

Abstract
The application of small molecules as catalysts for the diversification of natural product scaffolds is reviewed. Specifically, principles that relate to the selectivity challenges intrinsic to complex molecular scaffolds are summarized. The synthesis of analogs of natural products by this approach is then described as a quintessential “late-stage functionalization” exercise wherein natural products serve as the lead scaffolds. Given the historical application of enzymatic catalysts to the site-selective alteration of complex molecules, the focus of this review is on the recent studies of non-enzymatic catalysts. Reactions involving hydroxyl group derivatization with a variety of electrophilic reagents are discussed. C–H bond functionalizations that lead to oxidations, aminations, and halogenations are also presented. Several examples of site-selective olefin functionalizations and C–C bond formations are also included. Numerous classes of natural products have been subjected to these studies of site-selective alteration including polyketides, glycopeptides, terpenoids, macrolides, alkaloids, carbohydrates and others. What emerges is a platform for chemical remodeling of naturally occurring scaffolds that targets virtually all known chemical functionalities and microenvironments. However, challenges for the design of very broad classes of catalysts, with even broader selectivity demands (e.g., stereoselectivity, functional group selectivity, and site-selectivity) persist. Yet, a significant spectrum of powerful, catalytic alterations of complex natural products now exists such that expansion of scope seems inevitable. Several instances of biological activity assays of remodeled natural product derivatives are also presented. These reports may foreshadow further interdisciplinary impacts for catalytic remodeling of natural products, including contributions to SAR development, mode of action studies, and eventually medicinal chemistry.
Graphical Abstract
1. Introduction
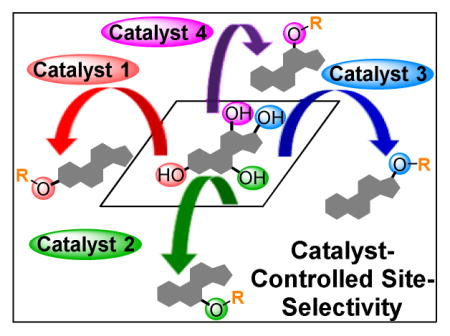
Complex natural products continue to be a quintessential interdisciplinary nexus. Whether through their isolation, characterization, or biosynthetic study, the exploration of natural products as bioactive agents has led to the development of a plethora of novel therapeutic agents. Additionally, the structural intricacy and diversity of these compounds have stimulated the development of new synthetic strategies and reactions. As such, natural products have inspired the development of new techniques and ideas throughout natural science.1,2 As structurally complex scaffolds, these molecules have also been the cornerstone of studies for substrate diversification. While this activity is often applications driven—for example seeking new, more biologically effective natural product-based medicine3—these efforts have also provided a significant test-bed for the identification of “site-selective” catalysts.4–9 For example, many complex natural products contain multiple occurrences of the same functional group for derivatization. Polyols (e.g., erythromycin A, 1),10 polyenes/polyols (e.g., amphotericin B, 2),11 polyamines/polyols (e.g., amino glycoside neomycin B, 3),12 and even mostly hydrocarbon, C–H bond-rich substrates (e.g., longifolene, 4)13 all contain numerous reactive functional groups that can be targeted to diversify the scaffolds for specific and selective analog generation (Figure 1A). Provided that targeted natural products can be isolated in sufficient quantity, this technique, now often described as “late-stage functionalization,” requires few synthetic steps to achieve the facile and direct functionalization of structurally complex molecules. However, while functional group repetition within complex molecules provides the opportunity to synthesize diverse derivatives, the question arises as to whether a comprehensive panel of catalysts can be discovered that selectively functionalize each functional group on a target molecule. For example, it would be desirable if five catalysts could be discovered that afford each of the potential monoesters derived from 1 (Figure 1B).
Figure 1.

(A) Selected natural products that present substantial challenges for site-selective functionalizations.10–13 (B) Challenges associated with the site-selective modificaiton of a natural product, such as the monoacylation of polyol 1.
Intrinsic to this challenge of catalytic derivatization of complex, naturally occurring molecules are numerous questions of stereoselectivity. In particular, as one targets stereochemically complex local environments, issues of diastereoselectivity and regioselectivity are pervasive in realizing the selective, catalytic functionalizations of intricate scaffolds. Inherent to these challenges is the reordering of the reactivity hierarchies of individual occurrences of the same or related functional groups in the natural product substrates.4 These issues amount to the multiplexing of the selectivity challenges that are encountered in the field of enantioselective catalysis. In the case of enantioselective catalysis, for example the desymmetrization shown in Figure 2A, the competing pathways to each enantiomer have the same energy of activation in the absence of a catalyst. Hence, a selective chiral catalyst that can preferentially reduce the energy of one activation barrier will subsequently result in the preferential formation of one enantiomer, with the magnitude of the ΔΔG‡ (1) dictating the degree of selectivity (Figure 2A, blue versus green pathway). However, in a more complex setting, the targeted functional groups will have different inherent reactivities. Hence, distinct energy profiles exist that yield various products in oftentimes unequal proportions (Figure 2B, red versus blue pathways). This may not be problematic if the lower energy, red pathway is desired. However, functionalizations of higher energy pathways, such as shown in blue, become substantially more difficult to favor. In these cases, depending on the initial differences in the activation barriers of competing pathways (ΔΔG‡ (2)), the catalyst-controlled, selective lowering of the ΔG‡ associated with one product by an increment of ΔΔG‡ commonly associated with high enantioselectivity (e.g., 2.7 kcal/mol, 95:5 er, at 298 K) may not result in high observed selectivities, since this energy difference may only account for the intrinsic differences in reactivity at competing sites, blue versus green pathways, ΔΔG‡ (3)). As seen in Figure 2B, even if a catalyst reduced the activation barrier of the inherent pathway to product B (ΔG‡2, blue) to half its original value (ΔG‡3, green), ΔG‡3 is still similar in energy to ΔG‡1 (red), resulting in an observed selectivity close to 1:1. Significantly larger ΔΔG‡ increments are thus required to dramatically overturn intrinsic selectivity (Figure 2B, blue versus purple pathways ΔΔG‡ (4)). And then, of course, when there are multiple competing pathways, the management of even more ΔΔG‡ increments is required, as the number of products that may be formed increases (Figure 2C). Consequently, the selective minimization of one reaction pathway can become exponentially more complicated as products of multiple functionalization accrue (Figure 2D).
Figure 2.

(A) Energy profiles of a desymmetrization reaction, where both potentially reacting alcohols are chemically equivalent. A catalyst that can selectively lower the activation barrier (
 pathway,
pathway,
 ) will result in high enantioselectivities,4
(B) Energy profiles of a site-selective transformation, where reactive groups are nonequivalent. Depending on the inherent energy profiles of the functional groups (
) will result in high enantioselectivities,4
(B) Energy profiles of a site-selective transformation, where reactive groups are nonequivalent. Depending on the inherent energy profiles of the functional groups (
 versus
versus
 pathways,
pathways,
 ), catalytic reduction of an energy barrier may not result in high observed selectivities (
pathway,
), catalytic reduction of an energy barrier may not result in high observed selectivities (
pathway,
 ). The achievement of highly selective functionalizations may require substantially more selective catalysts (
). The achievement of highly selective functionalizations may require substantially more selective catalysts (
 pathway,
pathway,
 ). (C) This problem is compounded by the addition of more reactive groups and (D) the ability for substrates to undergo multiple derivatization events.
). (C) This problem is compounded by the addition of more reactive groups and (D) the ability for substrates to undergo multiple derivatization events.
Despite these challenges, the last decade has witnessed a series of advances. First, numerous early reports use enzymes to synthesize analogs of complex molecules,14 and many impressive examples can be enumerated.15 In addition to enzyme-catalyzed solutions to late-stage modification, the application of non-enzymatic catalysts to natural products, while more nascent in development, has shown promise. Standing on the shoulders of four decades of remarkable progress in stereoselective catalysis of all types (enantioselective, diastereoselective, etc.),16,17 investigators increasingly have begun to apply non-enzymatic catalysts to natural product diversification.4–9 While it is fair to say that there are virtually no reports of a comprehensive set of “n” catalysts available for the selective derivatization of a reasonably complex molecule with “n” copies of the same functional group (i.e. 5 distinct catalysts for the derivatization of the 5 alcohols of Erythromycin, Figure 1B), there are now many precedents to build upon. This Review will endeavor to describe these advances. Included in our discussion are cases in which non-enzymatic catalysts have been applied to natural products above a certain arbitrarily defined threshold of complexity. Derivatizations of naturally occurring compounds such as glycerol, or other simple carbohydrates, are not included. As a note, in many cases we have included natural product substrates that have been converted to a minimally protected form. Additionally, for the most part, we focus on cases in which (a) non-enzymatic catalysts achieve divergent reactivity allowing more than one derivative to be synthesized selectively via catalyst control, (b) catalyst control influences the intrinsic reactivity of the substrate relative to a well-defined set of control reactions, and (c) the additives employed exhibit either bona fide catalytic turnover or at least appear to be unchanged by the reaction that alters the natural product substrate. That is, reagent-based derivatizations of natural products are not the emphasis of this Review, although some examples are included to highlight complementary approaches to access analogues distinct from catalyzed variants, in addition to addressing issues of inherent reactivity prevalent in the realm of site-selective catalysis.
What emerges is a foundation for the study of next-generation catalysts that manage complicated stereochemical and regiochemical problems in parallel. In some respects, this frontier is the manifestation of the “competition” experiment18 within a unimolecular substrate setting. As an intersection among synthetic chemistry, physical organic chemistry, inorganic and organometallic chemistry, and of course medicinal chemistry, natural products seem likely to retain their central place in molecular science for the foreseeable future.
This Review starts with a summary of catalyst-controlled group transfers to hydroxyl groups, one of the oft-studied fields in the realm of site-selective catalysis. A summary of selective C–H oxidations follows. While serving as an effective method for the diversification of natural products, only recently have catalyst-controlled C–H oxidations emerged to supplement the large breadth of substrate-controlled protocols. In a similar vein, the following section, the halogenation of natural products, is a field of growing interest and sophistication. Finally, a variety of other impressive, selective C–O, C–N, and C–C bond formations on natural products are reviewed here.
2. Group Transfer to Hydroxyl Groups
Alcohols are one of the most prevalent functional groups in natural products, and the propensity of these groups to react has been well studied since the inception of the field of organic synthesis.19,20 However, with their prevalence comes the challenge of discriminating between subtle differences in the reactivity of similar alcohols. One classical strategy to access analogs of polyols that are functionalized at a single alcohol is to use protecting groups, such that the most reactive alcohols can be masked and less reactive alcohols can be subsequently functionalized.21,22 While effective, this approach is often inefficient due to the amount of steps, time, and resources required to accomplish a selective transformation.23 It would be desirable if catalysts or reagents were developed that could distinguish between these extremely similar groups, thereby overturning the inherent reactivity of the native alcohols. Catalysts of this nature would enable the facile synthesis of biologically or pharmaceutically relevant natural product derivatives aimed at understanding the compounds’ mechanisms of action or heightening their activity.3 Of course, the total synthesis of numerous analogs is another approach, but can often become inefficient or costly given the natural products’ size and complexity. Nonetheless, landmark total syntheses of analog libraries have been reported.24 Catalytic site-selective modification of readily available natural products is a promising and complementary route to develop novel derivatives.
2.1. Enzyme-Catalyzed Group Transfer Reactions
Discussed only briefly here, protein kinases provide a quintessential and inspirational example of site-selective catalysis that can be found in nature.25–31 This ubiquitous class of enzymes acts as a pivotal regulator of cellular function, including but not limited to metabolism, cell division, and immune response.32 Kinases function by selectively phosphorylating alcohols (or other nucleophilic functional groups) within proteins, altering the proteins’ conformations and functions (Figure 3). With over 500 protein kinase genes identified in humans, kinases account for the fifth largest human gene family and the second largest class of human enzymes.28,29 It is truly remarkable, given the sheer number of proteins, carbohydrates, and small molecules that contain alcohols in a cell, combined with the number of alcohols on a singular substrate, that protein kinases are capable of reacting selectively at a single alcohol of a single protein.
Figure 3.
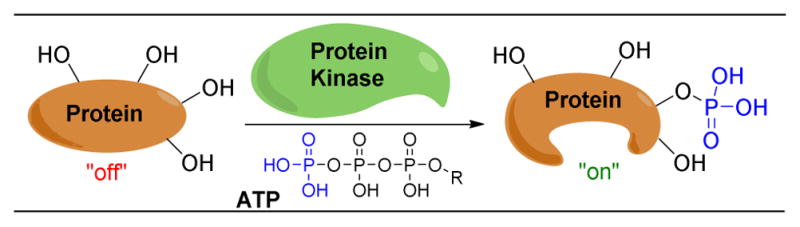
Protein kinases selectively phosphorylate protein hydroxyl groups. Among their many roles is serving in signal transduction pathways to turn the enzyme’s function “on” or “off”.25–31
The power of enzymes, not only in their impeccable selectivity, but also in their high turnover rate, has attracted many chemists to utilize enzymes in organic synthesis.14,15,33–35 However, this topic is beyond the focus of this Review.
2.2. Development of Peptides as Site-Selective Group Transfer Catalysts
The site-selective modification of polyols may be among the best-developed examples of non-enzymatic, catalytic modification of natural products. In one example, our laboratory explored an approach to the kinetic resolution of simple alcohols based on the incorporation of imidazole-containing π-methylhistidine (Pmh) residues into short peptides. One advantage of peptide catalysis lies in the ability to easily alter the peptide sequence appended to the reactive residue (Pmh in this case), thus potentially fine-tuning the catalysts’ selectivities toward various alcohols. The modularity of peptides is akin to nature’s ability to adjust the reactivity of a conserved catalytic residue through varying enzymes’ secondary and tertiary structures.36–38 Upon identifying a suitable peptide chain to append to the Pmh residue, the resulting catalytic peptide proved effective for selective acylation through a mechanism that likely involves the imidazole moiety as a nucleophilic or Lewis basic group in the catalytic mechanism (Figure 4A).39–42 The functionality of the peptide backbone has also been proposed to engage in favorable secondary interactions with the substrate, such as with an acetamide ((±)-5) to direct acyl transfer to particular hydroxyls (Figure 4B). The peptide sequence of 7 was chosen in large part due to its known bias towards adopting β-turns.43–45 As shown in Figure 4B, this C=Oi to NHi+3 intramolecular hydrogen bond (H-bond) can stabilize the secondary structure of the peptide and provide a relatively consistent structure for catalysis.46 This approach was also explored in reactions analogous to those catalyzed by kinases, culminating in the realization of peptide-catalyzed desymmetrizations of inositol derivatives (Figure 4C).47–51 Here, two distinct peptide scaffolds are utilized to access different phosphorylated derivatives of 8. These early reports of peptide-based nucleophilic catalysis helped establish a basis for the application of these concepts to the more complex substrates that are described below. In a related study, we showed that this class of catalysts could also be applied to the regioselective acylation of certain carbohydrates.52
Figure 4.

(A) Pmh-catalyzed acyl transfer. The imidazole (or other N-heterocycles) serves as a nucleophilic catalyst, decomposing the acid anhydride and delivering the acyl group to a substrate hydroxyl. Other functionality on the peptide can bind to the substrate and enforce selectivity. (B) The acetamide of 5 serves as a directing group for peptide 7, resulting in high levels of selectivity for this kinetic resolution by acylation.39 (C) Peptide-based phosphorylation can be accomplished on more complex substrates, such as 8. (D) Peptides utilized in this figure.48
2.2.1. Site-Selective Alcohol Derivatization of Erythromycin
One biologically relevant compound that is rich in alcohol groups is erythromycin A (13), which was first discovered and marketed by Eli Lilly as an antibiotic in the 1950s.10,53 However, given the widespread epidemic of drug-resistance, especially toward macrolide compounds such as erythromycin, novel compounds that can combat super bacteria are required.54,55 As such, numerous total syntheses have been reported for erythromycin and relevant derivatives.24,56–64 While these approaches allow for the derivatization of erythromycin’s structure at various stages, the syntheses of these macrolides can be challenging given their stereochemical complexity. The direct site-selective modification of erythromycin is hence particularly attractive, as the ability to target various locations on 13 would allow for the facile and expedient synthesis of numerous derivatives for structure–activity relationship (SAR) studies.
To approach the synthesis of novel derivatives of erythromycin A (13) by site-selective catalysis, our group first examined the inherent reactivity of 13 towards acylation (Figure 5).65 Using either a catalytic amount of N-methylimidazole (NMI) or using pyridine as a solvent, acetylation of the C2′–OH was observed to yield 14. Indeed, even without any added catalyst, reaction is observed at this position. After C2′–OH functionalization, C4″–OH is the next to react, as observed when adding 2.0 equivalents of acetic anhydride (Ac2O) to yield 15. It should be noted that the C2′–OAc is cleaved upon quenching the reaction with methanol. The inherent selectivity of 13 to undergo acylation on the appended glycoside alcohols, as opposed to on the hydroxyl groups present in the macrocycle, invites the challenge of reversing the intrinsic site-selectivity with catalysis.
Figure 5.

Selective acylation of erythromycin (13). Under NMI-catalyzed conditions, the C2′ and C4″ alcohols are the first and second most reactive functional groups (15 being isolated after methanol-induced cleavage of C2′–OAc). However, utilization of peptide 17 reveals a new product, 16.65
To target the functionalization of various positions, 137 peptide sequences were evaluated in an effort to find a different selectivity pattern from that of the achiral reagents. While C2′–OH was the most reactive alcohol towards acylation, the resulting acetyl group at this position could be cleaved by addition of meth-anol after completion of the reaction. Hence, by upon adding 2 equivalents of Ac2O, the selectivity of the second acylation event could be probed by catalyst screening. After acylation of C2′–OH, while most of the catalysts screened resulted in acylation of the C4″–OH, peptides like 17 that are biased to adopt β-turns43–46 revealed a new product, the C11-acylated adduct 16 (after cleavage with methanol). This reversal in selectivity is an intriguing result, especially given that the C11–OH is flanked by two vicinal β-methyl groups (Figure 5).
When reactions of 13 were carried out with different acid anhydrides, selectivities as high as 10:1 for the acylation of C11 (19) over C4″ (18) were recorded (Figure 6A). Note that no MeOH was added in these cases, leaving the C2′–OAc intact. Notably, upon acylation of the C-11 hydroxyl group, the structure underwent spontaneous hemiketalization.66 Intriguingly, higher selectivities were observed with the enantiomer of peptide 17 (ent-17).67 Here, the inherent chirality of substrate 13 plays an important role, as ent-17 must be more matched to interact with 13 in a manner that more productively leads to selective product formation, as opposed to 17. Additionally, upon treatment of diacylated product 19 with phosphorylating agent 20, the C4″–OH was functionalized (Figure 6B). Hence, by utilizing a combination of achiral reagents and specific and powerful small peptide catalysts, derivatives that are functionalized at three different positions of 13 can be accessed. Furthermore, the modest number of peptides that were screened in order to identify these selective peptides speaks to the tunability of peptide-based catalysts. Larger libraries of catalysts either rationally designed or randomly assigned sequences, could presumably be synthesized and combinatorially screened to target the functionalization of other positions on natural products.
Figure 6.

(A) Expansion of the scope of acylation of erythromycin (13) results in continued selectivity for C2″–OH acylation. (B) Upon isolation of diacylated product 19, the C4″–OH can be selectively phophorylated.67
Site-selective group transfers to hydroxyl groups are not only relevant for the synthesis of functionalized alcohols, but also for their deoxygenation. The removal of specific hydroxyl groups from 13 would allow for biological studies on the importance of the OH functionality at those particular positions.68–74 Toward this end, exchange of the simple acylating agents above for either a phosphoramidite75 or thiocar-bonylating agent76–79 can set up radical-mediated cleavage of its corresponding derivatives (Figure 7).80 Reaction conditions modeled after those of Koreeda were developed for phosphoramidite transfer in the absence of either hydrolysis or oxidation of the resulting P(III) species.75 Then, tetrazole-based catalysts were discovered that were compatible with and effective for delivering phosphoramidite 22 to certain alcohols of 14 (protected at the most reactive C2–OH position). As shown in Figure 7A, when C2′-acetylated erythromycin 14 is treated with 22 and phenyl tetrazole as a catalyst, clean phosphitylation at the C4″–OH is observed to yield 23. Upon addition of Bu3SnH and AIBN, the phosphite is easily cleaved to yield C4″-deoxygenated 24 in 83% yield.
Figure 7.

(A) The selective deoxygenation of 14 can be carried out at the C4″–OH via selective phosphitylation of this positon, followed by radical cleavage. (B) The selective deoxygnation of the C4″–OH of 13 can be carried out through the use of catalyst 26, as phenyl tetrazole in this case yields a complex mixture of phosphite products. The C2′–OH can be targeted for elimination through thiocarbonyl intermediate 28.80
When applying this system, however, toward fully unprotected erythromycin A (13), it was found that simple catalyst 5-phenyltetrazole yielded a complex mixture of various phophitylated compounds (Figure 7B). While one of the major products, upon radical cleavage, was C4″-deoxygenated 25, the overall reaction occurred with low yield. However, tetrazole-embedded, β-turn biased peptide 26 catalyzed the efficient formation of 25 as the only appreciable product in 50–60% yield. This selectivity is striking, especially when considering the previously reported results of the heightened reactivity of the C2′–OH when compared to C4″–OH under acylation conditions (see Figure 6). This reversal of selectivity is dictated through catalyst control, governed by secondary interactions between the catalyst and substrate that favor derivatization of a less reactive alcohol of 13. The C2′-deoxygenated product can also be accessed under different conditions, as NMI-catalyzed thiocarbonylation yields 28 as the major product. Here, 1,2,2,6,6-pentamethylpiperidine (PEMP) is utilized as a base in tandem with the NMI catalyst.79 Upon radical cleavage of the C2′–OH, deoxygenated 29 is obtained. The selectivity of the NMI-catalyzed process reveals the inherent reactivity of the C2′ position on 13, which is overturned when a small peptide catalyst is used. This demonstrates the remarkable ability of these small molecules to reverse substrate bias and favor functionalization of less reactive functional groups on natural products.
2.2.2. Site-Selective Acylation of Apoptolidin
The success of minimal-peptide-based catalysts in the site-selective acylation of erythromycin A led to the examination of whether this approach could be translated to other natural products, such as apoptolidin (30). This recently discovered natural product, isolated in 1997, was found to induce apoptosis in E1A-transformed cells.81–86 Inspired by this selective cytotoxicity, a collaboration between the Wender laboratory and ours set out to prepare unknown, site-selectively modified derivatives.87 Previous efforts by the Wender group to derivatize the alcohols native to 30 relied on the inherent reactivity of the substrate.86 As such, catalyst control was sought to achieve reactions at the intrinsically less reactive hydroxyl groups of 30 (Figure 8).
Figure 8.

(A) Acylation of apoptolidin A (30) with DMAP resulted information of 31 and 32. When using peptide ent-17 as catalyst, 31 was the primary acylation product. (B) Further acylated products 33 and 34 were observed in the presence of an excess of acylating agent.87
One of the most important considerations in pursuing this project was the quantity of apoptolidin available. Hence, screens were performed on quite small scale, employing only 20 μg of 30. The identification of highly selective reactions that favored predominantly one acylation product was thus a priority to facilitate structure identification. Specifically, targeting the functionalization of the C2′ alcohol was of interest, as derivatives modified at this position had yet to be isolated. When using 4-(dimethylamino)pyridine (DMAP) as a catalyst with Ac2O as the acylating reagent, the formation of two products, 31 and 32, were observed (Figure 8A). Following this observation, ~150 peptide sequences were screened, and peptide ent-17 was revealed to be a C2′-selective catalyst, resulting in preferential formation of 32. The reaction was then scaled up modestly to performance on 7.1 mg of 30. The identification of peptide 17 for the site-selective acylation of both erythromycin A (13, see Figure 5–6) and peptide ent-17 for apoptolidin (30) is of note for their applicability to two classes of presumably unrelated substrates.
Upon reacting 30 with a large excess of acylating agent, a complex mixture of products formed, from which two were isolated: triacylated 33 and 34. After isolation of sufficient material of products, these derivatives were screened against H292 human lung carcinoma cells. It was revealed that these mono-, di-, and triacylated compounds did not significantly perturb the cytotoxicity of this archetypal compound, and 30 remained the most active against the cancer cells.
2.2.3. Site-Selective Alcohol Derivatization of Vancomycin and Teicoplanin
Vancomycin (35) has also been studied as a template for peptide-catalyzed diversification. Reported by Eli Lilly in 1953, vancomycin is a heptapeptide with numerous oxidative cross-linkages and appended sugars.88,89 Vancomycin is renowned for its activity against gram-positive bacteria. It is now known to act through inhibition of bacterial cell wall biosynthesis, specifically targeting the formation of peptide cross-links between polysaccharide chains of the cell walls. The pendant peptide chains on these polysaccharide units contain terminal DAla-DAla residues, which are recognized by a transglycosylase enzyme. The enzyme then cleaves the final DAla residue and couples two polysaccharide-attached peptides together. By forming five H-bonds to the terminal DAla-DAla residues of these peptide components of the developing bacterial cell wall, vancomycin prevents the completion of the cell wall, resulting in the lysing of the attacked bacterial cells.88,89 However, bacteria are evolving to resist this mechanism of action via mutation of this DAla-DAla terminus.90–92 This bacterial resistance is particularly dangerous for humans, as vancomycin has served as one of the “antibiotics of last resort.” Such pharmaceuticals are reserved for the most dangerous and persistent bacterial infections; the drugs’ efficacy, however, has diminished due to resistance from numerous strains of bacteria. Hence, the synthesis of novel derivatives of 35 that are able to combat antibiotic resistance is essential.91,93–96
In order to combat this serious resistance to vancomycin, many scientists have turned to total synthesis,94–98 semi-synthesis,99–106 biosynthetic engineering,107,108 and enzymatic derivatization109–111 to access novel derivatives. An alternative approach to analog development is through the use of catalyst-controlled, site-selective, late-stage modification. Starting with easily accessible vancomycin would minimize the amount of synthesis necessary to access this complicated structure, enabling the facile and expedient development of countless derivatives for biological screening. However, targeting one motif on this functional group-rich natural product is incredibly challenging.
Our group set out to apply our peptide-based catalysts toward the synthesis of novel vancomycin derivatives. First, we started with the goal of selectively deoxygenating each of the six aliphatic alcohols of 35 (Figure 9).112 After protecting the secondary amine, the phenols, and the carboxylic acid to yield 36, we attempted to incorporate O-phenyl chlorothionoformate (37, PCTF) selectively into 36. Radical cleavage could then be performed, mediated by Bn3SnH and azobisisobutyronitrile (AIBN). Starting with NMI as a catalyst, moderate selectivity towards the Z6–OH (40) was observed, with a minor amount of G6–OH acylated product (39) forming as well (Figure 9A, entry 2). The reactivity of Z6–OH was somewhat unexpected due to the apparent steric accessibility of the G6–OH primary alcohol. When comparing Z6–OH to Z2–OH, the former is oriented towards the convex face of 36, while the latter is oriented in the concave binding pocket, making it less likely to react (Figure 10).
Figure 9.

(A) Deoxygenation of protected vancoymcin derivative 36 via site-selective acylation using a variety of Pmh-containing peptides. Reactivity is centered on the G6 and Z6 positions to yield 39 and 40 respectively. (B) Radical cleavage of the phenylthiocarbonyl intermediates results in deoxygenation products 47 and 48. (C) Pmh-containing peptide catalysts utilized in this study.112
Figure 10.

(A) Crystal structure of vancomycin (71, gray) binding to Ac-Lys(Ac)-DAla-DAla-O− (yellow), a mimic of 71’s native binding target. (B) Proposed binding strategy of catalyst 80 to deliver the phenyloxythiocarbonyl to the G6-hydroxyl selectively. Reproduced with permission from ref 112. Copyright 2012 American Chemical Society.
After assessing the inherent reactivity of the substrate, our group explored two approaches to developing short peptides to perturb the biased reactivity of vancomycin: (1) combinatorial screening of Pmh-containing libraries and (2) designing peptides that mimic the DAla-DAla motif of vancomycin’s biological binding target.
Evaluation of random peptides revealed two peptides in particular, 11 and ent-12, which were found to alter the innate selectivity profiles of 36. As a note, peptides 11 and 12 have been of previous interest to our group, given their induced selectivities in the phosphorylative desymmetrization of inositol (Figure 4C).48 Catalyst 11 reversed the inherent reactivity, favoring reaction at G6–OH (Figure 9A, entry 3); alternatively, peptide ent-12, reacted at Z6–OH over G6–OH with a 1:9 ratio (Figure 9A, entry 4). Peptide ent-41, in which the tert-butylhydroxyproline motif is deleted, showed similar reactivity to peptide ent-12, albeit with lower selectivity (Figure 9A, entry 5). The mode of action of these peptides is not currently known. One hypothesis involves a specific hydrogen-bonding network that might orient the reactive catalytic residue adjacent to the G6 or Z6 alcohols or block the dormant site from reacting.
Our efforts toward the rational design of a peptide-based catalyst were inspired by the well-known mechanism of action of vancomycin via binding the terminus of gram-positive bacterial peptidoglycan. This strong host-guest interaction is demonstrated by the crystal structure of the peptidoglycan mimic, Ac-Lys(Ac)-DAla-DAla-O−, and 35 (Figure 10A).112–114 We hypothesized that various alcohols of 36 could be targeted by strategically inserting Pmh-residues into peptides of this class. The crystal structure of Figure 10A served as a model for peptide design. Given the strong hydrogen bonding affinities of vancomycin to the DAla-DAla motif, it seems likely that the catalytically active imidazole ring is predictably and specifically positioned upon catalyst binding to 36. Thus, placing a Pmh-residue at the terminal position could potentially orient the reactive imidazole adjacent to G6–OH (Figure 10B). As such, peptides 42–44 were synthesized, in which the second DAla residue of this sequence was replaced with Pmh. While peptides 42 and 43, which each contain a protected C-terminus, resulted in minimal selectivity, free carboxylate-containing peptide 44 resulted in an excellent 47:1 selectivity for G6–OH (Figure 9A, entries 6–8). Even when lowering the catalytic loading from 100 mol% to 20 mol%, 27:1 selectivity is still retained (Figure 9A, entry 9). It is plausible that the free carboxylate at the C-terminus makes an essential H-bond(s) in order for it to be incorporated into the native binding pocket of vancomycin, as shown in Figure 10.
Indeed, the selectivities of ent-41 and 44 for the Z6 and G6 alcohols, respectively, enabled scale-up to 500 mg of 36, and while the selectivities were similar on both of these scales for catalyst 44, peptide ent-41 delivered 40 in a substantially higher ratio of 1:21 (Figure 9A, entries 10–11). At this scale, the deoxygenation using Bu3SnH and AIBN proceeded well and afforded vancomycin derivatives 44 and 45 (Figure 9B).
Following global deprotection to give 46 and 47, biological screening was conducted to compare their activities against resistant bacterial strains. The deletion of G6–OH (46) did not significantly affect the biological activity. However, removal of Z6–OH resulted in substantial erosion of pharmacological activity against bacterial strains. An HPLC and LC/MS analysis of 47 revealed two peaks, indicating that 47 was actually a mixture of two interconverting conformations at room temperature. This dependence of vanco-mycin’s activity on the Z6–OH could be ascribed to maintaining a particular conformation of vancomycin’s binding pocket essential for its binding to the bacteria’s DAla-DAla motif.
Although the novel analogues exhibited diminished biological activities, we were heartened by the success of peptide-catalysts in the site-selective modification of complex molecules. Hence, our group turned its attention towards teicoplanin (50), a naturally occurring antibiotic similar to vancomycin.115–120 Teicoplanin contains three distinct sugar motifs appended to various locations on the molecule, as well as an additional macrocycle defined by a diaryl ether moiety. Teicoplanin is more active against some bacterial strains, such as VanB, against which vancomycin has lost its efficacy. Hence, methods for diversifying this compound are essential to outcompete bacterial resistance. As such, we set out to selectively phosphorylate the hydroxyl groups of protected teicoplanin 50 (Figure 11).121 As a note, commercially available 49 contains a mixture of decanoylglucoasamine sugars appended to the backbone of teicoplanin (Figure 11A, red sugar), specifically in the variation of the length of the alkyl chains. Teicoplanin A2-2 (49) was purified from this mixture and used in this study.
Figure 11.

(A) Site-selective phosphorylation of protected teicoplanin A2-2 (50) reveals preferences for reaction at the three primary alcohols on the three sugars of 50. Three peptides were shown to selectively favor functionalization of each of these glycosides (B) Catalysts applied in these reactions. 121
Structurally, an essential distinction between 49 and vancomycin (35) is the presence of three primary hydroxyl groups on each of the three sugars. It was probable that these three positions would be the most reactive alcohols in the molecule. Furthermore, the Z6 alcohol of vancomycin (Figure 9), which was preferentially functionalized by peptide ent-41, is now capped with a glycoside.
Upon treatment of 50, a minimally protected variant of 49, with phosphorylating agent diphenyl chlorophosphate (51, DCPC) and NMI as an additive, compounds 52–54 are the three primary products observed, with a slight preference for phosphorylation of the red and blue sugars (Figure 11A, entry 2). In comparing the structures of vancomycin to teicoplanin, the most conserved reactive alcohol is the G6–OH, the primary alcohol on the red sugar. This position was previously targeted by “Xaa-DAla-DPmh” catalyst 44, which mimics the native binding target of vancomycin and teicoplanin. Subjecting 50 to the standard reaction conditions in the presence of peptide 44 reveals a preference for reactivity at the red sugar to yield 52, highlighting the power of mimicking strong biological host-guest interactions in small molecule synthetic catalysts (Figure 11A, entry 3).
With the precedent of the DAla-DAla mimicking strategy, we wondered whether incorporating the Pmh residue at a different position in the peptide sequence would favor reactivity at the blue sugar. As shown in Figure 10B, the DAla residue’s methyl group is pointed directly towards this part of the natural product. However, screening peptide 55 revealed an unselective mixture of 52 and 54 (Figure 11A, entry 4). To demonstrate the rate acceleration afforded by the matched phosphorylation peptide, 1.0 equivalent of both peptide 44 and 55 were added into the reaction, and despite the presence of a non-selective peptide (55), peptide 44’s selectivity towards the red sugar dominated (Figure 11A, entry 5). However, the challenge of targeting the blue sugar still remained, and it was found that phosphorylation to yield 54 could be accomplished through the use of peptide 11 (Figure 11A, entry 6). Previously, this peptide had been shown to favor acylation of the G6–OH position of vancomycin (the red sugar). It is noteworthy that peptide 11 is able to mediate two fundamentally distinct product formations for both vancomycin and teicoplanin, overturning the inherent substrate reactivity in both cases. Yet, the origin of this divergent reactivity is still unknown.
In order to target the least reactive of the glycoside primary alcohols, the green sugar, a number of peptide sequences were screened, including 42, 56, and 57, with no success (Figure 11A, entries 7–9). It is most intriguing that peptide ent-41, which was previously shown to be a selective catalyst for vancomycin acylation at Z6–OH (Figure 9, the alcohol to which the green sugar is appended), is not effective here. At this stage, we wondered whether adding the Pmh-residue onto the opposite side of the DAla-DAla motif might orient this catalytic residue adjacent to the green sugar. This hypothesis proved to be effective, as peptide 57 phosphorylated the green sugar (53) selectively (Figure 11A, entry 10).
To assess this selectivity for the least reactive primary alcohol of 50, we attempted to obtain a crystal structure of peptide catalyst 57 docked to teicoplanin (Figure 12).122 Building on previously reported results in which appending a carrier-protein to the DAla-DAla-containing peptide123 enabled more facile X-ray crystal structure determinations, complexation between 57 and teicoplanin was observed. As hypothesized, the Pmh-residue was oriented directly adjacent to the green sugar and the primary alcohol owing to the specificity of the teicoplanin-binding pocket for coordinating to DAla-DAla sequences.
Figure 12.
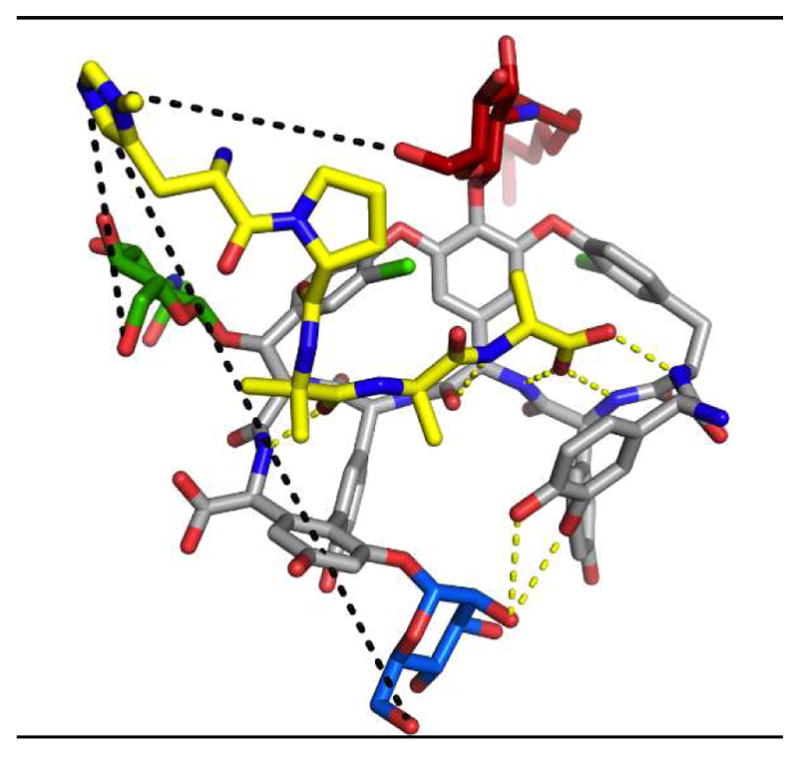
Crystal structure of teicoplanin A2-2 (50, C-atoms in gray) binding to Pmh-DPro-Aib-DAla-DAla, showing the Pmh residue is pointed directly towards the green sugar. Reproduced with permission from ref 122. Copyright 2014 American Chemical Society.
While biological screening of these novel teicoplanin derivatives revealed comparable or lower biological activities to teicoplanin (except a minor improvement against vancomycin- and teicoplanin-resistant bacterial strain VanA), the strategy of peptide-based group transfer of natural products continued to prove effective for functionalizing complex molecules.
Upon proving the efficacy of peptides as catalysts for both phosphorylation and thiocarbonyl incorporation geared towards deoxygenation, our group turned its attention to lipidation of 35.124 Due to the observation that lipophilic antibiotics are more active against bacteria, potentially due to their increased membrane permeability, lipidation is a useful technique to functionalize to natural products and pharmaceuticals.125–129
Indeed, upon undertaking the site-selective lipidation of vancomycin, it was revealed that the background reaction rate is decelerated for lipidation with decanoic anhydride (59, Figure 13) in comparison to other acylation reactions previously discussed. As shown in Figure 13A.i, there is no non-catalyzed background rate; yet, even with NMI, the conversion is still extremely low (Figure 13A.ii). However, upon treatment of 36 with catalyst 66, a large rate acceleration is observed and two products are revealed, with lipidation at the G6 (60) and G4 (61) positions (Figure 13A.iii). The rate enhancement of peptide-catalysts toward acylation is commonly observed in these systems.130
Figure 13.

(A) Site-selective lipidation of protected vancomycin 36 results in functionalization of G4, G6, or Z6 alcohols. Three different peptide sequences are able to achieve selective lipidation at these three positions.(B) Numerous lipidated analogues of vancomycin display heightened biological activity, especially important against VanA and VanB, which are vancomycin resistant.124
In expanding the peptide library that was screened, catalyst 66, which had previously been reported as a selective acylating peptide for the kinetic resolution of formamides,131 favored lipidation at the G4 position to yield 61 (Figure 13A.iv). Additionally, β-turn-containing catalyst ent-41, which had previously been discovered as a Z6–OH selective acylating agent (see Figure 9),112 indeed yielded the same selectivity pattern here to afford 62 (Figure 13A.v). Finally, “Xaa-DAla-DPmh” catalyst 44, which had previously been discovered as an active thiocarbonylation and phosphorylation catalyst for the G6 position of vancomycin and teicoplanin (see Figures 9112 and 10121), also showed similar levels of selectivity here to yield 60 (Figure 13A.vi). This catalyst is believed to function via mimicking vancomycin’s native binding target, as shown in Figures 10 and 12, positioning the active imidazole ring adjacent to the G6–OH. It is noteworthy that despite differences in the actual transformations occurring, catalysts ent-41 and 44 are able to show reproducibility in the sites for which they select.
The globally deprotected lipidation products (63–65) showed increases in pharmacological action, especially for vancomycin-resistant bacterial strains VanA and VanB. Other lipid derivatives (67–72) that were synthesized using the same methods did not show the same levels of enhancement against resistant bacteria, indicating the reduction in the lipid chain length is important for biological activity.
These three examples of catalyst-controlled, site-selective modification of complex molecules demonstrate the strengths of peptide-based catalysis. By making use of one conserved catalytic residue, in addition to very subtle changes in the associated minimal peptide sequence, different positions on complex natural products can be targeted. In this way, these minimal peptide-based catalysts are able to emulate the ability of nature to tune the reactivity and selectivity of a particular reactive functional group by varying the structure of the enzyme around the active site. In the case of smaller peptides, selectivity can be achieved through the reliance of strong secondary interactions between catalysts and substrates, which oftentimes are conserved regardless of the actual reaction. This strategy has also enabled access to a number of derivatives of vancomycin and teicoplanin, an important consideration given the widespread challenge of antibiotic resistance today.132–135
2.3. 4-Pyrrolidinopyridine Catalysts for Saccharide Functionalization
The site-selective acylation of complex molecules has not been limited to Pmh-containing peptides, as other highly effective nucleophilic catalysts have been developed to accomplish these transformations. As previously described, DMAP is a well-established catalyst used in acylation reactions, as the electron-rich pyridine ring serves as a strong nucleophile to activate the acylating agent. However, using a simple achiral reagent often renders it difficult to gear reactivity towards an inherently less favored position on a substrate. For example, in the functionalization of simple monosaccharides, the primary alcohol, being the least sterically hindered, is normally the most reactive position (Figure 14). In an attempt to exert catalyst-derived chemo- and site-selective acylations of saccharides, the Kawabata group developed a novel class of amino acid substituted 4-pyrrolidinopyridines (73).136–139 These pyridines are functionalized with amino acids that provides a number of handles for secondary interactions with the substrate and chiral base, allowing for strong binding between catalyst and substrate. In addition, the large number of commercially available amino acids allows for facile diversification and screening of large catalyst libraries. Indeed, these 4-pyrrolidinopyridine catalysts were proven to be effective in the selective acylation of the secondary C4-alcohols of monosaccharides (Figure 14A). This selectivity, despite the presence of three additional alcohol groups on the second saccharide unit, is presumed to occur through a pivotal hydrogen bond between the peptide-pyrrolidine backbone to the primary alcohol of the saccharide (Figure 14B). Hence, in spite of the higher inherent reactivity of the primary C6-alcohol, the C4-alcohol is positioned to intercept the incipient acylpyridinium adduct. It is notable that the diversification of these sugars is accomplished without the use of protecting groups, an uncommon and useful advance given the presence of a number of similarly reactive alcohols in these saccharides.
Figure 14.

(A) Site-selective acylation of the C4–OH of monosaccharides using 4-pyrrolidinopyridine catalyst 73. DMAP provides an unselective mixture of multiple products. (B) Proposed transition state. The catalyst’s amide carbonyl hydrogen bonds to the C6 primary alcohol, orienting C4–OH for selective acylation. (C) Scope of selective acylation reactions. (D) Scope of acylating agents utilized.136–139
Indeed, this strong catalyst-substrate binding is further demonstrated in the derivatization of more complex molecules. When examining disaccharide 76, 78–94% regioselectivity was observed for the same C4–OH, and product was isolated in 38–92% yield (Figure 14C).137 Since the additional saccharide unit does not contain a free primary alcohol, the catalyst is not directed here. The Kawabata group has also applied this catalytic approach in total synthesis, beginning with the syntheses of the multifidosides A–C, which requires an early-stage selective acylation of the C4 alcohol of a similar monosaccharide (77).139 They accomplish this transformation in 59–78% yield for monoacylation and 87–91% selective for the C4 position.
The total synthesis of monosaccharides of the ellagitannins class (78 shown as a representative example) was also accomplished through this protocol (Figure 15).140 Upon treatment of saccharide 79 with anhydride 80 in the presence of catalyst 73, selective C4′ acylation yields 81 in 91% yield. At this stage, the C6′–OH is the most reactive alcohol, and this can be coupled to carboxylic acid 82 in a site-selective ester-ification to yield 83. Following hydrogenative deprotection of the benzylic hydroxyls, copper catalyzed oxidative phenol coupling yields biaryl 85. Global deprotection resulted in 78 in a concise and high-yielding synthesis.
Figure 15.

Total synthesis of strictinin (78) involving a key site-selective acylation, catalyzed by 4-pyrrolidinopyridine catalyst 73, site-selective esterification, and oxidative phenol coupling.140
The excellent selectivity afforded by catalyst 73 is not limited to smaller saccharide-containing molecules, as Kawabata and coworkers also employed this catalyst to selectively derivatize digitoxin (86, Figure 16).141 Upon reacting 86 in the presence of DMAP as a catalyst and Ac2O as the acylating agent, a strong preference for reaction of C4‴ was observed, as product 87 was obtained in 66% yield of the C4‴ acylated product. However, 18% of the C3‴–C4‴ diacylated species (88) was also isolated. Despite the challenge that digitoxin does not contain a primary alcohol to direct reactivity to an adjacent position, catalyst 73 is able to minimize overreaction, resulting in 90–98% yield of 87 depending on the anhydride.
Figure 16.

Selective acylation of digitoxin (86). Only one product is observed when using 4-pyrrolidinopyridine catalyst 73.141
Upon successfully modifying a complex molecule with their catalysts, the Kawabata group turned their attention to lanatoside C (89), which contains an additional saccharide unit (Figure 17).142 Unlike digitoxin (86), the terminal saccharide of 89 contains a primary alcohol. Upon evaluating the acylation of 89 using DMAP as a catalyst, of 10 accessible hydroxyl groups, C3‴′ acylated 92 is observed as the predominant product (Figure 17, entry 1). Secondary C3‴′–OAc formation was a particularly surprising result, given the presence of a primary alcohol on the terminal saccharide. However, upon calculating the lowest energy structure of 89 utilizing MacroModel with the MM3* force field and a GB/SA solvation model in CHCl3, C6‴′–OH and C3‴–OAc are shown to be within 2.9 Å of each other, potentially indicating that a hydrogen bond exists between these two alcohols. This H-bond could block catalyst complexation at this primary alcohol.
Figure 17.

Site-selective acylation of lanatoside C (89). While DMAP offers selectivity for the 3‴′ secondary alcohol, catalyst 73 favors reactivity at C4‴′–OH. Variation of the chirality of catalyst 73 or 93 leads to different selectivity patterns, implying while the Trp-stereochemistry is not as important to observed trends, the chirality at the pyrrolidine ring is essntial for matched interactions between the catalyst and substrate.142 a20 °C, 48 h. b−20 °C, 96 h. cDMF
Having discovered a catalyst with high selectivity for the formation of 92, the authors turned their focus to accessing 91. Using catalyst 73, and upon lowering the temperature of the reaction from 20 °C to −60 °C, high selectivity for C4‴′–OH acylation was observed to yield 91 (Figure 17, entries 2–4). This result resonates with previously observed results with catalyst 73, which prefers to react with the C4 position of saccharides via formation of a complex with the primary C6-alcohol, which is adjacent to the primary alcohol, as the catalyst forms an H-bond with the C6-alcohol (Figure 14B).
Examination of the stereoisomers of catalyst 73 revealed that the chirality of the catalyst is pivotal to selectivity. Replacing the LTrp with DTrp in the catalyst results in slightly lower selectivity for 91 (Figure 17, entry 5). However, upon combining LTrp with inversion of chirality at the pyrrolidine ring (ent-93), 92 is the predominant product (Figure 17, entry 6). This trend continues with catalyst ent-73, where the inversion of stereochemistry at the pyrrolidine ring again results in substantial selectivity for 92, and downplays the importance of the stereochemical configuration of the Trp residues (Figure 17, entry 7). These data indicate that catalysts ent-93 or ent-73 are reverting to substrate-control, akin to the DMAP-catalyzed reaction, and implies that the pyrrolidine stereochemistry is essential to forming a matched complex with saccharide substrates. Lastly, modification of the C-terminal protecting groups of the catalyst (94) results in a slightly higher distribution for product 91 (Figure 17, entry 8).
Finally, in an effort to achieve reactivity at C6‴′–OH of 89, the authors sought to perturb the inherent intramolecular H-bond between this alcohol and C3‴–OAc. Calculations on the ground state structure of 89 in H2O, as opposed to chloroform which is the more typical solvent for this chemistry, showed a longer 5.7 Å distance between C6‴′–OH and the C3‴–OAc. Hence, DMF was chosen to mimic this effect of water and to disrupt the C6‴′–OH to C3‴–OAc intramolecular H-bond; indeed, application of DMAP or 73 facilitated the predominant formation of 90 in DMF (Figure 17, entries 9–10). The reversal to primary alcohol acylation indicates that both the intramolecular H-bonding network of 89 and the intermolecular complexation between catalyst 73 and 89 are likely disrupted.
Following this impressive example of catalyst-controlled saccharide functionalization, the 4-pyrrolidinopyridine catalyst system was next applied towards the selective acylation of avermectin B2a (95, Figure 18).143 This compound is part of an intriguing class of bioactive natural products that were first discovered in 1979 and have since been used to treat a number of parasitic infections, such as those causing river blindness or lymphatic filariasis.144–147 Hence, the development of novel analogues of this class of pharmaceuticals for SAR studies is of significant importance, and the authors targeted the four hydroxyl groups on 95 for diversification.
Figure 18.

Selective acylation of avermectin B2a (95).143
Upon reacting 95 in the presence of Ac2O and DMAP as a catalyst, C4″–OH and C5–OH were found to be the two most reactive alcohols, resulting in a relatively unselective mixture of products 96 and 97 (Figure 18, entry 1). However, using catalyst 73 resulted in a 15.5:1 ratio of 97/96, with only a small amount of diacylated 98 formed (Figure 18, entry 2). This selectivity is a particularly powerful result, given that 96 is dissimilar to most of the previous substrates to which this catalytic system has been applied, namely saccharides with a primary alcohol group adjacent to the C4 position. Similar to previous studies described above, screening of stereoisomers of catalyst 73 resulted in diminished selectivity for 97 (Figure 18, entries 3–5). However, lowering the reaction temperature to −65 °C resulted in even higher selectivities for 97 (Figure 18, entry 6). Altering the acylating reagent to bulky isobutyric anhydride or electron-deficient dichloroacetic anhydride resulted in similar reactivity patterns for both DMAP (unselective) and catalyst 73, which is highly 97-selective (Figure 18, entries 7–10). The authors proposed a model for this selectivity, hypothesizing that the C7–OH serves as a directing group for acylation at C5–OH. The 1,3-relationship between the directing group and the alcohol to be acylated is the same situation for the saccharide functionalizations shown above, in which the C6–OH directed reactivity to the C4–OH alcohol.
However, upon changing to trichloroacetic anhydride as the acylating agent, substantial changes in the normal product distribution were observed. With DMAP as the catalyst, 96 was now highly favored (Figure 18, entry 11). Utilization of catalyst 73 resulted in substantial perturbation of this selectivity and resulted in a 1.0:1.9 mixture of 97/96. While this ratio may look like poor selectivity, it is a substantial advance, considering that the inherent reactivity with an achiral catalyst is heavily geared towards 96 (Figure 18, entry 12). Yet, upon switching to catalyst 93, the inherent selectivity for 96 is enhanced (Figure 18, entry 14). Similar reactivity was observed in the case of trifluoroacetic anhydride, however yields were substantially lower due to the thermodynamic instability of the acetates (Figure 18, entries 15–17). An explanation as to the origins of this excellent perturbation of selectivity is currently unknown, yet it represents an orthogonal method for selectively functionalizing two different alcohols on 95.
Moving away from saccharide-containing molecules, taxol derivative 10-deacetylbaccatin III (99) was subjected to this 4-pyrrolidinopyridine-catalyzed acylation (Figure 19).148 Upon reacting 99 with anhydrides in the presence of DMAP, a slight preference for C10-acylation to yield 101 was observed (Figure 19, entry 1). However, this inherent selectivity was reinforced through the use of catalyst 73, which afforded a 93:7 ratio of 101:100 (Figure 19, entry 2). Similar to their previous reports, the authors screened all the potential diastereomers of catalyst 73 and found that while 93 and ent-93 gave similar product distributions, they reacted at substantially slower rates compared to 73 (Figure 19, entries 2–5). Additionally, this catalytic system tolerated a range of anhydrides, directing them to react at the C10–OH in >90:10 selectivity (Figure 19, entries 9–14). Intriguingly, upon using trichloroacetic anhydride, the selectivity is completely shifted to the C7–OH to yield 100. Previous reports seem to indicate that trichloroacetylation is favored at this position (Figure 19, entry 15). The authors suggest that this reaction hence illustrates true substrate control, and that the addition of a DMAP or DMAP-like catalyst favored reactivity instead at the C10 position. Basing the inherent reactivity of a substrate on the site-selectivity afforded by achiral catalysts can be troublesome sometimes, as they have the potential themselves to perturb reactivity patterns in complex stereochemical environments.
Figure 19.

Selective acylation of 10-Deacetylbaccatin III (99). Catalyst 73 enhances the inherent selectivity for 101 over 100 as afforded by DMAP. Functionalization of C10–H to yield 100 can be accessed by using (CCl3CO)2O as the anhydride source. a5.0 equiv. Bz2O. b3.0 equiv (CCl3CO)2O.148
2.4. Cis-1,2-Diol-Selective, Imidazole-Containing 2-Methoxyoxazolidine Catalysts
A common structural motif in saccharides and other natural products is the cis-1,2-diol. Diols are a useful functional group as one of the alcohols can promote the functionalization of the adjacent alcohol. Targeting this motif, the Tan group engineered a catalyst that contains both an imidazole ring to serve as a Lewis base for acyl transfer and a 2-methoxyoxazolidine that could act as a latent electrophile to which an additional, adjacent alcohol on the substrate could bind (102 and 103, Figure 20B and 20D).149 Indeed, for a number of small saccharides, the authors successfully demonstrated this concept. For example, while rhamnose (104) contains three hydroxyl groups, only two are in the cis relationship to each other (Figure 20A). Upon mesylating 104 in the presence of NMI as a catalyst, a mixture of the three potential products (105–107) results. However, two pseudo-enantiomeric catalysts, (+)-102 and (−)-103, enable access to both 106 and 105, respectively, in high selectivity. It is particularly noteworthy that catalyst (−)-103 is able to overturn the inherent substrate bias for 106 to afford product 105 instead. To probe the mechanism of this transformation, the authors synthesized catalysts omitting the anomeric methoxy group, which resulted in a stunning loss of selectivity and indicating that covalent bond formation to 102 or 103 is essential for catalytic activity (red in Figure 20B).
Figure 20.

(A) Site-selective mesylation of rhamnose (104). The catalyst is selective for cis-1,2-diols. (B) Imidazole-2-methoxyoxazolidine catalysts. (C) Selectivity patterns for the mesylation of 104 with various catalysts. (D) A proposed intermediate that forms upon addition of one of the cis-1,2-alcohols into the oxazolidine. The imidazole will next deliver the mesylate via to the free alcohol. (E) Site-selective mesylation of mupirocin methyl ester (108). (F) Site-selective modification of digoxin (111).149
The selectivity of the catalytic system for cis-1,2-diols was further evaluated on more complex substrates, such as mupirocin methyl ester (108, Figure 20E) and digoxin (111, Figure 20F). In both cases, despite the number of potentially reactive alcohols, the cis-diol was always the most reactive when treated with 102 or 103. Furthermore, the divergent selectivity between the pseudo-enantiomeric catalysts, 102 and 103, is observed in both of these cases, suggesting each alcohol of the cis-1,2-diol is matched with one enantiomer of the catalyst.
2.5. Site-Selective Acylation of Amphotericin B
In site-selective catalysis, it is desirable for a reaction to be under kinetic control. Variation of the catalysts, reagents, or substrates can selectively raise or lower the activation barriers for reactions at particular sites within a compound of interest. Hence, subtle chemical differences among the reaction inputs can potentially cause large deviations in the energies of transition states associated with particular pathways. Targeting these concepts, Burke and coworkers turned to variation in electrophile structure to achieve site-selective acylation of the antifungal agent amphotericin B (AmB; Figure 21A).150
Figure 21.

(A) Site-selective acylation of protected amphotericin B (114). (B) The more sterically hindered the acyl transfer reagent, the higher the C2′-selectivity. (C) The more electron-rich the benzoyl chloride, the less reactive the reagent is and more C2′ selectivity is observed. (D) Tuning of acid anhydrides results in two additional site-selective reactions. (E) Optimized acylating agent. p-tertbutylbenzoyl chloride.150
After applying a protecting group strategy, the authors arrived at AmB derivative 114, which contains five secondary hydroxyl groups and represents an excellent platform to examine the possibility of selectivity as a function of electrophile variation (Figure 21A). The selective functionalization of the C2′ alcohol was of particular interest, as this position is proposed to provide an essential hydrogen bond as part of its mode of biological activity.151 However, reactions catalyzed by DMAP or Hünig’s base as additives produced a mixture of products from reactions occurring at C2′–OH, C4′–OH, and C15–OH. When initially using Ac2O as the acyl transfer reagent, the authors only analyzed the amount of reactivity at the C2′ position, and indeed, low selectivity was observed at that position (Figure 21B). While a collection of lipase enzymes appeared promising in biasing reactivity towards C2′-OH, methods based on this strategy suffered from problems of reproducibility, scalability, and conversion. In contrast, Burke and coworkers found that varying the steric bulk of the acyl group could modulate both the rate of acyl transfer and the associated site-selectivity. Isobutyryl chloride resulted in 48% site-selectivity for acylation of the C2′ position to yield 115. However, it was still tremendously difficult to separate the desired product from the complex mixture that ensued.
Hence, a method that was substantially more site-selective was desired in order to limit the amount of unwanted byproducts. The authors thus probed the possibility of changing the electronics of the acylating agent. A number of p-substituted benzoyl chlorides were screened, and interestingly, a correlation between electron-releasing potential of the p-substituent and C2′-selectivity was observed (Figure 21C). For example, when utilizing electron-donating p-N,N′-dimethylaminobenzoyl chloride, 72% C2′-selectivity was observed.
After optimizing the acylating agent, Burke and coworkers also explored how the electronic tuning of the dissociated carboxylate counterion, which is believed to play a role in the rate-limiting acyl transfer step, affected reactivity.152 Thus, two additional products were achieved selectively through deliberate choice of the counterion (Figure 21C). When sterically hindered and electron-rich p-tert-butyl benzoic anhydride was screened, C4′–OH was selectively acylated (116, Figure 21D). However, utilization of electron deficient p-nitro benzoic anhydride resulted in functionalization of the C15′–OH (117).
Inspired by these results and returning to C2′-OH derivatization, when p-tert-butyl benzoyl chloride was utilized, site-selectivity for C2′ was 66%, with 68% conversion (Figure 21E). The product was easily isolated by silica chromatography, and 3 grams of 115 was isolated in 45% yield.
2.6. Site-Selective Glycosylation
The glycosylation of complex substrates is an essential task in organic synthesis. However, despite the classic nature of the transformation,153,154 achieving selectivity with unprotected substrates remains difficult. This challenge is due to the number of hydroxyl groups in saccharides, in addition to the high reactivity of oxocarbenium ion-like intermediates. While nature has evolved enzymes with excellent selectivity for glycosylation of a myriad of substrates,17,155–157 performing these reactions in vitro and controlling the reactivity of various sites is difficult. Despite these challenges, many methods have been developed for site-selective glycosylation, including some that involve minimal protection. However, many rely on complex protecting group strategies that often require a substantial number of steps, depending on the desired glycoside acceptor.158,159
With the task of developing a mild, protecting group-free glycosylation method in mind, the Miller and Schepartz groups set out to selectively functionalize saccharides, such as sucrose (118), with α-fluoroglucose (119) as a glycoside donor, and without the use of protecting groups (Figure 22).160 Furthermore, the absence of protecting groups enables the solubility of sugars in water. Derivatization reactions of oligosaccharides in aqueous conditions are scarce, as water itself can serve as a nucleophile to quench any glycoside donor, such as 119. We hypothesized that the rate of sucrose glycosylation could be accelerated through the use of a combined Lewis acid/Lewis base strategy. Indeed, it was found that calcium triflate and trimethylamine were able to selectively glycosylate unprotected glucose at the C3′–OH with a variety of glycosyl fluorides (Figure 22A). With this system, potential side products, such as hydrolysis of 119, rearrangement of 119 to fructose, addition of trimethylamine to 119, or intramolecular cyclization of the primary alcohol of 119 to the α-position were suppressed. The proposed mechanism of action of this Lewis acid/base pair was shown to rely on the inherent H-bonding network of sucrose, in addition to selective complexation of a number of alcohols to the calcium ions. Given this mild and site-selective transformation, the substrate scope was expanded to more complicated oligosaccharide substrates, two examples of which are shown in Figure 22B. Here, despite the presence of 18 or 17 hydroxyl groups, only one is selectively glycosylated to give 121 or 122, respectively, in high yields.
Figure 22.

(A) Aqueous site-selective glycosylation of sucrose with Ca(OTf)2 and trimethyl amine. (B) Expansion of scope of glycosylation to extremely complex substrates. Lactosyl fructofuranoside is the starting oligosaccharide for 121, stachyose for 122.160
Site-selective glycosylation has also been accomplished for a number of other complex substrates, including glycoside-substituted steroids (Figure 23).161 One such complex molecular target is digitoxin (86), which contains a trisaccharide appended to the C3-position of a steroid core. Reagents must choose between five hydroxyl groups to glycosylate. The Taylor group has reported numerous accomplishments in this field, exploiting their previously developed diarylborinic acids (124) as catalysts for regioselective glycosylations and other alcohol functionalizations.162–165 Key to the success of this catalyst is the presence of a cis-1,2-diol motif. The aminoalcohol ligand on the borinic acid will detach and cis-diols can coordinate to the boron atom. Upon abstraction of the glycosyl halide (123) with silver oxide, the activated diol can selectively attack the glycoside donor. Catalyst 124 was able to deliver a variety of different glycosyl donors to 123, resulting in high yields of C4‴-functionalized product 125.
Figure 23.

Site-selective glycosylation of digitoxin (87) in the presence of diphenylborinic acid catalyst 124, which proceeds via a cis-1,2-diol coordinating to the borinic acid.161
2.7. Rh(II)-Catalyzed Site-Selective Diazo Transfer
While the utilization of N-heterocycles as acyl transfer catalysts has been shown to be an effective method for the selective incorporation of diverse functionality into natural products, other powerful strategies have been reported for the derivatization of alcohols. Rh(II)-carbenoids are well known for their propensities to insert into X–H bonds, including those of hydroxyl groups (Figure 24, X = heteroatom).166 Furthermore, the utility of rhodium (II) catalysts for a myriad of stereoselective reactions, such as cyclopropanations and C–H functionalizations, has resulted in the development of a number of well-defined and highly active dirhodium catalysts.167–171 Given the diverse ligands and differing reactivities of rhodium catalysts, Romo and coworkers applied these privileged catalysts towards the site-selective modification of polyol containing natural products.
Figure 24.
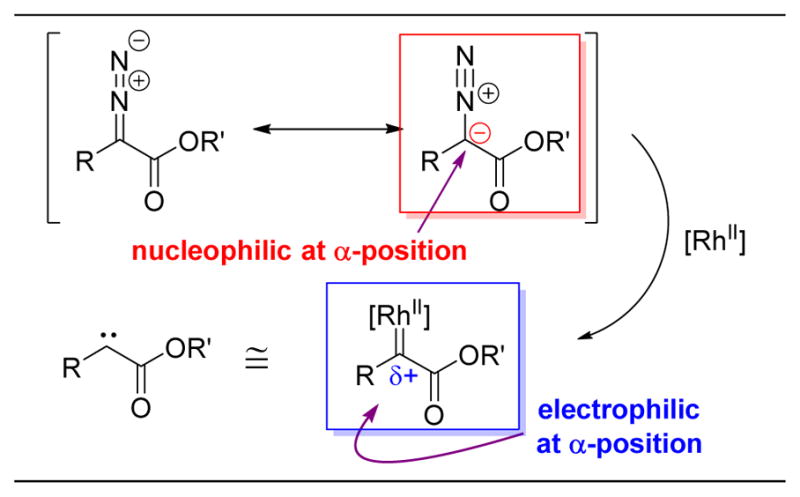
Diazo esters are nucleophilic at the α-position. Upon rhodim carbenoid formation, the now electrophilic the α-position is susceptible to nucleophilic attack (e.g. alcohols or C–H bonds).166–171
Starting with gibberellic acid methyl ester (126), which contains two alcohols, the authors screened a number of diazo esters (127a–g) with Rh2(OAc)4 (131) as a catalyst (Figure 25A).172 The relative rates of decomposition of diazo esters, along with their later productive or unproductive reaction pathways, are chiefly determined by the identity of the α-substituent to the diazo compound (the R group of 127a–g). While diazo esters themselves have nucleophilic character at the α-position, upon reaction with dirhodium species, the incipient rhodium carbenoids become electrophilic (Figure 24). Davies and Nikolai have pioneered the use of donor/acceptor diazo esters.168–170 These species contain an aromatic ring as the R group, which serves as an electron donor to stabilize the electrophilic Rh-carbenoid species. This donor stabilization allows for tuning the reactivity of the carbenoids species, lowering its reactivity enough to become selective.
Figure 25.

(A) Site selective O–H insertion of 126 with Rh2(OAc)4. 4-bromophenyl-substituted diazo esters were found to give the best selectivity for mono O–H insertion. (B) Expansion of complex molecule substrate scope for site-selective O–H insertion. All compounds give one mono insertion product, with the exception of 147, which gives primarly diether 149. Note: numbers in parantheses represent %RSM. (C) Alteration of dirhodium catalyst perturbs the ratio of the two mono insertion products. Abbrev: HBPA: 5-hexynyl-(α-4-bromophenyl)acetate.172
Hence, Romo and coworkers probed the effect of various diazo compounds on the selective O–H insertion of gibberellic acid methyl ester (126). Starting with reagent 127a, in which R=H, dimerization of the diazo compound was found to be the prevalent product (Figure 25A, entry 1). Styryl and phenyl diazo esters resulted in highly reactive species that favored diether formation (129a–g, both alcohols reacting) (Figure 25A, entries 2–3). The donor substituents on these diazo esters presumably accelerate diazo decomposition and hence over-accelerate reaction rates. However, reducing the electron-donating character of the donor substituent of the diazo compound, either by removing resonance or incorporating EWGs on the phenyl rings, high selectivities for the formation of mono-substituted product were observed (Figure 25A, entries 4–7), presumably due to decelerating the rate of carbenoid formation. A 4-bromophenyl substituted diazo compound was found to be the best compromise between selectivity and reactivity, and product could be isolated in 55% yield in this case (Figure 25A, entry 7).
In applying this method toward other natural products, the most nucleophilic hydroxyl group was normally the most reactive under these conditions (Figure 25B). As such, the primary alcohol of 137 is functionalized to yield 138, along with the aliphatic alcohol of 139 to yield 140. Paclitaxel (141) was also found to undergo O–H insertion at the most sterically accessible 2′ position to yield 142. The remaining alcohols are buried within the complex ring structure of 141, and the amide NH must be deactivated via resonance. A similar pattern emerges for 143, which reacts at a secondary alcohol over an indole NH, which is less nucleophilic due to the aromaticity of that ring, to yield 144. This rhodium O–H insertion method is also able to differentiate between subtle steric differences, as 145 undergoes selective reaction at the cyclopentyl alcohol to yield 146, as opposed to the allylic hydroxyl, presumably due to the slightly enhanced steric accessibility of the former. Finally, FK506 (147) was the only compound of this set to exhibit heightened reactivity of two of the alcohols, as both the C32 and C24 alcohols undergo reaction to yield 149, with C32 being slightly faster (with 148 also observed).
Upon O–H functionalization, the Romo group was particularly interested not only in SAR studies, but also in arming these compounds with bioprobes. All of these diazo esters that were utilized contained a pendant alkyne that can undergo a Sharpless–Huisgen cycloaddition with a suitable azide to append a bioprobe, such as biotin or a radiolabel. The inclusion of further sites for elaboration enables the exploration of the affinity of these natural products for various proteomes. Furthermore, the Romo group’s C–H insertion strategy is also amenable to complex molecules where the structure has not been rigorously determined. If by conventional methods it is known that X–H bonds are present in recently discovered natural products, they can be submitted to these rhodium carbenoid conditions and appended with a bioprobe. The compound could then be screened for biological activity and binding affinities, with biologically interesting natural products being studied further. Hence, Romo’s simultaneous SAR and probe arming strategy is a powerful technique for the facile and expedient screening of novel and known natural products for biological activity.
Finally, functionalization of an inherently less reactive alcohol group on a natural product can also be targeted, with an eye to the study of SAR. Utilizing a variety of different dirhodium and copper catalysts, the Romo group screened the O–H insertion of gibberellic acid methyl ester (126) for the site-selectivity between the two hydroxyl groups to yield 128g and 130 (Figure 25C). While the clear preference of this system was to react with the C3 alcohol (128g), utilizing either Rh2(NHCOCF3)4 or Rh2(esp)2 resulted in a 1:1 ratio of 128g and 130. While a 1:1 ratio at first glance is not normally an impressive level of selectivity, when considering that the inherent reactivity of this complex is heavily weighted towards C3, obtaining such a large amount of C13 functionalized product is a significant success. These catalysts hence meet the energetic requirements of reordering the functional group reactivity hierarchy at least to some extent.
However, one problem with the original method was the necessity for a bulky 4-bromophenyl substituent near the natural product.173 Having this very bulky group could affect the biological properties of the natural product. Thus, it would be desirable to incorporate a sterically less demanding R-group on the diazo compound, which produces a compound with minimal perturbation of the native biological activity of the compound. A method using a different donor/acceptor archetype was developed utilizing α-trifluoroethyl substituted carbenoid precursors and led to the observation of high yields and selectivities (Figure 26A), comparable to what was previously observed.172 The authors tuned the reactivity of this diazo ester with various dirhodium catalysts and found similarly to above that the C3 position was most favored for O–H insertion. However, when using catalysts such as Rh2(OCOCF3)4 or Rh2(4S-MEOX)4, a 1:1 ratio of 151 and 152 could be obtained, which marks a substantial deviation from the inherent substrate reactivity.
Figure 26.

(A) Site-selective O–H insertion of 126 with α-trifluoroethyl-substituted diazo esters. Various dirhodium catalysts gave different ratios of mono and difunctionalized products. (B) Expansion of scope for site-selective O–H insertion.175
Upon applying this method to other complex molecule substrates, similar selectivities were observed as above for 141 and 145 (Figure 26B). However, surprisingly, upon treatment of 147 under these new conditions, only C32 functionalization was observed with no overreacted diether product observed.
3. C–H Oxidation
One of the most difficult challenges associated with the site-selective modification of complex molecules is the competitive reactivity of similar functional groups. Even simplified goals of designing a catalyst that can discriminate between as few as two groups of the same functionality or that can react preferentially with a single type of functional group can be difficult. C–H functionalization is therefore a daunting task, given the prevalence of this ubiquitous group in nearly all organic molecules.7,8,174–180 Furthermore, aliphatic C–H bonds are one of the least reactive functional groups in organic chemistry, as their stability to most reaction conditions has informed the strategies of chemical synthesis, in targeting more traditionally reactive chemical handles, for centuries.177 Hence, it is a substantial challenge to override a reagent’s or catalyst’s propensity to react with customarily more activated functional groups, such as alkenes, alcohols, or carbonyls, in order to achieve C–H bond functionalizations. Moreover, owing to their low polarity, most C–H bonds are chemically similar, which requires catalysts to differentiate between extremely subtle electronic and steric variations among a myriad of comparable bonds.7
Nonetheless, the potential advantages of site-selective C–H activation are substantial. For example, a major impediment to the total synthesis of natural products or biologically interesting derivatives can be the functionalization of unreactive positions. Achieving reactivity at these inactive positions can entail additional synthetic steps or integrating a functional group handle at the targeted positions from smaller building blocks, which can oftentimes be a laborious and inefficient process.175 Hence, the conversion of traditionally “dormant” C–H bonds into useful and accessible chemical handles has and will continue to have a tremendous effect on synthesis and the late-stage modification of complex natural products. Towards this end, the development of techniques to understand and control the complex reactivity patterns of C–H bonds in complex molecules is critical for the fields of chemical synthesis and pharmaceutical development.
3.1. Enzyme Catalyzed C–H Oxidation
Despite the immense challenge afforded by the abundance of C–H bonds, most chemical substrates, and especially in complex natural products, nature achieves highly selective C–H functionalization reactions by way of highly evolved enzymes that oxidize specific C–H bonds as part of natural biosynthetic pathways.181–183 The ability of enzymes to selectively decorate simple alkanes and construct valuable complex molecules is a process central to life. The impressive rate-acceleration that enzymes are able to achieve for the functionalization of particular C–H bonds is accomplished via exquisitely engineered binding pockets that bind specific substrates in specific orientations.184 This exceptional molecular recognition allows enzymes to override the inherent order of reactivity of the C–H bonds present in the substrates and instead oxidize less reactive C–H bonds due to their proximity to the active site. Indeed, to date, many C–H oxidation enzymes have been discovered in nature, most of which use either iron or copper in their active sites.185 The most prodigious class of these oxidation-active enzymes is the superfamily of cytochrome P450s, comprising ~12,000 enzymes characterized by a highly active Fe-porphyrin.184,186
Perhaps one of the most stunning examples of the enhanced selectivity and reactivity afforded by P450 enzymes is the biosynthesis of paclitaxel, also known as taxol (141, Figure 27).187–189 Originally discovered in the bark of the evergreen tree Taxus brevifolia, 141 is currently used as a treatment for a number of cancers.188 In the biosynthesis of taxol, cyclization of geranylgeranyl diphosphate (162) to yield taxadiene (163) is followed by the highly selective insertion of eight additional oxygen atoms into 163, catalyzed by eight distinct P450 enzymes.189 The level of control the P450s afford in these oxidations is extraordinary, especially given the presence of 2 alkenes, 2 tertiary C–H bonds, and 14 secondary C–H bonds, all of which are potentially reactive in the presence of an oxidant. However, the question is raised as to whether analogous levels and modes of selectivity are possible with synthetic catalysts.
Figure 27.
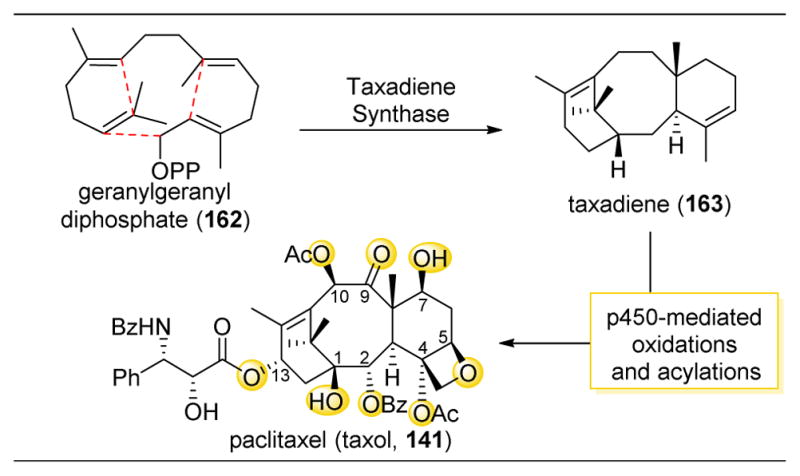
Biosynthesis of paclitaxel. After initial cyclization, a number of enzymes of the P450 family catalyze the site- and stereoselective oxidations to yield 141.189
While a rigorous review of in vivo and in vitro enzymatic C–H activation, including laboratory directed evolution for abiological site-selective catalysis, is beyond the scope of this Review, these biological processes inspire the goal to mimic their superb selectivity with synthetic catalysts.184,185,190,191
3.2. Tether-Assisted C–H Oxidation
An early strategy to realize nature’s level of selectivity in C–H functionalizations that has had a tremendous amount of success is the use of an oxidant that is covalently attached to a substrate. Depending on the length and location of the tether, the site of C–H functionalization can be varied (Figure 28).192–194 However, this topic will not be covered here, as the primary focus of this Review is the catalyst-controlled site-selective modification of natural products.
Figure 28.
Site-selective tether-assisted oxidation of steroids. A shorter tether length forces oxidation at C15, while a longer tether allows the benzophenone oxidant to reach C17–H.192–194
3.3. Site-Selective C–H Oxidation of Natural Products for O-Atom Incorporation
3.3.1. Factors that Influence Site-Selectivity in C–H Oxidation
While the structural intricacies of enzymes allow for extremely specific substrate–catalyst interactions that give rise to high selectivity, catalysts of this size are not necessary to affect site-selective C–H oxidations on complex molecules. Different reagents, depending on their sterics, electronics, and mechanisms of action, can afford substantially different reactivity patterns.195 Furthermore, careful analysis of the steric and electronic profiles of individual substrates can allow for predictability in which positions on a substrate or more likely to react over others.196
Figure 29 depicts the principle factors that control the reactivity of C–H bonds for oxidations.7,197,198 One of the most important factors controlling C–H oxidation is the electron density intrinsic to these bonds. C–H bonds generally serve as the nominal nucleophile in these transformations, hence the most electron-rich C–H bonds typically react first (Figure 29A). Consequently, the rate of C–H oxidation follows the trend: tertiary (3°) > secondary (2°) ≫> primary (1°). Due to the similar electronics of most C–H bonds, even remote electron withdrawing groups (EWG) can provide substantial deactivation of C–H bonds (Figure 29B). In addition to electronic control, reagents can also be sensitive to the steric environments surrounding the site of reaction. If all the 3° C–H bonds of a particular substrate are sterically shielded, 2° C–H bond oxidation is oftentimes observed. Additionally, the discussion of sterics is not limited to 3° versus 2° C–H bonds alone, as the three-dimensional structures of molecules can allow or impede reactivity at many sites. This interplay between electronics and sterics plays a pivotal role in most C–H oxidations.
Figure 29.
(A) Interplay between hyperconjugative stabilization of intermediates from native substrate functionality and steric hinderance. 3° and 2° C–H bonds are often competitive for oxidation, 1° C–H bonds are disfavored. (B) Given the near equivalence of most C–H bonds, remote EWGs can have large effects.7,197,198 (C) α-Hyperconjugative donation from oxygen lone pairs to the C-H antibonding orbital greatly enhances the nucleophilicity of these bonds.7,199 (D) Oftentimes, the mechanism of C–H oxidation involves full or partial planarization of targeted bonds. C–H bonds where this deviation from ideal bond angles is accompanied with strain release are favored for oxidation.200 (E) Directing groups can outcompete other considersations.201–202
However, local sterics and electronics alone do not control the selectivity of C–H oxidations, as a number of stereoelectronic effects can also play substantial roles in influencing substrate biases exerting an inductive effect over several bonds. First, the nucleophilicity of C–H bonds can be greatly enhanced through hyperconjugative donation from an electron-rich species, such as an ether, into α-σ*C–H orbitals (Figure 29C).7,199 In another case, C–H bonds that are in highly strained ground state conformations, such as those that interact with two axial methyl groups within a cyclohexane scaffold, are more susceptible to oxidation, as reaction relieves this strain (Figure 29D).200 Finally, despite all of these factors controlling selectivity, they can be superseded by the presence of a directing group (Figure 29E).201,202 If a certain functional group, such as a carboxylic acid, interacts strongly enough with the catalyst or reagent, it can become nominally equivalent to a tethered oxidation (see Figure 28), and the reaction will be directed towards a position near this location.
3.3.2. Various Reagent-Controlled Site-Selectivity in C–H Oxidation
To exemplify all these competing factors, Figure 30 presents a number of studies in site-selective C–H oxidations involving steroid derivatives. Steroids are some of the most common substrates for C–H functionalizations, as their abundance of aliphatic C–H bonds provide a good benchmark in selectivity for various oxidation strategies. Furthermore, the oxidation of steroids is a common and powerful technique in total synthesis.175 While this Review is not able to be comprehensive given the extensive history and rapidly expanding present, the examples herein represent the essential factors that govern substrate-controlled, site-selective C–H oxidations.
Figure 30.

(A–J) Assorted site- and stereoselective C–H oxidations using a variety of stoichiometric or catalytic oxidations. Selectivity is miainly governed by substrate bias.
,
, and
orbs represent dominance of electronic, steric, and stereoelectronic factors respectively.99,200,203–212
Exploiting the idea that the most electron-rich C–H bonds will react preferentially, Adam and coworkers achieved selectivity in the oxidation of brassinosteroid derivative 164 (Figure 30B).203 Due to the four acetyl groups, in addition to the B ring lactone, the steroid ring system is deactivated, and reactivity is observed with methyl(trifluoromethyl)dioxirane (TFDO) at C25–H. Considering that no oxidation products are observed at the 3° C14–H and C17–H bonds, these positions are presumably deactivated due to either subtle and remote electron withdrawing effects or the larger steric profile of the ring system. The Du Bois group observed similar selectivity when using η3-Ru(V) complex 166 as a catalyst in tandem with the oxidants cerium ammonium nitrate (CAN) and AgClO4, achieving oxidation at the C25 position of steroid 165, with the three acetyl groups presumably deactivating the steroid ring C–H bonds, leaving C25–H as the most sterically accessible 3° C–H bond (Figure 30C).204
In contrast, Bovicelli and coworkers observed sterically driven selectivity for the functionalization of coprostane steroid derivative 167, with the C5 oxidation product formed preferentially (Figure 30D).205 This selectivity is notable given the proximity of the C5 position to the lone acetyl group in the molecule. The C5–H bond must be the most sterically accessible C–H bond, even more so than C25–H, due to its location at the fusion of the cis-decalin of the A,B ring system. It is intriguing that cis-decalin 165 shows no reactivity at C5–H, though this could be due to either the more bulky metal reagent or the fewer number of EWG on substrate 167 (Figure 30C). The same reactivity pattern is shown on substrate 168, using per-fluorinated oxaziridine 169 (Figure 30E).206
Additionally, the directing effect of cyclic ethers leading to α-functionalization is demonstrated in the oxidation of steroid 170, using a combination of RuCl3 and NaIO4, which occurs selectively at the C16 position (Figure 30F).207 This stereoelectronic effect is not reserved to steroid substrates, as the site-selective oxidation of bryostatin analogue (171) is also reported. Here, Wender and coworkers were able to selectively hydroxylate the C9 position of 171 in the presence of both a primary and a secondary alcohol. These C26 and C3 positions benefit from similar hyperconjugative donation from adjacent oxygen lone pairs, though their proximity to a number of EWGs presumably deactivates the C–H bonds (Figure 30G).208 The reactivity of C9 over other α-ethereal positions is excellent. In comparing C9–H and C5–H, the latter is likely deactivated due to the remote electron withdrawing nature of the C1 ester, and C11–H and C15–H are less favorable to oxidize due to the oxocarbenium character of both ethers in this ring. Making subtle chemical alterations to substrates can drastically affect the observed reactivities. This concept is demonstrated by Fuchs and coworkers with the selective oxidations of three steroidal ethers (Figure 31H).199 In the case of steroid 172, oxidation is favored at the C16 or C22 positions due to hyperconjugative activation of nO to σ *C–H (Figure 29C). While both are 3° C–H bonds, C22–H is adjacent to an additional tertiary center, which makes this position less sterically accessible, resulting in oxidation at C16–H. However, by adding an electron withdrawing acetate to C15 (173), the reaction at C16–H is electronically deactivated and oxidation of 173 occurs exclusively at C22–H. Thus, choice of substrate and protecting group strategy can have a large effect on selectivity.
Figure 31.
(A) Hartwig’s Ir-catalyzed C–H silylation/oxidation pathway favors formation of 1,3-diols. (B) The oxidation of 184 results in a primary alcohol at the C23 position. The previous state of the art to access oxidized derivatives at the position was a 10 step synthesis. (C) After Ru-catalyzed silylation of the C3 alcohol, a similar Ir-catalyzed rearrangement/oxidation yields C23 oxidized 188.202
Additionally, strain release has been exploited to achieve the selective functionalization of steroids (Figure 29D). As reported by the Eschenmoser and Baran groups, the 2° C11–H bonds adjacent to multiple axial methyl groups on steroids such as 174 show proclivity towards oxidation (Figure 30I).200,209 Oxidation at this position occurs preferentially to oxidation at six 3° C–H bonds and twenty four other 2° C–H bonds due to the presumed partial planarization of the C11 atom in the transition state, which relieves this axial strain. This selectivity is particularly noteworthy given that there are no EWG or activating groups on substrate 174, making most of the C–H bonds in the molecule of similar polarity.
One consideration that was briefly alluded to in the oxidation of bryostatin analogue 171 (Figure 31G) is the potential competitive reactivity of non C–H bonds. In an approach aimed to be tolerant of functional groups, Hilinski and coworkers were able to oxidize C25–H of 176 in the presence of an unprotected alcohol using iminium catalyst 177, which forms an oxaziridine in situ (Figure 30J).210 Indeed, unlike in the case of 171, which contains a number of EWGs adjacent to the native hydroxyl groups, the C3–H of 176 is fairly activated. This broad functional group selectivity for C–H hydroxylation over the more common alcohol oxidation is attributed to a hydrophobic effect on the activated oxaziridine catalyst. When utilizing aqueous hydrogen peroxide as the stoichiometric oxidant and hexafluoroisopropanol (HFIP) as a solvent, the face–face packing of the hydrophobic oxaziridine catalyst and the steroid ring is favorable for the exclusion of water.211 Bringing the secondary alcohol near the catalyst would hence disrupt this hydrophobic packing, disfavoring this pathway. Intriguingly, the polarity of the medium is necessary to achieve this C–H oxidation selectively over alcohol oxidation, as utilization of more nonpolar dichloromethane (DCM) results in less selective oxidation.
Finally, most of the steroid substrates examined so far have lacked aromatic groups. Arenes can engage in various modes of reactivity other than oxidation that can be difficult to control. Despite this challenge, upon reacting estrone derivative 178 with oxaziridine 179, Du Bois and coworkers observe oxidation at the C11 methylene (Figure 30K).212 However, the authors posit that this occurs via initial oxidation of the 3° C9–H bond due to resonance stabilization, followed by elimination to form the C9–C11 alkene. This alkene is subsequently transformed into the C11 ketone. This reaction exemplifies the challenge of chemoselectivity in C–H oxidations.
3.3.3. Directing Group-Assisted C–H Oxidations
While probing the inherent reactivity of certain substrates can be powerful, and modifying the choice of oxidant or catalyst can have large effects on selectivity, these approaches limit natural product derivatization to the most reactive positions. Hence, if the functionalization of a less reactive position is desired, a different strategy must be undertaken. In these cases, one avenue for achieving high levels of selectivity could be the inclusion of a directing group on the substrate. If the catalyst can bind to or direct a reagent to this added functionality, proximity-induced assistance can enable the C–H oxidation of these normally unreactive positions.
Making use of the extremely common alcohol functionality, Hartwig and coworkers revealed an effective hydroxyl-directed, 1,3-oxidation using this strategy (Figure 31A).202 Here, upon silylation of alcohol 180, an iridium complex can catalyze the 1,3-selective C–H bond silylation to yield 182, after which Fleming–Tamao–Kumada oxidation with H2O2 results in 1,3-diol 183. Under normal oxidation conditions, given the number of 3° and 2° C–H bonds present, hydroxylation of a 1° C–H bond would be highly disfavored. When this method was applied to steroids 184 or 187, selectivity towards oxidation of the C23–1° methyl group was achieved due to the directing power of the C3 oxygen atom (Figures 31B–C). With the C3 alcohol being in the equatorial position, it cannot geometrically access the 5-membered ring transition state to obtain products from oxidation at the C1 position. Only the C23–H or C24–H remain as potentially reactive bonds, between which the catalyst differentiates effectively. Intriguingly, the concern of stereoselectivity in this case is an additional factor beyond standard site-selectivity that can be a challenge in the selective functionalization of natural products. It should be noted that the previous state of the art for the oxidation of C23–H of 184 went through a 10 step process culminating in 36% overall yield.213 Finally, owing to the mechanism of this iridium-catalyzed silylation/oxidation method, oxidation of the alcohols and alkenes present in these substrates to ketones or epoxides is unlikely, a trait often uncommon for other oxidation methods.
Another strategy for directed C–H hydroxylation at the C12 position of steroids was studied by the Schönecker and Baran groups utilizing pyridyl-imines (Figure 32A).214,215 Starting with steroids containing C17 D-ring ketones, pyridyl imines are condensed (189) to direct Cu-catalyzed oxidations to occur at the C12–H to yield 190 (Figure 32). The achievement of selectivity at the C12 position is striking, given the electron withdrawing nature of the imine, which limits the inherent nucleophilicity of C12–H. Furthermore, the proximity of the C18-methyl group also makes this site less sterically accessible than others. However, as shown in Figure 32, the pyridyl-imine serves as a ligand for the copper, which generates the incipient oxygen diradical close to the C12 position. Notably, in addition to leaving 4 tertiary and 7 secondary positions intact, both alcohols and alkenes are tolerated with these reaction conditions.
Figure 32.
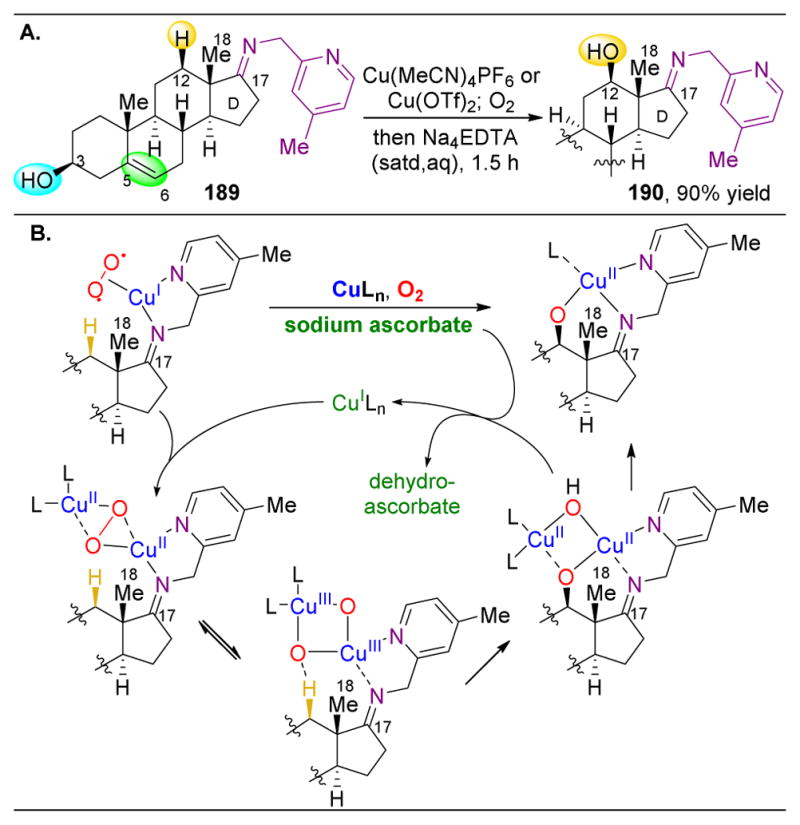
(A) Pyridyl-appended imines can function as directing groups for Cu-catalyzed aerobic oxidations of C12–H in steroids. This method tolerates both alcohols and alkenes. (B) The proposed mechanism proceeds via bimetallic activation of O2 to activate the C12–H bond.214
3.3.4 Site-Selective Allylic C–H Oxidation
Another common strategy that enables site-selective oxidations is allylic C–H oxidation. Transformations that are chemoselective toward allylic C–H positions are possible due to the unique reactivity of this substructure. However, some current methods for allylic oxidation utilize toxic reagents or can lead to oxidation of more than one allylic C–H bond within the same allylic system. Progress towards addressing these limitations were reported by the Baran group, in which they make use of an effective electrochemical oxidation method for the site-selective C–H oxidation of small molecules and natural products (Figure 33).216
Figure 33.

Site-selective C–H oxidation of the allylic positions of a variety of natural products.216
Utilizing reticulated vitreous carbon for their electrodes, Baran and coworkers found that a combination of Cl4NHPI and tBuOOH was a most effective electrochemical mediator/oxidant pair when pyridine was used as a base. In addition to successfully applying this system to the allylic oxidation of a variety of small molecule substrates, the authors utilize this transformation to modify a large number of complex molecules; six are presented here as representative examples (Figure 33A). This method normally obtained good levels of regioselectivity, in addition to the chemoselective targeting of allylic C–H bonds. Indeed, even molecules with multiple alkenes only reacted at one site. The site-selectivity of this method for particular alkenes could be linked to the mechanism of this transformation, which is believed to proceed via an allylic radical. The potential primary radical, as in the cases of 191–193, would be less stable than those radicals derived from the internal alkene. A proposed mechanism is presented in Figure 33B. Here, upon deprotonation, Cl4NHPI is oxidized by the anode. The O-centered radical then abstracts an allylic C–H bond and this radical can be quenched by tBuOOH and later converted to the ketone.
3.3.5. Orthogonal Functionalization of Betulin
Given the number of oxidation systems that have been developed, the question arises as to whether a number of different sites on one substrate can be targeted selectively for modification through the use of a variety of orthogonal oxidation techniques. Baran and coworkers addressed this challenge as they reported an effective display of complementary selectivity for the oxidation of betulin (197) and betulinic acid (198, Figure 34).195 First, the C20 C30 alkene of 197 was protected as an alcohol or peroxide using Carreira’s cobalt-catalyzed hydration conditions in order to prevent epoxidation of the alkene.217 Next, acetylation of the C3 and C28 alcohols generated two substrates, 199 and 200.
Figure 34.

(A) Oxidation of betulin derivatives 199 and 200 with TFDO and PIDA to selectively target the C16 and C12 positions. C16–H is the most electron rich and sterically accessible secondary C–H bond in the molecule. C12-oxidation is achieved via direction from the C20 alcohol or peroxide. (B) Directed oxidation of betulin derivative 204 with Pb(OAc)4 followed by treatment with AgOAc reveals a C13 oxidized product that has undergone skeletal rearrangement via a 1,2-shift. (C) The Hartwig [Ir]-catalyzed 1,3-diol oxidation protocol was utilized to target the C23–H, directed by the C3 alcohol. (D) Summary of all oxidation methods used to target different positions of betulin (197) and betulinic acid (198).195
The authors sought a way to determine which C–H bonds on 199 were the most electron-rich. Using 13C NMR and 13C 13C INADEQUATE NMR spectroscopy, the electronic character of each C–H bond in the molecule was analyzed, and it was shown that the C6 and C16 positions are the most electron-rich 2° C–H bonds. Calculations at the B3LYP level of theory revealed that the activation enthalpy (ΔH‡) of C16–H oxidation is 1.6 kcal/mol lower than the barrier to oxidation of C6–H. While the other tertiary C–H bonds could be more electronically activated, their increased steric profiles, especially in congested steroid rings, make them difficult targets. Secondary C–H bonds hence offer an intermediate in both steric and electronic considerations. Indeed, it was found that trifluoromethyldioxirane (TFDO) reacted to form a ketone at C16 (201) (Figure 34A).
It is also possible to reverse this innate reactivity towards C16–H by inclusion of a well-placed directing group, such as the C20-alcohol that is engineered into 199. Using Suarez’s procedure, phenyliodine(III) diacetate (PIDA) can form an O-centered radical on the C20 alcohol, which can selectively abstract the C12–H, forming a radical that is quenched by I2.218 This C12–I can then be displaced to form tetrahydropyran 202 in 30% yield (Figure 34A). The configuration of the C19 atom places it in close proximity only to C12, C18, and C21. Perhaps the observed selectivity can be explained via preferential reactivity at the sterically less demanding 2° C–H bonds. Alternatively, when replacing the C20 alcohol with a peroxide (200), alkene 203 is formed as the major product in 34% yield. Product 203 is assumedly formed by hydrogen abstraction at C12, followed by oxidation of the incipient carbon-centered radical, and finally elimination of a proton from C13.
As opposed to designing a directing group into the substrate at the C20 position, the C28-alcohol of 197 can also be explored as a directing group by selectively acetylating the C3–OH over the C28–OH (Figure 34B). Subsequent reduction of the C20–C30 alkene yields monoacetylated 204. The remaining free C28 alcohol can be used to direct oxidation towards the 3° C13–H bond with Pb(OAc)4 to install an iodine at C13. Given difficulties purifying this product, the authors opted to treat this intermediate with AgOAc to promote a skeletal rearrangement via a 1,2-shift of the C13–C14 bond to reveal 205 in 50% yield.218 This site-selectivity is impressive given that the axial directing group here is positioned proximally to C–H bonds at C13, C15, and C19–C22.
Additionally, given the native C3–OH of 197, the C23–H could be targeted through Hartwig’s 1,3-alcohol-directed oxidation method. The Baran group began with betulinic acid (198) so that the C26 position, now a carboxylate, could be easily protected as a benzyl ester (Figure 34C).202 The ester underwent an Ir-catalyzed intramolecular C23–H silylation followed by subsequent Fleming–Tamao–Kumada oxidation to reveal 206.
Finally, while not the topic of this Review, two microorganisms were also used to oxidize 197. Streoptomyces fragilis reacted to form the di-oxidized product at C2 and C7, while Bacillus megaterium formed the di-oxidized product at C7 and C15 (Figure 34D).
Figure 34D presents a summary of all the positions for which selective oxidation was achieved. By subtly changing the sterics, electronics, chemical reactivity, and directing groups of substrates like 198 and 199, in addition to varying the reagents used based on their properties and reactivity, eight different C–H bonds can be oxidized. This example highlights the power of simple stoichiometric reagents, in addition to catalysts that operate via diverse mechanisms in enabling various derivatives to be synthesized through orthogonal reactivity. The considerations enumerated here will have a large influence over catalyst-controlled site-selective reactions and will be addressed thoroughly in later sections.
3.3.6. Cyclodextrin-Assisted C–H Oxidation
Returning to the idea of enzyme-catalyzed transformations, many C–H oxidations carried out by enzymes involve the Fe-containing P450 family. The site of reactivity is controlled by the enzymes’ structures and the nature of the docking interactions between particular substrates and active sites. This catalyst-controlled selectivity is opposed to the results presented in the previous sections, where the inherent sterics and electronics of substrates, among other considerations, dictated the location of C–H oxidation. However, the question arises as to whether the catalyst-directed selectivity that enzymes afford in functionalizing positions that are inherently less reactive can be emulated using smaller, synthetic catalysts.
One of the earliest methods to carry out catalyst-controlled oxidations was through the use of cyclodextrin as a binding pocket to orient substrates in a selective manner near the catalytically active porphyrin (Figure 35).219–221 Here, Mn-porphyrin catalysts such as 207 and 208 that mimic the activity of cytochrome P450 enzymes are appended with four cyclodextrin units (Figure 35D). As the oxidations using these catalysts are conducted in water, the cyclodextrins act as hydrophobic cavities to bind to the hydrophobic tert-butyl groups on the substrates (Figure 35E). In the presence of substrates with tethers of appropriate length, the catalyst can hold the steroid substrate at both ends, positioning the steroid ring system directly over the manganese active site. Depending on the location of these tert-butyl receptors, different C–H bonds will sit over the Mn=O.
Figure 35.

(A–C) The use of cyclodextrin-containing porphyrin catalysts (D) and receptor-containing substrates to achieve site-selective oxidations of steroids. (E) The cyclodextrin units of the catalyst bind to two (A) or three (B) of the receptor arms, positioning the steroid ring and a specific C–H bond directing over the Mn-active site.219–221
There have been numerous reports using this strategy,219–221 and this Review will not exhaustively cover this extensive literature. However, three examples are presented here. Breslow and coworkers pursued the oxidation of substrate 209, with the linkers placed on the C3 and C17 positions. In this case, the sulfonate groups on the linker are added to the substrates to enhance their water solubility. Remarkably, upon reacting 209 with Mn catalysts 207 or 208, good yields of C6-hydroxylated 210 are observed (Figure 35A). Further selective transformations can be accomplished by appending an additional receptor on the C6-alcohol (211, Figure 35B). In this case, 208 can bind to all three of the receptors with three of the four cyclodextrin units on the catalyst, resulting in selective C9 oxidation to yield 212. When one of the receptor arms is replaced with a simple alkane chain (213), the catalyst’s selectivity is degraded, resulting in a complex mixture of numerous oxidized products (Figure 35C). Hence, the extraordinary selectivity afforded by the porphyrin catalysts is the result of strong multipoint binding between the substrate and catalyst. Placing only one C–H bond in close proximity to the active metal is analogous to the mechanism of action of many enzymes.
3.3.7. Development of Selective Iron-Catalyzed C–H Oxidation
Catalyst-control over C–H oxidations with more diverse classes of substrates, and particular by those without directing groups, has also been pursued. While the incorporation of linker and receptor groups into substrates has led to impressive selectivities for steroid oxidations, this strategy may not be amenable towards other substrates. Furthermore, it would be desirable to exploit the inherent functionality of substrates that are only minimally derivatized, in combination with small molecule catalysts, to mediate selective transformations. Indeed, in 2007, the White group identified a smaller and simpler iron complex (214), which contains a N,N′-Bis(2-pyridylmethyl)]-2,2′-bipyrrolidine (PDP or BPBP) ligand, that could catalyze highly selective C–H oxidations in the presence of hydrogen peroxide and acetic acid (Figure 36A–B).197 These oxidations are notable due to their mild reaction conditions, short reaction times, and high levels of observed selectivities. Furthermore, the use of iron, in addition to mimicking P450 enzymes, engages an inexpensive and abundant alternative to precious metal catalysts.222 Grounded in the considerations presented above in Figure 29, the White group applied their catalyst towards the oxidation of artemisinin (+)-215, a powerful antimalarial agent.223–225 Derived from the leaves of sweet wormwood (Artemisia annua), it had been used from antiquity to treat fevers. However, it was not until the 1970s where the active ingredient, (+)-215, was isolated. Since then, it has served as one of the most important anti-malarial pharmaceuticals. However, resistance to one of the world’s most essential drugs has raised concerns about its efficacy going forward, and hence the synthesis of novel derivatives becomes essential. The White group pursued the derivatization (+)-215 and found oxidations occurred preferentially at the C10–H bond, yielding alcohol (+)-216 selectively in 54% yield (Figure 36C). To achieve this yield, three batches of catalyst, H2O2, and AcOH were iteratively added over the course of the reaction time, with recovered starting material being recycled and resubmitted to the same reaction conditions twice more. This selectivity is also observed when (+)-215 is oxidized to (+)-216 using microbial cultures of Cunninghamella echinulate, but the reaction takes four days to deliver only 47% yield. To mimic enzymatic selectivity with a small molecule catalyst over a substantially shorter reaction time is an impressive result, especially when considering the presence of four other 3° C–H bonds, eight 2° C–H bond, and nine 1° C–H bonds, in addition to the endoperoxide motif in (+)-215, which is sensitive to Fe(II)-mediated cleavage. The observed selectivity is explained by the fact that C10–H is the 3° C–H that is furthest away from the electron withdrawing lactone, acetal, and endoperoxide. Given that the other 3° C–H bonds, especially C7–H, are fairly remote from EWGs, this demonstrates how suitable catalyst 214 is for selectively discriminating between the minute electronic differences in these bonds.
Figure 36.

(A) Fe-PDP catalysts. (B) Summary of iron-catalyzed, 3°-selective C–H oxidations on small molecule substrates. (C) The oxidation of (+)-artemisinin (215) with 214a favors the most electron rich C10–H bond. (D) The presence of a free carboxylate at C6 replaces the AcOH ligands, overiding the inherent selectivity and favoring lactone formation at C15–H instead. (E) 219 is too sterically encumbered to oxidize C12–H. The remaining C–H bonds are too electronically deactivated by the lactones and epoxide to be oxidized.197
The White group subsequently targeted the oxidation of tetrahydrogibberellic acid (+)-217, which contains a free carboxylic acid (Figure 36D). They hypothesized that the carboxylate could coordinate to the catalyst and direct oxidation to an adjacent C–H bond to form a lactone. This theory was based on the efficacy of carboxylates as ligands in non-heme iron complexes226 and their own use of acetic acid in these transformations. When substrate (+)-217 was subjected to the standard reaction conditions, but with the exclusion of acetic acid in order to favor ligand association of the carboxylate of 217, C15–H was found to react preferentially, giving lactone (+)-218 in 52% yield despite the presence of four other 3° C–H bonds. Carboxylate directing groups in catalysis are particularly enticing given their native appearance in many natural products and as directing groups that can help override inherent substrate electronics and sterics to install oxygens at desired positions.
Finally, α-dihydropicrotoxinin, (−)-222, was exposed to the same conditions and starting material was recovered in 92% yield (Figure 38E). The most electronically favored position, C12–H is sterically encumbered as it is buried in the tricyclic core of (−)-222. In order to be exposed to the convex side of the molecule, the C4–C12 bond would have to be rotated, causing strain between the geminal dimethyl groups and the tricyclic core of (−)-222. The remaining C–H bonds, including three 3° C–H bonds, are presumed to be too electronically deactivated by the two lactones and epoxide to be oxidized by 214a.
Figure 38.
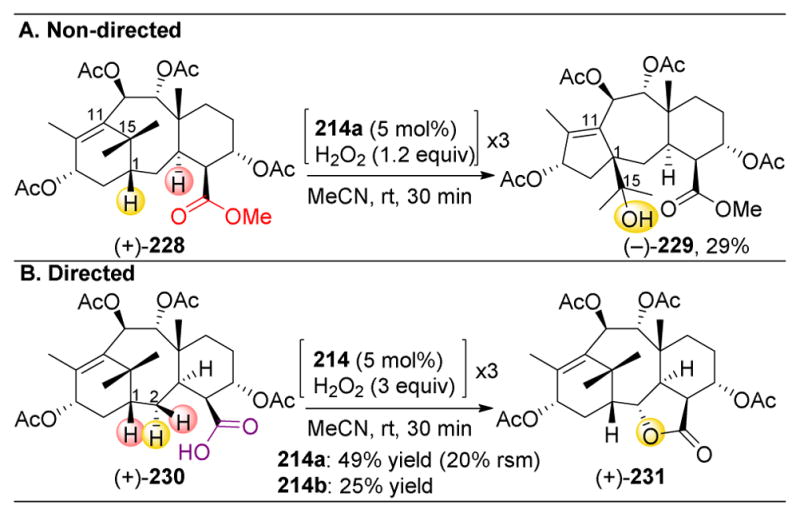
(A) With a methyl ester, the innate substrate reactivity for C1–H atom abstraction followed by rearrangement and oxidation predominates. (B) The free carboxylate directs oxidation to C2–H.201
There were several persistent challenges for this method, including: (1) the requirement for iterative addition of catalyst 214, acetic acid, and hydrogen peroxide, three times over 30 minutes; (2) the need to recycle starting material and resubject it to the reaction conditions again to increase yields; and (3) the occurrence of catalyst decomposition to a species that suppresses oxidation.227 The White group thus developed a slow addition process that afforded comparable yields as shown above, eliminating the necessity for recycling the starting material and minimizing catalyst degradation (51% yield for both (+)-215 to (+)-216 and (+)-217 to (+)-218).
In probing the mechanism of this non-heme iron catalyst, the authors suspected that 214 proceeded via a radical formation (Figure 37A).228 Studies on small molecule substrates continued to validate this hypothesis. The oxidation of taxane (220) was performed under standard conditions and indeed, a surprising rearrangement and oxidation product (222) was observed. This process is believed to first proceed by H-atom abstraction to 223, followed by a Wagner–Meerwein-type rearrangement to 224, and hydroxylation to 222. C1-oxidized 221, synthesized independently, was also submitted to the reaction conditions, and this possible intermediate did not proceed to yield product, implying that the reaction of 220 to 222 does not occur by hydroxylation followed by cationic rearrangement. The authors therefore propose a radical-based mechanism as shown in Figure 37B for all 214-catalyzed oxidations.
Figure 37.

(A) The conversion of 220 to 222 is known to proceed by a prescedented radical rearrangement and oxidation, supporting the hypothesis that catalyst 214 operates via a H-atom abstraction and radical based mechanism. (B) Proposed general radical mechanism. (C) Oxidation of picrotoxinin derivative 225 reveals two lactone products.228
White and coworkers further hypothesized, as previously seen in Figure 36, that carboxylate ligands play an essential role in the transformation. To further demonstrate this principle, the authors performed the oxidation of picrotoxinin derivative 225 and the carboxylic acid was found to direct to the reaction to the 3° C–H bond to form a γ-lactone (226, Figure 37C). An over-oxidation product (227) was also observed in 39% yield, where the 1° C–H bond adjacent to the incipient lactone was oxidized. This over-reactivity is believed to proceed through an alkene intermediate, which is epoxidized and then ring-opened by the carboxylate. The mechanism of formation of this alkene presumably occurs via a radical C–H abstraction/oxidation mechanism.
Furthermore, when analyzing the structure of taxane (220), White and coworkers hypothesized that 220 would be an excellent substrate class to test carboxylate directed C–H oxidation.201 Hence, substrates 228 and 230 were synthesized and tested under normal 214-catalyzed conditions. With methyl ester 228, the C1–H atom is still favored for abstraction, followed by rearrangement and oxidation to 229 (Figure 38A). However, the incorporation of a free acid alters selectivity to C2–H, providing lactone 231 in 49% yield (Figure 38B). These results are strikingly similar to those of Figure 36D and 37C, though it is particularly compelling that the catalyst is able to differentiate between the two secondary C2–H bonds. This example showcases the selectivity of this catalyst when used in tandem with a directing group.
In addition to developing a system that effectively target 3° C–H bonds, White and coworkers assessed whether 2° C–H bonds could be selectively oxidized using the same catalyst.198 While 3° C–H bonds are normally more electronic rich than 2° C–H bonds, numerous approaches can be imagined to favor the reactivity of the latter. First, as already seen with 217 (Figure 36D) and 230 (Figure 37C), a directing group such as a carboxylic acid could favor an otherwise less reactive C–H bond. Second, if all 3° C–H bonds in the substrate were sterically encumbered or near an electron withdrawing group, then an activated 2° C–H bond could react preferentially (see Figure 29A). Third, placing an electron donating group, such as an ester, adjacent to 2° C–H bonds could activate them via hyperconjugation (see Figure 29C). Finally, destabilization of a particular 2° C–H bond with a steric or stereoelectronic effect could outcompete 3° C–H bonds (Figure 29D).
In initial experiments to apply their catalytic system to oxidize 2° C–H bonds, White and coworkers observed that methylenes were oxidized to ketones under the reaction conditions. In the case of (−)-232, derived from (+)-manool, it was found that C2 was the most reactive methylene, outcompeting two 3° C–H bonds and twelve 2° C–H bonds (Figure 39A).198 The carbonyl appears to deactivate the A ring, including the 3° C–H bonds on C5 and C9. These bonds are also fairly sterically encumbered due to the steroid ring system. In the case of the secondary C1–H, C2–H, and C3–H bonds, C2–H is oxidized preferentially to (−)-233 in 52% yield due to the strain between this position and two axial methyl groups on B ring that is relieved during oxidation, as the reaction likely proceeds via a planar radical intermediate (see Figure 29D). An additional 28% yield of C3-oxidized product 234 is also observed. In these reactions, oxidation to ketones instead of to alcohols presumably occurs due to hyperconjugative activation of the adjacent C–H bond by the newly installed alcohol, activating the adjacent C–H bond via hyperconjugation, in analogy to the effect of ethers as previously discussed (Figure 29C).
Figure 39.

(A) Secondary C2–H bonds are favored due to strain release of C2 due to the axial methyl groups at C4 and C6, while the tertiary C–H bonds are deactivated via sterics and electronics. (B) Secondary C2–H bonds are more electron rich due to hyperconjugative activation from an adjacent cyclopropane. (C) Iterative oxidation of 237. The first oxidation is driven by the electron-richness of C–H bonds adjacent to ethers and the second by strain release. (D) Oxidation C7-H relieves strain in the starting material. The tertiary C–H bonds are deactivated via steric and electronics. Intriguingly, the unprotected primary alcohol at C22 is not oxidixed. (E) Examples of π-rich cyclopropane’s hyperconjugation with adjacent C–H bonds and of hyperconjugation from oxygen lone pairs to adjacent C–H bonds.198
Hyperconjugative activation of 2° C–H bonds, as opposed to steric considerations, was also explored by the White group. The C–C bonds in cyclopropane rings, due to the strain associated with accessing this 60° geometry, have a large amount of p-orbital character,229,230 allowing them to be involved in hyperconjugation with adjacent C–H bonds (Figure 39E). Evaluating the potential to use these hyperconjugative effects for selective C–H oxidations, substrate (−)-235 was oxidized at C3 exclusively to form (−)-236 in 45% yield (Figure 39B). It is again noteworthy that the catalyst is able to differentiate between C3–H and C6–H simply due to the subtle electron withdrawing effect of a carbonyl that is two bonds away.
Continuing to exploit this hyperconjugative effect, White and coworkers explored the oxidation of (−)-ambroxide (237). This molecule contains the C2–H bond that is strained by di-axial interactions, two additional 3° C–H bonds at C5 and C9, and α-ethereal C12–H. Despite these numerous, potentially reactive sites, the first oxidation proceeds exclusively to C12–H affording the product in 80% yield (Figure 39C). When the product (+)-238 is exposed to the same oxidation conditions, oxidation occurs at both C2 (239) and C3 (240) positions, as shown with analogous substrate (−)-232. Comparison of these results to those obtained with enzymes231,232 reveals that iron catalyst 214 is substantially faster, though with complementary reactivity patterns for the oxidations of both (−)-237 and (+)-238.
Finally, White and coworkers selectively oxidized (−)-dihydropleuromutilone (−)-243 at the C7 position (both alcohol and ketone products in combined 62% yield, Figure 39D). The C7–H bond is probably favored over other C–H bonds due to the strain release from the large axial substituents of C10 and C14. The fact that a substantial amount of hydroxylation product 244 is observed is potentially due to the steric encumbrance associated with the catalyst accessing the axial C7–H bond. The overall selectivity is noteworthy due to the presence of an unprotected primary alcohol in the molecule.
However, despite the multitude of recent advancements in small molecule-catalyzed C–H oxidations, especially in Fe(PDP)-mediated transformations, White and coworkers noted that true catalyst control has yet to be achieved.233 This catalyst control is pivotal to C–H oxidations in nature, in which enzymes can select nearly any C–H bond in a molecule for oxidation given the proper active site. White and coworkers therefore embarked to modify catalyst 214 to increase its ability to discriminate subtly distinct C–H bonds on substrates. Indeed, catalyst 246 was developed, which contains additional ortho-CF3 aryl rings on the pyridine ligands’ 5 and 5′ positions (Figure 40). These ortho-functionalities serve to partially block the Fe-active site, decreasing the potential trajectories of approach of substrates and making the catalysts more sensitive to minor steric and electronic differences in substrates.
Figure 40.
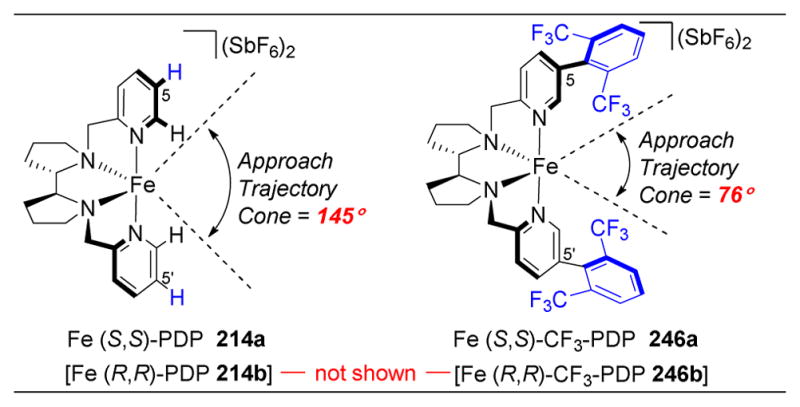
The inclusion of bulimy groups on the pyridine 5 and 5′ position reduces the cone angle, making the catalyst even more susceptible to minor steric effects.233
With the discriminating nature of catalyst 246, the White group examined whether catalyst tuning could be an effective strategy for C–H oxidation. First, catalyst 214a was shown to be able to oxidize tricalysiolide B triacetate (−)-247 with moderate selectivity towards the C6 and C7 positions (248 and 249, Figure 41).196 The observed selectivity arises from the electron-withdrawing acetates and lactone, in addition to the the concave shape of the substrate, which blocks reaction at other positions. From here, a parameterization model was developed in order to assess both the electronics and sterics of particular substrates in an effort to predict reactivity for each catalyst.233 As shown in Figure 41B, each equatorial C–H of the molecule is assigned a value, the lower value indicating higher reactivity. Using this model to assess (−)-247, the C6, C7, C12, and C14 positions were found to be the most favorable positions for oxidation. Indeed, catalysts 214b and 246b both favor the formation of these oxidation products. After assessing the substrate alone, computations were performed with both catalyst and substrate to predict each catalyst’s reactivity. The results highlighted in Figure 41 show strong correlation between observed and predicted results. Catalyst 214b is fairly unselective, whereas catalyst 246b highly favors oxidation of C6. The enhanced selectivities observed with 246 compared to 214b imply that 246 is more sensitive to the steric hindrance of the bicycle in 247, preventing 246 from accessing the C7 position.
Figure 41.

(A) Oxidation of (−)-247 reveals preference for C6 and C7. (B) The substrate was analyzed with computations, comparing the C–H atoms’ relative electronic and steric parameters in order to predict reactivity. Catalyst 246 is better abie to respond to minor steric effects and yields substantaily higher selectivity than 214b. Predicted results correlated well with experimental findings. (C) The electron withdrawing lactone and acetates, along with the sterically hindered concave side of the molecule favor the C6 and C7 atoms for oxidation.233
Applying catalyst 246 towards (+)-artemisinin (215), however, revealed catalyst-controlled selectivity that overrides the innate reactivity of the substrate (Figure 42A).233 While catalyst 214a favored oxidation at C10 (216, as previously seen in Figure 36C), the bulkier catalyst 246a favored the 2° C9–H, which is still distant from the electron-withdrawing groups on the molecules, to yield 260. This significant reversal in selectivity shows that tuning of the catalyst can have a large effect on which position reacts. Furthermore, White and coworkers again used their parameterization method to show that their model successfully predicted the reactivity of both catalysts.
Figure 42.

(A) Artemisinin was subjected to oxidation with both 214a and 246a, revealing that tertiary C10–H is too hindered for oxidation by 246a, which instead favors C9–H. Predicted results from paramaterization correlate well with observed results. (B) Catalyst 214a favors tertiary C11–H while more hindered catalyst 246a favors C10–H.233
This selectivity of 246 toward 2° C–H bonds rather than 3° C–H bonds is also observed in the oxidation of (+)-261 (Figure 42B). While catalyst 214a gives a near 1:1 ratio of products due to the oxidation of C10 (263) and C11 (262), catalyst 246a preferentially forms a mixture of alcohol and ketone products at C10, as predicted by their parameterization method.
Moving on to other substrate classes, one potential challenge that can arise is the functionalization of molecules containing amine functionality (Figure 43).234 Electron-rich nitrogen atoms can both bind and deactivate metals, as well as present chemoselectivity concerns due to their facile oxidation to N-oxides. However, if amines are protonated by tetrafluoroboric acid (HBF4), thus precluding coordination or oxidation, high levels of selective C–H oxidation on a variety of N-containing natural products can be achieved.
Figure 43.

(A) Oxidation of dextrometliorphan derivative (+)-254 revealed preferences for oxidation of the C6–H and one of the C7–H bonds, the most distal groups from the protonated piperazine ring. (B) Oxidation of abiraterone acetate analogue (+)-257 shows preference for the C6–H bond, which is distant from the EWGs on the A and D rings. This is aided by the strain release that is afforded in the planarization of C6 from the axial methyl group at C10. The catalyst is also able to select for C6–H against tertiary C–H bonds at C5, C8, and C14. (C) Cycloheximine derivative (+)-260 is oxidized at C19–H, which is furthest away from the multiple EWGs of the molecule.234
As shown in Figure 43A, 246 is an effective catalyst when applied to functionalization of the dextromethorphan derivative (+)-254, which contains a piperidine ring. Using HBF4 protonates the nitrogen atom, converting the incipient ammonium adduct into an EWG, thus favoring oxidation at the C6 and C7 positions. Intriguingly, while the C7 position is oxidized to ketone (+)-255, the C6 position is only oxidized to alcohol (+)-256. Given the sensitivity of 246 to minute steric differences, it is possible that C6–H is in a more hindered environment compared to C7–H, limiting the catalyst’s ability to react at C6 a second time.
The approach established by White and coworkers is also amenable to pyridine-containing molecules, like abiraterone acetate analogue (+)-257 (Figure 43B). With an electron withdrawing acetate and a protonated pyridine at opposite ends of the molecule, oxidation at C6 is favored. This position is also more reactive due to strain between the C6–H bond and the C10–Me, which is relieved upon planarization of C6 (Figure 29D).
Finally, cycloheximide derivative (+)-260 is also tolerated under these acidic conditions, resulting in (+)-261 as the sole product in reactions catalyzed by 214 (Figure 43C). Most of the C–H bonds in the molecule are deactivated by a number of EWGs, making reaction at the C19 position highly favored. This approach was used in the late stage synthesis of streptovitacin (262). It should be noted that the 1°, α-keto chloride of (+)-260 is tolerated under these reaction conditions.
This significant change in selectivity with catalyst 246 showcases a new and underexplored mode of selectivity reversal in C–H oxidation, tuning the catalyst. Similarly, Costas and coworkers synthesized a variety of derivatives of non-heme iron catalysts for C–H oxidation (Figure 44A).235,236 Here, three main changes are made to the catalysts compared to 214 and 246. First, the backbone diamine is changed from the PDP scaffold in the case of 214 and 246 to an N,N′-bis(2-pyridylmethyl)-N,N′-dimethyl-1,2-cyclohexanediamine (mcp) ligand, which plays a critical role in orienting the pyridine rings and determining the steric environment near the open site on the iron catalyst (264). Second, similar to the White group’s approach of adding ortho-CF3-aryl rings to the 5 and 5′ positions of the pyridine rings, Costas and coworkers appended (+)-pinene-derived functionality on the pyridines. These bulky chiral auxiliaries add additional stereocenters near the active site and provide a smaller range of trajectories from which the substrate can approach the catalyst (264 and 265). Finally, the acetonitrile ligands at iron were replaced by triflates, precluding the need for counterions (263–265).
Figure 44.

(A) Catalyst derivatives with X-type ligands, removing the need for counterions, modification of the aliphatic chiral diamine, and substitution of the pyridine rings with chiral bulky (+)-pinene. (B) Oxidation of (−)-ambroxide (237) reveals a strong preferene for C–H bond adjacent to the cyclic ether due to strong hyperconjugative activation. Increased yields are observed at lower catalytic loading with novel catalysts. Oxidation of (+)-cedryl acetate (266) favors the tertiary C–H bond distal from the electron withdrawing acetate. Yields are substantial lower, though catalyst modification increases the yields. (C) Oxidation of (+)-sclareolide (238) reveals a mixture of three predominant oxidation products. Alteration of the catalysts is shown to substantially alter product ratios, revealing conditions that favor all three substrates in moderate yields.236
One of the key features of these modifications is the increased steric hindrance at the iron active site, which stabilizes the catalysts from possible degradation pathways. This enables the use of lower catalyst loadings (3 mol%), as the increased steric bulk does not lower the catalysts’ reactivities towards C–H oxidation. When (−)-ambroxide (237) was first targeted, it was found to react preferentially at the C–H bond adjacent to the cyclic ether (Figure 44B). This hyperconjugative effect has previously been described (Figure 29C). Changing from 214 with non-associating counterions to the associated triflates of 263 increases the yields of this transformation. Additionally, utilization of catalyst 264 improves the yield to 73%. Similar results are observed when oxidizing (+)-cedryl acetate (266), where a 3° C–H that is distant from the electron withdrawing acetate is favored (Figure 44B). Yields are significantly lower for this substrate, probably the result of increased steric bulk around the 3° C–H bond. However, catalyst 265, containing the bpbp backbone with addition of (+)-pinene-derived scaffold, gives a 57% yield.
The final showcase of enhanced catalyst selectivity with these catalysts is the oxidation of (+)-sclareolide (238). Here, the cyclic ether has already been oxidized to the lactone, providing an electron withdrawing group that deactivates one portion of the molecule. However, there are limited driving forces in the substrate alone to differentiate between the sites shown in yellow, green, and blue in Figure 44C (most notably the strain release that might favor the green site). Indeed, early catalyst screens revealed that products from the oxidation of all three sites are formed in various yields. However, subtle changes in the catalyst have large effects on observed product ratios. Catalyst Δ-264 reacts with a strong preference to form 269 while Λ-265 favors formation of 270 instead. Amazingly, when reactions containing this same catalyst, Λ-265, are conducted at −35 °C, it instead favors product 268. It should be noted that this position on steroid scaffolds had yet to be targeted by these Fe catalysts (as the green and blue positions have been), making it even more significant. Furthermore, C. lunata, has been known to perform the oxidation of 238 to 268, yet takes 14 days and only yields 18%, while catalyst Λ-265 only requires 30 minutes to obtain 36% yield. These reversals of regioselectivity resulting from changes to the catalysts are extraordinary, especially considering the similarities of the steric and electronic profiles of all the C–H bonds in 238.
In the future, further modulation of the structure of the catalyst could be performed in order to achieve entirely different selectivity patterns for various systems, overriding the inherent steric and electronic preferences of substrates, or differentiating between numerous similar C–H bonds (Figure 29).196 Overall, the advances made in predicting the reactivity patterns of catalysts 214 and 246 and controlling their reactivity among numerous C–H bonds is profound, and the future applications of this Fe catalyst system are vast.
3.3.8. Site-Selective C–H Oxidation in the Total Synthesis of Natural Products
In addition to their utility in the site-selective modification of complex molecules, C–H oxidations are also useful in the synthesis of natural products, and a number of elegant examples are present in the literature.175 While this topic is beyond the purview of this Review, one example is presented here that eloquently demonstrates the utility of the selective C–H oxidations of late-stage intermediates.237 As shown in Figure 27, nature often utilizes a two-pronged approach in the synthesis of complex molecules. First, a cyclase phase assembles the complex ring structures and carbon-based framework of the molecule. The establishment of a steroidal backbone is followed by an oxidase phase, which installs the various oxidized functionality into the molecule. Inspired by this biosynthetic strategy, the Baran group utilized this approach in their total syntheses of two oxidized derivatives of taxol: decinnamoyltaxinine E (271) and taxabaccatin III (272, Figure 45).
Figure 45.

The total syntheses of two highly oxidized taxanes. Inspired by nature’s use of distinct cylcase and oxidase phases, the synthetic strategy features a number of selective oxidation reactions.237
Starting with taxadienone (273), which already contains the taxol-derived carbon framework, Baran and coworkers undertook the selective oxidization the molecule. After achieving 274, vanadium-mediated epoxidation yields 275, which is then opened in the presence of NaOH, followed by acylation to yield 276. Selective allylic oxidation of 276 at the C13 position as opposed to C10 can next be achieved when utilizing a Cr-V reagent to yield 277. The C10 position can next be hydroxylated via allylic bromination followed by subsequent hydrolysis and protection to yield 278. After elimination of the C4–OH, reduction of the C13 ketone, Mom protection, and oxidation of the C10 alcohol with 2-iodoxybenzoic acid (IBX), 279 is obtained. The α-hydroxylation of the C9 position can be achieved by subjecting 279 to Et2NLi and Vedejs reagent (MoOPh), followed by oxidation to the C9 ketone with Cu(OAc)2 to yield 280. After modulation of the oxidation states and protection strategies of 281, both 271 and 272 can be accessed efficiently in good yields.
3.4. Site-Selective C–H Bond Functionalization of Natural Products for Halide Insertion
3.4.1. Site-Selective, Aliphatic C–H Halogenation
In addition to the site-selective oxidation of C–H bonds in natural products, the introduction of halogens into complex molecules is also a tremendously valuable transformation. A growing number of novel drugs have been reported that contain halogens due to their intriguing biological and pharmacological properties.238–240 Furthermore, halogens are a versatile functional group, enabling further transformations such as cross-coupling.241 Despite their utility, less work has been reported on site-selective halogenation as compared to C–H oxidation. The presence of a myriad of halogenated natural products makes the discovery of novel and selective techniques to halogenate compounds a worthy endeavor.
In 2010, Groves and coworkers reported a Mn-porphyrin that catalyzes selective chlorination of steroids.242 Treatment of 5α-cholestane (283) with Mn(TMP)Cl (282) revealed a preference for chlorination at the C2 and C3 positions, forming both axial and equatorial products (Figure 46A). Selective chlorination of C2 and C3 is noteworthy given the lack of functionality or deactivating groups in the molecule. Furthermore, it is intriguing that no chlorination at the C25 position is observed. This position is a less sterically encumbered position at which C–H oxidations are commonly observed (see Figures 29D and 30I). Additionally, applying this Mn catalyst to sclareolide (238) revealed that the catalyst has a 7:1 preference for chlorination of the equatorial C2–H over C3, yielding 286 in 42% yield (Figure 46B). The B and C rings of 238 are deactivated due to the γ-lactone, and the C4 geminal dimethyl groups make the approach of the bulky catalyst 282 to the C3 position more difficult than the approach to the C2 position. C2 is also adjacent to two axial methyl groups, resulting in activation by strain release. The proposed mechanism for this transformation is presented in Figure 46C, with initial oxidation of the precatalyst to MnV followed by radical abstraction of the reactive C–H bond. After hypochlorite delivery to the Mn, the incipient substrate radical is quenched via chloride abstraction.
Figure 46.

(A) Chlorination of 5α-cholestane (283) with Mn-porphyrin 282 results in the selective formation of 284 and 285. (B) Chlorination of sclareolide (238) results in preferential formation of the equatorial C2-chlorination product (286). (C) Proposed mechanism for the Mn-porphyrin catalyzed chlorinations. (D) Fluorination of 5α-androstan-17-one (288) results in preferential formation of 289 and 290. (E) Fluorination of sclareolide (238) with Mn-porphyrin 282 results in the selective formation of 291 and 292. (F) Proposed mechanism for the Mn-porphyrin catalyzed fluorination. (G) Structure of Mn(TMP)CI 284. Abrev. TMP: tetramesityporphyrin.242,243
After developing this Mn-porphyrin-catalyzed chlorination protocol, Groves and coworkers next applied this strategy towards fluorination (Figure 47D and 47E),243 which is an area of intense research due to the intriguing pharmacological properties of organofluorine compounds.238–240 They proposed that upon a similar radical C–H abstraction by a MnV complex, fluorine could be delivered to the substrate-centered radical through a Mn–F bond (Figure 47F). Indeed upon reacting two steroids (288 and 238) with AgF and TBAF, in the presence of catalyst 282, similar site-selectivity patterns are observed to the chlorination technique, as fluorinated products are observed at the C2 and C3 positions (289, 290, 291 and 292, Figure 47D and 47E). Intriguingly, in the case of the fluorination of 288, C2-fluorination slightly predominates compared to C3-fluorination, which is opposite for chlorination.
Figure 47.
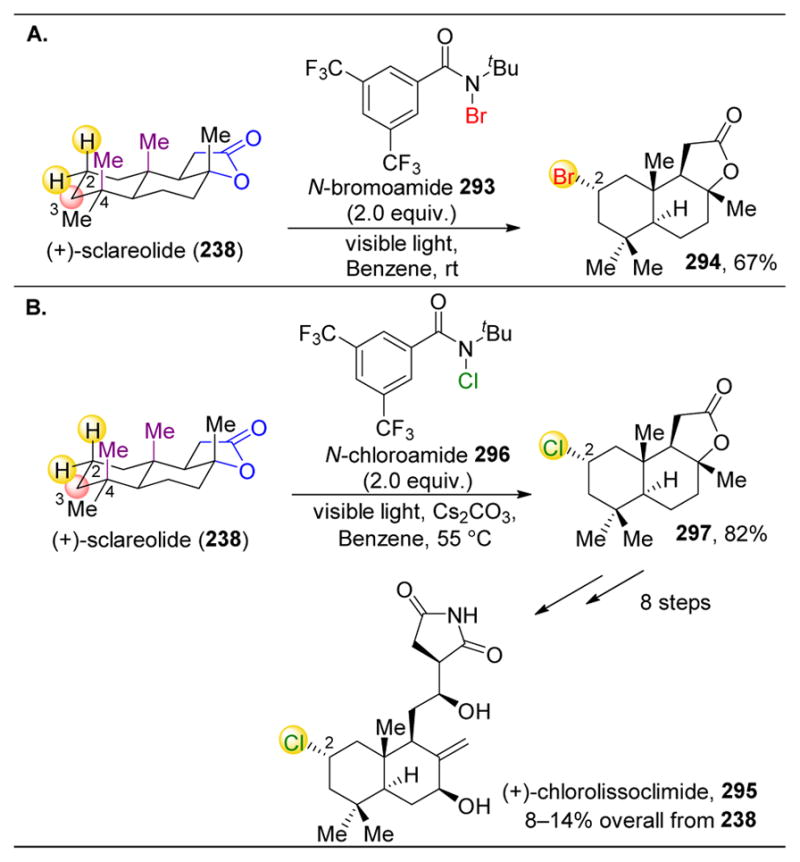
(A) Bromination of 238 by N-chloroamide (293) under visible light irradiation conditions revealed preference for C2-equatorial bromination (294). (B) This C2-equatorial selectivity is also observed for chlorination to yield 297, which is further utilized in the total synthesis of 295.244,246
While much progress has been made in the realm of selective aliphatic C–H halogenation, oftentimes methods can still be inefficient and require an excess of substrate, especially for intermolecular C–H brominations. As such, the Alexanian group undertook the development of a novel, simple, and scalable C–H bromination method.244 Inspired by the Hofmann–Löffler–Freytag process, which performs remote, intramolecular C–H halogenations via heteroatom-centered radicals that facilitate a 1,5-H-atom abstraction.245 Alexanian and coworkers postulated that this type of mechanism that proceeds through an N-centered radical could be rendered intermolecular using the correct type of reagent (Figure 47A). Indeed, this can be accomplished through the use of N-bromoamides 293 in tandem with visible light irradiation. Intriguingly, the choice of reagent could override the inherent substrate selectivity and deliver reagent-controlled bromination results. Upon reacting sclareolide (238) under these conditions, the authors found that selective equatorial bromination of the C2 position could be achieved in 67% yield to give 294. This could be attributed to the relative electron-rich nature of this C–H bond compared to others in 238, in addition to the benefit of strain release in the transition state upon initial H-atom abstraction.
Additionally, the Vanderwal and Alexanian groups targeted sclareolide for chlorination, aiming to devise a synthetic strategy to access (+)-chlorolissoclimide (295), an intriguing anti-cancer agent (Figure 47B).246 The chiral chloride-substituted C2 position in 295 is remote from the functionality of the molecule, making the installation of this group a challenge. However, treatment of 238 with N-chloroamide 296 under visible light irradiation revealed the desired chlorination product 297 in 82% yield. This transformation was possible on gram scale, with only trace amounts of any byproducts observed. This selectivity, as above, could be ascribed to the electron withdrawing nature of the γ-lactone, in addition to the strain release afforded by the axial methyl groups adjacent to C2.
3.4.2. Site-Selective Halogenation of Arenes
The insertion of halogens into natural products is not limited to aliphatic C–H functionalization. Arene C–H bonds, especially those on electron-rich aromatic rings, are susceptible to halogenation. Romo and coworkers pursued the site-selective iodination of a variety of arene-containing natural products (Figure 48).247 Specifically, the authors were most interested in using aryl iodides as handles for adding tags to these natural products for SAR studies. A combination of N-iodosuccinimide (NIS) and a mild Lewis acid, In(OTf)3, was found to give high yields of mono-iodinated products at room temperature. As shown in Figure 48, the site-selectivity of the iodination method is excellent given the number of similar C–H bonds and arenes in these molecules. As a general trend, the more electron-rich rings react first, with positions ortho to electron donating groups (EDGs) being the most activated. In the cases of 298 and 302, it is noteworthy that in the presence of two extremely similar ortho C–H bonds adjacent to the phenol hydroxyl, only one mono-iodinated product is observed. Upon addition of a second EDG into a phenol ring, such as in substrates 300 and 310, iodination occurs ortho to both of these groups. It is intriguing that no para-iodination is ever observed, perhaps due to a directing effect of the nearby heteroatoms.
Figure 48.

Site-selective iodination of a number of natural products. aDMF as used as solvent. bTfOH (0.1 equiv) w/as added. cNo ln(OTf)3 was utilized.247
In competition with C–H iodination, aliphatic C–H functionalization can also occur at C–H bonds adjacent to carbonyls, as demonstrated by substrate 304. To exemplify the utility of site-selective halogenation, the authors carried out several Suzuki-couplings to generate analogues for SAR studies.
Similarly, Snyder and coworkers applied site-selective halogenation to target the oligomers (314b–c, 316b, 318b, 319b, 320b) of resveratrol (310) that exhibit biological activity relevant to preventing heart disease (Figure 49).248 However, isolation of these molecules has been quite difficult, making total synthesis a viable option to access sufficient quantities of these materials for biological testing. Using the model compound 313, in which the core ring structure was intact, the authors examined conditions for the selective bromination of these molecules, with the hopes of incorporating additional dihydrofuran rings on the arenes.
Figure 49.

(A) NBS-mediated selective bromination of HA of 313. (B) NBS-mediated bromination of HA and the alkene of 314. The incipient brominum intermediate undergoes EAS. (C) Substrate and reagent controlled bromination. While most brominating sources functionalize HA, treatment of 317 with BDSB results in bromination of HB. (D) Brominating agents.248 Abrev. NBS: N-bromosuccinimide; NBSac: N-bromosaccharin; TBCO: tetrabromocyclohexadienone; NBA: N-bromoacetamide; TCCA: trichlorocyanuric acid; BDSB: Bromodiethylsulphide bromopentachloroantimonate.
In the case of substrate 313, DFT calculations revealed that HA and HB were the most electronically favored sites for halogenation (Figure 49A). It should be noted that there are two HA and HB atoms in this molecule due to the compound’s symmetry. All brominating reagents, including N-bromosuccinimide (NBS), revealed that bromination occurred at the desired HA positions and dibromide product 314a could be transformed to both natural 314b and unnatural 314c. However, controlling for bromination of a single arene ring of 315 in order to access 316b was challenging. An alternative strategy would be the desymmetrization of the two HA bromides, instead of selective bromination. When unsymmetrical 315 was submitted to NBS-mediated bromination, an additional bromide was added onto the alkene to yield 316a (Figure 49B). Alkene bromination is presumed to occur through a bridged bromonium intermediate that reacts with the adjacent arene ring in an electrophilic aromatic substitution (EAS) reaction. While the initial reactivity of both HA atoms is difficult to control, upon isolation of 316a, the two arene rings are in substantially distinct chemical environments. To access 316c, both aryl bromides had to be removed, leaving the aliphatic Br. Upon treatment of this with NBS, one bromide is selectively incorporated onto the desired and more sterically accessible arene ring of the molecule (316b).
The authors next turned their attention to bridged substrate 317, which contains four, non-equivalent, electron-rich C–H bonds (Figure 49C). It was experimentally found that bromination of HA was facile, as most brominating reagents preferentially favored this site, perhaps due to the steric accessibility of that side of the compound. Upon addition of more equivalents of NBS, HB can also be brominated second to yield 319b. However, no methods yielded preferential bromination of HB over HA until a recently developed brominating source,249 BDSB, was screened and yielded 320C in 78% yield. This observed selectivity is a profound example of reagent-controlled differentiation of extremely similar aryl C–H bonds for bromination.
3.4.3 Site-Selective Halogenation of Vancomycin and Teicoplanin
As previously discussed, bacterial resistance is a serious problem for both pharmaceutical development and society at large. This minimized effectiveness of drugs is particularly alarming for this “antibiotic of last resort,” such as vancomycin (35) and teicoplanin (49), against which certain strands of bacteria have evolved to resist. As shown in Section 2.2.3, in order to synthesize novel derivatives of vancomycin and teicoplanin, 71 and 99 have been subjected to site-selective acylation, thiocarbonylation, and phosphorylation using a variety of N-heterocycle-containing peptide-based catalysts.112,121,124 As described above, some of these mimic the native DAla-DAla binding target of these antibiotics (Figure 50A). This interaction orients the catalytic residue proximal to the desired reactive positions. Inspired by these observations, site-selective bromination of 35 and 49 was also explored by us. The notion of achieving site-selective bromination catalysis began with studies of atropisomer-selective arene halogenation.250–255 As with our original studies of group transfer reactions, lessons learned in explorations of enantioselectivity provided a helpful prelude to investigations in complex molecule settings.
Figure 50.

(A) Model of vancomycin (35) binding to a DAla-DAla containing peptide. The Asn side chain may then deliver a bromine atom to the 7-ring. (B) The bromination of vancomycin (35) with no catalyst or in the presence of a achiral promoter offer no selectivity for either 322 or 323. Hit catalyst 321 preferentially favors bromination to 322 under various conditions. (C) Guanidine is able to reverse selectivity for 322 and favor bromination to 323. Catalyst 325 can deliver tribromide 326 with good selectivity.256
Alternatively to the catalytic motifs present in our investigation of bromination on small molecule substrates, it was envisioned that an asparagine residue containing a protected N,N-dimethylacetamide side chain (Asn(Me)2), could also be a catalyst for N-bromophthalimide (NBP)-mediated bromination.256 Indeed, when vancomycin (35) was first submitted for bromination without a catalyst, low reactivity and selectivity was observed (Figure 50B, entry 1). The simple additive N,N-dimethylacetamide yielded a mixture of bromination products 322 and 323 in addition to the over-brominated product 324 (Figure 50B, entry 2). Ring 7 is the most electron-rich arene due to the presence of two phenolic hydroxyl groups and hence the most likely to undergo bromination. Utilization of a peptide containing an Asn(Me)2 residue linked to the DAla-DAla binding agent (321) resulted in significant selectivity however, affording a 14.6:1.0:2.6 distribution among 322:323:324 (Figure 50B, entries 3–5). When performed on a preparative scale, product 322 was isolated in 41% yield (Figure 50C). This useful level of reactivity and selectivity is likely facilitated by the five key vancomycin (35) hydrogen bonds to the DAla-DAla sequence, which may position the reactive bromonium-delivering carbonyl of the asparagine side chain in proximity to the 7dH position, which then undergoes amide-catalyzed bromination (Figure 50A). It was also found that utilization of achiral guanidine as an additive could yield a mixture favoring 323 and over-brominated 324 as a complementary method to 321-mediated reactions. In addition, 55% yield of 324 could be obtained using peptide 321 with an excess of brominating agent. Finally, an additional peptide sequence, Boc-Leu-DGln(Me2)-DAla-OH (325), was found to selectively form tribromide 326. The divergent regiochemical outcomes for these reactions illustrate the potential of combined approaches for the diversification of a complex scaffold like vancomycin.
Upon discovering the viability of peptide catalysts in selectively brominating vancomycin, we next turned our attention to teicoplanin A2-2 (49), which contains seven aromatic rings compared to vancomycin’s (35) five.257 Similar to vancomycin (35), non-catalyzed reactions brominate ring 7 selectively, producing 332 with high selectivity (Figure 51A–i). On preparative scale with no catalyst, reacting 50.0 mg of 49 results in the isolation of 10.0 mg of 332 (19%) and 6.0 mg of recovered starting material (49, 12%). Product 7dBr is not observed here, perhaps due to its proximity to the sterically demanding sugar appended to the 7-ring phenol. When peptide 321 was used as a catalyst of the reaction, the resultant HPLC traces revealed broad peaks, presumably due to the effectively irreversible association of the peptide bound to the teicoplanin derivatives. After consulting literature concerning binding studies of DXaa-DXaa peptides with vancomycin-like glycopeptides, a DAla was exchanged for DLeu to reduce the catalyst’s affinity for the substrate.123,258 Indeed, this strategy was effective, as peptide 327 resulted in selective bromination to yield 330 (Figure 51A-ii), and peptide-free products could now be observed and isolated by HPLC. This result showed that the peptide can not only enforce the location of bromination on the most reactive arene ring alone (Figure 50), but also alter the inherent substrate selectivity of the reaction to occur at a less reactive site on the molecule. It is also noteworthy that the catalytically active Asn(Me2) residue still assumedly binds as shown in Figure 50A, placing it in close proximity to ring 7. Furthermore, changing the location of the Asn(Me2), as in peptide 328 with Asn(Me2) as the central residue, did not affect the selectivity patterns (Figure 51A-iii). Hence, upon binding to teicoplanin (49), catalysts 327 and 328 may induce a conformational change in 49 that substantially accelerates the inherent background reactivity of ring 3 in comparison to both the background and catalyzed reactivities at ring 7. Performing the bromination of 160 mg 49 with catalyst 328 on a preparative scale resulted in isolation of 47.0 mg 330 (28%) yield, 35.0 mg of recovered starting material (49, 22%), and 16 mg of dibrominated 331 (9%). This observation also represents, in a sense, mimicry of nature’s enzymes, which can effect reactivity at particular sites on a molecule, overcoming substrates’ innate reactivities. Changing a DLeu in peptide 328 to Leu (peptide 330), however, reverted the reactivity preference to ring 7 (Figure 51A-iv). The importance of stereochemical configuration at that position is probably the result of poorer binding of the catalyst to teicoplanin, as LXaa-DXaa is a mismatched system. It remains unclear what the exact mechanism of bromination is in these reactions.
Figure 51.

(A) Site-selective bromination of teicoplanin (49). The inherent reactivity reveals a preference for 333, while use of either catalyst 328 or 329 favors bromination of ring 3. (B) Derivatization of the aryl bromides and aryl chlorides of the teicoplanin derivatives via Suzuki cross-couplings. The 2CCl reacts faster than the 7fBr and 3CBr. (C) Screening of teicoplanin derivatives against bacterial strains. Compounds 334, 337, 339, and 340 are particularly effective against vancomycin resistant VanA.257
Bromination was chosen as a transformation to examine on vancomycin (35) and teicoplanin (49), as aryl bromide products are excellent substrates in cross-coupling reactions. Consequently, after one selective bromination, a myriad of derivatives of 35 and 49 can be synthesized. However, these palladium-catalyzed cross-coupling reactions can still be challenging given the presence of an aryl chloride on ring 2 that could react competitively. Indeed, when 49, 330, and 332 were subjected to aqueous Suzuki–Miyaura cross-coupling conditions using 50 mol% Pd(OAc)2 and 100 mol% of water soluble-SPhos, the 2CCl position was found to react fastest and products 334–339 were isolated (Figure 51B). Finally, 340, with a mono-coupled furanyl group at the 3b position, was the only product found to be derived from the faster reaction of the 3cBr as opposed to the 2CCl.
All these novel derivatives were tested against five bacterial strains, including methicillin resistant S. aureaus (MRSA) and vancomycin-resistant Enterococcus (VRE; VanA is resistant to both vancomycin and teicoplanin; VanB is resistant to vancomycin, while teicoplanin is effective against this strain). Intriguingly, products 334, 337, 339, and 340 all revealed increased activity towards all strains except VanA (Figure 51C). Furthermore, compounds 335 and 336 began showing heightened activity towards VanA, revealing the importance of a biphenyl or alkenyl substituent on the 2C position. These findings highlight the utility of site-selective modifications. The ability to modify complex molecules at specific sites and expediently create numerous derivatives for testing is an effective method for the discovery of novel therapeutics. This approach prevents the need for long and expensive total syntheses to piece together large target molecules.
Finally, given the exceptional activities of 2C coupled products against vancomycin-resistant bacterial strains, our group next looked at derivatizing the 6C aryl chloride of 71.259 Given the reactivity of the 2C position, in order to access cross-coupling at ring 6C, removal of 2cCl would be required. Using Pd/C and H2, the 2cCl could be removed selectively on vancomycin (35) to yield 341 selectively in 21% yield (Figure 52). Further Suzuki–Miyaura cross-coupling on 341 yielded seven derivatives in 11–52 % yield (343–349). Biological testing of these derivatives against various bacterial strains revealed a decrease in activity for all cases, potentially due to steric hindrance of the DAla-DAla binding pocket by placing larger functional groups at the 2C position.
Figure 52.

(A) Selective removal of the 3CCl of 35. (B) Selective cross-coupleavariety of boronic acids on the 6C position.259
The combination of site-selective arene C–H halogenation with site-selective cross-coupling represents a powerful strategy for the diversification of arene-containing natural products. It is clear that advances in many sub-fields of catalysis can contribute to the advancement of this challenging activity in the heart of complex molecule modification.
3.5. Site-Selective Epoxidation of Farnesol
The ability to oxidize complex molecules in a site-selective fashion is not limited to the reactions of C–H bonds. Natural products can contain a number of other oxidizable functional groups susceptible to oxidation, such as alkenes. Just as natural enzymes mediate site- and stereoselective C–H oxidations, enzymes have also evolved to catalyze site-selective epoxidations of alkenes.260,261 Indeed, the biosynthetic pathway of steroids proceeds from a variety of terpenes, such as squalene (350), and includes the quintessential selective epoxidation of the terminal olefin to yield 351 (Figure 53).260 This selective epoxidation is truly remarkable given that all six alkenes of squalene are locally similar, but six positionally or stereochemically unique monoepoxides can be envisioned. After the selective epoxidation, a cascade cyclization forms the core rings of the steroids to yield after further steps, a variety of steroids, such as cholesterol (352).
Figure 53.

Biosynthesis of cholesterol from squalene, featuring a site-selective epoxidation.260,261
Inspired by this exquisite selectivity demonstrated by squalene epoxidase, our group set out to emulate this reactivity using small molecule catalysts.262,263 Specifically, we wondered whether minimal peptide sequences could mimic the impressive reactivity of much larger enzymes (Figure 54A). To create a peptide with activity for epoxidation, we considered aspartic acid as a potential catalytic residue. Asp-containing peptides had previously been discovered to be effective oxidation catalysts when using hydrogen peroxide as a terminal oxidant.264–269 The proposed catalytic cycle of Asp-catalyzed epoxidations is presented in Figure 54B.266 Initial oxidation of the side-chain carboxylic acid with N, N′-diisopropylcarbodiimide (DIC) (mediated by hydroxybenzotriazole (HOBt) and 4-dimethylaminopyridine (DMAP)) is followed by addition of hydrogen peroxide to the activated ester. The resultant activated peroxy acid species hence serves as the electrophilic oxidant for epoxidation. Indeed, we set out to find short peptide chains to append to the Asp that could control the selectivity of this catalytically active residue. To explore this question, farnesol (353) was chosen as a simplified substrate because it contains only three potential alkenes and because the alcohol could provide a handle for discrimination of the olefins. Upon treatment of 353 with mCPBA, a mixture of the three monoepoxide products is formed. Combinatorial screening of peptides containing Asp at the N-terminus revealed peptide 357 that catalyzed the same reaction with good levels of site-selectivity and enantioselectivity for the 2,3-olefin (354, >100:1:1 site-selectivity, 86% ee). Subsequent studies on this peptide revealed that it is a highly selective allylic alcohol-directed epoxidation catalyst.266 Furthermore, an additional peptide (358) was also discovered from the library that favored oxidation at the internal 6,7-olefin (355). It is believed that upon hydrogen bonding to the terminal alcohol, the same catalytically active aspartyl peracid is positioned close to the internal olefin. Possible mechanistic proposals for the actions of peptides 357 and 358 are presented in Figure 54C. Efforts to uncover a 10,11-olefin (356)-selective catalyst were unsuccessful. However, it remains encouraging that by simply changing short peptide sequences attached to a common reactive species, the selectivity for epoxidation can be significantly perturbed.
Figure 54.

(A) Site-selective epoxidation of famesol (353). Peptides that yielded 2,3-epoxide (354) and 6,7-epoxide (355) were discovered. (B) Mechanism of Asp-catalyzed epoxidation. (C) Proposed mechanistic models for 2,3- and 6,7-peptide catalyzed epoxidation.262,263,266
3.6. Site-Selective Insertion of N-Atoms into Natural Products
In addition to the site-selective incorporation of oxygen atoms into natural products, the insertion of nitrogen atoms into natural products and pharmaceuticals, either through amination, aziridination, or azidation, is an area of great importance given the valuable biological and pharmacological properties of amines.
Recently, Baran and coworkers reported the selective amination of sclareolide (238) utilizing Rh(II) catalyst 359 and 361 as the nitrogen source, a method developed in the Du Bois group (Figure 55).202,270 The proposed mechanism proceeds via rhodium-catalyzed insertion of a nitrene into a C–H bond. In this case, the equatorial C2–H of 238 is aminated (362). As shown previously with sclareolide (see Figures 39C, 44C, 46, and 47), the C2 position is beneficially placed distally to the electron-withdrawing lactone of the C-ring. The presence of two axial methyl groups adjacent to the C2 positions provide strain release in the transition state during amination at this position, and 362 is formed in 98% yield.
Figure 55.
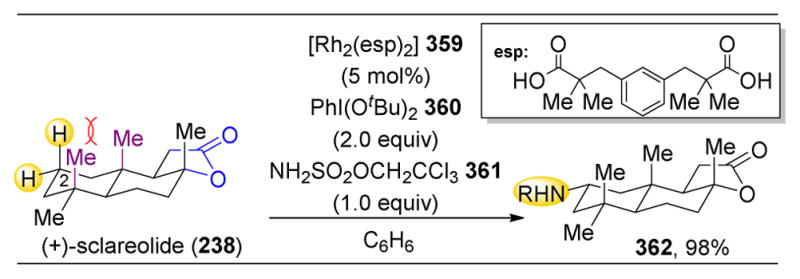
Site-selective amination of sclareolide (238) using dirhodium catalyst 359. The C2 position (362) is favored due to strain release in the transition state of the amination.202
Additionally, Du Bois and co-workers reported an intriguing and selective C–H amination of 3° C–H bonds in a variety of natural products using Rh2(esp)2 (359, Figure 56).271 The choice of amine source was critical, as decomposition of the amine is a competing pathway. Indeed, the authors specifically found that amine sources containing alkyl groups were incompatible with the reaction conditions, as these alkyl C–H bonds on the amine were the first to be oxidized. Using substrate 363 with DfsNH2, the authors were able to obtain 364 in 57% yield. Presumably, the other tertiary C–H bonds in 363 are deactivated due to the presence of three EWGs on the molecule.
Figure 56.
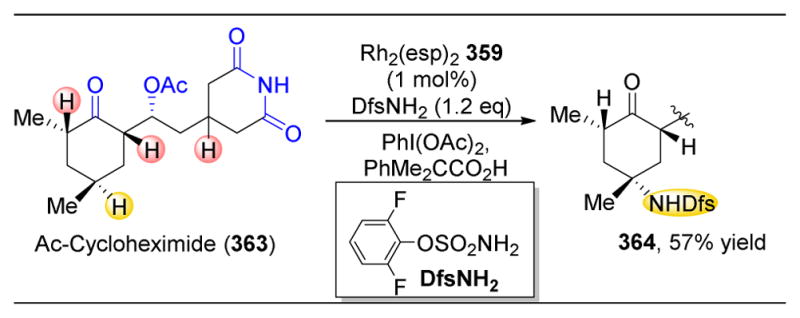
3°C–H selective amination of cycloheximide derivative 363.271
However, in analogy to oxidation chemistry, in which alkene epoxidation and C–H functionalization can be competing pathways, metal nitrenes can engage in both olefin aziridination and C–H amination. Romo and coworkers sought to address this problem to enable selective incorporation of amines into natural products, specifically to allow for the selective incorporation of fluorescent tags into the molecules for SAR studies (Figure 57A).272 Hence, sulfonamide 366 that contains an alkyne handle was used as a substrate.
Figure 57.

(A) Competition between aziridination and C–H amination of aikene-containing substrates. (B) Common conditions for amination reactions. (C) Representative aminations of natural products. Benzylic and allylic amination, in addition to aziridination are the common reaction pathways. (D) Rearrangement pathway for aziridination product 126.272
Starting with steroid 367, which does not contain any alkenes, dirhodium catalyst 359 resulted in C–H amination at the C6 position (368), presumably because of its benzylic character (Figure 57C). This C–H functionalization strategy also enables allylic 2° C–H amination, as shown in the reactions of 369 and 371. However, in the event that there are no secondary allylic or benzylic positions present in the molecule, aziridination can occur, as shown with substrate 373. Here, the exocyclic alkene is selectively aziridinated to form 374. The alkene conjugated with the ester is presumably more electron-rich than the alkene of the enone.
In another case, treatment of gibberellic acid methyl ester 126 with 365 using PhMe as the solvent, yields 55% of an abnormal rearrangement product (376). This product is presumed to result from ring cleavage and concomitant 1,2-pinacol rearrangement of 375 to 376 after initial aziridination. However, upon switching to DCM as the solvent, 40% of aziridinated 375 is observed, in addition to 23% of 376. It is intriguing that 126 does not undergo any C15–H amination at its 2° allylic C–H bond. The selectivity for the exocyclic alkene is perhaps driven by sterics, as both allylic C–H bonds are fairly hindered.
Additionally, Groves and coworkers, building off their previous Mn-catalyzed C–H chlorinations and fluorinations (see Figure 46), sought to expand their protocol to include azidation (Figure 58).273 Azides are a versatile functional group that can be transformed into a myriad of other functional groups through reduction, Huisgen cycloadditions, Staudinger ligation, and more. Postulating the azide would behave similar to the fluorine rebound mechanism postulated previously, the authors undertook the challenge of selective C–N bond formation. Indeed, two complexes (282 and 378) were found to both be active C–H azidating agents, while often giving different results. Applying this method towards natural products, the Groves group found that similar to previous results, these Mn catalysts target the C2 position of sclareolide (238) to yield 379. However, Mn(salen)Cl (378) was also able to afford intriguing selectivities for the azide insertion into estrone derivative 380 and artemisinin derivative 382.
Figure 58.
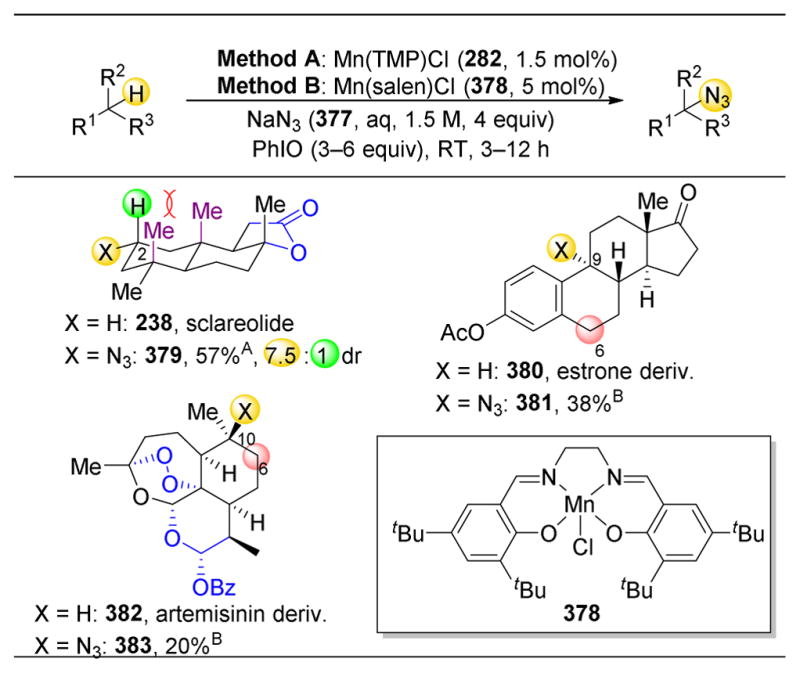
Mn-porphyrin-catalyzed azidation of aliphatic C–H bonds. Selective reactions on sclareolide, estrone, and artemisinin derivatives have been achieved.273
In addition, the Hartwig group pursued the development of a mild and selective azidation strategy, relying on inexpensive Fe(OAc)2 in tandem with hypervalent iodine species 384.274,275 The authors discovered that PyBox ligand 385 was capable of facilitating selective C–H azidation in high yields on numerous model substrates (Figure 59). On small molecule substrates, the authors observed many similar trends to those observed for iron-catalyzed C–H oxidations as shown above. First, electronically activated C–H bonds, such as tertiary C–H bonds, or those remote from EWGs, were first to react. Second, benzylic positions were found to be highly active.
Figure 59.

Selective azidation of natural products using Fe(OAc)2 and a chiral ligand source.274,275
Applying these conditions to more complex settings, the Hartwig group observed similar selectivity patterns (Figure 59). The proclivity of this Fe-catalyzed system for derivatization of benzylic positions was observed in podocarpic acid (386) and estrone derivatives (388 and 391). No reactions at electron-rich tertiary C–H bonds were observed, and differentiation of the various benzylic positions was accomplished due to EDGs on the arene. Benzylic positions para to protected alcohol substituents were favored for C–H azidation. However, intriguingly, incorporation of a less electron-rich ether, present in substrate 391, results in higher levels of both site- and diastereoselectivity. The authors propose this to be accomplished via a change in mechanism, where in the cases of 386 and 388, a stabilized cationic intermediate is achieved, whereas for 391, a benzylic radical is achieved. However, detailed mechanistic studies have yet to be carried out.
Continuing to probe the selectivity for benzylic positions over other electron-rich C–H bonds, such as tertiary C–H bonds, Hartwig and coworkers carried out the azidations of totarol derivative 393 and δ-tocopherol derivative 395. In the case of 395, the benzylic position is even more activated than the secondary C–H bond adjacent to an ether.
Upon targeting natural products devoid of benzylic positions, intriguing observations on the interplay between sterics and electronics for this catalytic system were observed. Similar to the trends discussed previously, this iron catalyst favors the functionalization at sterically accessible and electron-rich C–H bonds. For cycloheximide substrate 363, given the number of EWGs on the eastern section of the molecule as shown, azidation is favored at the C–H bond shown in gold (Figure 59). For a number of steroid derivatives, C–H bonds at trans-decalin ring junctions were unreactive, presumably due to the lesser steric accessibility. In the reaction of dihydrodipterocarpol 398, azidation reactions occur at C25–H. Furthermore, betulin derivative 400 undergoes facile azidation of the tertiary C–H bond on the E-ring. While trans-decalins are unreactive, cis-decalin-containing digoxigenin diacetate 402 favored reactivity at the ring junction, inverting to the more stable trans-decalin over the course of the reaction.
The sensitivity of this azidation method to sterics is further observed in the reaction of artemisinic acid derivative (404) under standard conditions. Despite the tertiary C6–H bond being most remote from any EWGs, it does not undergo azidation due to being on the concave face of the molecule. The three tertiary C–H bonds on the convex face of 404 are reactive, despite being fairly proximal to the lactone. Similarly, dihydrocaryophyllene oxide (408) contains three similar tertiary C–H bonds, yet upon azidation under the previously mentioned conditions, only one product is observed (409). The nonreactive C–H bonds could be sterically deactivated by the geminal dimethyl functionality.
Additionally, a complementary strategy for the modification of gibberellic acid derivative 410 arose from this method. Whereas White and coworkers were able to show high selectivity for the C–H oxidation of the C15 position of gibberellic acid derivative 217 via direction from the nearby carboxylate (Figure 36D), the Romo group was able to achieve aziridination of derivative 375 at a C16 alkene, which can later rearrange to yield 377 (Figure 57D). Additionally, the Hartwig group applied their azidation system to gibberellic acid derivative 410, revealing azidation is favored at the C16 position. It should be noted in comparing substrates 217 and 410, the C17 alcohol group is protected in the former and not in the latter. Hence, the acetylated alcohol of 217 is deactivating, while in the case of 410, α-ethereal activation can result. Regardless, depending on the catalytic system, a variety of derivatives of gibberellic acid can be accessed with a minimal protecting group strategy.
One additional consideration that the authors investigated was the relative reactivity of alkenes compared to either allylic or non-allylic C–H bonds. While 402 contains an alkene, it is electronically deactivated and unlikely to undergo azidation. However, madecassic acid derivative 412 contains an electron-rich trisubstituted alkene and indeed, azidation is observed to occur at the allylic, secondary C–H bond to yield 413a (the allylic, tertiary C–H bond is presumably deactivated by its proximity to the ester) (Figure 60).275 As a note, 16% of diene side product 413b is observed, which the authors suggest arises from oxidation of the incipient allylic secondary radical to a carbocation, followed by elimination. However, upon oxidizing gibberellic acid derivative 414a, which contains a 1,1-disubstituted olefin and a 1,2-disubstituted alkene, diazidiation of the terminal olefin is observed. This diazide is presumed to form via initial addition of an azidyl radical to the olefin.
Figure 60.
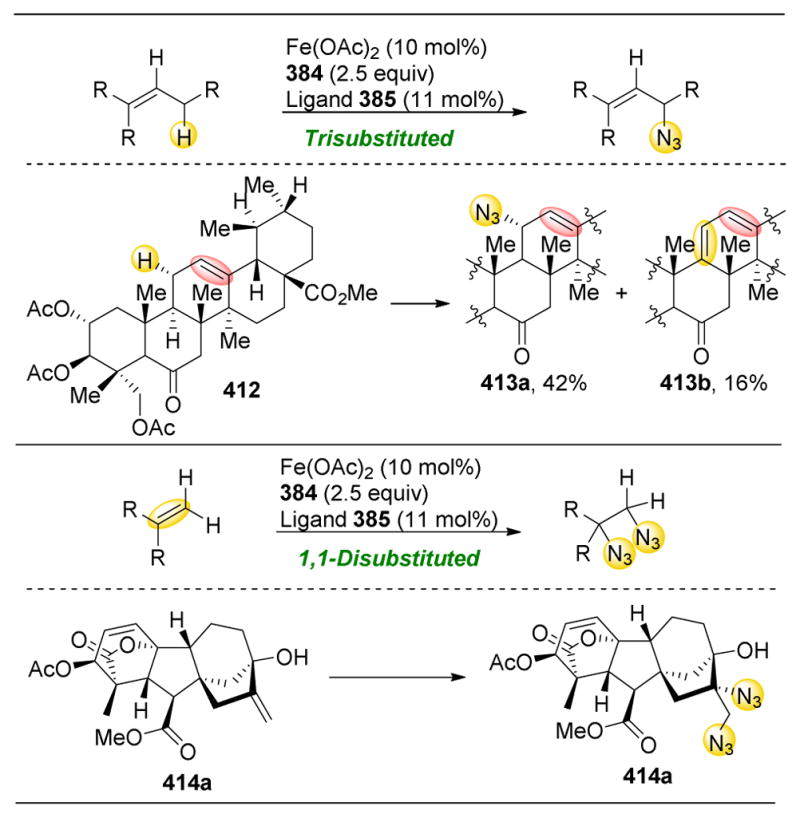
Allylicazidation versus diazidation.275
With this success in the di-azidations of olefins, the authors expanded the catalytic method to include nonequivalent difunctionalizations, such as trifluoromethylazidation reactions (Figure 61).275 In this case, trimethylsilyl azide (TMSN3) and Togni’s reagent (415) were used and indeed, selective formation of products was observed. In all cases, the products contained trifluoromethyl groups at the less substituted position and azides at the most substituted site. This regioselectivity indicates that a CF3 radical adds into the olefin initially, followed by trapping with the azide.
Figure 61.
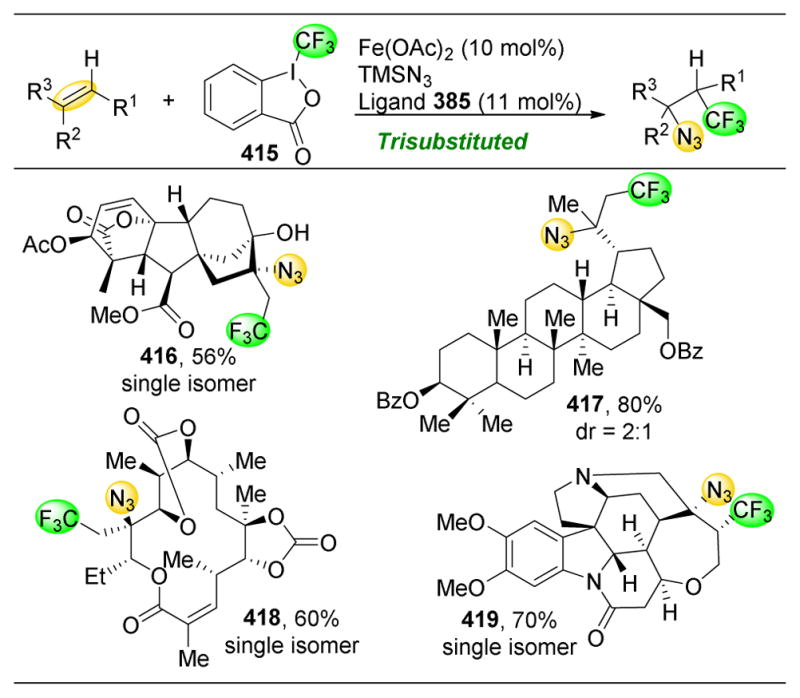
Selective trifluoromethylation-azidation reactions.275
After the development of an effective method for site-selective C–H azidation and regioselective trifluoromethylazidation reactions, Hartwig and coworkers utilized their system, in tandem with other reported transformations, to quickly access numerous derivatives of the cholesterol scaffold (420, Figure 62). 275 Nucleophilic displacement of the C3–OH with MeSO2Cl and NaN3 revealed product 421. A two-step hydroboration/azidation of 420 could access the anti-Markovnikov product 422. The Hartwig group’s trifluoromethylazidation method was amenable to cholesterol, as 423 was accessed in 60% yield. With the inclusion of TMSN3 and Togni’s reagent (415), the other electron-rich C–H bonds remained untouched. However, upon hydrogenation of the alkene, the incipient steroid could be functionalized at the tertiary C25–H with Fe(OAc)2 and azide-containing hypervalent iodine 384, the most electron-rich and sterically unencumbered, to yield 424. Finally, allylic azidation product 425 is produced under the same conditions without initial hydrogenation of the alkene of 420.
Figure 62.

Divergent selectivity in the derivatization of the scaffold of cholesterol derivative 420.275
Notably, the authors presented a number of molecules that were difficult to functionalize under their reaction conditions. Monensin derivative 426 gave a complex mixture of numerous products, while artemisinin (215) and oxymatrine (427) were unreactive (Figure 63).
Figure 63.

Examples of ineffective substrates for Fe-catalyzed azidation
Over the past two decades, there has been a tremendous amount of progress in the site-selective oxidation of complex molecules. The advances in C–H functionalization are particularly exciting, as further efforts in the activation of these normally inert bonds could lead to a major reversal in synthetic thinking and strategy. So far, most of the reactions in this area have focused on the inherent reactivity of substrates; however, there has recently been an increasing number of examples of catalyst-controlled C–H oxidations. One can anticipate future advances in this field of site-selective reactions catalyzed by small molecule catalysts, eventually mimicking the extraordinary rate acceleration and selectivity that enzymes afford.
4. Miscellaneous Site-Selective Transformations
4.1. Site-Selective C–H Xanthylation
Building off their success with C–H brominations and chlorinations utilizing N-haloamides, the Alexanian group additionally undertook a similar C–H xanthylation strategy (Figure 64).276 The selective xanthylation of complex molecules would be a facile method to expediently derivatize a molecule of interest given the ease of transforming xanthates into a variety of other functional groups, ranging from carbon-heteroatom to carbon-carbon bond formations.277 Indeed, upon screening a number of aliphatic C–H bond rich natural products with xanthylating agent, N-xanthylamide 428, selective reactivity was achieved. The reactions of sclareolide (238) and ambroxide (237) gave similar selectivities to those previous observed for these substrates in various other C–H functionalizations, with reaction at the C2 position and α-ethereal C12 position respectively. Xanthlyation of steroids 431 and 434 occurred at the C6 position, between A and D ring electron-withdrawing groups. Furthermore, despite the lack of functionality on both longifolene (4) and 5α-cholestane (283), selective xanthylation was observed. For longifolene, this appears to be driven by sterics, as reactivity occurs at the most accessible ring, away from the quaternary carbon centers of the core structure. For 283, impressively only C2 and C3 xanthylation is observed with no C25–S bond formation occurring, as is sometimes seen in the case of C–H oxidation.
Figure 64.

Method for the site-selective C–H xanthylation of aliphatic C–H bonds, including on a number of natural products. Xanthates can be further derivatized into a variety of different functional groups.276
4.2. Site-Selective C–H Trifluoromethylthiolation
An emerging approach to activating C–H bonds for natural product modification employs photoredox catalysis. For example, the selective trifluoromethylthiolation of various natural products is shown in Figure 66.278 Utilizing an IrIII photocatalyst (439), benzoate co-catalyst 440, trifluoromethylthiolating agent 441, and visible light, the authors observed selective C–H functionalizations on a variety of small molecule substrates (Figure 65A). Normally, these exhibited preferential reactivity at tertiary C–H bonds that were the most sterically accessible and electron-rich. Moving on to more complex substrates, the authors next pursued the selective derivatization of ambroxide (237, Figure 65C). As previously observed, the C12–H bond is most electron-rich due to the α-ethereal effect. However, a small amount of C5 functionalization product was also observed. Furthermore, the authors also accomplished selective C–H trifluoromethylthiolations on two steroid derivatives (443 and 445), targeting the tertiary C–H bonds in each. The mechanism of this transformation is believed to proceed via initial photoexcitation of the Ir-photoredox catalyst to provide the excited state oxidant, which can then oxidize the benzoate co-catalyst through a single electron transfer (SET, Figure 65D). This radical can perform a hydrogen atom abstraction (HAT) on the substrate, which can then be trapped with trifluoromethylating agent 441. This area is poised for rapid growth.
Figure 66.

Site-selective deoxyfluorination of natural products. Using 447 asasource of nucleophilic fluorine, the least sterically hindered alcohols are displaced onarange of complex molecules.281
Figure 65.

(A) Trifluoromethylthiolation of C–H bonds c an be accomplished via visible-light promoted and Ir-catalyzed photoredox catalysis.(B) Structure of Ir-photoredox catalyst.(C) Natural products targeted by this method. The most electron rich and sterically accessible 3° C–H bonds are functionalized here. (D) Proposed mechanism. After excitation of the Ir-photocatalyst can be coupled to oxidation of the benzoate co-catalyst, which facilitates the C–H abstraction on the substrate. This c an be trapped with the trifluoromethylthiolating agent. Abrev. HAT=hydrogen atom transfer; SET=single electron transfer.278
4.3. Site-Selective Deoxyfluorination
The fluorination of natural products and pharmaceuticals is an area of intense interest, as fluorinated drugs often exhibit heightened metabolic stability in addition to greater penetration of the blood–brain barrier.238–240,279–280 However, strategies to incorporate fluorine atoms into biologically relevant molecules in an economical manner at a late-stage in the synthesis are scarce. This challenge is especially true in the realm of aliphatic fluorination.
Ritter and coworkers hence undertook the development of a late-stage, site-selective fluorination of complex natural products (Figure 66).281 Here, they devised a strategy of deoxyfluorination, in which they utilized a source of nucleophilic fluorine to selectively displace alcohols on substrates. While the deoxyfluorination of simple alcohols is known, these methods often lack functional group tolerance and undesired side products are often observed due to elimination.282,283 Furthermore, common reagents for this transformation can be unstable and even explosive. However, the Ritter group had previously developed a facile and mild method for the deoxyfluorination of phenols using PhenoFluor (419),284 and indeed, application of this reagent to aliphatic hydroxyl displacement was a success. High yields were observed for SN2-like displacement of simple alcohols. Two additives are beneficial to this transformation: Hünig’s base, which accelerates the reaction rate, and KF, which reduces the elimination side-products.
The authors next evaluated this method to functionalize more complex natural products. Starting with everolimus (420), deoxyfluorination was observed at the least sterically hindered primary alcohol to yield 421. Next, when applying this method towards ivermectin B1a (422), fluorine incorporation was observed at the allylic position to yield 423, as opposed to occurring at either the free secondary position on the cyclohexane derivative or the tertiary position in the macrocycle. It is intriguing here that only SN2 products are isolated, as no SN2′ pathways were observed. Finally, the authors tested their deoxyfluorination protocol on oligomycin A (424), which contains four secondary alcohols and one tertiary alcohol. However, the four hydroxyl groups on the top of the molecule (C24–C32) are both adjacent to β-substitution and are also engaged in a stabilizing hydrogen bonding network. These two factors are believed to explain the selectivity of PhenoFluor (419) for the C2–OH, as 425 is isolated in 71% yield.
Hence, the selectivity of this method appears to follow general trends in the reactivity for nucleophilic substitution reactions: less sterically hindered positions are prone to reactions and leaving groups on allylic positions are activated for attack.
4.4. Site-Selective C–H Functionalization with Rhodium Carbenoids
Presently, it appears that the majority of small molecule-catalyzed alterations of natural products have focused on selective formation of carbon-heteroatom bonds. However, some attention has also been directed to transformations of C–H to C–C bonds. Given the growing utility of dirhodium complexes for C–H functionalization reactions, especially using donor/acceptor diazo complexes,170–172 Davies and coworkers accomplished the site selective C–H functionalization of complex molecules by Rh-catalyzed insertion of carbenes into C–H bonds (Figure 67).285 Starting with brucine (454), the authors screened a number of dirhodium catalysts and they found that subtle variations of the electronic and steric properties of these complexes had tremendous effects on the selectivity of the C–H functionalization (Figure 67A). These Rh(II) catalysts all functionalized the α-positions of the tertiary tricyclic amine, yet depending on the catalyst, any of the three α-positions could be targeted. Achiral Rh2(TPA)4 (456) and chiral Rh2(S-BTPCP)4 (457) effected C–H activation at distinct C–H bonds to yield products 459 and 460, respectively. Additionally, when using Rh2(Oct)4, (458) an intriguing insertion product was observed (461). This abnormal product is believed to occur via transfer of the diazo compound to the tertiary nitrogen atom itself, followed by a selective Stevens rearrangement to form 461. While the nature of the catalysts and the sources of their selectivity differences are not known, it is an excellent display of the importance that ligand sets can play in the control of transition metal-catalyzed C–H activation of complex molecules. The scope of diazo compounds was expanded from 455, however all remained in the traditional donor-acceptor class.
Figure 67.

(A) Three different dirhodium catalysts reveal three different selective functionalizations of 454. (B) Application of these rhodium carbenoid reactions towards other natural products results in similar observed selectivitles.285
Additionally, the authors probed the efficacy of this method towards other natural products, including securinine (462) and apovincamine (464, Figure 67B). For 462, despite the three adjacent sites to the tertiary nitrogen, only one product is observed. While the diastereomeric ratio (dr) is more modest than in the previous cases, it was found that the choice of rhodium complex could tune the dr. In the case of 464, it was found that instead of insertion adjacent to the nitrogen atom, benzylic insertion product 465 was observed instead.
The generality of this process was next applied to a number of other natural products and a pattern of activation of α-positions adjacent to tertiary nitrogen atoms has been observed (Figure 67C). Despite a number of benzylic, ethereal, and 3° C–H bonds (all highlighted in red), the delivery of diazo compounds to α-nitrogen positions is a highly selective transformation. It appears that in the case of these additional substrates, the most sterically accessible C–H bond is activated, despite the fact that it is oftentimes a 1° C–H bond.
4.5. Site-Selective Bromotrifluoromethoxylation
Given the importance of the incorporation of halogens into natural products, especially fluorine, 238–240,279,280 Tang and coworkers envisioned a method of selectively halogenating alkenes in complex molecules (Figure 68A).286 Specifically, they targeted the addition of the trifluoromethoxide group, which has excellent lipophilicity values compared to other halogen sources, such as CF3. Indeed, the authors chose to work with 477, which is a stable source of F3CO− and allowing for the in situ generation of this nucleophile with AgF. DMDBH was chosen as a source of electrophilic bromine to initially activate targeted alkenes for attack by trifluoromethoxide. Finally, after screening, (DHQD)2PHAL was found to be an effective chiral ligand to mediate this transformation with high levels of selectivity. When applying this system towards taxol derivative 478, which contains two double bonds, Markovnikov attack at the terminal alkene is favored. Applying this towards 480, where the regioselectivity of addition is less predictable, a good regiomeric ratio (rr) of 4.6:1 is observed with this protocol (Figure 68B). Finally, a non-intuitive product is observed in the bromotrifluoromethoxylation of 482, though this system again affords products with good rr and dr. A tentative mechanistic hypothesis is proposed, where the chiral ligand coordinates to both bromine and silver, with the silver cation delivering the trifluoromethoxide to the alkene (Figure 68D).
Figure 68.

(A) Site-selective bromotrifluoromethyoxylation of taxol derivative 478. (B) Selective reactions on other natural products. (C) The structure of (DHQD)2PHAL (476). (D) Proposed mechanism for the bromotrifluoromethoxylation.286 aUsing 2.0 equiv DBDMH
4.6. Site-Selective Cyclopropanation
In continuation of their SAR studies of natural products, the Romo group explored the late-stage and selective incorporation of alkyne-containing moieties into natural products.287 This technique allows for the rapid evaluation of the effects of derivatization on pharmacological activity and the incorporation of bioprobes for binding affinity studies. Towards this end, one technique they examined was the site-selective cyclopropanation of natural products. Specifically, this presented a challenge on whether both electron-rich and electron-poor olefins could be selectively targeted depending on the method of cyclopropanation that was utilized.
A common method for electrophilic cyclopropanation, in which the alkene serves as the nucleophile, is via Rh(II)-catalysis (Figure 69). As previously discussed in Section 2.7, upon decomposition of a diazo ester by a dirhodium catalyst, the incipient rhodium carbenoids are susceptible to attack by nucleophiles (Figure 24). The nucleophiles include X–H bonds, C–H bonds, or very commonly, alkenes. Indeed, this approach was discovered to work well in the case of vitamin K (486) and a gibberellic acid derivative (488), providing cyclopropane-containing products 487 and 489 in high yield and moderate diastereoselectivity, with attack taking place at the most electron-rich and sterically unencumbered position. As a note, all X–H bonds were protected to prevent insertion, however, less reactive C–H bonds did not undergo competitive insertion
Figure 69.

Site-selective cyclopropanation of alkene-containing natural products. Diazo esters derived Rh-carbenoids selectively react with electron rich olefins, while alkynyl sulfonium ylides prefer to add to electron-deficient alkenes.
In order to target electron deficient alkenes, Romo and coworkers turned instead to alkynyl sulfonium ylides (485, Figure 69). Containing a fairly acidic α-C–H bond, these species can be deprotonated to form a C-centered nucleophile for addition into a conjugated system. The incipient enolate can displace the sulfonium group to form a cyclopropane in a nominal Corey–Chaykovsky reaction. Likewise, upon application of this method to complex substrates (490, 492, 494, and 486), cyclopropanation of electron-poor, often α,β-unsaturated alkenes were shown to predominate over reactivity at electron-rich olefins. Given the change of mechanism, this method tolerates free O–H and N–H bonds.
These examples represent the specificity of this method for the targeting of various types of alkenes in natural products. It also enables, upon incorporation of these alkyne-arms into natural products, the Sharpless–Huisgen cycloadditions to append molecular probes for global protein profiling.
4.7. Site-Selective Diels–Alder Cycloaddition
One final example of site-selective modification of natural products is the use of Diels- Alder cycloaddition on complex molecules. While there are numerous potential diene and dienophile substrates that could be targeted, one of the most prolific dienophiles in this area are nitroso compounds, which have been utilized in a myriad of site- and stereoselective Diels–Alder reactions.288 For example, rapamycin (498) contains one triene motif that could undergo a variety of site-, regio-, and stereoselective cycloadditions (Figure 70). Despite this, upon reaction with nitroso compound 499, only one product is observed (500).289 This Review will not go into detail on the number of other reports of this type of reaction.
Figure 70.
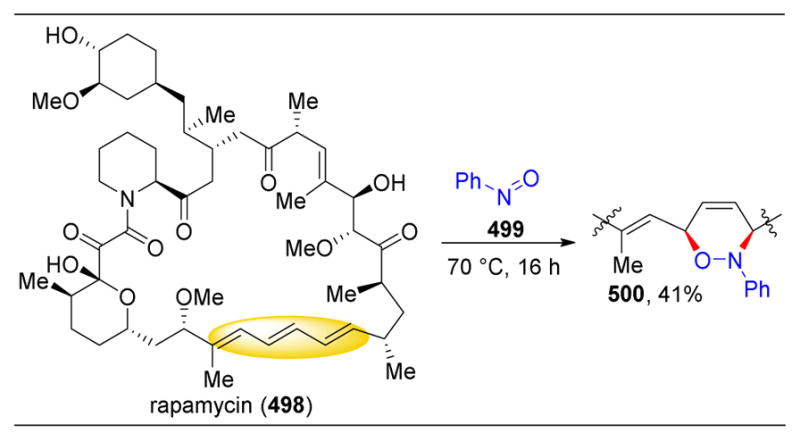
Site-, regio-, and stereoselective nitroso Diels-Aider cycioaddition of rapamycin.
5. Conclusion
The application of non-enzymatic catalysts to the selective alteration of complex molecules is a field that is gaining momentum. The challenges associated with this approach include virtually all types of selectivity that one might address in a study of catalysts in the context of synthetic methodology. Issues of stereoselectivity are omnipresent. While enantioselectivity does not typically present itself in this context since the substrates mined from nature tend to be homochiral, the triumphs of the field of “asymmetric catalysis” are now serving as platforms for the development of catalysts that address all manner of chemical selectivity issues. These challenges include diastereoselectivity, regioselectivity, site-selectivity, chemoselectivity, and even functional group selectivity.269 Perhaps these are terms whose specific meanings will also undergo refinement in the future.
Given the ambition to fully diversify complex natural products and assemble natural product-like libraries,89 one can also see a need for the development of not only effective and perhaps even privileged catalysts,290–292 but also privileged catalyst libraries. Figure 71 shows several selected cases where natural products have been directly converted to analogs, with colored spheres highlighting the sites successfully targeted by catalysts. But of course, there are not enough spheres shown, as there are not enough catalysts yet known to achieve direct modification of all the potential sites to access all the possible analogs of these complex molecules. Perhaps this is one gauntlet thrown down by the discovery and characterization of new, biologically active natural products.
Figure 71.

Molecules that have been the target for catalyst-controlled selective group transfer and oxidation chemistry.
To the extent that selective biological function remains an opportunity at the interfaces of chemistry, biology, and medicine, the role of catalysis seems poised to emerge as central to the achievement of effective agents. Alongside of venerable approaches such as total chemical synthesis, biosynthetic discovery and engineering, and of course, the pursuit of non-natural drugs, further studies of catalysis in the context of complex natural products—with both enzymatic and non-enzymatic catalysts—could emerge as an important tool for both natural product science and medicinal chemistry.
Acknowledgments
We would like to thank Tyler J. Wadzinski for assistance in both framing the content and focus of and in the preparation of this Review. C.R.S. thanks the National Science Foundation Graduate Research Fellowship Program for funding (2014176660). Research in the group of S.J.M. that addresses these general topics is supported by the National Institute of General Medical Sciences of the NIH (GM-068649 and GM-096403), for which we are grateful.
Biographies
Christopher R. Shugrue was born in Hartford, Connecticut in 1991. He obtained a B.A. in Chemistry from the College of the Holy Cross in 2013, carrying out research under the tutelage of Professor Brian R. Linton on both the development of enantioselective organocatalytic transformations and more effective teaching demonstrations. He simultaneously pursued an NSF-REU at the University of Texas at Austin in Professor Stephen F. Martin’s laboratory in 2012, focusing on asymmetric iodolactonizations. In 2013, he began graduate studies at Yale University and was awarded an NSF Graduate Research Fellowship. Under the supervision of Professor Scott J. Miller, he has explored the development of phosphothreonine-embedded peptides as a novel class of chiral phosphoric acid catalysts. His current research interests include the study and advancement of dynamic catalytic systems.
Scott J. Miller received his B.A. (1989), M.A. (1989) and Ph.D. (1994) from Harvard University, where he worked in the laboratory of Professor David Evans as a National Science Foundation Predoctoral Fellow. Subsequently, he traveled to the California Institute of Technology where he was a National Science Foundation Postdoctoral Fellow in the laboratory of Robert Grubbs until 1996. For the following decade, he was a member of the faculty at Boston College, until joining the faculty at Yale University in 2006. In 2008, he was appointed as the Irénée duPont Professor of Chemistry, and in 2009, the Chairperson of the Chemistry Department, a position he held for two consecutive terms, concluding in 2015. His research program focuses on problems in catalysis, employing strategies that include catalyst design, the development of combinatorial techniques for catalyst screening, and the application of these approaches to the preparation of biologically active agents. Three particular interests of his laboratory are (a) the selective functionalization of complex molecules, (b) the exploration of potential analogies between synthetic catalysts and enzymes and (c) the discovery of molecules that are effective antibiotics despite increasing resistance challenges.
Footnotes
Notes
The authors declare no competing financial interests.
References
- 1.Nicolaou KC, Montagnon T. Molecules That Changed the World. Wiley-VCH; 2008. [Google Scholar]
- 2.Corey EJ, Kürti L, Czako B. Molecules and Medicine. Wiley-VCH; 2007. [Google Scholar]
- 3.Newman DJ, Cragg GM. Natural Products as Sources of New Drugs from 1981 to 2014. J Nat Prod. 2016;79:629–661. doi: 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- 4.Giuliano MW, Miller SJ. Site-Selective Reactions with Peptide-Based Catalysts. Top Curr Chem. 2016;372:157–202. doi: 10.1007/128_2015_653. [DOI] [PubMed] [Google Scholar]
- 5.Robles O, Romo D. Chemo- and Site-Selective Derivatizations of Natural Products Enabling Biological Studies. Nat Prod Rep. 2014;31:318–334. doi: 10.1039/c3np70087a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahatthananchai J, Dumas AM, Bode JW. Catalytic Selective Synthesis. Angew Chem Int Ed. 2012;51:10954–10990. doi: 10.1002/anie.201201787. [DOI] [PubMed] [Google Scholar]
- 7.Newhouse T, Baran PS. If C–H Bonds Could Talk: Selective C–H Bond Oxidation. Angew Chem Int Ed. 2011;50:3362–3374. doi: 10.1002/anie.201006368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brückl TBR, Baxter RD, Ishihara Y, Baran PS. Innate and Guided C–H Functionalization Logic. Acc Chem Res. 2012;45:826–839. doi: 10.1021/ar200194b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davis HJ, Phipps RJ. Harnessing Non-Covalent Interactions to Exert Control Over Regioselectivity and Site-Selectivity in Catalytic Reactions. Chem Sci. 2017;8:864–877. doi: 10.1039/c6sc04157d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wiley PF, Gerzon K, Flynn EH, Sigal MV, Jr, Weaver O, Quarck UC, Chauvette RR, Monahan R. Erythromycin. X. 1 Structure of Erythromycin. J Am Chem Soc. 1957;79:6062–6070. [Google Scholar]
- 11.Dutcher JD. The Discovery and Development of Amphotericin B. Dis Chest. 1968;54:296–298. doi: 10.1378/chest.54.supplement_1.296. [DOI] [PubMed] [Google Scholar]
- 12.Umezawa H, Hooper IR. Aminoglycoside Antibiotics. Springer-Verlag; 1982. [Google Scholar]
- 13.Simonsen J. L LXI.—The constituents of Indian turpentine from Pinus longifolia, Roxb. Part I. J Chem Soc Trans. 1920;117:570–578. [Google Scholar]
- 14.Toscano MD, Woycechowsky KJ, Hilvert D. Minimalist Active-Site Redesign: Teaching Old Enzymes New Tricks. Angew Chem Int Ed. 2007;46:3212–3236. doi: 10.1002/anie.200604205. [DOI] [PubMed] [Google Scholar]
- 15.Lutz S, Bornscheuer UT. Protein Engineering Handbook. 1–2 Wiley-VCH; 2011. [Google Scholar]
- 16.Corey EJ, Kürti L. Enantioselective Chemical Synthesis: Methods, Logic and Practice. Direct Book Publishing; Dallas: 2010. [Google Scholar]
- 17.Kadokawa JI. Precision Polysaccharide Synthesis Catalyzed by Enzymes. Chem Rev. 2011;111:4308–4345. doi: 10.1021/cr100285v. [DOI] [PubMed] [Google Scholar]
- 18.Mullins RJ, Vedernikov A, Viswanathan R. Competition Experiments as a Means of Evaluating Linear Free Energy Relationships. An Experiment for the Advanced Undergraduate Organic Chemistry Lab. J Chem Educ. 2004;81:1357–1361. [Google Scholar]
- 19.Tojo G, Fernández M. Oxidation of Alcohols to Aldehydes and Ketones. Springer; 2006. [Google Scholar]
- 20.Smith MB. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure. Wiley; Hoboken: 2013. [Google Scholar]
- 21.Kocienski PJ. Protecting Groups. 3. Thieme; 2005. [Google Scholar]
- 22.Wuts PGM, Greene TW. Greene’s Protecting Groups in Organic Synthesis. 4. Wiley; Hoboken: 2007. [Google Scholar]
- 23.Young IS, Baran PS. Protecting-Group-Free Synthesis as an Opportunity for Invention. Nat Chem. 2009;1:193–205. doi: 10.1038/nchem.216. [DOI] [PubMed] [Google Scholar]
- 24.Seiple IB, Zhang Z, Jakubec P, Langlois-Mercier A, Wright PM, Hog DT, Yabu K, Allu SR, Fukuzaki T, Carlsen PN, et al. A Platform for the Discovery of New Macrolide Antibiotics. Nature. 2016;533:338–345. doi: 10.1038/nature17967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krebs EG, Fischer EH. Phosphorylase Activity of Skeletal Muscle Extracts. J Biol Chem. 1955;216:113–120. [PubMed] [Google Scholar]
- 26.Hunter T. Signaling—2000 and Beyond. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- 27.Yaffe MB, Elia AEH. Phosphoserine/threonine-binding domains. Curr Opin Cell Biol. 2001;13:131–138. doi: 10.1016/s0955-0674(00)00189-7. [DOI] [PubMed] [Google Scholar]
- 28.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The Protein Kinase Complement of the Human Genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 29.Cohen P. Protein Kinases—the Major Drug Targets of the Twenty-First Century? Nat Rev Drug Discov. 2002;1:309–315. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- 30.Alonso A, Sasin J, Bottini N, Friedberg II, Friedberg II, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T. Protein Tyrosine Phosphatases in the Human Genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 31.Tarrant MK, Cole PA. The Chemical Biology of Protein Phosphorylation. Ann Rev Biochem. 2009;78:797–825. doi: 10.1146/annurev.biochem.78.070907.103047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roskoski R., Jr A Historical Overview of Protein Kinases and Their Targeted Small Molecule Inhibitors. Pharmacol Res. 2015;100:1–23. doi: 10.1016/j.phrs.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 33.Schmid A, Dordick JS, Hauer B, Kiener A, Wubbolts M, Witholt B. Industrial biocatalysis today and tomorrow. Nature. 2001;409:258–268. doi: 10.1038/35051736. [DOI] [PubMed] [Google Scholar]
- 34.Schoemaker HE, Mink D, Wubbolts MG. Dispelling the Myths—Biocatalysis in Industrial Synthesis. Science. 2003;299:1694–1697. doi: 10.1126/science.1079237. [DOI] [PubMed] [Google Scholar]
- 35.Koeller KM, Wong CH. Enzymes for Chemical Synthesis. Nature. 2001;409:232–240. doi: 10.1038/35051706. [DOI] [PubMed] [Google Scholar]
- 36.Anantharaman V, Aravind L, Koonin EV. Emergence of Diverse Biochemical Activities in Evolutionarily Conserved Structural Scaffolds of Proteins. Curr Opin Chem Biol. 2003;7:12–20. doi: 10.1016/s1367-5931(02)00018-2. [DOI] [PubMed] [Google Scholar]
- 37.Mahadevi AS, Sastry GN. Cooperativity in Noncovalent Interactions. Chem Rev. 2016;116:2775–2825. doi: 10.1021/cr500344e. [DOI] [PubMed] [Google Scholar]
- 38.Lairson LL, Henrissat B, Davies GJ, Withers SG. Glycosyltransferases: Structures, Functions, and Mechanisms. Annu Rev Biochem. 2008;77:521–555. doi: 10.1146/annurev.biochem.76.061005.092322. [DOI] [PubMed] [Google Scholar]
- 39.Copeland GT, Jarvo ER, Miller SJ. Minimal Acylase-Like Peptides. Conformational Control of Absolute Stereospecificity. J Org Chem. 1998;63:6784–6785. doi: 10.1021/jo981642w. [DOI] [PubMed] [Google Scholar]
- 40.Miller SJ, Copeland GT, Papaioannou N, Horstmann TE, Ruel EM. Kinetic Resolution of Alcohols Catalyzed by Tripeptides Containing the N-Alkylimidazole Substructure. J Am Chem Soc. 1998;120:1629–1630. [Google Scholar]
- 41.Copeland GT, Miller SJ. A Chemosensor-Based Approach to Catalyst Discovery in Solution and on Solid Support. J Am Chem Soc. 1999;121:4306–4307. [Google Scholar]
- 42.Copeland GT, Miller SJ. Selection of Enantioselective Acyl Transfer Catalysts from a Pooled Peptide Library through a Fluorescence-Based Activity Assay: An Approach to Kinetic Resolution of Secondary Alcohols of Broad Structural Scope. J Am Chem Soc. 2001;123:6496–6502. doi: 10.1021/ja0108584. [DOI] [PubMed] [Google Scholar]
- 43.Haque TS, Little JC, Gellman SH. “Mirror Image” Reverse Turns Promote β-Hairpin Formation. J Am Chem Soc. 1994;116:4105–4106. [Google Scholar]
- 44.Wilmot CM, Thornton JM. Analysis and Prediction of the Different Types of β-Turn in Proteins. J Mol Biol. 1988;203:221–232. doi: 10.1016/0022-2836(88)90103-9. [DOI] [PubMed] [Google Scholar]
- 45.Haque TS, Little JC, Gellman SH. Stereochemical Requirements for β-Hairpin Formation: Model Studies with Four-Residue Peptides and Depsipeptides. J Am Chem Soc. 1996;118:6975–6985. [Google Scholar]
- 46.Metrano AJ, Abascal NC, Mercado BQ, Paulson EK, Hurtley AE, Miller SJ. Diversity of Secondary Structure in Catalytic Peptides with β-Turn-Biased Sequences. J Am Chem Soc. 2017;139:492–516. doi: 10.1021/jacs.6b11348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sculimbrene BR, Miller SJ. Discovery of a Catalytic Asymmetric Phosphorylation through Selection of a Minimal Kinase Mimic: A Concise Total Synthesis of D-Myo-Inositol-1-Phosphate. J Am Chem Soc. 2001;123:10125–10126. doi: 10.1021/ja016779+. [DOI] [PubMed] [Google Scholar]
- 48.Sculimbrene BR, Morgan AJ, Miller SJ. Enantiodivergence in Small-Molecule Catalysis of Asymmetric Phosphorylation: Concise Total Syntheses of the Enantiomeric D-Myo-Inositol-1-Phosphate and D-Myo-Inositol-3-Phosphate. J Am Chem Soc. 2002;124:11653–11656. doi: 10.1021/ja027402m. [DOI] [PubMed] [Google Scholar]
- 49.Sculimbrene BR, Xu Y, Miller SJ. Asymmetric Syntheses of Phosphatidylinositol-3-Phosphates with Saturated and Unsaturated Side Chains through Catalytic Asymmetric Phosphorylation. J Am Chem Soc. 2004;126:13182–13183. doi: 10.1021/ja0466098. [DOI] [PubMed] [Google Scholar]
- 50.Xu Y, Sculimbrene BR, Miller SJ. Streamlined Synthesis of Phosphatidylinositol (PI), PI3P, PI3, 5P2, and Deoxygenated Analogues as Potential Biological Probes. J Org Chem. 2006;71:4919–4928. doi: 10.1021/jo060702s. [DOI] [PubMed] [Google Scholar]
- 51.Fiori KW, Puchlopek ALA, Miller SJ. Enantioselective Sulfonylation Reactions Mediated by a Tetrapeptide Catalyst. Nat Chem. 2009;1:630–634. doi: 10.1038/nchem.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Griswold KS, Miller SJ. A Peptide-Based Catalyst Approach to Regioselective Functionalization of Carbohydrates. Tetrahedron. 2003;59:8869–8875. [Google Scholar]
- 53.Washington JA, II, Wilson WR. Erythromycin: A Microbial and Clinical Perspective After 30 Years of Clinical Use (First of Two Parts) Mayo Clin Proc. 1985;60:271–278. doi: 10.1016/s0025-6196(12)60322-x. [DOI] [PubMed] [Google Scholar]
- 54.McDaniel R, Welch M, Hutchinson CR. Genetic Approaches to Polyketide Antibiotics. 1. Chem Rev. 2005;105:543–558. doi: 10.1021/cr0301189. [DOI] [PubMed] [Google Scholar]
- 55.Bax R, Griffin D. Introduction to Antibiotic Resistance. Chem Rev. 2005;105:391–393. doi: 10.1021/cr030100y. [DOI] [PubMed] [Google Scholar]
- 56.Corey EJ, Kim S, Yoo SE, Nicolaou KC, Melvin LS, Jr, Brunelle DJ, Falck JR, Trybulski EJ, Lett R, Sheldrake PW. Total Synthesis of Erythromycins. 4. Total Synthesis of Erythronolide B1. J Am Chem Soc. 1978;100:4620–4622. [Google Scholar]
- 57.Woodward RB, Logusch E, Nambiar KP, Sakan K, Ward DE, Au-Yeung BW, Balaram P, Browne LJ, Card PJ, Chen CH, et al. Asymmetric Total Synthesis of Erythromycin. 1. Synthesis of an Erythronolide a Seco Acid-Derivative via Asymmetric Induction. J Am Chem Soc. 1981;103:3210–3213. [Google Scholar]
- 58.Masamune S, Hirama M, Mori S, Ali SA, Garvey DS. Total Synthesis of 6-Deoxyerythronolide B. J Am Chem Soc. 1981;103:1568–1571. [Google Scholar]
- 59.Paterson I, Mansuri MM. Tetrahedron Report Number 190. Recent Developments in the Total Synthesis of Macrolide Antibiotics. Tetrahedron. 1985;41:3569–3624. [Google Scholar]
- 60.Paterson I, Rawson DJ. Studies in Macrolide Synthesis: A Highly Stereoselective Synthesis of (+)-(9S)-Dihydroerythronolide a Using Macrocyclic Stereocontrol. Tetrahedron Lett. 1989;30:7463–7466. [Google Scholar]
- 61.Myles DC, Danishefsky SJ, Schulte G. Development of a Fully Synthetic Stereoselective Route to 6-Deoxyerythronolide B by Reiterative Applications of the Lewis Acid Catalyzed Diene Aldehyde Cyclocondensation Reaction: A Remarkable Instance of Diastereofacial Selectivity. J Org Chem. 1990;55:1636–1648. [Google Scholar]
- 62.Evans D, Kim AS, Metternich R, Novack VJ. General Strategies toward the Synthesis of Macrolide Antibiotics. The Total Synthesis of 6-Deoxyerythronolide B and Oleandolide. J Am Chem Soc. 1998;120:5921–5942. [Google Scholar]
- 63.Crimmins MT, Slade DJ. Formal Synthesis of 6-Deoxyerythronolide B. Org Lett. 2006;8:2191–2194. doi: 10.1021/ol0607241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Breton P, Hergenrother PJ, Hida T, Hodgson A, Judd AS, Kraynack E, Kym PR, Lee WC, Loft MS, Yamashita M, Martin SF. Total Synthesis of Erythromycin B. Tetrahedron. 2007;63:5709–5729. [Google Scholar]
- 65.Lewis CA, Miller SJ. Site-Selective Derivatization and Remodeling of Erythromycin A by Using Simple Peptide-Based Chiral Catalysts. Angew Chem Int Ed. 2006;45:5616–5619. doi: 10.1002/anie.200601490. [DOI] [PubMed] [Google Scholar]
- 66.Everett JR, Hunt E, Tyler JW. Ketone-Hemiacetal Tautomerism in Erythromycin A in Non-Aqueous Solutions. An NMR Spectroscopic Study. J Chem Soc, Perkin Trans 2. 1991:1481–1487.
- 67.Lewis CA, Merkel J, Miller SJ. Catalytic Site-Selective Synthesis and Evaluation of a Series of Erythromycin Analogs. Bioorg Med Chem Lett. 2008;18:6007–6011. doi: 10.1016/j.bmcl.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Baker R, Kulagowski JJ, Billington DC, Leeson PD, Lennon IC, Liverton N. Synthesis of 2- and 6-Deoxyinositol 1-Phosphate and the Role of the Adjacent Hydroxy Groups in the Mechanism of Inositol Monophosphatase. J Chem Soc, Chem Commun. 1989:1383–1385. [Google Scholar]
- 69.Baker R, Leeson PD, Liverton NJ, Kulagowski JJ. Identification of (1S)-Phosphoryloxy-(ZR,4S)-Dihydroxycyclohexane. J Chem Soc, Chem Commun. 1990:462–464. [Google Scholar]
- 70.Kozikowski AP, Qiao L, Tuckmantel W, Powis G. Synthesis of 1D-3-Deoxy- and -2,3-Dideoxyphosphatidylinositol. Tetrahedron. 1997;53:14903–14914. [Google Scholar]
- 71.Miller DJ, Beaton MW, Wilkie J, Gani D. The 6-OH Group of D-Inositol 1-Phosphate Serves as an H-Bond Donor in the Catalytic Hydrolysis of the Phosphate Ester by Inositol Monsphosphatase. ChemBioChem. 2000;1:262–271. doi: 10.1002/1439-7633(20001117)1:4<262::AID-CBIC262>3.0.CO;2-%23. [DOI] [PubMed] [Google Scholar]
- 72.Morgan AJ, Wang YK, Roberts MF, Miller SJ. Chemistry and Biology of Deoxy-Myo-Inositol Phosphates: Stereospecificity of Substrate Interactions within an Archaeal and a Bacterial IMPase. J Am Chem Soc. 2004;126:15370–15371. doi: 10.1021/ja047360x. [DOI] [PubMed] [Google Scholar]
- 73.Croatt MP, Carreira EM. Probing the Role of the Mycosamine C2′-OH on the Activity of Amphotericin B. Org Lett. 2011;13:1390–1393. doi: 10.1021/ol2000765. [DOI] [PubMed] [Google Scholar]
- 74.Wilcock BC, Endo MM, Uno BE, Burke MD. C2′-OH of Amphotericin B Plays an Important Role in Binding the Primary Sterol of Human Cells but Not Yeast Cells. J Am Chem Soc. 2013;135:8488–8491. doi: 10.1021/ja403255s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang L, Koreeda M. Radical Deoxygenation of Hydroxyl Groups via Phosphites. J Am Chem Soc. 2004;126:13190–13191. doi: 10.1021/ja0462777. [DOI] [PubMed] [Google Scholar]
- 76.Barton DHR, McCombie SW. A New Method for the Deoxygenation of Secondary Alcohols. J Chem Soc, Perkin Trans 1. 1975:1574–1585. [Google Scholar]
- 77.Barton DHR, Blundell P, Dorchak J, Jang DO, Jaszberenyi JC. The Invention of Radical Reactions. Part XXI. Simple Methods for the Radical Deoxygenation of Primary Alcohols. Tetrahedron. 1991;47:8969–8984. [Google Scholar]
- 78.Wiberg KB, Wang YG, Miller SJ, Puchlopek ALA, Bailey WF, Fair JD. Disparate Behavior of Carbonyl and Thiocarbonyl Compounds: Acyl Chlorides vs Thiocarbonyl Chlorides and Isocyanates vs Isothiocyanates. J Org Chem. 2009;74:3659–3664. doi: 10.1021/jo9004316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sánchez-Roselló M, Puchlopek ALA, Morgan AJ, Miller SJ. Site-Selective Catalysis of Phenyl Thionoformate Transfer as a Tool for Regioselective Deoxygenation of Polyols. J Org Chem. 2008;73:1774–1782. doi: 10.1021/jo702334z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jordan PA, Miller SJ. An Approach to the Site-Selective Deoxygenation of Hydroxy Groups Based on Catalytic Phosphoramidite Transfer. Angew Chem Int Ed. 2012;51:2907–2911. doi: 10.1002/anie.201109033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hayakawa Y, Kim JW, Adachi H, Shin-Ya K, Fujita KI, Seto H. Structure of Apoptolidin, a Specific Apoptosis Inducer in Transformed Cells. J Am Chem Soc. 1998;120:3524–3525. [Google Scholar]
- 82.Salomon AR, Zhang Y, Seto H, Khosla C. Structure-Activity Relationships within a Family of Selectively Cytotoxic Macrolide Natural Products. Org Lett. 2001;3:57–59. doi: 10.1021/ol006767d. [DOI] [PubMed] [Google Scholar]
- 83.Wender PA, Gulledge AV, Jankowski OD, Seto H. Isoapoptolidin: Structure and Activity of the Ring-Expanded Isomer of Apoptolidin. Org Lett. 2002;4:3819–3822. doi: 10.1021/ol0266222. [DOI] [PubMed] [Google Scholar]
- 84.Pennington JD, Williams HJ, Salomon AR, Sulikowski GA. Toward a Stable Apoptolidin Derivative: Identification of Isoapoptolidin and Selective Deglycosylation of Apoptolidin. Org Lett. 2002;4:3823–3825. doi: 10.1021/ol026829v. [DOI] [PubMed] [Google Scholar]
- 85.Wender PA, Jankowski OD, Tabet EA, Seto H. Facile Synthetic Access to and Biological Evaluation of the Macrocyclic Core of Apoptolidin. Org Lett. 2003;5:2299–2302. doi: 10.1021/ol0346335. [DOI] [PubMed] [Google Scholar]
- 86.Wender PA, Jankowski OD, Longcore K, Tabet EA, Seto H, Tomikawa T. Correlation of F0F1-ATPase Inhibition and Antiproliferative Activity of Apoptolidin Analogues. Org Lett. 2006;8:589–592. doi: 10.1021/ol052800q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lewis CA, Longcore KE, Miller SJ, Wender PA. An Approach to the Site-Selective Diversification of Apoptolidin A with Peptide-Based Catalysts. J Nat Prod. 2009;72:1864–1869. doi: 10.1021/np9004932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Williams DH, Bardsley B. The Vancomycin Group of Antibiotics and the Fight against Resistant Bacteria. Angew Chem Int Ed. 1999;38:1172–1193. doi: 10.1002/(SICI)1521-3773(19990503)38:9<1172::AID-ANIE1172>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 89.Nicolaou KC, Boddy CNC, Bräse S, Winssinger N. Chemistry, Biology, and Medicine of the Glycopeptide Antibiotics. Angew Chem Int Ed. 1999;38:2096–2152. doi: 10.1002/(sici)1521-3773(19990802)38:15<2096::aid-anie2096>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 90.Neu HC. The Crisis in Antibiotic Resistance. Science. 1992;257:1064–1073. doi: 10.1126/science.257.5073.1064. [DOI] [PubMed] [Google Scholar]
- 91.James RC, Pierce JG, Okano A, Xie J, Boger DL. Redesign of Glycopeptide Antibiotics: Back to the Future. ACS Chem Biol. 2012;7:797–804. doi: 10.1021/cb300007j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Butler MS, Hansford KA, Blaskovich MAT, Halai R, Cooper MA. Glycopeptide Antibiotics: Back to the Future. J Antibiot (Tokyo) 2014;67:631–644. doi: 10.1038/ja.2014.111. [DOI] [PubMed] [Google Scholar]
- 93.Ashford PA, Bew SP. Recent Advances in the Synthesis of New Glycopeptide Antibiotics. Chem Soc Rev. 2012;41:957–978. doi: 10.1039/c1cs15125h. [DOI] [PubMed] [Google Scholar]
- 94.Nicolaou KC, Mitchell HJ, Jain NF, Winssinger N, Hughes R, Bando T. Total Synthesis of Vancomycin. Angew Chem Int Ed. 1999;38:240–244. [Google Scholar]
- 95.Crowley BM, Boger DL. Total Synthesis and Evaluation of [Ψ[CH2NH]Tpg4]Vancomycin Aglycon: Reengineering Vancomycin for Dual D-Ala-D-Ala and D- Ala-D-Lac Binding. J Am Chem Soc. 2008;128:2885–2892. doi: 10.1021/ja0572912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Crane CM, Boger DL. Synthesis and Evaluation of Vancomycin Aglycon Analogues That Bear Modifications in the N-Terminal D-Leucyl Amino Acid. J Med Chem. 2009;52:1471–1476. doi: 10.1021/jm801549b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Xie J, Pierce JG, James RC, Okano A, Boger DL. A Redesigned Vancomycin Engineered for Dual D-Ala- D-Ala and D-Ala-D-Lac Binding Exhibits Potent Antimicrobial Activity Against Vancomycin-Resistant Bacteria. J Am Chem Soc. 2011;133:13946–13949. doi: 10.1021/ja207142h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Okano A, Nakayama A, Schammel AW, Boger DL. Total Synthesis of [Ψ[C(=NH)NH]Tpg4]Vancomycin and Its (4-Chlorobiphenyl)methyl Derivative: Impact of Peripheral Modifications on Vancomycin Analogues Redesigned for Dual D-Ala-D-Ala and D-Ala-D-Lac Binding. J Am Chem Soc. 2014;136:13522–13525. doi: 10.1021/ja507009a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Harris CM, Fesik SW, Thomas AM, Kannan R, Harris TM. Iodination of Vancomycin, Ristocetin A, and Ristocetin Pseudoaglycon. J Org Chem. 1986;43:1509–1513. [Google Scholar]
- 100.Ge M, Chen Z, Onishi HR, Kohler J, Silver LL, Kerns R, Fukuzawa S, Thompson C, Kahne D. Vancomycin Derivatives That Inhibit Peptidoglycan Biosynthesis without Binding D-Ala-D-Ala. Science. 1999;284:507–511. doi: 10.1126/science.284.5413.507. [DOI] [PubMed] [Google Scholar]
- 101.Kerns R, Dong SD, Fukuzawa S, Carbeck J, Kohler J, Silver L, Kahne D. The Role of Hydrophobic Substituents in the Biological Activity of Glycopeptide Antibiotics. J Am Chem Soc. 2000;122:12608–12609. [Google Scholar]
- 102.Nicolaou KC, Cho SY, Hughes R, Winssinger N, Smethurst C, Labischinski H, Endermann R. Solid- and Solution-Phase Synthesis of Vancomycin and Vancomycin Analogues with Activity against Vancomycin-Resistant Bacteria. Chem Eur J. 2001;7:3798–3823. doi: 10.1002/1521-3765(20010903)7:17<3798::aid-chem3798>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 103.Ritter TK, Mong KKT, Liu H, Nakatani T, Wong CH. A Programmable One-Pot Oligosaccharide Synthesis for Diversifying the Sugar Domains of Natural Products: A Case Study of Vancomycin. Angew Chem Int Ed. 2003;42:4657–4660. doi: 10.1002/anie.200351534. [DOI] [PubMed] [Google Scholar]
- 104.Griffin JH, Linsell MS, Nodwell MB, Chen QQ, Pace JL, Quast KL, Krause KM, Farrington L, Wu TX, Higgins DL, et al. Multivalent Drug Design. Synthesis and in Vitro Analysis of an Array of Vancomycin Dimers. J Am Chem Soc. 2003;125:6517–6531. doi: 10.1021/ja021273s. [DOI] [PubMed] [Google Scholar]
- 105.Crane CM, Pierce JG, Leung SSF, Tirado-Rives J, Jorgensen WL, Boger DL. Synthesis and Evaluation of Selected Key Methyl Ether Derivatives of Vancomycin Aglycon. J Med Chem. 2010;53:7229–7235. doi: 10.1021/jm100946e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pinchman JR, Boger DL. Probing the Role of the Vancomycin E-Ring Aryl Chloride: Selective Divergent Synthesis and Evaluation of Alternatively Substituted E-Ring Analogues. J Med Chem. 2013;56:4116–4124. doi: 10.1021/jm4004494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Weist S, Bister B, Puk O, Bischoff D, Pelzer S, Nicholson GJ, Wohlleben W, Jung G, Süssmuth RD. Fluorobalhimycin - A New Chapter in Glycopeptide Antibiotic Research. Angew Chem Int Ed. 2002;41:3383–3385. doi: 10.1002/1521-3773(20020916)41:18<3383::AID-ANIE3383>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 108.Weist S, Kittel C, Bischoff D, Bister B, Pfeifer V, Nicholson GJ, Wohlleben W, Süssmuth RD. Mutasynthesis of Glycopeptide Antibiotics: Variations of Vancomycin’s AB-Ring Amino Acid 3,5-Dihydroxyphenylglycine. J Am Chem Soc. 2004;126:5942–5943. doi: 10.1021/ja0499389. [DOI] [PubMed] [Google Scholar]
- 109.Kruger RG, Lu W, Oberthür M, Tao J, Kahne D, Walsh CT. Tailoring of Glycopeptide Scaffolds by the Acyltransferases from the Teicoplanin and A-40, 926 Biosynthetic Operons. Chem Biol. 2005;12:131–140. doi: 10.1016/j.chembiol.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 110.Oberthür M, Leimkuhler C, Kruger RG, Lu W, Walsh CT, Kahne D. A Systematic Investigation of the Synthetic Utility of Glycopeptide Glycosyltransferases. J Am Chem Soc. 2005;127:10747–10752. doi: 10.1021/ja052945s. [DOI] [PubMed] [Google Scholar]
- 111.Thayer DA, Wong CH. Vancomycin Analogues Containing Monosaccharides Exhibit Improved Antibiotic Activity: A Combined One-Pot Enzymatic Glycosylation and Chemical Diversification Strategy. Chem Asian J. 2006;1:445–452. doi: 10.1002/asia.200600084. [DOI] [PubMed] [Google Scholar]
- 112.Fowler BS, Laemmerhold KM, Miller SJ. Catalytic Site-Selective Thiocarbonylations and Deoxygenations of Vancomycin Reveal Hydroxyl-Dependent Conformational Effects. J Am Chem Soc. 2012;134:9755–9761. doi: 10.1021/ja302692j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nitanai Y, Kikuchi T, Kakoi K, Hanamaki S, Fujisawa I, Aoki K. Crystal Structures of the Complexes between Vancomycin and Cell-Wall Precursor Analogs. J Mol Biol. 2009;385:1422–1432. doi: 10.1016/j.jmb.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 114.Protein Database: 1FVM.
- 115.Howden BP, Davies JK, Johnson PDR, Stinear TP, Grayson ML. Reduced Vancomycin Susceptibility in Staphylococcus Aureus, Including Vancomycin-Intermediate and Heterogeneous Vancomycin-Intermediate Strains: Resistance Mechanisms, Laboratory Detection, and Clinical Implications. Clin Microbiol Rev. 2010;23:99–139. doi: 10.1128/CMR.00042-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Higgins DL, Chang R, Debabov DV, Leung J, Wu T, Krause KM, Sandvik E, Hubbard JM, Schmidt DE, Jr, Gao Q, et al. Telavancin, a Multifunctional Lipoglycopeptide, Disrupts Both Cell Wall Synthesis and Cell Membrane Integrity in Methicillin-Resistant Staphylococcus Aureus. Antimicrob Agents Chemother. 2005;49:1127–1134. doi: 10.1128/AAC.49.3.1127-1134.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mainardi JL, Shlaes DM, Goering RV, Shlaes JH, Acar JF, Goldstein FW. Decreased Teicoplanin Susceptibility of Methicillin-Resistant Strains of Staphylococcus Aureus. J Infect Dis. 1995;171:1646–1650. doi: 10.1093/infdis/171.6.1646. [DOI] [PubMed] [Google Scholar]
- 118.Schmit JL. Efficacy of Teicoplanin for Enterococcal Infections: 63 Cases and Review. Clin Infect Dis. 1992;15:302–306. doi: 10.1093/clinids/15.2.302. [DOI] [PubMed] [Google Scholar]
- 119.Liassine N, Frei R, Jan I, Auckenthaler R. Characterization of Glycopeptide-Resistant Enterococci from a Swiss Hospital. J Clin Microbiol. 1998;36:1853–1858. doi: 10.1128/jcm.36.7.1853-1858.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Park IJ, Lee WG, Shin JH, Lee KW, Woo GJ. VanB Phenotype-vanA Genotype Enterococcus Faecium with Heterogeneous Expression of Teicoplanin Resistance. J Clin Microbiol. 2008;46:3091–3093. doi: 10.1128/JCM.00712-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Han S, Miller SJ. Asymmetric Catalysis at a Distance: Catalytic, Site-Selective Phosphorylation of Teicoplanin. J Am Chem Soc. 2013;135:12414–12421. doi: 10.1021/ja406067v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Han S, Le BV, Hajare HS, Baxter RHG, Miller SJ. X-Ray Crystal Structure of Teicoplanin A2-2 Bound to a Catalytic Peptide Sequence via the Carrier Protein Strategy. J Org Chem. 2014;79:8550–8556. doi: 10.1021/jo501625f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Economou NJ, Nahoum V, Weeks SD, Grasty KC, Zentner IJ, Townsend TM, Bhuiya MW, Cocklin S, Loll PJ. A Carrier Protein Strategy Yields the Structure of Dalbavancin. J Am Chem Soc. 2012;134:4637–4645. doi: 10.1021/ja208755j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yoganathan S, Miller SJ. Structure Diversification of Vancomycin through Peptide-Catalyzed, Site-Selective Lipidation: A Catalysis-Based Approach to Combat Glycopeptide-Resistant Pathogens. J Med Chem. 2015;58:2367–2377. doi: 10.1021/jm501872s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cochrane SA, Lohans CT, Brandelli JR, Mulvey G, Armstrong GD, Vederas JC. Synthesis and Structure-Activity Relationship Studies of N-Terminal Analogues of the Antimicrobial Peptide Tridecaptin A1. J Med Chem. 2014;57:1127–1131. doi: 10.1021/jm401779d. [DOI] [PubMed] [Google Scholar]
- 126.Yarlagadda V, Akkapeddi P, Manjunath GB, Haldar J. Membrane Active Vancomycin Analogues: A Strategy to Combat Bacterial Resistance. J Med Chem. 2014;57:4558–4568. doi: 10.1021/jm500270w. [DOI] [PubMed] [Google Scholar]
- 127.Nakama Y, Yoshida O, Yoda M, Araki K, Sawada Y, Nakamura J, Xu S, Miura K, Maki H, Arimoto H. Discovery of a Novel Series of Semisynthetic Vancomycin Derivatives Effective against Vancomycin-Resistant Bacteria. J Med Chem. 2010;53:2528–2533. doi: 10.1021/jm9017543. [DOI] [PubMed] [Google Scholar]
- 128.Kahne D, Leimkuhler C, Lu W, Walsh C. Glycopeptide and Lipoglycopeptide Antibiotics. Chem Rev. 2005;105:425–448. doi: 10.1021/cr030103a. [DOI] [PubMed] [Google Scholar]
- 129.Baltz RH, Miao V, Wrigley SK. Natural Products to Drugs: Daptomycin and Related Lipopeptide Antibiotics. Nat Prod Rep. 2005;22:717–741. doi: 10.1039/b416648p. [DOI] [PubMed] [Google Scholar]
- 130.Jarvo ER, Copeland GT, Papaioannou N, Bonitatebus PJ, Miller SJ. A Biomimetic Approach to Asymmetric Acyl Transfer Catalysis. J Am Chem Soc. 1999;121:11638–11643. [Google Scholar]
- 131.Fowler BS, Mikochik PJ, Miller SJ. Peptide-Catalyzed Kinetic Resolution of Formamides and Thioformamides as an Entry to Nonracemic Amines. J Am Chem Soc. 2010;132:2870–2871. doi: 10.1021/ja9107897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fischbach MA, Walsh CT. Antibiotics for Emerging Pathogens. Science. 2009;325:1089–1093. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wright GD. Antibiotics: A New Hope. Chem Biol. 2012;19:3–10. doi: 10.1016/j.chembiol.2011.10.019. [DOI] [PubMed] [Google Scholar]
- 134.Clardy J, Walsh C. Lessons from Natural Molecules. Nature. 2004;432:829–837. doi: 10.1038/nature03194. [DOI] [PubMed] [Google Scholar]
- 135.Thaker MN, Wright GD. Opportunities for Synthetic Biology in Antibiotics: Expanding Glycopeptide Chemical Diversity. ACS Synth Biol. 2015;4:195–206. doi: 10.1021/sb300092n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kawabata T, Furuta T. Nonenzymatic Regioselective Acylation of Carbohydrates. Chem Lett. 2009;38:640–647. doi: 10.1021/jo901569v. [DOI] [PubMed] [Google Scholar]
- 137.Mishiro W, Furuta K, Kawabata T. Functional Group Tolerance in Organocatalytic Regioselective Acylation of Carbohydrates. J Org Chem. 2009;74:8802–8805. doi: 10.1021/jo901569v. [DOI] [PubMed] [Google Scholar]
- 138.Kawabata T, Muramatsu W, Nishio T, Shibata T, Schedel H. A Catalytic One-Step Process for the Chemo- and Regioselective Acylation of Monosaccharides. J Am Chem Soc. 2007;129:12890–12895. doi: 10.1021/ja074882e. [DOI] [PubMed] [Google Scholar]
- 139.Ueda Y, Furuta T, Kawabata T. Final-Stage Site-Selective Acylation for the Total Syntheses of Multifidosides A-C. Angew Chem Int Ed. 2015;54:11966–11970. doi: 10.1002/anie.201504729. [DOI] [PubMed] [Google Scholar]
- 140.Takeuchi H, Mishiro K, Ueda Y, Fujimori Y, Furuta T, Kawabata T. Total Synthesis of Ellagitannins through Regioselective Sequential Functionalization of Unprotected Glucose. Angew Chem Int Ed. 2015;54:6177–6180. doi: 10.1002/anie.201500700. [DOI] [PubMed] [Google Scholar]
- 141.Yoshida K, Furuta T, Kawabata T. Perfectly Regioselective Acylation of a Cardiac Glycoside, Digitoxin, via Catalytic Amplification of the Intrinsic Reactivity. Tetrahedron Lett. 2010;51:4830–4832. [Google Scholar]
- 142.Ueda Y, Mishiro K, Yoshida K, Furuta T, Kawabata T. Regioselective Diversification of a Cardiac Glycoside, Lanatoside C, by Organocatalysis. J Org Chem. 2012;77:7850–7857. doi: 10.1021/jo301007x. [DOI] [PubMed] [Google Scholar]
- 143.Yamada T, Suzuki K, Hirose T, Furata T, Ueda Y, Kawabata T, Ômura S, Sunazuka T. Organocatalytic Site-Selective Acylation of Avermectin B2a, a Unique Endectocidal Drug. Chem Pharm Bull. 2016;64:856–864. doi: 10.1248/cpb.c16-00205. [DOI] [PubMed] [Google Scholar]
- 144.Egerton JR, Ostlind DA, Blair LS, Eary CH, Suhayda D, Cifelli S, Riek RF, Campbell WC. Avermectins, New Family of Potent Anthelmintic Agents: Efficacy of the B1a Component. Antimicrob Agents Chemother. 1979;15:372–378. doi: 10.1128/aac.15.3.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Õmura S, Crump A. Timeline: The Life and Times of Ivermectin — A Success Story. Nat Rev Microbiol. 2004;2:984–989. doi: 10.1038/nrmicro1048. [DOI] [PubMed] [Google Scholar]
- 146.Hertweck C. Natural Products as Source of Therapeutics against Parasitic Diseases. Angew Chem Int Ed. 2015;54:14622–14624. doi: 10.1002/anie.201509828. [DOI] [PubMed] [Google Scholar]
- 147.The 2015 Nobel Prize in Physiology or Medicine was awarded for efforts towards the discovery of this class of therapeutics.
- 148.Yanagi M, Ninomiya R, Ueda Y, Furuta T, Yamada T, Sunazuka T, Kawabata T. Organocatalytic Site Selective Acylation of 10-Deacetylbaccatin III. Chem Pharm Bull. 2016;64:907–912. doi: 10.1248/cpb.c16-00037. [DOI] [PubMed] [Google Scholar]
- 149.Sun X, Lee H, Lee S, Tan KL. Catalyst Recognition of Cis-1,2-Diols Enables Site-Selective Functionalization of Complex Molecules. Nat Chem. 2013;5:790–795. doi: 10.1038/nchem.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wilcock BC, Uno BE, Bromann GL, Clark MJ, Anderson TM, Burke MD. Electronic Tuning of Site-Selectivity. Nat Chem. 2012;4:996–1003. doi: 10.1038/nchem.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Matsumori N, Sawada Y, Murata M. Mycosamine Orientation of Amphotericin B Controlling Interaction with Ergosterol: Sterol-Dependent Activity of Conformation-Restricted Derivatives with an Amino-Carbonyl Bridge. J Am Chem Soc. 2005;127:10667–10675. doi: 10.1021/ja051597r. [DOI] [PubMed] [Google Scholar]
- 152.Lutz V, Glatthaar J, Würtele C, Serafin M, Hausmann H, Schreiner PR. Structural Analyses of N-Acetylated 4-(Dimethylamino)pyridine (DMAP) Salts. Chem Eur J. 2009;15:8548–8557. doi: 10.1002/chem.200901379. [DOI] [PubMed] [Google Scholar]
- 153.Fischer E. Ueber Die Glucoside Der Alkohole. Eur J Inorg Chem. 1893;26:2400–2412. [Google Scholar]
- 154.Lemieux RU, Hendriks KB, Stick RV, James K. Halide Ion Catalyzed Glycosidation Reactions. Syntheses of Alpha-Linked Disaccharides. J Am Chem Soc. 1975;97:4056–4062. [Google Scholar]
- 155.Wang LX, Davis BG. Realizing the Promise of Chemical Glycobiology. Chem Sci. 2013;4:3381–3394. doi: 10.1039/C3SC50877C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bojarová P, Rosencrantz RR, Elling L, Křen V. Enzymatic Glycosylation of Multivalent Scaffolds. Chem Soc Rev. 2013;42:4774–4797. doi: 10.1039/c2cs35395d. [DOI] [PubMed] [Google Scholar]
- 157.Kobayashi S, Makino A. Enzymatic Polymer Synthesis: An Opportunity for Green Polymer Chemistry. Chem Rev. 2009;109:5288–5353. doi: 10.1021/cr900165z. [DOI] [PubMed] [Google Scholar]
- 158.Hanessian S, Lou B. Stereocontrolled Glycosyl Transfer Reactions with Unprotected Glycosyl Donors. Chem Rev. 2000;100:4443–4463. doi: 10.1021/cr9903454. [DOI] [PubMed] [Google Scholar]
- 159.Lee D, Taylor M. Catalyst-Controlled Regioselective Reactions of Carbohydrate Derivatives. Synthesis. 2012;44:3421–3431. doi: 10.1007/128_2015_656. [DOI] [PubMed] [Google Scholar]
- 160.Pelletier G, Zwicker A, Allen CL, Schepartz A, Miller SJ. Aqueous Glycosylation of Unprotected Sucrose Employing Glycosyl Fluorides in the Presence of Calcium Ion and Trimethylamine. J Am Chem Soc. 2016;138:3175–3182. doi: 10.1021/jacs.5b13384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Beale TM, Taylor MS. Synthesis of Cardiac Glycoside Analogs by Catalyst-Controlled, Regioselective Glycosylation of Digitoxin. Org Lett. 2013:1358–1361. doi: 10.1021/ol4003042. [DOI] [PubMed]
- 162.Lee D, Williamson CL, Chan L, Taylor MS. Regioselective, Borinic Acid-Catalyzed Monoacylation, Sulfonylation and Alkylation of Diols and Carbohydrates: Expansion of Substrate Scope and Mechanistic Studies. J Am Chem Soc. 2012;134:8260–8267. doi: 10.1021/ja302549c. [DOI] [PubMed] [Google Scholar]
- 163.Lee D, Taylor MS. Borinic Acid-Catalyzed Regioselective Acylation of Carbohydrate Derivatives. J Am Chem Soc. 2011;133:3724–3727. doi: 10.1021/ja110332r. [DOI] [PubMed] [Google Scholar]
- 164.Chan L, Taylor MS. Regioselective Alkylation of Carbohydrate Derivatives Catalyzed by a Diarylborinic Acid Derivative. Org Lett. 2011;13:3090–3093. doi: 10.1021/ol200990e. [DOI] [PubMed] [Google Scholar]
- 165.Gouliaras C, Lee D, Chan L, Taylor MS. Regioselective Activation of Glycosyl Acceptors by a Diarylborinic Acid-Derived Catalyst. J Am Chem Soc. 2011;133:13926–13929. doi: 10.1021/ja2062715. [DOI] [PubMed] [Google Scholar]
- 166.Gillingham D, Fei N. Catalytic X–H Insertion Reactions Based on Carbenoids. Chem Soc Rev. 2013;42:4918–4931. doi: 10.1039/c3cs35496b. [DOI] [PubMed] [Google Scholar]
- 167.Maas G. Ruthenium-Catalysed Carbenoid Cyclopropanation Reactions with Diazo Compounds. Chem Soc Rev. 2004;33:183–190. doi: 10.1039/b309046a. [DOI] [PubMed] [Google Scholar]
- 168.Davies HML, Denton JR. Application of Donor/Acceptor-Carbenoids to the Synthesis of Natural Products. Chem Soc Rev. 2009;38:3061–3071. doi: 10.1039/b901170f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Davies HML, Morton D. Guiding Principles for Site Selective and Stereoselective Intermolecular C–H Functionalization by Donor/acceptor Rhodium Carbenes. Chem Soc Rev. 2011;40:1857–1869. doi: 10.1039/c0cs00217h. [DOI] [PubMed] [Google Scholar]
- 170.Davies HML, Beckwith REJ. Catalytic Enantioselective C–H Activation by Means of Metal-Carbenoid-Induced C–H Insertion. Chem Rev. 2003;103:2861–2903. doi: 10.1021/cr0200217. [DOI] [PubMed] [Google Scholar]
- 171.Ball ZT. Designing Enzyme-like Catalysts: A Rhodium(II) Metallopeptide Case Study. Acc Chem Res. 2013;46:560–570. doi: 10.1021/ar300261h. [DOI] [PubMed] [Google Scholar]
- 172.Peddibhotla S, Dang Y, Liu JO, Romo D. Simultaneous Arming and Structure/Activity Studies of Natural Products Employing O–H Insertions: An Expedient and Versatile Strategy for Natural Products-Based Chemical Genetics. J Am Chem Soc. 2007;129:12222–12231. doi: 10.1021/ja0733686. [DOI] [PubMed] [Google Scholar]
- 173.Chamni S, He QL, Dang Y, Bhat S, Liu JO, Romo D. Diazo Reagents with Small Steric Footprints for Simultaneous Arming/SAR Studies of Alcohol-Containing Natural Products via O–H Insertion. ACS Chem Biol. 2011;6:1175–1181. doi: 10.1021/cb2002686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Shul’pin GB. Selectivity Enhancement in Functionalization of C–H Bonds: A Review. Org Biomol Chem. 2010;8:4217–4228. doi: 10.1039/c004223d. [DOI] [PubMed] [Google Scholar]
- 175.Gutekunst WR, Baran PS. C–H functionalization logic in total synthesis. Chem Soc Rev. 2011;40:1976–1991. doi: 10.1039/c0cs00182a. [DOI] [PubMed] [Google Scholar]
- 176.Lu H, Zhang XP. Catalytic C–H Functionalization by Metalloporphyrins: Recent Developments and Future Directions. Chem Soc Rev. 2011;40:1899–1909. doi: 10.1039/c0cs00070a. [DOI] [PubMed] [Google Scholar]
- 177.McMurray L, O’Hara F, Gaunt MJ. Recent Developments in Natural Product Synthesis Using Metal-Catalysed C–H Bond Functionalisation. Chem Soc Rev. 2011;40:1885–1898. doi: 10.1039/c1cs15013h. [DOI] [PubMed] [Google Scholar]
- 178.White MC. Adding Aliphatic C–H Bond Oxidations to Synthesis. Science. 2012;335:807–809. doi: 10.1126/science.1207661. [DOI] [PubMed] [Google Scholar]
- 179.Dick AR, Sanford MS. Transition Metal Catalyzed Oxidative Functionalization of Carbon-Hydrogen Bonds. Tetrahedron. 2006;62:2439–2463. [Google Scholar]
- 180.Hartwig JF, Larsen MA. Undirected, Homogeneous C–H Bond Functionalization: Challenges and Opportunities. ACS Cent Sci. 2016;2:281–292. doi: 10.1021/acscentsci.6b00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Groves JT. Enzymatic C–H Bond Activation: Using Push to Get Pull. Nat Chem. 2014;6:89–91. doi: 10.1038/nchem.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Munro AW, Girvan HM, McLean KJ. Variations on a (T)heme - Novel Mechanisms, Redox Partners and Catalytic Functions in the Cytochrome P450 Superfamily. Nat Prod Rep. 2007;24:585–609. doi: 10.1039/b604190f. [DOI] [PubMed] [Google Scholar]
- 183.Denisov IG, Makris TM, Sligar SG, Schlichting I. Structure and Chemistry of Cytochrome P450. Chem Rev. 2005;105:2253–2277. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 184.Fasan R. Tuning P450 Enzymes as Oxidation Catalysts. ACS Catal. 2012;2:647–666. [Google Scholar]
- 185.Lewis JC, Coelho PS, Arnold FH. Enzymatic Functionalization of Carbon-Hydrogen Bonds. Chem Soc Rev. 2011;40:2003–2021. doi: 10.1039/c0cs00067a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Nelson DR. The Cytochrome p450 Homepage. Hum Genomics. 2009;4:59–65. doi: 10.1186/1479-7364-4-1-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Ajikumar PK, Xiao WH, Tyo KEJ, Wang Y, Simeon F, Leonard E, Mucha O, Phon TH, Pfeifer B, Stephanopoulos G. Isoprenoid Pathway Optimization for Taxol Precursor Overproduction in Escherichia Coli. Science. 2010;330:70–74. doi: 10.1126/science.1191652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Croteau R, Ketchum REB, Long RM, Kaspera R, Wildung MR. Taxol Biosynthesis and Molecular Genetics. Phytochem Rev. 2006;5:75–97. doi: 10.1007/s11101-005-3748-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Chau M, Jennewein S, Walker K, Croteau R. Taxol Biosynthesis: Molecular Clong and Characterization of a Cytochrome P450 Taxoid 7B-Hydroxylase. Chem Biol. 2004;11:663–672. doi: 10.1016/j.chembiol.2004.02.025. [DOI] [PubMed] [Google Scholar]
- 190.Ortiz De Montellano PR. Hydrocarbon Hydroxylation by Cytochrome P450 Enzymes. Chem Rev. 2010;110:932–948. doi: 10.1021/cr9002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kille S, Zilly FE, Acevedo JP, Reetz MT. Regio- and Stereoselectivity of P450-Catalysed Hydroxylation of Steroids Controlled by Laboratory Evolution. Nat Chem. 2011;3:738–743. doi: 10.1038/nchem.1113. [DOI] [PubMed] [Google Scholar]
- 192.Breslow R, Baldwin S, Flechtner T, Kalicky P, Liu S, Washburn W. Remote Oxidation of Steroids by Photolysis of Attached Benzophenone Groups. J Am Chem Soc. 1973;95:3251–3262. doi: 10.1021/ja00791a031. [DOI] [PubMed] [Google Scholar]
- 193.Breslow R. Biomimetic Control of Chemical Selectivity. Acc Chem Res. 1980;13:170–177. [Google Scholar]
- 194.Wong MK, Chung NW, He L, Wang XC, Yan Z, Tang YC, Yang D. Investigation on the Regioselectivities of Intramolecular Oxidation of Unactivated C–H Bonds by Dioxiranes Generated in Situ. J Org Chem. 2003;68:6321–6328. doi: 10.1021/jo0347011. [DOI] [PubMed] [Google Scholar]
- 195.Michaudel Q, Journot G, Regueiro-Ren A, Goswami A, Guo Z, Tully TP, Zou L, Ramabhadran RO, Houk KN, Baran PS. Improving Physical Properties via C–H Oxidation: Chemical and Enzymatic Approaches. Angew Chem Int Ed. 2014;53:12091–12096. doi: 10.1002/anie.201407016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Bigi MA, Liu P, Zou L, Houk KN, White MC. Cafestol to Tricalysiolide B and Oxidized Analogues: Biosynthetic and Derivatization Studies Using Non-Heme Iron Catalyst Fe(PDP) Synlett. 2012;23:2768–2772. doi: 10.1055/s-0032-1317708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Chen MS, White MC. A Predictably Selective Aliphatic C–H Hydroxylation Reaction. Science. 2007;318:783–787. doi: 10.1126/science.1148597. [DOI] [PubMed] [Google Scholar]
- 198.Chen MS, White MC. Combined Effects on Selectivity in Fe-Catalyzed Methylene Oxidation. Science. 2010;327:566–571. doi: 10.1126/science.1183602. [DOI] [PubMed] [Google Scholar]
- 199.Lee S, Fuchs PL. An Efficient C–H Oxidation Protocol for α-Hydroxylation of Cyclic Steroidal Ethers. Org Lett. 2004;6:1437–1440. doi: 10.1021/ol049712a. [DOI] [PubMed] [Google Scholar]
- 200.Chen K, Eschenmoser A, Baran PS. Strain Release in C–H Bond Activation? Angew Chem Int Ed. 2009;48:9705–9708. doi: 10.1002/anie.200904474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Bigi MA, Reed SA, White MC. Directed Metal (Oxo) Aliphatic C–H Hydroxylations: Overriding Substrate Bias. J Am Chem Soc. 2012;134:9721–9726. doi: 10.1021/ja301685r. [DOI] [PubMed] [Google Scholar]
- 202.Simmons EM, Hartwig JF. Catalytic Functionalization of Unactivated Primary C–H Bonds Directed by an Alcohol. Nature. 2012;483:70–73. doi: 10.1038/nature10785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Voigt B, Porzel A, Golsch D, Adam W, Adam G. Regioselective Oxyfunctionalization of Brassinosteroids by Methyl(trifluoromethyl)dioxirane: Synthesis of 25-Hydroxy-Brassinolide and 25-Hydroxy-24-Epibrassinolide by Direct C–H Insertion. Tetrahedron. 1996;52:10653–10658. [Google Scholar]
- 204.McNeill E, Du Bois J. Catalytic C–H Oxidation by a Triazamacrocyclic Ruthenium Complex. Chem Sci. 2012;3:1810–1813. [Google Scholar]
- 205.Bovicelli P, Gambacorta A, Lupattelli P, Mincione E. A Highly Regio- and Stereoselective C5 Oxyfunctionalization of Coprostane Steroids by Dioxiranes: An Improved Access to Progestogen and Androgen Hormones. Tetrahedron Lett. 1992;33:7411–7412. [Google Scholar]
- 206.Amone A, Cavicchioli M, Montanan V, Resnati G. Direct Oxyfunctionalization at Unactivated Sites. Synthesis of 5b-Hydroxysteroids by Perfluorodialkyloxaziridines. J Org Chem. 1994;59:5511–5513. [Google Scholar]
- 207.Lee JS, Cao H, Fuchs PL. Ruthenium-Catalyzed Mild C–H Oxyfunctionalization of Cyclic Steroidal Ethers. J Org Chem. 2007;72:5820–5823. doi: 10.1021/jo070382s. [DOI] [PubMed] [Google Scholar]
- 208.Wender PA, Hilinski MK, Mayweg AVW. Late-Stage Intermolecular CH Activation for Lead Diversification: A Highly Chemoselective Oxyfunctionalization of the C-9 Position of Potent Bryostatin Analogues. Org Lett. 2005;7:79–82. doi: 10.1021/ol047859w. [DOI] [PubMed] [Google Scholar]
- 209.Schreiber JV, Eschenmoser A. Uber Die Relative Geschwindigkeit Der Chromosaureoxydation Sekundarer, Alicyclischer Alkohole. Helv Chim Acta. 1955;38:1529–1536. [Google Scholar]
- 210.Wang D, Shuler WG, Pierce CJ, Hilinski MK. An Iminium Salt Organocatalyst for Selective Aliphatic C–H Hydroxylation. Org Lett. 2016;18:3826–3829. doi: 10.1021/acs.orglett.6b01832. [DOI] [PubMed] [Google Scholar]
- 211.Biscoe MR, Breslow R. Oxaziridinium Salts as Hydrophobic Epoxidation Reagents: Remarkable Hydrophobically-Directed Selectivity in Olefin Epoxidation. J Am Chem Soc. 2005;127:10812–10813. doi: 10.1021/ja053660i. [DOI] [PubMed] [Google Scholar]
- 212.Adams AM, Du Bois J. Organocatalytic C–H Hydroxylation with Oxone® Enabled by an Aqueous Fluoroalcohol Solvent System. Chem Sci. 2014;5:656–659. [Google Scholar]
- 213.García-Granados A, López PE, Melguizo E, Parra A, Simeó Y. Remote Hydroxylation of Methyl Groups by Regioselective Cyclopalladation. Partial Synthesis of Hyptatic Acid-A. J Org Chem. 2007;72:3500–3509. doi: 10.1021/jo070116e. [DOI] [PubMed] [Google Scholar]
- 214.See YY, Herrmann AT, Aihara Y, Baran PS. Scalable C–H Oxidation with Copper: Synthesis of Polyoxypregnanes. J Am Chem Soc. 2015;137:13776–13779. doi: 10.1021/jacs.5b09463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Schönecker B, Lange C, Zheldakova T, Günther W, Görls H, Vaughan G. Copper-mediated regio- and stereoselective 12β-hydroxylation of steroids with molecular oxygen and an unexpected 12β-chlorination. Tetrehedron. 2005;61:103–114. [Google Scholar]
- 216.Horn EJ, Rosen BR, Chen Y, Tang J, Chen K, Eastgate MD, Baran PS. Scalable and Sustainable Electrochemical Allylic C–H Oxidation. Nature. 2016;533:77–81. doi: 10.1038/nature17431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Waser J, Gaspar B, Nambu H, Carreira EM. Hydrazines and Azides via the Metal-Catalyzed Hydrohydrazination and Hydroazidation of Olefins. J Am Chem Soc. 2006;128:11693–11712. doi: 10.1021/ja062355+. [DOI] [PubMed] [Google Scholar]
- 218.Concepción JI, Francisco CG, Hernández R, Salazar JA, Suárez E. Intramolecular Hydrogen Abstraction. Iodosobenzene Diacetate, an Efficient and Convenient Reagent for Alkoxy Radical Generation. Tetrahedron Lett. 1984;25:1953–1956. [Google Scholar]
- 219.Breslow R, Zhang X, Huang Y. Selective Catalytic Hydroxylation of a Steroid by an Artificial Cytochrome P-450 Enzyme. J Am Chem Soc. 1997;119:4535–4536. [Google Scholar]
- 220.Yang J, Breslow R. Selective Hydroxylation of a Steroid at C-9 by an Artificial Cytochrome P-450. Angew Chem Int Ed. 2000;39:2692–2694. doi: 10.1002/1521-3773(20000804)39:15<2692::aid-anie2692>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 221.Fang Z, Breslow R. Metal Coordination-Directed Hydroxylation of Steroids with a Novel Artificial P-450 Catalyst. Org Lett. 2006;8:251–254. doi: 10.1021/ol052589i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Su B, Cao ZC, Shi ZJ. Exploration of Earth-Abundant Transition Metals (Fe, Co, and Ni) as Catalysts in Unreactive Chemical Bond Activations. Acc Chem Res. 2015;48:886–896. doi: 10.1021/ar500345f. [DOI] [PubMed] [Google Scholar]
- 223.Noedl H, Se Y, Schaecher K, Smith BL, Socheat D, Fukuda MM. Evidence of Artemisinin-Resistant Malaria in Western Cambodia. N Engl J Med. 2008;359:2619–2620. doi: 10.1056/NEJMc0805011. [DOI] [PubMed] [Google Scholar]
- 224.Paddon CJ, Westfall PJ, Pitera DJ, Benjamin K, Fisher K, McPhee D, Leavell MD, Tai A, Main A, Eng D, et al. High-Level Semi-Synthetic Production of the Potent Antimalarial Artemisinin. Nature. 2013;496:528–532. doi: 10.1038/nature12051. [DOI] [PubMed] [Google Scholar]
- 225.Faurant C. From Bark to Weed: The History of Artemisinin. Parasite. 2011;18:215–218. doi: 10.1051/parasite/2011183215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Tshuva EY, Lippard SJ. Synthetic Models for Non-Heme Carboxylate-Bridged Diiron Metalloproteins: Strategies and Tactics. Chem Rev. 2004;104:987–1012. doi: 10.1021/cr020622y. [DOI] [PubMed] [Google Scholar]
- 227.Vermeulen NA, Chen MS, Christina White M. The Fe(PDP)-Catalyzed Aliphatic C–H Oxidation: A Slow Addition Protocol. Tetrahedron. 2009;65:3078–3084. [Google Scholar]
- 228.Bigi MA, Reed SA, White MC. Diverting Non-Haem Iron Catalysed Aliphatic C–H Hydroxylations towards Desaturations. Nat Chem. 2011;3:216–222. doi: 10.1038/nchem.967. [DOI] [PubMed] [Google Scholar]
- 229.Walsh AD. Structures of Ethylene Oxide and Cyclopropane. Nature. 1947;159:712–713. doi: 10.1038/159712a0. [DOI] [PubMed] [Google Scholar]
- 230.Wiberg KB. Bent Bonds in Organic Compounds. Acc Chem Res. 1996;29:229–234. [Google Scholar]
- 231.Farooq A, Taharaa S. Oxidative Metabolism of Ambrox and Sclareolide by Botrytis Cinerea. Z Naturforsch C. 2015;55:341–346. doi: 10.1515/znc-2000-5-608. [DOI] [PubMed] [Google Scholar]
- 232.Ata A, Conci LJ, Betteridge J, Orhan I, Sener B. Novel Microbial Transformations of Sclareolide. Chem Pharm Bull. 2007;55:118–123. doi: 10.1248/cpb.55.118. [DOI] [PubMed] [Google Scholar]
- 233.Gormisky PE, White MC. Catalyst-Controlled Aliphatic C–H Oxidations with a Predictive Model for Site-Selectivity. J Am Chem Soc. 2013;135:14052–14055. doi: 10.1021/ja407388y. [DOI] [PubMed] [Google Scholar]
- 234.Howell JM, Feng K, Clark JR, Trzepkowski LJ, White MC. Remote Oxidation of Aliphatic C–H Bonds in Nitrogen–Containing Molecules. J Am Chem Soc. 2015;137:14590–14593. doi: 10.1021/jacs.5b10299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Gómez L, Garcia-Bosch I, Company A, Benet-Buchholz J, Polo A, Sala X, Ribas X, Costas M. Stereospecific C–H Oxidation with H2O2 Catalyzed by a Chemically Robust Site-Isolated Iron Catalyst. Angew Chem Int Ed. 2009;48:5720–5723. doi: 10.1002/anie.200901865. [DOI] [PubMed] [Google Scholar]
- 236.Gomez L, Canta M, Font D, Prat I, Ribas X, Costas M. Regioselective Oxidation of Nonactivated Alkyl C–H Groups Using Highly Structured Non-Heme Iron Catalysts. J Org Chem. 2013;78:1421–1433. doi: 10.1021/jo302196q. [DOI] [PubMed] [Google Scholar]
- 237.Yuan C, Jin Y, Wilde NC, Baran PS. Short, Enantioselective Total Synthesis of Highly Oxidized Taxanes. Angew Chem Int Ed. 2016;55:8280–8284. doi: 10.1002/anie.201602235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Wagner C, Omari ME, König GM. Biohalogenation: Nature’s Way to Synthesize Halogenated Metabolites. J Nat Prod. 2009;72:540–553. doi: 10.1021/np800651m. [DOI] [PubMed] [Google Scholar]
- 239.Müller K, Faeh C, Diederich F. Fluorine in Pharmaceuticals: Looking beyond Intuition. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 240.Furuya T, Kamlet AS, Ritter TS. Catalysis for Fluorination and Trifluoromethylation. Nature. 2011;473:470–477. doi: 10.1038/nature10108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Chung WJ, Vanderwal CD. Stereoselective Halogenation in Natural Product Synthesis. Angew Chem Int Ed. 2016;55:4396–4434. doi: 10.1002/anie.201506388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Liu W, Groves JT. Manganese Porphyrins Catalyze Selective C–H Bond Halogenations. J Am Chem Soc. 2010;132:12847–12849. doi: 10.1021/ja105548x. [DOI] [PubMed] [Google Scholar]
- 243.Liu W, Huang X, Cheng MJ, Nielsen RJ, Goddard WA, III, Groves JT. Oxidative Aliphatic C–H Fluorination with Fluoride Ion Catalyzed by a Manganese Poyrphyrin. Science. 2012;337:1322–1325. doi: 10.1126/science.1222327. [DOI] [PubMed] [Google Scholar]
- 244.Schmidt VA, Quinn RK, Brusoe AT, Alexanian EJ. Site-Selective Aliphatic C–H Bromination Using N-Bromoamides and Visible Light. J Am Chem Soc. 2014;136:14389–14392. doi: 10.1021/ja508469u. [DOI] [PubMed] [Google Scholar]
- 245.Majetich G, Wheless K. Remote Intramolecular free Radical Functionalizations: An Update. Tetrahedron. 1995;51:7095–7129. [Google Scholar]
- 246.Quinn RK, Könst ZA, Michalak SE, Schmidt Y, Szklarski AR, Flores AR, Nam S, Horne DA, Vanderwal CD, Alexanian EJ. Site-Selective Aliphatic C–H Chlorination Using N-Chloroamides Enables a Synthesis of Chlorolissoclimide. J Am Chem Soc. 2016;138:696–702. doi: 10.1021/jacs.5b12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Zhou CY, Li J, Peddibhotla S, Romo D. Mild Arming and Derivatization of Natural Products via an In(OTf) 3-Catalyzed Arene Iodination. Org Lett. 2010;12:2104–2107. doi: 10.1021/ol100587j. [DOI] [PubMed] [Google Scholar]
- 248.Snyder SA, Gollner A, Chiriac MI. Regioselective Reactions for Programmable Resveratrol Oligomer Synthesis. Nature. 2011;474:461–466. doi: 10.1038/nature10197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Snyder SA, Treitler DS, Brucks AP. Simple Reagents for Direct Halonium-Induced Polyene Cyclizations. J Am Chem Soc. 2010;132:14303–14314. doi: 10.1021/ja106813s. [DOI] [PubMed] [Google Scholar]
- 250.Gustafson JL, Lim D, Barrett KT, Miller SJ. Synthesis of Atropisomerically Defined, Highly Substituted Biaryl Scaffolds through Catalytic Enantioselective Bromination and Regioselective Cross-Coupling. Angew Chem Int Ed. 2011;50:5125–5129. doi: 10.1002/anie.201101147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Barrett KT, Miller SJ. Enantioselective Synthesis of Atropisomeric Benzamides through Peptide-Catalyzed Bromination. J Am Chem Soc. 2013;135:2963–2966. doi: 10.1021/ja400082x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Diener ME, Metrano AJ, Kusano S, Miller SJ. Enantioselective Synthesis of 3-Arylquinazolin-4(3H)-Ones via Peptide-Catalyzed Atroposelective Bromination. J Am Chem Soc. 2015;137:12369–12377. doi: 10.1021/jacs.5b07726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Metrano AJ, Abascal NC, Mercado BQ, Paulson EK, Miller SJ. Structural Studies of β-Turn-Containing Peptide Catalysts for Atroposelective Quinazolinone Bromination. Chem Commun. 2016;52:4816–4819. doi: 10.1039/c6cc01428c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Mori K, Ichikawa Y, Kobayashi M, Shibata Y, Yamanaka M, Akiyama T. Enantioselective Synthesis of Multisubstituted Biaryl Skeleton by Chiral Phosphoric Acid Catalyzed Desymmetrization/Kinetic Resolution Sequence. J Am Chem Soc. 2013;135:3964–3970. doi: 10.1021/ja311902f. [DOI] [PubMed] [Google Scholar]
- 255.Miyaji R, Asano K, Matsubara S. Bifunctional Organocatalysts for the Enantioselective Synthesis of Axially Chiral Isoquinoline N-Oxides. J Am Chem Soc. 2015;137:6766–6769. doi: 10.1021/jacs.5b04151. [DOI] [PubMed] [Google Scholar]
- 256.Pathak TP, Miller SJ. Site-Selective Bromination of Vancomycin. J Am Chem Soc. 2012;134:6120–6123. doi: 10.1021/ja301566t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Pathak TP, Miller SJ. Chemical Tailoring of Teicoplanin with Site-Selective Reactions. J Am Chem Soc. 2013;135:8415–8422. doi: 10.1021/ja4038998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Perkins HR, Hill M, Chicago N. The Specificity of Combination between Ristocetins and Peptides Related to Bacterial Cell Wall Mucopeptide Precursors. Biochem J. 1971;124:845–852. doi: 10.1042/bj1240845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Wadzinski TJ, Gea KD, Miller SJ. A Stepwise Dechlorination/Cross-Coupling Strategy to Diversify the Vancomycin “in-Chloride”. Bioorg Med Chem Lett. 2016;26:1025–1028. doi: 10.1016/j.bmcl.2015.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Spencer T. The Squalene Dioxide Pathway of Steroid Biosynthesis. Acc Chem Res. 1994;57:83–90. [Google Scholar]
- 261.Thibodeaux CJ, Chang WC, Liu HW. Enzymatic Chemistry of Cyclopropane, Epoxide, and Aziridine Biosynthesis. Chem Rev. 2012;112:1681–1709. doi: 10.1021/cr200073d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Lichtor PA, Miller SJ. Combinatorial Evolution of Site- and Enantioselective Catalysts for Polyene Epoxidation. Nat Chem. 2012;4:990–995. doi: 10.1038/nchem.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Lichtor PA, Miller SJ. Experimental Lineage and Functional Analysis of a Remotely Directed Peptide Epoxidation Catalyst. J Am Chem Soc. 2014;136:5301–5308. doi: 10.1021/ja410567a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Peris G, Jakobsche CE, Miller SJ. Aspartate-Catalyzed Asymmetric Epoxidation Reactions. J Am Chem Soc. 2007;9:8710–8711. doi: 10.1021/ja073055a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Kolundzic F, Noshi MN, Tjandra M, Movassaghi M, Miller SJ. Chemoselective and Enantioselective Oxidation of Indoles Employing Aspartyl Peptide Catalysts. J Am Chem Soc. 2011;133:9104–9111. doi: 10.1021/ja202706g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Abascal NC, Lichtor PA, Giuliano MW, Miller SJ. Function-Oriented Investigations of a Peptide-Based Catalyst That Mediates Enantioselective Allylic Alcohol Epoxidation. Chem Sci. 2014;5:4504–4511. doi: 10.1039/C4SC01440E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Mercado-Marin EV, Garcia-Reynaga P, Romminger S, Pimenta EF, Romney DK, Lodewyk MW, Williams DE, Andersen RJ, Miller SJ, Tantillo DJ, et al. Total Synthesis and Isolation of Citrinalin and Cyclopiamine Congeners. Nature. 2014;509:318–324. doi: 10.1038/nature13273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Romney DK, Colvin SM, Miller SJ. Catalyst Control over Regio- and Enantioselectivity in Baeyer-Villiger Oxidations of Functionalized Ketones. J Am Chem Soc. 2014;136:14019–14022. doi: 10.1021/ja508757g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Alford JS, Abascal NC, Shugrue CR, Colvin SM, Romney DK, Miller SJ. Aspartyl Oxidation Catalysts That Dial In Functional Group Selectivity, along with Regio- and Stereoselectivity. ACS Cent Sci. 2016;2:733–739. doi: 10.1021/acscentsci.6b00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Fiori KW, Du Bois J. Catalytic Intermolecular Amination of C–H Bonds: Method Development and Mechanistic Insights. J Am Chem Soc. 2007;129:562–568. doi: 10.1021/ja0650450. [DOI] [PubMed] [Google Scholar]
- 271.Roizen JL, Zalatan DN, Du Bois J. Selective Intermolecular Amination of C–H Bonds at Tertiary Carbon Centers. Angew Chem Int Ed. 2013;52:11343–11346. doi: 10.1002/anie.201304238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Li J, Cisar JS, Zhou CY, Vera B, Williams H, Rodríguez AD, Cravatt BF, Romo D. Simultaneous Structure-Activity Studies and Arming of Natural Products by C–H Amination Reveal Cellular Targets of Eupalmerin Acetate. Nat Chem. 2013;5:510–517. doi: 10.1038/nchem.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Huang X, Bergsten TM, Groves JT. Manganese-Catalyzed Laste-Stage Aliphatic C–H Aziation. J Am Chem Soc. 2015;137:5300–5303. doi: 10.1021/jacs.5b01983. [DOI] [PubMed] [Google Scholar]
- 274.Sharma A, Hartwig JF. Metal-Catalysed Azidation of Tertiary C–H Bonds Suitable for Late-Stage Functionalization. Nature. 2015;517:600–604. doi: 10.1038/nature14127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Karimov RR, Sharma A, Hartwig JF. Late Stage Azidation of Complex Molecules. ACS Cent Sci. 2016;2:715–724. doi: 10.1021/acscentsci.6b00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Czaplyski WL, Na CG, Alexanian EJ. C–H Xanthylaton: A Synthetic Platform for Alkane Functionalization. J Am Chem Soc. 2016;138:13854–13857. doi: 10.1021/jacs.6b09414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Quiclet-Sire B, Zard SZ. Powerful Carbon-Carbon Bond Forming Reactions Based on a Novel Radical Exchange Process. Chem Eur J. 2006;12:6002–6016. doi: 10.1002/chem.200600510. [DOI] [PubMed] [Google Scholar]
- 278.Mukherjee S, Maji B, Tlahuext-Aca A, Glorius F. Visible Light-Promoted Activation of Unactivated C(sp3) H Bonds and Its Selective Trifluoromethylthiolation. J Am Chem Soc. 2016;138:16200–16203. doi: 10.1021/jacs.6b09970. [DOI] [PubMed] [Google Scholar]
- 279.Böhm HJ, Banner D, Bendels S, Kansy M, Kuhn B, Müller K, Obst-Sander U, Stahl M. Fluorine in Medicinal Chemistry. ChemBioChem. 2004;5:637–643. doi: 10.1002/cbic.200301023. [DOI] [PubMed] [Google Scholar]
- 280.Swallow S. Fluorine in Medicinal Chemistry. Prog Med Chem. 2015;54:65–133. doi: 10.1016/bs.pmch.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 281.Sladojevich F, Arlow SI, Tang P, Ritter T. Late-Stage Deoxy Fluorination of Alcohols with PhenoFluor. J Am Chem Soc. 2013;135:2470–2473. doi: 10.1021/ja3125405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Kirk KL. Fluorination in Medicinal Chemistry: Methods, Strategies, and Recent Developments. Org Process Res Dev. 2008;12:305–321. [Google Scholar]
- 283.Singh RP, Shreeve JM. Recent Advances in Nucleophilic Fluorination Reactions of Organic Compounds- Using Deoxofluor and DAST. Synthesis. 2002:2561–2578.
- 284.Tang P, Wang W, Ritter T. Deoxyfluorination of Phenols. J Am Chem Soc. 2011;133:11482–11484. doi: 10.1021/ja2048072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.He J, Hamann LG, Davies HML, Beckwith REJ. Late-Stage C–H Functionalization of Complex Alkaloids and Drug Molecules via Intermolecular Rhodium-Carbenoid Insertion. Nat Commun. 2015;6:5943. doi: 10.1038/ncomms6943. [DOI] [PubMed] [Google Scholar]
- 286.Guo S, Cong F, Wang L, Tang P. Asymmetric Silver-Catalysed Intermolecular Bromotrifluoromethoxylation of Alkenes with a New Trifluoromethoxylation Reagent. Nat Chem. 2017 doi: 10.1038/nchem.271. [DOI] [PubMed]
- 287.Robles O, Serna-Saldívar SO, Gutiérrez-Uribe JA, Romo D. Cyclopropanations of Olefin-Containing Natural Products for Simultaneous Arming and Structure Activity Studies. Org Lett. 2012;14:1394–1397. doi: 10.1021/ol300105q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Carosso S, Miller MJ. Nitroso Diels-Alder (NDA) Reaction as an Efficient Tool for the Functionalization of Diene-Containing Natural Products. Org Biomol Chem. 2014;12:7445–7468. doi: 10.1039/c4ob01033g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Ruan B, Pong K, Jow F, Bowlby M, Crozier Ra, Liu D, Liang S, Chen Y, Mercado ML, Feng X, et al. Binding of Rapamycin Analogs to Calcium Channels and FKBP52 Contributes to Their Neuroprotective Activities. Proc Natl Acad Sci U S A. 2008;105:33–38. doi: 10.1073/pnas.0710424105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Yoon TP, Jacobsen EN. Privileged Chiral Catalysts. Science. 2003;299:1691–1694. doi: 10.1126/science.1083622. [DOI] [PubMed] [Google Scholar]
- 291.Burke MD, Schreiber SL. A Planning Strategy for Diversity-Oriented Synthesis. Angew Chem Int Ed. 2004;43:46–58. doi: 10.1002/anie.200300626. [DOI] [PubMed] [Google Scholar]
- 292.Schreiber SL. Target-Oriented and Diversity-Oriented Organic Synthesis in Drug Discovery. Science. 2000;287:1964–1969. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]