevalence or severity, and have developmental origins. Currently, the roles of both sex hormones/sex chromosome complement and microglia in most of these disorders is poorly or incompletely understood.
Autism Spectrum Disorder
Autistic individuals have significantly elevated blood levels of numerous inflammatory cytokines (Molloy and others 2006; Schwarz and others 2011), especially those with the most severe behavioral symptoms (Ashwood and others 2011). The brains of autistic individuals also show increased inflammatory gene expression and cytokine levels in postmortem analysis (X. Li and others 2009), and inflammation and immune processes often appear as the top entry in genetic pathway analysis studies on post-review (Saxena and others 2012; Voineagu and others 2011). Perinatal inflammation and infection studies in animal models, using the bacterial endotoxin, lipopolysaccharide, or the viral mimetic, poly I:C induce many behavioral signatures of autism, including decreased social behavior and communication (Kirsten and others 2010; Malkova and others 2012; Taylor and others 2012), increased anxiety (Lin and others 2012), increased stereotyped or repetitive behavior (Kirsten and others 2010; Malkova and others 2012), and sensory disturbances (Patterson 2009). Perinatal inflammation also alters the trajectory of normal developmental processes that are hypothesized to play a role in the ontogeny of neurodevelopmental disorders, including cell genesis (Forrest and others 2012), cell survival and programmed cell death (Järlestedt and others 2013; Nimmervoll and others 2013), synaptogenesis (Bilousova and others 2009), synaptic pruning (Tremblay and others 2010), and myelination (Lieblein-Boff and others 2013; Rousset and others 2006).
Microglia specifically have been linked to the etiology of autism or endophenotypes for autism in animal models (Fig. 4). Increased microglial numbers and reactivity are apparent in the postmortem brains of autistic individuals (Tetreault and others 2012; Vargas and others 2005). In vivo positron emission tomography–based brain imaging of human microglia with a radiotracer shows autistic individuals have increased microglial activity throughout the brain, but particularly strikingly in the cerebellum, the brain region that is most strongly implicated in autism (Suzuki and others 2013). Microglia-neuron crosstalk during development may be one means through which inflammation confers risk for autism. When microglial-neuronal interaction is disrupted via fractalkine-receptor knockout, mice show impaired social behavior, increased repetitive behavior, and synaptic mis-wiring that make up core abnormalities of autism (Zhan and others 2014).
Schizophrenia
As discussed above, the incidence of schizophrenia is higher in children of women who experienced inflammation or illness during pregnancy. As in autism, peripheral inflammatory cytokines are higher in schizophrenics, particularly those with the most treatment resistance (Lin and others 1998). Inflammatory genes are up-regulated in postmortem brains of people diagnosed with schizophrenia (Saetre and others 2007), and increased numbers of activated microglia are also found throughout the cortex and hippocampus (Radewicz and others 2000; Steiner and others 2006). Early life inflammation in the brain produces schizophrenic-like behaviors in animal models, including deficits in working memory (Takao and others 2013) and sensorimotor gating (Borrell and others 2002; Q. Li and others 2009; Romero and others 2010) as well as alterations in dopaminergic and glutamatergic neurotransmission (Meyer and others 2008; Meyer and Feldon 2009).
Sex Differences in Neurodevelopmental Disorders
For several of these disorders, including autism, early-onset forms of schizophrenia, and early-onset bipolar disorder, males are at significantly higher risk than females of developing the disorder (Fig. 4). Autism is four to eight times more common in males (Baron-Cohen and others 2009; Fombonne 2002) and has even been characterized as having a hypermasculinized phenotype, behaviorally and physiologically (Baron-Cohen 2002). Matching data on human sex biases in prevalence, male rodents show larger disruptions in social behavior than females following prenatal immune challenge (Taylor and others 2012). Research on the sex bias in autism and autistic-like behavior has largely focused on the factors and processes that may make males more vulnerable than females, including fetal testosterone levels (Geier and others 2012). We hypothesize this male vulnerability occurs at least in part because microglia and inflammatory molecules are involved in the normal developmental process of sexual differentiation (Lenz and others 2013) and also because males have more activated innate immune cells in the developing brain under normal developmental conditions (Schwarz and others 2012). Although the research focus has largely been on male-specific genetic and physiological factors that confer vulnerability to neurodevelopmental disorders, a recent study of familial patterns of autism shows that females with autism have a greater etiological load, including clear environmental factors or large genetic changes (Jacquemont and others 2014; Robinson and others 2013). This sex difference in etiological load argues that there is a female protective factor that prevents a large majority of females from reaching threshold for autism diagnosis unless risk penetrance is very high (Robinson and others 2013). Therefore it is essential that we study female-specific biology to determine the factors that may confer resilience (Werling and Geschwind 2013), especially if the goal is to counteract the environmental factors that increase vulnerability in males.
Further complicating matters is that males and females with autism exhibit different physiological abnormalities and behavioral phenotypes from one another (Werling and Geschwind 2013). Characterization of peripheral biomarkers for autism revealed several immune and growth factors in blood were dysregulated in individuals with Asperger’s disorder relative to controls, and yet there was little overlap between the markers that were dysregulated in males versus females (Schwarz and others 2011). What is most intriguing in this data set is that males show alterations in levels of cytokines and other inflammatory molecules, whereas females show alterations in growth factors and hormone levels (Schwarz and others 2011). This study not only argues that both inflammatory and endocrine factors may influence the etiology of autism but also that these mechanisms may be sex specific.
Other disorders of brain development, including learning disabilities, ADHD, dyslexia, and cerebral palsy, are all more common in males, but inflammation has not (yet) been implicated in the etiology of these disorders. Together, these gender differences suggest the male brain may be more vulnerable to early life perturbations and/or that females may be more resilient.
Sex Differences in Adult Disorders
Mood Disorders
Substantial evidence implicates immune processes in the etiology of affective disorders (Fig. 4), particularly major depressive disorder and anxiety disorders (Dantzer and others 2008; Dunn and others 2005; O’Brien and others 2004). Peripheral cytokines are high in individuals suffering from depression and anxiety (Raison and others 2006; Schmidt and others 2011). Administration of endotoxin to humans induces key emotional symptoms of major depressive disorder, including anhedonia and depressed mood (Eisenberger and others 2010). Similarly, administration of inflammatory cytokines during cancer treatment, such as interferon-γ, induces sleep disruption, anhedonia, apathy and depressed mood, especially in individuals previously predisposed to depressive episodes (Walker and others 1997; Capuron and Ravaud 1999). Administration of endotoxins or cytokines to rodents similarly induces depressive-like behaviors, including decreased sucrose preference and increased immobility on the forced swim test (Henry and others 2008; Makino and others 2000; Tonelli and others 2008; Wu and Lin 2008). Inflammatory challenge not only increases proinflammatory cytokines in the periphery and brain but also leads to altered serotonin synthesis, possibly through microglial-mediated activation of the enzyme, IDO, which diverts tryptophan from the synthetic pathway for serotonin (Müller and Schwarz 2008; Steiner and others 2011). Inflammation also influences serotonin transporter function and thus reuptake (Baganz and Blakely 2013) and may thereby contribute to the etiology of depression through multiple routes. At this stage, there is clear evidence of a relationship between depression and inflammation, yet it remains unclear whether neuroinflammation leads to depression, depression leads to neuroinflammation, or both.
Unlike neurodevelopmental disorders, which have a male-biased preponderance, mood disorders, including depression, anxiety, and posttraumatic stress disorder, are diagnosed at approximately double the rate in women than in men (Angst and others 2002; Breslau and others 1997; Nolen-Hoeksema 1987). Ovarian hormones are thought to play a role, with rates of depression increasing during the transition to menopause (Freeman and Sammel 2006) and also during states of low or rapidly changing estrogen levels, such as the postpartum period (Ahokas and others 2001). Similarly, individuals with menstrual cycle–associated mood disorders show decreased estrogen sensitivity (Huo and others 2007). The research on hormonal modulation of mood is rife with contradiction, likely due to the heterogeneity in symptoms of mood disorders themselves. In humans, conclusions also remain hard to make because of high variability in women’s hormonal status that results from both intrinsic biological variability as well as different exogenous hormonal treatments (both hormone replacement and hormonal contraceptives). An interesting possibility is that the immune system, particularly the innate immune system in the brain, plays a role in this sex bias in mood disorder prevalence. While males have more microglia and greater inflammatory tone in the developing rodent brain (Lenz and others 2013; Schwarz and others 2012), the opposite is the case in adulthood, when females have more activated microglia in the brain and higher levels of pro-inflammatory cytokines (Schwarz and others 2012). Estrogens are argued to have largely anti-inflammatory effects in the brain (Czlonkowska and others 2006), and the correlation between low estrogen levels in females and mood disorders may be mediated by changes in microglia, cytokines, and inflammation in the brain.
Autoimmune Disorders
Autoimmune disorders are a group of up to 80 disorders in which the body’s immune system is over activated and attacks endogenously derived proteins as if they were foreign. Many such disorders, including systemic lupus erythematosus, multiple sclerosis, thyroid disorders, and rheumatoid arthritis are all at least twice as common in women as in men, and in total, nearly 80% of autoimmune disorder patients are women (Jacobson and others 1997; Whitacre 2001). This relatively higher incidence in females is largely attributed to increased sensitivity of the peripheral immune system (Whitacre 2001). As with mood disorders, ovarian hormones are likely to play a role. Both pregnancy and menopause can modulate symptom severity in autoimmune disorders (Selmi 2008). In fact, the alleviation of multiple sclerosis symptoms during pregnancy is so dramatic that clinical trials have been pursued to use estriol, an estrogen only made in substantial quantities in the body during pregnancy, as a multiple sclerosis therapeutic (Sicotte and others 2002; Soldan and others 2003). Elegant rodent work suggests that estrogens modulate multiple sclerosis symptom severity by inhibiting microglial over activation, but in complicated ways that depend on both which particular estrogens are administered and which receptors they act upon (Saijo and others 2011). Given that our current understanding of basic sex differences in innate immune cells and their signaling in the brain is limited at this point, we remain hampered in our ability to use hormonal therapeutics to effectively treat and even prevent brain disorders in which the innate immune system plays a central role.
Integrating Hormones, Genetics, and the Environment
While hormones are a potent and pervasive force in brain development, it is also true that every cell in the brain is either XX or XY in chromosome complement. Recent years have seen a dawning appreciation for the importance of sex chromosome complement as a direct source of variation in brain function. This new appreciation has its origins in two sources. First was an acceptance of the fact that hormones could not explain everything when it came to sex differences in the brain, and second was the advent of new animal models and genomic sequencing technologies. The identification of Sry as the initiating gene for testicular development lent itself to relocation on an autosome in genetically modifiable mice, and thus the ability to generate genetic females with testis (i.e., XX but Sry+) and genetic males with ovaries (i.e., XY but Sry−). This arrangement provided for a separation of gonadal sex, meaning hormones, from chromosome complement, meaning genetics. Multiple neuroanatomical and behavioral endpoint sex differences reflect a genetic contribution, sometimes in combination with hormonal modulation (Arnold and Chen 2009). These range from cognitive tasks such as habit formation (Quinn and others 2007), to social behaviors like aggression (Gatewood and others 2006) and disease susceptibility using an animal model of multiple sclerosis (Du and others 2014).
In retrospect, a contribution of the X chromosome to neuroanatomical sex differences seems self evident in light of the now emerging evidence that this chromosome is heavily enriched in genes that impact brain function. Up to 3.5 times as many genes affecting cognitive ability are found on the human X compared with autosomes (Zechner and others 2001). More surprising and tantalizing is that the X chromosome is equally enriched in in genes of the immune system, including multiple interleukin and Toll-like receptors (Fish 2008). The preponderance of immune related genes on the X chromosome is the basis for the increased vulnerability of males to several X-linked immunological diseases, but may also be the source of the increased vulnerability of females to autoimmune disorders (Libert and others 2010). This is due to the three unique ways in which the X chromosome differs in males and females. First, males receive only a maternal X, and thus any mutated or imprinted genes on that X are unopposed. Second, females receive both a maternal and paternal X but one copy is randomly inactivated in each cell to achieve dosage compensation. However, inactivation is also subject to skewing, and the degree of heteroscedasticity can vary by tissue or even with age (Wu and others 2014). The skewing of X chromosome inactivation can be either protective or damaging. Lastly, up to 10% to 15% of genes on the human X chromosome escape X inactivation, and even this may be an underestimate as X-gene silencing has turned out to be far less frequent and more variable than previously thought (Carrel and Willard 2005). Thus there are multiple opportunities for twice the protein dose in females (Pinheiro and others 2011; Spolarics 2007).
An additional distinguishing feature of the X chromosome is its higher density of miRNAs, small non-coding RNAs believed to regulate the expression of 30% to 50% of all genes. These ubiquitous molecules have been of intense interest in the field of cancer due to their gene repression effects, escape from which can release oncogenes (Garzon and others 2009). Several X chromosome miRNAs also have fundamental roles in immunity, down regulating both pro-and anti-inflammatory mediators. A single miRNA can regulate the translation of multiple genes and some of those implicated in regulation of the immune system also target the α-isoform of the estrogen receptor (Pinheiro and others 2011).
To date there are no clear connections between immune genes of the X chromosome and brain sex differences, but this seems likely to be more from lack of looking than lack of evidence. New tools that allow for quick, precise and affordable sequencing of large numbers of samples will surely open new vistas into which genes are being regulated by or regulate the sex of the brain. Thus we are rapidly approaching a time when an integrated synthesis of hormonal, genetic, and ultimately environmental factors will expand our understanding of the origins and impact of brain sex differences. With the emerging importance of immune system mediators in normal brain development, particularly in males, the impact of environmental variables that include injury or inflammation may be a nodal point for sex differences in both health and disease.
Summary and Conclusions
First described almost a century ago, microglia had been largely considered only in the context of injury and inflammation. Recent findings have fueled a renaissance in the study and interpretation of all non-neuronal cells in the brain and continue to reveal surprising and unexpected roles for these versatile partners. The ubiquitous and relatively uniform distribution of microglia throughout the brain positions them as surveyors of the microenvironment and rapid responders to local change. Previously, the nature of the response was thought to be restricted to evocation by major disruptions of homeostasis and consisting of the mechanical clearing of debris and the release of pro- or anti-inflammatory signaling molecules. We now know that the role of microglia is far more complex and at times subtle yet profoundly important. The regulation of cell number and synaptic patterning are two of the most fundamental components of neural development and microglia perform central functions in both by selective phagocytosis. We have discovered yet another novel role for microglia in regulating synaptic patterning selectively in males versus females that involves an intimate partnership with neighboring neurons and astrocytes. Steroid hormone signaling initiated in neurons is amplified by microglial release of prostaglandins and is an essential contributor to the induction and maintenance of dendritic spine excitatory synapses in the developing male brain. Whether the dual nature of microglia as responders to injury and mediators of brain masculinization contributes to the greater risk of males for developmental disorders remains an open but intriguing question.
Acknowledgments
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Insitute for Neurological Disorders and Stroke (F32NS076327), the National Institute for Mental Health (R01MH052716), and The Ohio State University startup funds.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- Aguzzi A, Barres BA, Bennett ML. Microglia: scapegoat, saboteur, or something else? Science. 2013;339:156–61. doi: 10.1126/science.1227901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahokas A, Kaukoranta J, Wahlbeck K, Aito M. Estrogen deficiency in severe postpartum depression: successful treatment with sublingual physiologic 17β-estradiol: a preliminary study. J Clin Psychiatry. 2001;62:332–6. doi: 10.4088/jcp.v62n0504. [DOI] [PubMed] [Google Scholar]
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10:1538–43. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- Alliot F, Godin I, Pessac B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res Dev Brain Res. 1999;117:145–52. doi: 10.1016/s0165-3806(99)00113-3. [DOI] [PubMed] [Google Scholar]
- Amateau SK, McCarthy MM. A novel mechanism of dendritic spine plasticity involving estradiol induction of prostaglandin-E2. J Neurosci. 2002a;22:8586–96. doi: 10.1523/JNEUROSCI.22-19-08586.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amateau SK, McCarthy MM. Sexual differentiation of astrocyte morphology in the developing rat preoptic area. J Neuroendocrinol. 2002b;14:904–10. doi: 10.1046/j.1365-2826.2002.00858.x. [DOI] [PubMed] [Google Scholar]
- Amateau SK, McCarthy MM. Induction of PGE2 by estradiol mediates developmental masculinization of sex behavior. Nat Neurosci. 2004;7:643–50. doi: 10.1038/nn1254. [DOI] [PubMed] [Google Scholar]
- Angst J, Gamma A, Gastpar M, Lépine J-P, Mendlewicz J, Tylee A. Gender differences in depression. Epidemiological findings from the European DEPRES I and II studies. Eur Arch Psychiatry Clin Neurosci. 2002;252:201–9. doi: 10.1007/s00406-002-0381-6. [DOI] [PubMed] [Google Scholar]
- Arnold AP, Chen X. What does the “four core genotypes” mouse model tell us about sex differences in the brain and other tissues? Front Neuroendocrinol. 2009;30:1–9. doi: 10.1016/j.yfrne.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah I, Van de Water J. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav Immun. 2011;25:40–5. doi: 10.1016/j.bbi.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atladóttir HO, Thorsen P, Schendel DE, Østergaard L, Lemcke S, Parner ET. Association of hospitalization for infection in childhood with diagnosis of autism spectrum disorders: a Danish cohort study. Arch Pediatr Adolesc Med. 2010;164:470–7. doi: 10.1001/archpediatrics.2010.9. [DOI] [PubMed] [Google Scholar]
- Baganz NL, Blakely RD. A dialogue between the immune system and brain, spoken in the language of serotonin. ACS Chem Neurosci. 2013;4:48–63. doi: 10.1021/cn300186b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron-Cohen S. The extreme male brain theory of autism. Trends Cogn Sci. 2002;6:248–254. doi: 10.1016/s1364-6613(02)01904-6. [DOI] [PubMed] [Google Scholar]
- Baron-Cohen S, Scott FJ, Allison C, Williams J, Bolton P, Matthews FE, et al. Prevalence of autism-spectrum conditions: UK school-based population study. Br J Psychiatry. 2009;194:500–9. doi: 10.1192/bjp.bp.108.059345. [DOI] [PubMed] [Google Scholar]
- Barron KD. The microglial cell. A historical review. J Neurol Sci. 1995;134(Suppl):57–68. doi: 10.1016/0022-510x(95)00209-k. [DOI] [PubMed] [Google Scholar]
- Bialas AR, Stevens B. TGF-β signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat Neurosci. 2013;16:1773–82. doi: 10.1038/nn.3560. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Bilousova TV, Dansie L, Ngo M, Aye J, Charles JR, Ethell DW, et al. Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J Med Genet. 2009;46:94–102. doi: 10.1136/jmg.2008.061796. [DOI] [PubMed] [Google Scholar]
- Borrell J, Vela JM, Arévalo-Martin A, Molina-Holgado E, Guaza C. Prenatal immune challenge disrupts sensorimotor gating in adult rats. Implications for the etiopathogenesis of schizophrenia. Neuropsychopharmacology. 2002;26:204–15. doi: 10.1016/S0893-133X(01)00360-8. [DOI] [PubMed] [Google Scholar]
- Breslau N, Davis GC, Andreski P, Peterson EL, Schultz LR. Sex differences in posttraumatic stress disorder. Arch Gen Psychiatry. 1997;54:1044–8. doi: 10.1001/archpsyc.1997.01830230082012. [DOI] [PubMed] [Google Scholar]
- Brown CM, Mulcahey TA, Filipek NC, Wise PM. Production of proinflammatory cytokines and chemokines during neuroinflammation: novel roles for estrogen receptors alpha and beta. Endocrinology. 2010;151:4916–25. doi: 10.1210/en.2010-0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capuron L, Ravaud A. Prediction of the depressive effects of interferon α therapy by the patient’s initial affective state. N Engl J Med. 1999;340:1370. doi: 10.1056/NEJM199904293401716. [DOI] [PubMed] [Google Scholar]
- Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434:400–4. doi: 10.1038/nature03479. [DOI] [PubMed] [Google Scholar]
- Collaer ML, Hines M. Human behavioral sex differences: a role for gonadal hormones during early development? Psychol Bull. 1995;118:55–107. doi: 10.1037/0033-2909.118.1.55. [DOI] [PubMed] [Google Scholar]
- Costello DA, Lyons A, Denieffe S, Browne TC, Cox FF, Lynch MA. Long term potentiation is impaired in membrane glycoprotein CD200-deficient mice: a role for Toll-like receptor activation. J Biol Chem. 2011;286:34722–32. doi: 10.1074/jbc.M111.280826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crain JM, Nikodemova M, Watters JJ. Microglia express distinct M1 and M2 phenotypic markers in the postnatal and adult central nervous system in male and female mice. J Neurosci Res. 2013;91:1143–51. doi: 10.1002/jnr.23242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CL, Martínez-Cerdeño V, Noctor SC. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J Neurosci. 2013;33:4216–33. doi: 10.1523/JNEUROSCI.3441-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czlonkowska A, Ciesielska A, Gromadzka G. Gender differences in neurological disease. Endocrine. 2006;29:243–56. doi: 10.1385/ENDO:29:2:243. [DOI] [PubMed] [Google Scholar]
- Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du S, Itoh N, Askarinam S, Hill H, Arnold AP, Voskuhl RR. XY sex chromosome complement, compared with XX, in the CNS confers greater neurodegeneration during experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2014;111:2806–11. doi: 10.1073/pnas.1307091111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn AJ, Swiergiel AH, de Beaurepaire R. Cytokines as mediators of depression: what can we learn from animal studies? Neurosci Biobehav Rev. 2005;29:891–909. doi: 10.1016/j.neubiorev.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Ehninger D, Kempermann G. Regional effects of wheel running and environmental enrichment on cell genesis and microglia proliferation in the adult murine neocortex. Cereb Cortex. 2003;13:845–51. doi: 10.1093/cercor/13.8.845. [DOI] [PubMed] [Google Scholar]
- Eisenberger NI, Berkman ET, Inagaki TK, Rameson LT, Mashal NM, Irwin MR. Inflammation-induced anhedonia: endotoxin reduces ventral striatum responses to reward. Biol Psychiatry. 2010;68:748–54. doi: 10.1016/j.biopsych.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish EN. The X-files in immunity: sex-based differences predispose immune responses. Nat Rev Immunol. 2008;8:737–44. doi: 10.1038/nri2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fombonne E. Epidemiological trends in rates of autism. Mol Psychiatry. 2002;7(Suppl 2):S4–6. doi: 10.1038/sj.mp.4001162. [DOI] [PubMed] [Google Scholar]
- Forest MG, Cathiard AM, Bertrand JA. Evidence of testicular activity in early infancy. J Clin Endocrinol Metab. 1973;37:148–51. doi: 10.1210/jcem-37-1-148. [DOI] [PubMed] [Google Scholar]
- Forrest CM, Khalil OS, Pisar M, Smith RA, Darlington LG, Stone TW. Prenatal activation of Toll-like receptors-3 by administration of the viral mimetic poly(I:C) changes synaptic proteins, N-methyl-D-aspartate receptors and neurogenesis markers in offspring. Mol Brain. 2012;5:22. doi: 10.1186/1756-6606-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman E, Sammel M, Lin H, Nelson DB. Associations of hormones and menopausal status with depressed mood in women with no history of depression. Arch Gen Psychiatry. 2006;63:375–82. doi: 10.1001/archpsyc.63.4.375. [DOI] [PubMed] [Google Scholar]
- Garzon R, Calin GA, Croce CM. MicroRNAs in cancer. Annu Rev Med. 2009;60:167–79. doi: 10.1146/annurev.med.59.053006.104707. [DOI] [PubMed] [Google Scholar]
- Gatewood JD, Wills A, Shetty S, Xu J, Arnold AP, Burgoyne PS, et al. Sex chromosome complement and gonadal sex influence aggressive and parental behaviors in mice. J Neurosci. 2006;26:2335–42. doi: 10.1523/JNEUROSCI.3743-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geier D, Kern J, King P, Sykes LK, Geier MR. An evaluation of the role and treatment of elevated male hormones in autism spectrum disorders. Acta Neurobio Exp (Wars) 2012;72:1–17. doi: 10.55782/ane-2012-1876. [DOI] [PubMed] [Google Scholar]
- Gendrel D, Chaussain JL, Roger M, Job JC. Simultaneous postnatal rise of plasma LH and testosterone in male infants. J Pediatr. 1980;97:600–2. doi: 10.1016/s0022-3476(80)80018-7. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–5. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodfellow PN, Lovell-Badge R. SRY and sex determination in mammals. Annu Rev Genet. 1993;27:71–92. doi: 10.1146/annurev.ge.27.120193.000443. [DOI] [PubMed] [Google Scholar]
- Gorski RA, Harlan RE, Jacobson CD, Shryne JE, Southam AM. Evidence for the existence of a sexually dimorphic nucleus in the preoptic area of the rat. J Comp Neurol. 1980;193:529–39. doi: 10.1002/cne.901930214. [DOI] [PubMed] [Google Scholar]
- Gregory CD, Pound JD. Cell death in the neighbourhood: direct microenvironmental effects of apoptosis in normal and neoplastic tissues. J Pathol. 2011;223:177–94. doi: 10.1002/path.2792. [DOI] [PubMed] [Google Scholar]
- Hanisch U-K, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–94. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Henry CJ, Huang Y, Wynne A, Hanke M, Himler J, Bailey MT, et al. Minocycline attenuates lipopolysaccharide (LPS)-induced neuroinflammation, sickness behavior, and anhedonia. J Neuroinflammation. 2008;5:15. doi: 10.1186/1742-2094-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey WF, Vass K, Lassmann H. Bone marrow-derived elements in the central nervous system: an immunohistochemical and ultrastructural survey of rat chimeras. J Neuropathol Exp Neurol. 1992;51:246–56. doi: 10.1097/00005072-199205000-00002. [DOI] [PubMed] [Google Scholar]
- Honda S, Harada N, Ito S, Takagi Y, Maeda S. Disruption of sexual behavior in male aromatase-deficient mice lacking exons 1 and 2 of the cyp19 gene. Biochem Biophys Res Commun. 1998;252:445–9. doi: 10.1006/bbrc.1998.9672. [DOI] [PubMed] [Google Scholar]
- Huo L, Straub RE, Roca C, Schmidt PJ, Shi K, Vakkalanka R, et al. Risk for premenstrual dysphoric disorder is associated with genetic variation in ESR1, the estrogen receptor alpha gene. Biol Psychiatry. 2007;62:925–33. doi: 10.1016/j.biopsych.2006.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson DL, Gange SJ, Rose NR, Graham NM. Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin Immunol Immunopathol. 1997;84:223–43. doi: 10.1006/clin.1997.4412. [DOI] [PubMed] [Google Scholar]
- Jacquemont S, Coe BP, Hersch M, Duyzend MH, Krumm N, Bergmann S, et al. A higher mutational burden in females supports a “female protective model” in neurodevelopmental disorders. Am J Hum Genet. 2014;94:415–25. doi: 10.1016/j.ajhg.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Järlestedt K, Naylor AS, Dean J, Hagberg H, Mallard C. Decreased survival of newborn neurons in the dorsal hippocampus after neonatal LPS exposure in mice. Neuroscience. 2013;253:21–8. doi: 10.1016/j.neuroscience.2013.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil M, Ronda J, Weintraub M, Jain K, Silver R, Silverman A-J. Brain mast cell relationship to neurovasculature during development. Brain Res. 2007;1171:18–29. doi: 10.1016/j.brainres.2007.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–44. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsten TB, Taricano M, Flório JC, Palermo-Neto J, Bernardi MM. Prenatal lipopolysaccharide reduces motor activity after an immune challenge in adult male offspring. Behav Brain Res. 2010;211:77–82. doi: 10.1016/j.bbr.2010.03.009. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–8. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Kudwa AE, Bodo C, Gustafsson J-A, Rissman EF. A previously uncharacterized role for estrogen receptor beta: defeminization of male brain and behavior. Proc Natl Acad Sci U S A. 2005;102:4608–12. doi: 10.1073/pnas.0500752102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambracht-Hall M, Dimitriadou V, Theoharides TC. Migration of mast cells in the developing rat brain. Brain Res Dev Brain Res. 1990;56:151–9. doi: 10.1016/0165-3806(90)90077-c. [DOI] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39:151–70. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- Lenz KM, Nugent BM, Haliyur R, McCarthy MM. Microglia are essential to masculinization of brain and behavior. J Neurosci. 2013;33:2761–72. doi: 10.1523/JNEUROSCI.1268-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz KM, Nugent BM, McCarthy MM. Sexual differentiation of the rodent brain: dogma and beyond. Front Neurosci. 2012;6:26. doi: 10.3389/fnins.2012.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz KM, Wright CL, Martin RC, McCarthy MM. Prostaglandin E2 regulates AMPA receptor phosphorylation and promotes membrane insertion in preoptic area neurons and glia during sexual differentiation. PLoS One. 2011;6:e18500. doi: 10.1371/journal.pone.0018500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenzlinger PM, Hans VH, Jöller-Jemelka HI, Trentz O, Morganti-Kossmann MC, Kossmann T. Markers for cell-mediated immune response are elevated in cerebrospinal fluid and serum after severe traumatic brain injury in humans. J Neurotrauma. 2001;18:479–89. doi: 10.1089/089771501300227288. [DOI] [PubMed] [Google Scholar]
- Li Q, Cheung C, Wei R, Hui ES, Feldon J, Meyer U, et al. Prenatal immune challenge is an environmental risk factor for brain and behavior change relevant to schizophrenia: evidence from MRI in a mouse model. PLoS One. 2009;4:e6354. doi: 10.1371/journal.pone.0006354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Chauhan A, Sheikh AM, Patil S, Chauhan V, Li X-M, et al. Elevated immune response in the brain of autistic patients. J Neuroimmunol. 2009;207:111–6. doi: 10.1016/j.jneuroim.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libert C, Dejager L, Pinheiro I. The X chromosome in immune functions: when a chromosome makes the difference. Nat Rev Immunol. 2010;10:594–604. doi: 10.1038/nri2815. [DOI] [PubMed] [Google Scholar]
- Lieblein-Boff JC, McKim DB, Shea DT, Wei P, Deng Z, Sawicki C, et al. Neonatal E. coli infection causes neurobehavioral deficits associated with hypomyelination and neuronal sequestration of iron. J Neurosci. 2013;33:16334–45. doi: 10.1523/JNEUROSCI.0708-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A, Kenis G, Bignotti S, Tura GJ, De Jong R, Bosmans E, et al. The inflammatory response system in treatment-resistant schizophrenia: increased serum interleukin-6. Schizophr Res. 1998;32:9–15. doi: 10.1016/s0920-9964(98)00034-6. [DOI] [PubMed] [Google Scholar]
- Lin Y-L, Lin S-Y, Wang S. Prenatal lipopolysaccharide exposure increases anxiety-like behaviors and enhances stress-induced corticosterone responses in adult rats. Brain Behav Immun. 2012;26:459–68. doi: 10.1016/j.bbi.2011.12.003. [DOI] [PubMed] [Google Scholar]
- Loram LC, Sholar PW, Taylor FR, Wiesler JL, Babb JA, Strand KA, et al. Sex and estradiol influence glial pro-inflammatory responses to lipopolysaccharide in rats. Psychoneuroendocrinology. 2012;37:1688–99. doi: 10.1016/j.psyneuen.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino M, Kitano Y, Komiyama C, Hirohashi M, Kohno M, Moriyama M, et al. Human interferon-α induces immobility in the mouse forced swimming test: involvement of the opioid system. Brain Res. 2000;852:482–4. doi: 10.1016/s0006-8993(99)02235-0. [DOI] [PubMed] [Google Scholar]
- Malkova NV, Yu CZ, Hsiao EY, Moore MJ, Patterson PH. Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav Immun. 2012;26:607–16. doi: 10.1016/j.bbi.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín-Teva JL, Dusart I, Colin C, Gervais A, van Rooijen N, Mallat M. Microglia promote the death of developing Purkinje cells. Neuron. 2004;41:535–47. doi: 10.1016/s0896-6273(04)00069-8. [DOI] [PubMed] [Google Scholar]
- McCarthy MM. Estradiol and the developing brain. Physiol Rev. 2008;88:91–124. doi: 10.1152/physrev.00010.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Feldon J. Neural basis of psychosis-related behaviour in the infection model of schizophrenia. Behav Brain Res. 2009;204:322–34. doi: 10.1016/j.bbr.2008.12.022. [DOI] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Schwendener S, Knuesel I, Yee BK, Feldon J. Relative prenatal and postnatal maternal contributions to schizophrenia-related neurochemical dysfunction after in utero immune challenge. Neuropsychopharmacology. 2008;33:441–56. doi: 10.1038/sj.npp.1301413. [DOI] [PubMed] [Google Scholar]
- Molloy CA, Morrow AL, Meinzen-Derr J, Schleifer K, Dienger K, Manning-Courtney P, et al. Elevated cytokine levels in children with autism spectrum disorder. J Neuroimmunol. 2006;172:198–205. doi: 10.1016/j.jneuroim.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Müller N, Schwarz MJ. COX-2 inhibition in schizophrenia and major depression. Curr Pharm Des. 2008;14:1452–65. doi: 10.2174/138161208784480243. [DOI] [PubMed] [Google Scholar]
- Nakanishi M, Niidome T, Matsuda S, Akaike A, Kihara T, Sugimoto H. Microglia-derived interleukin-6 and leukaemia inhibitory factor promote astrocytic differentiation of neural stem/progenitor cells. Eur J Neurosci. 2007;25:649–58. doi: 10.1111/j.1460-9568.2007.05309.x. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–8. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Nimmervoll B, White R, Yang J-W, An S, Henn C, Sun J-J, et al. LPS-induced microglial secretion of TNFα increases activity-dependent neuronal apoptosis in the neonatal cerebral cortex. Cereb Cortex. 2013;23:1742–55. doi: 10.1093/cercor/bhs156. [DOI] [PubMed] [Google Scholar]
- Nolen-Hoeksema S. Sex differences in unipolar depression: evidence and theory. Psychol Bull. 1987;101:259–82. [PubMed] [Google Scholar]
- O’Brien SM, Scott LV, Dinan TG. Cytokines: abnormalities in major depression and implications for pharmacological treatment. Hum Psychopharmacol. 2004;19:397–403. doi: 10.1002/hup.609. [DOI] [PubMed] [Google Scholar]
- Ogawa S, Chester AE, Hewitt SC, Walker VR, Gustafsson JA, Smithies O, et al. Abolition of male sexual behaviors in mice lacking estrogen receptors α and β (αβERKO) Proc Natl Acad Sci U S A. 2000;97:14737–41. doi: 10.1073/pnas.250473597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olah M, Ping G, De Haas AH, Brouwer N, Meerlo P, Van Der Zee EA, et al. Enhanced hippocampal neurogenesis in the absence of microglia T cell interaction and microglia activation in the murine running wheel model. Glia. 2009;57:1046–61. doi: 10.1002/glia.20828. [DOI] [PubMed] [Google Scholar]
- Paolicelli R, Bolasco G, Pagani F, Maggi L. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456–8. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- Parboosing R, Bao Y, Shen L, Schaefer CA, Brown AS. Gestational influenza and bipolar disorder in adult offspring. JAMA Psychiatry. 2013;70:677–85. doi: 10.1001/jamapsychiatry.2013.896. [DOI] [PubMed] [Google Scholar]
- Patterson PH. Immune involvement in schizophrenia and autism: etiology, pathology and animal models. Behav Brain Res. 2009;204:313–21. doi: 10.1016/j.bbr.2008.12.016. [DOI] [PubMed] [Google Scholar]
- Pinheiro I, Dejager L, Libert C. X-chromosome-located microRNAs in immunity: might they explain male/female differences? The X chromosome-genomic context may affect X-located miRNAs and downstream signaling, thereby contributing to the enhanced immune response of females. Bioessays. 2011;33:791–802. doi: 10.1002/bies.201100047. [DOI] [PubMed] [Google Scholar]
- Quinn JJ, Hitchcott PK, Umeda EA, Arnold AP, Taylor JR. Sex chromosome complement regulates habit formation. Nat Neurosci. 2007;10:1398–400. doi: 10.1038/nn1994. [DOI] [PubMed] [Google Scholar]
- Radewicz K, Garey LJ, Gentleman SM, Reynolds R. Increase in HLA-DR immunoreactive microglia in frontal and temporal cortex of chronic schizophrenics. J Neuropathol Exp Neurol. 2000;59:137–50. doi: 10.1093/jnen/59.2.137. [DOI] [PubMed] [Google Scholar]
- Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 2006;27:24–31. doi: 10.1016/j.it.2005.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravichandran KS. Beginnings of a good apoptotic meal: the find-me and eat-me signaling pathways. Immunity. 2011;35:445–55. doi: 10.1016/j.immuni.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes FI, Winter JSD, Faiman C. Studies on human sexual development. I. Fetal gonadal and adrenal sex steroids. J Clin Endocrinol Metab. 1973;37:74–8. doi: 10.1210/jcem-37-1-74. [DOI] [PubMed] [Google Scholar]
- Robinson EB, Lichtenstein P, Anckarsäter H, Happé F, Ronald A. Examining and interpreting the female protective effect against autistic behavior. Proc Natl Acad Sci U S A. 2013;110:5258–62. doi: 10.1073/pnas.1211070110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JT, Morganti JM, Bachstetter AD, Hudson CE, Peters MM, Grimmig BA, et al. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J Neurosci. 2011;31:16241–50. doi: 10.1523/JNEUROSCI.3667-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero E, Guaza C, Castellano B, Borrell J. Ontogeny of sensorimotor gating and immune impairment induced by prenatal immune challenge in rats: implications for the etiopathology of schizophrenia. Mol Psychiatry. 2010;15:372–83. doi: 10.1038/mp.2008.44. [DOI] [PubMed] [Google Scholar]
- Rousset CI, Chalon S, Cantagrel S, Bodard S, Andres C, Gressens P, et al. Maternal exposure to LPS induces hypomyelination in the internal capsule and programmed cell death in the deep gray matter in newborn rats. Pediatr Res. 2006;59:428–33. doi: 10.1203/01.pdr.0000199905.08848.55. [DOI] [PubMed] [Google Scholar]
- Saetre P, Emilsson L, Axelsson E, Kreuger J, Lindholm E, Jazin E. Inflammation-related genes up-regulated in schizophrenia brains. BMC Psychiatry. 2007;7:46. doi: 10.1186/1471-244X-7-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo K, Collier JG, Li AC, Katzenellenbogen JA, Glass CK. An ADIOL-ERβ-CtBP transrepression pathway negatively regulates microglia-mediated inflammation. Cell. 2011;145:584–95. doi: 10.1016/j.cell.2011.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena V, Ramdas S, Ochoa CR, Wallace D, Bhide P, Kohane I. Structural, genetic, and functional signatures of disordered neuroimmunological development in autism spectrum disorder. PLoS One. 2012;7:e48835. doi: 10.1371/journal.pone.0048835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt HD, Shelton RC, Duman RS. Functional bio-markers of depression: diagnosis, treatment, and pathophysiology. Neuropsychopharmacology. 2011;36:2375–94. doi: 10.1038/npp.2011.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz E, Guest PC, Rahmoune H, Wang L, Levin Y, Ingudomnukul E, et al. Sex-specific serum biomarker patterns in adults with Asperger’s syndrome. Mol Psychiatry. 2011;16:1213–20. doi: 10.1038/mp.2010.102. [DOI] [PubMed] [Google Scholar]
- Schwarz JM, Sholar PW, Bilbo SD. Sex differences in microglial colonization of the developing rat brain. J Neurochem. 2012;120:948–63. doi: 10.1111/j.1471-4159.2011.07630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmi C. The X in sex: how autoimmune diseases revolve around sex chromosomes. Best Pract Res Clin Rheumatol. 2008;22:913–22. doi: 10.1016/j.berh.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Shigemoto-Mogami Y, Hoshikawa K, Goldman JE, Sekino Y, Sato K. Microglia enhance neurogenesis and oligodendrogenesis in the early postnatal subventricular zone. J Neurosci. 2014;34:2231–43. doi: 10.1523/JNEUROSCI.1619-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicotte NL, Liva SM, Klutch R, Pfeiffer P, Bouvier S, Odesa S, et al. Treatment of multiple sclerosis with the pregnancy hormone estriol. Ann Neurol. 2002;52:421–8. doi: 10.1002/ana.10301. [DOI] [PubMed] [Google Scholar]
- Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, et al. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010;7:483–95. doi: 10.1016/j.stem.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver R, Curley JP. Mast cells on the mind: new insights and opportunities. Trends Neurosci. 2013;36:513–21. doi: 10.1016/j.tins.2013.06.001. [DOI] [PubMed] [Google Scholar]
- Silverman AJ, Millar RP, King JA, Zhuang X, Silver R. Mast cells with gonadotropin-releasing hormone-like immunoreactivity in the brain of doves. Proc Natl Acad Sci U S A. 1994;91:3695–9. doi: 10.1073/pnas.91.9.3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman AJ, Sutherland AK, Wilhelm M, Silver R. Mast cells migrate from blood to brain. J Neurosci. 2000;20:401–8. doi: 10.1523/JNEUROSCI.20-01-00401.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldan SS, Alvarez Retuerto AI, Sicotte NL, Voskuhl RR. Immune modulation in multiple sclerosis patients treated with the pregnancy hormone estriol. J Immunol. 2003;171:6267–74. doi: 10.4049/jimmunol.171.11.6267. [DOI] [PubMed] [Google Scholar]
- Spolarics Z. The X-files of inflammation: cellular mosaicism of X-linked polymorphic genes and the female advantage in the host response to injury and infection. Shock. 2007;27:597–604. doi: 10.1097/SHK.0b013e31802e40bd. [DOI] [PubMed] [Google Scholar]
- Steiner J, Mawrin C, Ziegeler A, Bielau H, Ullrich O, Bernstein H-G, et al. Distribution of HLA-DR-positive microglia in schizophrenia reflects impaired cerebral lateralization. Acta Neuropathol. 2006;112:305–16. doi: 10.1007/s00401-006-0090-8. [DOI] [PubMed] [Google Scholar]
- Steiner J, Walter M, Gos T, Guillemin GJ, Bernstein H-G, Sarnyai Z, et al. Severe depression is associated with increased microglial quinolinic acid in sub-regions of the anterior cingulate gyrus: evidence for an immune-modulated glutamatergic neurotransmission? J Neuroinflammation. 2011;8:94. doi: 10.1186/1742-2094-8-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Sugihara G, Ouchi Y, Nakamura K, Futatsubashi M, Takebayashi K, et al. Microglial activation in young adults with autism spectrum disorder. JAMA Psychiatry. 2013;70:49–58. doi: 10.1001/jamapsychiatry.2013.272. [DOI] [PubMed] [Google Scholar]
- Takao K, Kobayashi K, Hagihara H, Ohira K, Shoji H, Hattori S, et al. Deficiency of schnurri-2, an MHC enhancer binding protein, induces mild chronic inflammation in the brain and confers molecular, neuronal, and behavioral phenotypes related to schizophrenia. Neuropsychopharmacology. 2013;38:1409–25. doi: 10.1038/npp.2013.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor PV, Veenema AH, Paul MJ, Bredewold R, Isaacs S, de Vries GJ. Sexually dimorphic effects of a prenatal immune challenge on social play and vasopressin expression in juvenile rats. Biol Sex Differ. 2012;3:15. doi: 10.1186/2042-6410-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetreault NA, Hakeem AY, Jiang S, Williams BA, Allman E, Wold BJ, et al. Microglia in the cerebral cortex in autism. J Autism Dev Disord. 2012;42:2569–84. doi: 10.1007/s10803-012-1513-0. [DOI] [PubMed] [Google Scholar]
- Tonelli LH, Holmes A, Postolache TT. Intranasal immune challenge induces sex-dependent depressive-like behavior and cytokine expression in the brain. Neuropsychopharmacology. 2008;33:1038–48. doi: 10.1038/sj.npp.1301488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay M, Lowery R, Majewska A. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 2010;8:e1000527. doi: 10.1371/journal.pbio.1000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Beek C, Thijssen JHH, Cohen-Kettenis PT, van Goozen SHM, Buitelaar JK. Relationships between sex hormones assessed in amniotic fluid, and maternal and umbilical cord serum: what is the best source of information to investigate the effects of fetal hormonal exposure? Horm Behav. 2004;46:663–9. doi: 10.1016/j.yhbeh.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 2005;57:67–81. doi: 10.1002/ana.20315. [DOI] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–4. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vukovic J, Colditz MJ, Blackmore DG, Ruitenberg MJ, Bartlett PF. Microglia modulate hippocampal neural precursor activity in response to exercise and aging. J Neurosci. 2012;32:6435–43. doi: 10.1523/JNEUROSCI.5925-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci. 2009;29:3974–80. doi: 10.1523/JNEUROSCI.4363-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakselman S, Béchade C, Roumier A, Bernard D, Triller A, Bessis A. Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor. J Neurosci. 2008;28:8138–43. doi: 10.1523/JNEUROSCI.1006-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LG, Walker MB, Heys SD, Lolley J, Wesnes K, Eremin O. The psychological and psychiatric effects of rIL-2 therapy: a controlled clinical trial. Psychooncology. 1997;6:290–301. doi: 10.1002/(SICI)1099-1611(199712)6:4<290::AID-PON283>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Walton N, Sutter B, Laywell E, Levkoff LH, Kearns SM, Marshall GP, 2nd, et al. Microglia instruct subventricular zone neurogenesis. Glia. 2006;54:815–25. doi: 10.1002/glia.20419. [DOI] [PubMed] [Google Scholar]
- Werling DM, Geschwind DH. Sex differences in autism spectrum disorders. Curr Opin Neurol. 2013;26:146–53. doi: 10.1097/WCO.0b013e32835ee548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitacre CC. Sex differences in autoimmune disease. Nat Immunol. 2001;2:777–80. doi: 10.1038/ni0901-777. [DOI] [PubMed] [Google Scholar]
- Wohleb ES, McKim DB, Shea DT, Powell ND, Tarr AJ, Sheridan JF, et al. Re-establishment of anxiety in stress-sensitized mice is caused by monocyte trafficking from the spleen to the brain. Biol Psychiatry. 2013 Dec 11; doi: 10.1016/j.biopsych.2013.11.029. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright CL, Burks SR, McCarthy MM. Identification of prostaglandin E2 receptors mediating perinatal masculinization of adult sex behavior and neuroanatomical correlates. Dev Neurobiol. 2008;68:1406–19. doi: 10.1002/dneu.20665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright CL, McCarthy MM. Prostaglandin E2-induced masculinization of brain and behavior requires protein kinase A, AMPA/kainate, and metabotropic glutamate receptor signaling. J Neurosci. 2009;29:13274–82. doi: 10.1523/JNEUROSCI.3603-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Luo J, Yu H, Rattner A, Mo A, Wang Y, et al. Cellular resolution maps of X chromosome inactivation: implications for neural development, function, and disease. Neuron. 2014;81:103–19. doi: 10.1016/j.neuron.2013.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T-H, Lin C-H. IL-6 mediated alterations on immobile behavior of rats in the forced swim test via ERK1/2 activation in specific brain regions. Behav Brain Res. 2008;193:183–91. doi: 10.1016/j.bbr.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40:274–88. doi: 10.1016/j.immuni.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechner U, Wilda M, Kehrer-Sawatzki H, Vogel W, Fundele R, Hameister H. A high density of X-linked genes for general cognitive ability: a run-away process shaping human evolution? Trends Genet. 2001;17:697–701. doi: 10.1016/s0168-9525(01)02446-5. [DOI] [PubMed] [Google Scholar]
- Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci. 2014;17:400–6. doi: 10.1038/nn.3641. [DOI] [PubMed] [Google Scholar]
- Zhong Y, Zhou L-J, Ren W-J, Xin W-J, Li Y-Y, Zhang T, et al. The direction of synaptic plasticity mediated by C-fibers in spinal dorsal horn is decided by Src-family kinases in microglia: the role of tumor necrosis factor-α. Brain Behav Immun. 2010;24:874–80. doi: 10.1016/j.bbi.2010.01.007. [DOI] [PubMed] [Google Scholar]