

Abstract
Embryonic stem cells maintain pluripotency through countless mitoses. A recent report shows that the transcription factor Esrrb remains bound to chromatin during mitosis, including at regulatory regions that support pluripotency. Mitotic chromatin occupancy by Esrrb might stabilize the defining transcriptional programmes of embryonic stem cells through cell division.
During mitosis the metazoan nucleus is disassembled, chromosomes condense, transcription is transiently silenced, and many — but not all — transcription factors dissociate from DNA1. However, developmental processes often require that cells stably propagate cell-type-specific gene expression programmes through cell division. This raises the question of how cells maintain these programmes in the face of such mitotic disruption to nuclear processes.
One potential answer to this question is that certain molecular features associated with transcriptional regulation, such as open chromatin configurations at gene promoters2,3, histone modifications4 and some transcription factors1 remain present on mitotic chromosomes. These features of mitotic chromatin have been hypothesized to serve as molecular ‘bookmarks’ that direct the cell-type-appropriate ‘reading’ of the genome by the transcription machinery after mitosis. Transcription factors that can bind the genome during mitosis include those that are required for tissue-specific development5,6, leading to the hypothesis that their mitotic occupancy may preserve memory of tissue-specific transcriptional control through mitosis5,6. However, testing whether the binding of any transcription factor specifically during mitosis has any influence on cell phenotype (beyond the functions of such factors during interphase) remains an unmet challenge in the field. Embryonic stem cells in particular are remarkable in their ability to preserve transcriptional patterns through mitosis as they can self-renew to maintain their pluripotency throughout countless cell divisions. However, how this is accomplished has remained largely unknown. In this issue of Nature Cell Biology, Festuccia et al.7 bring us one step closer to understanding this process, by reporting that the transcription factor Esrrb, an orphan nuclear receptor that supports pluripotency8,9, retains its bulk association with chromosomes during all phases of mitosis. In contrast, Nanog, another pluripotency transcription factor, is excluded from mitotic chromatin.
The authors demonstrated the mitotic chromatin association of Esrrb with various techniques, including immunofluorescence microscopy, live-cell imaging of Esrrb fusion proteins using different fluorophore-conjugated tags, and tagging Esrrb at its endogenous locus. Together, these results exclude the possibility of localization artefacts due to chemical fixation, overexpression, or the specific type of fluorescent tag used, and highlight the specificity of mitotic chromatin association by Esrrb. The authors showed that mutating the DNA-binding domain of Esrrb reduced, but did not completely eliminate, its microscopic colocalization with mitotic chromatin. Thus, although these data support that Esrrb directly contacts DNA during mitosis, they also indicate that some Esrrb molecules are enriched on mitotic chromatin independently of direct DNA binding through an as yet unexplored mechanism. Esrrb molecules in the vicinity of chromatin were more mobile during mitosis than in interphase, as measured by fluorescence recovery after photobleaching (FRAP) experiments. Increased mobility near mitotic chromatin appears to be a property shared by other transcription factors previously examined by FRAP6,10,11 and further supports the idea that mitotic chromosome condensation does not render DNA inaccessible to macromolecules2,3.
To identify the genomic regions to which Esrrb binds during mitosis Festuccia et al. used chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq), and found that Esrrb retains some degree of mitotic binding at ~10–15% of its interphase binding sites, with reduced signals in mitosis. This result is very similar to other studies showing genome-wide transcription factor binding during mitosis5,6,11–13. An important question is what provides the specificity that distinguishes sites bound by Esrrb in mitosis, from those that are not. The answer to this remains elusive for Esrrb, as well as for all transcription factors examined so far. However, Festuccia et al.7 uncovered an important clue by showing that ectopically integrated arrays of Esrrb binding sites are sufficient to reproduce Esrrb mitotic chromatin binding. This finding may facilitate future attempts at testing the contribution of DNA sequence in the discrimination between mitotically bound and vacated sites. Of the chromatin features examined, the authors found that the levels of histone 3 lysine 27 acetylation (H3K27ac) and of RNA polymerase II binding in asynchronous cells were the most strongly associated with Esrrb mitotic occupancy. It is likely that other chromatin features also contribute to the specificity of Esrrb mitotic binding.
To examine the gene regulatory consequences of mitotic Esrrb binding, embryonic stem cells were grown in the presence or absence of inducible Esrrb for 24 h, and approximately 500 genes were identified to be responsive to Esrrb induction. The authors isolated cells from the early G1, late G1 and G2 phases and determined the genes that are up- or downregulated specifically in each cell cycle stage. They found that Esrrb mitotic binding sites tend to reside near genes that are upregulated by Esrrb in early G1, and include genes that are important for embryonic stem cell identity and pluripotency. This lends correlative support to the idea that mitotic Esrrb binding contributes to stabilizing stemness and self-renewal (Fig. 1), perhaps by counterbalancing the known propensity of embryonic stem cells to differentiate in G114. Related to these findings, most of the genes in a somatic cell line were found to exhibit a spike in absolute transcriptional activity during the earliest rounds of transcription immediately following reversal of mitotic silencing15. Of note, this early G1 transcriptional spike is best predicted by locally elevated levels of H3K27ac in mitotic chromatin15. The potential causal roles of the diverse features retained on mitotic chromatin in transcriptional control, particularly how they might be used in similar or distinct ways to preserve cell identity across cell types, remain unexplored frontiers in this field.
Figure 1.
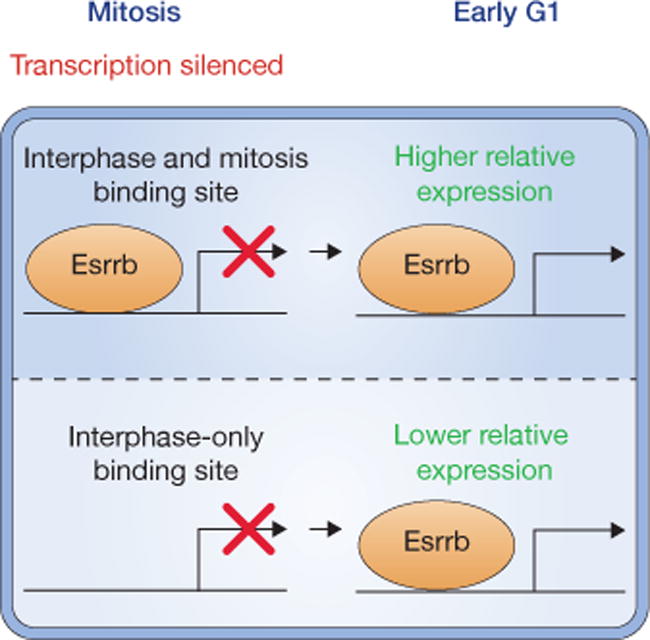
Partial retention of Esrrb on mitotic chromatin. Esrrb was discovered to bind the mitotic genome at ~10–15% of its interphase binding sites. Esrrb mitotic genome binding correlates with upregulation of nearby genes in early G1, including genes that support pluripotency.
Esrrb appears to be the first pluripotency factor examined in such depth for its chromatin association in mitotic embryonic stem cells. As for any important study, this report raises many questions, some specific to embryonic stem cells, and some of general importance regarding the role of transcription factors in transcriptional memory during mitosis. For example, why is Nanog excluded from mitotic chromatin, and what are the mitotic behaviours of the Oct4 and Sox2 transcription factors, which are absolutely required for maintaining embryonic stem cells in an undifferentiated state? More broadly, does transcription factor binding specifically during mitosis, or the presence of any other types of bookmarks, have any impact on the stability of cell fate? Can cells dispense with such bookmarks during mitosis and still preserve the appropriate phenotype? Addressing these overarching questions requires the development of tools to perturb potential bookmarks without interfering with their interphase functions. Conventional perturbations, such as knock-down approaches, do not achieve this type of cell cycle stage specificity. Mitosis-specific interference with transcription factor function has been attempted through the use of mitosis-specific protein destruction modules5. Identifying protein mutants that impair function only in mitosis has also been challenging, and needs to be pursued in greater depth. Analogous experiments to address the mitosis-specific functions of other types of putative bookmarks, such as histone modifications and chromatin accessibility, are likely to be even more challenging. The rapid progress in reagent design and genome editing are expected to deliver better approaches that will deepen our understanding of the mechanisms of transcriptional memory through mitosis.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Contributor Information
Chris C.-S. Hsiung, Division of Hematology, The Children’s Hospital of Philadelphia, Philadelphia, PA 19104, USA
Gerd A. Blobel, Division of Hematology, The Children’s Hospital of Philadelphia, Philadelphia, PA 19104, USA. blobel@email.chop.edu
References
- 1.Kadauke S, Blobel GA. Epigenetics Chromatin. 2013;6:6. doi: 10.1186/1756-8935-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martínez-Balbás MA, Dey A, Rabindran SK, Ozato K, Wu C. Cell. 1995;83:29–38. doi: 10.1016/0092-8674(95)90231-7. [DOI] [PubMed] [Google Scholar]
- 3.Hsiung CC, et al. Genome Res. 2015;25:213–225. doi: 10.1101/gr.180646.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang F, Higgins JMG. Trends Cell Biol. 2013;23:175–184. doi: 10.1016/j.tcb.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 5.Kadauke S, et al. Cell. 2012;150:725–737. doi: 10.1016/j.cell.2012.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caravaca JM, et al. Genes Dev. 2013;27:251–260. doi: 10.1101/gad.206458.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Festuccia N, et al. Nat Cell Biol. 2016;18:1139–1148. doi: 10.1038/ncb3418. [DOI] [PubMed] [Google Scholar]
- 8.Martello G, et al. Cell Stem Cell. 2012;11:491–504. doi: 10.1016/j.stem.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Festuccia N, et al. Cell Stem Cell. 2012;11:477–490. doi: 10.1016/j.stem.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen D, et al. J Cell Biol. 2005;168:41–54. doi: 10.1083/jcb.200407182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lake RJ, Tsai PF, Choi I, Won KJ, Fan HY. PLoS Genet. 2014;10:e1004204. doi: 10.1371/journal.pgen.1004204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Follmer NE, Wani AH, Francis NJ. PLoS Genet. 2012;8:e1003135. doi: 10.1371/journal.pgen.1003135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang J, Sung E, Donlin-Asp PG, Corces VG. Nat Commun. 2013;4:1464. doi: 10.1038/ncomms2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dalton S. Trends Cell Biol. 2015;25:592–600. doi: 10.1016/j.tcb.2015.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsiung CC, et al. Genes Dev. 2016;30:1423–1439. doi: 10.1101/gad.280859.116. [DOI] [PMC free article] [PubMed] [Google Scholar]