

Abstract
Cancer cachexia, a wasting syndrome characterized by skeletal muscle depletion, contributes to increased patient morbidity and mortality. While the intricate balance between protein synthesis and breakdown regulates skeletal muscle mass, the suppression of basal protein synthesis may not account for the severe wasting induced by cancer. Therefore, recent research has shifted to the regulation of “anabolic resistance,” which is the impaired ability of nutrition and exercise to stimulate protein synthesis. Emerging evidence suggests that oxidative metabolism can regulate both basal and induced muscle protein synthesis. While disrupted protein turnover and oxidative metabolism in cachectic muscle have been examined independently, evidence suggests a linkage between these processes for the regulation of cancer-induced wasting. The primary objective of this review is to highlight the connection between dysfunctional oxidative metabolism and cancer-induced anabolic resistance in skeletal muscle. First, we review oxidative metabolism regulation of muscle protein synthesis. Second, we describe cancer-induced alterations in the response to an anabolic stimulus. Finally, we review a role for exercise to inhibit cancer-induced anabolic suppression and mitochondrial dysfunction.
1. Introduction
Cachexia, a complex wasting syndrome characterized by skeletal muscle mass depletion, is prevalent in many cancers types and cannot be reversed by standard nutritional treatment [1, 2]. Approximately 20–40% of all cancer-related deaths have been attributed to cachexia, and skeletal muscle mass loss is directly associated with cancer patient morbidity and mortality [3, 4]. Skeletal muscle depletion reduces chemotherapy adherence, increases susceptibility to treatment toxicity, decreases physical function, and impairs psychosocial ability [1, 5–7]. Given the importance of muscle mass for systemic metabolic health and survival, an improved understanding of the mechanisms associated with cachexia development and progression should serve to enhance cancer patient treatment.
Skeletal muscle responds to both systemic and local environmental stimuli. Inflammation, insulin resistance, and hypogonadism are systemic perturbations associated with cancer that can disrupt skeletal muscle energy metabolism and proteostasis during cancer cachexia [8, 9]. While cancer-induced suppression of basal protein synthesis has been reported in patients and preclinical cachexia models [10–12], this suppression does not appear to fully account for the severe muscle wasting observed with cachexia. Furthermore, the importance of the cyclic anabolic stimulation of protein synthesis by nutrients and exercise has been firmly established [13]. Conversely, the inability to stimulate protein synthesis has been termed “anabolic resistance” and has an established role in the regulation of muscle mass loss with aging [13, 14]. While our mechanistic understanding of cachexia-induced protein synthesis suppression, protein degradation activation, and disrupted oxidative metabolism in skeletal muscle continues to develop [8, 15, 16], we still have a limited understanding of how these processes are interconnected for regulating the skeletal muscle response to the cancer environment.
There is considerable evidence that a muscle's capacity for oxidative metabolism can influence wasting susceptibility [8, 17]. Furthermore, muscle oxidative metabolism and protein turnover are disrupted in cachectic muscle [8]. While these two critical processes have historically been examined independently during skeletal muscle wasting, recent evidence has started to establish a linkage between these processes in the regulation of cancer-induced muscle wasting. Therefore, the primary objective of this review is to emphasize dysfunctional oxidative metabolism's connection with anabolic resistance during cancer-induced muscle wasting. First, we review oxidative metabolism regulation of skeletal muscle protein synthesis. Then, we describe cancer-induced alterations in the response to an anabolic stimulus. Finally, we review a role for exercise to inhibit cancer-induced anabolic suppression and mitochondrial dysfunction.
2. Muscle Oxidative Metabolism Regulation of Protein Synthesis
Skeletal muscle's metabolic capacity provides the basis for many functions that are critical for health and quality of life. Muscle fiber types have been linked to both contractile and metabolic properties [18], which are highly plastic and can be influenced by the systemic and local microenvironment. Classically, fiber types are classified by myosin heavy chain isoform expression (e.g., “slow” type I and the “fast” types IIA, IIX, and IIB), which can differ substantially in mitochondrial content and metabolic enzyme capacities [18, 19]. Generally, the rate and capacity for protein synthesis are greater in oxidative muscles when compared to glycolytic muscles, which has been related to total RNA capacity and a higher rate of protein turnover in oxidative muscles [20]. Additionally, rates of protein synthesis demonstrate fiber type differences in rodent and human skeletal muscle [21, 22]. While a causal relationship exists between oxidative phenotype and protein turnover, the disruption of oxidative metabolism can negatively impact both basal and induced protein synthesis in muscle. The following section highlights the current understanding of muscle oxidative metabolism regulation of basal protein synthesis.
2.1. Overview of Basal Protein Synthesis Regulation
Translational efficiency (polypeptide synthesis per ribosome) and capacity (total number of ribosomes) serve as critical control points for muscle protein synthesis. Translation efficiency is regulated at initiation, elongation, and termination steps [23]. Significant advances have been made in our understanding of the mechanisms regulating initiation in skeletal muscle. In brief, translation initiation includes (1) binding of the GTP-bound form of eukaryotic initiation factor 2 (eIF2) and initiator methionyl-tRNA (met-tRNAi) complex to the small (40S) ribosomal subunit to form the 43S preinitiation complex (PIC), (2) binding of the 43S PIC to the 7-methylguanosine (m7G) cap of the 5′ end of messenger RNA (mRNA), and (3) 43S PIC scanning of the mRNA for an AUG initiation codon [24, 25]. Upon AUG recognition, the large (60S) ribosomal subunit joins the 40S subunit to form an 80S initiation complex capable of promoting elongation [25]. Several eukaryotic initiation factors (e.g., eIF2, eIF3, and eIF5) and binding proteins (e.g., poly(A)-binding protein (PABP)) have critical roles in each one of these processes and can be regulated by cellular environments such as stress or nutrient availability [23]. Indeed, eIF2 activity is thought to be the rate-limiting step for translation initiation [24, 26]. The phosphorylation of eIF2α on Ser51 can prevent the formation of the eIF2•GTP•Met-tRNAi complex and inhibit global protein synthesis through competitive inhibition of the 5-subunit guanine nucleotide exchange factor (GEF), eIF2B [26]. Several nutrient and energy sensitivity kinases such as general control nonderepressible 2 (GCN2), protein kinase RNA- (PKR-) activated, and protein kinase R- (PKR-) like endoplasmic reticulum kinase (PERK) regulate eIF2α phosphorylation in response to atrophic stimuli [27]. Collectively, translation initiation regulation has emerged as a highly dynamic and coordinated process that is tightly linked to nutrient availability and energy stress.
The mammalian target of rapamycin (mTOR) is an established regulator of cellular processes related to metabolism and growth (Figure 1). mTOR is a serine/threonine kinase that interacts with several proteins to form two distinct complexes, mTOR complex 1 (mTORC1) and 2 (mTORC2). mTORC1 has been investigated most widely for its role in the regulation of processes related to protein synthesis through the control of translational efficiency and capacity in muscle. In contrast, mTORC2 has been implicated in cell survival, cytoskeletal organization, and metabolism [28]. mTORC1 serves as a critical integration point for anabolic stimuli such as growth factors, nutrients, and mechanical load (Figure 2) [28, 29]. Upon activation, mTORC1 directly phosphorylates the eukaryotic translation initiation factor 4E (eIF4E) binding protein 1 (4E-BP1) and S6 kinase 1 (S6K1) to promote protein synthesis [30]. The hyperphosphorylation of 4E-BP1 prevents binding to eIF4E and the formation of 4E-BP1-eIF4E complex, resulting in the assembly of the eIF4F complex and translation initiation. The activation of mTORC1 has also been implicated in cap-dependent translation, translation elongation, and ribosomal biogenesis. Indeed, mTORC1 signaling has been implicated in the inhibition of eukaryotic elongation factor 2 kinase (eEF2K) activity [31]. The eukaryotic elongation factor 2 (eEF2) is responsible for the translocation of peptidyl-tRNA at the ribosome during translation, and eEF2 phosphorylation by eEF2K inhibits its activity [32]. Related to ribosomal capacity, mTORC1 can regulate translational machinery components by promoting synthesis of mRNAs encoding ribosomal proteins (RPs) and the transcription of ribosomal RNAs (rRNAs) [28, 33]. Thus, mTORC1 signaling serves as a critical regulatory point for growth and metabolic alterations that could be targeted in muscle wasting conditions.
Figure 1.
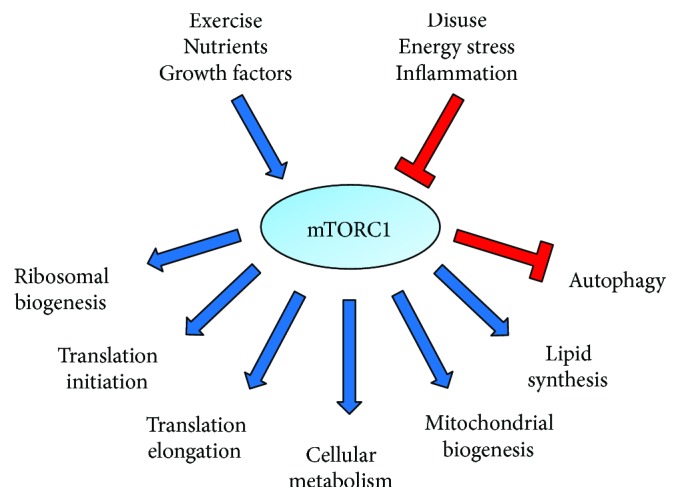
mTORC1-regulated cellular processes. mTORC1 has been shown to stimulate cellular processes related to protein synthesis, cellular metabolism, mitochondrial biogenesis and function, and lipid synthesis. mTORC1 has also been described in the inhibition of autophagy. Positive regulators of mTORC1 signaling include exercise, nutrients (e.g., amino acids), and growth factors (e.g., insulin and insulin-like growth factor-1). Negative regulators of mTORC1 signaling include disuse, energy stress (e.g., nutrient deprivation and unfolded protein response), and inflammation.
Figure 2.
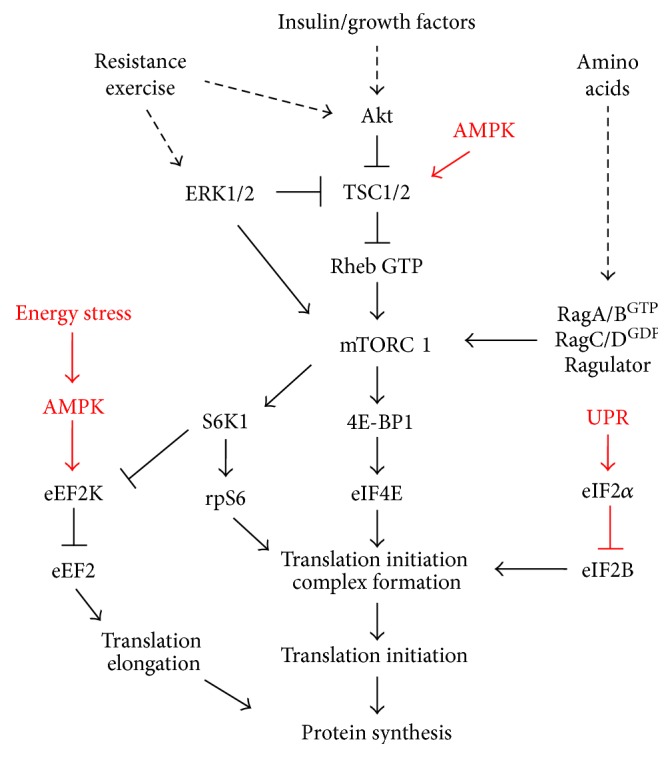
mTORC1 signaling regulation by acute anabolic stimuli. Exercise, growth factors, and nutrients can positively regulate muscle mTORC1 signaling. Cancer cachexia can suppress mTORC1 signaling through disrupted oxidative capacity and mitochondrial quality control. Disrupted muscle homeostasis can promote energy stress and an unfolded protein response, which negatively regulate mTORC1 signaling at several levels. Black lines: positive regulation. Red lines: negative regulation. ERK, extracellular signal-related kinases; Akt, protein kinase B; TSC1/2, tuberous sclerosis complex 1 and 2; Rheb, ras homolog enriched in brain; mTORC1, mammalian target of rapamycin complex 1; RagA/B, RagA and RagB heterodimer; RagC/D, RagC and RagD heterodimer; AMPK, AMP-activated protein kinase; S6K1, ribosomal protein S6 kinase beta-1; S6, ribosomal protein S6; 4E-BP1, eukaryotic translation initiation factor binding protein 1; eIF4E, eukaryotic initiation factor 4E; eEF2K, eukaryotic elongation factor 2 kinase; eEF2, eukaryotic elongation factor 2; eIF2α, eukaryotic initiation factor alpha; eIF2B, eukaryotic initiation factor 2B; UPR, unfolded protein response.
2.2. Role of Oxidative Metabolism in Protein Synthesis and mTORC1 Signaling
Mitochondrial respiration and ATP synthesis are intimately linked to cellular energy utilization, and thus mitochondria exert control over ATP-utilizing processes [34]. Therefore, it is not surprising that mitochondrial function has an established role in the regulation of cellular processes related to protein synthesis and growth [35]. Protein synthesis is an energy-demanding process that accounts for approximately 20–30% of mammalian ATP consumption [34, 36]. While specific synthesis rates can vary between muscle fractions (e.g., myofibrillar, sarcoplasmic, and mitochondria) [37], the energy required for protein metabolism has been linked to de novo protein synthesis. Moreover, many nonribosomal energy-consuming enzymes such as ATP-dependent RNA helicases, AAA-ATPases, GTPases, and kinases are required for the synthesis, assembly, and maintenance of ribosomes [38, 39]. Collectively, the overall energetic cost of protein synthesis implicates a role for mitochondrial integrity and function in this important anabolic process.
The energy-sensing molecule 5′-AMP-activated protein kinase (AMPK) regulates energy homeostasis in response to metabolic stress [40] and is well suited to couple cellular energy status to protein synthesis. AMPK is a highly conserved heterotrimeric kinase complex composed of a catalytic α-subunit (α1 and α2) and two regulatory (β- and γ-) subunits [41]. The β subunit contains a glycogen-binding domain (GBD), whereas the γ subunit is responsible for nucleotide binding [42]. Several upstream kinases have been identified that regulate AMPK activity: the serine-threonine liver kinase B1 (LKB1) and calcium/calmodulin kinase kinase-β (CaMKKβ) [41]. AMPK is activated under conditions of energy stress when intracellular ATP levels decline and intracellular AMP increases (high intracellular AMP/ATP ratio). AMPK activation stimulates ATP synthesis while concurrently inhibiting ATP consumption [43]. Thus, AMPK has been implicated in the regulation of mTORC1 [44], ubiquitin E3 ligase expression [45], autophagy/mitophagy [46, 47], and mitochondrial biogenesis [48]. AMPK inhibits mTORC1 signaling through the direct phosphorylation of TSC2 and Raptor [49–51]. TSC2 phosphorylation inhibits mTORC1 signaling through the maintenance of Ras homologue enriched in brain (Rheb) in its GDP-bound inactive state [50, 52]. Moreover, Raptor phosphorylation at two highly conserved serine residues (S722 and S792) can also suppress mTORC1 activity [51]. Thus, AMPK activation in response to mitochondrial dysfunction could serve to suppress mTORC1 activation of protein synthesis.
2.3. Mitochondrial Dysfunction and Energetic Stress
Mitochondrial dysfunction impairs the ability to convert nutrients (carbohydrate, fatty acids, etc.) into energy while concomitantly producing reactive oxygen species (ROS) that can inhibit biosynthetic pathways required for metabolic homeostasis [53]. Moreover, oxidative stress-induced accumulation of damaged and misfolded proteins can cause endoplasmic reticulum (ER) stress and an unfolded protein response (UPR). Thus, global protein synthesis is downregulated to resolve dysfunctional protein accumulation. The following sections highlight signaling pathways that link disrupted mitochondrial function to suppressed protein synthesis.
2.3.1. ROS
Subsarcolemma and intermyofibrillar mitochondria are prominent sources of skeletal muscle ROS generation [54–56]. While ROS can serve as signaling molecules to regulate cellular processes [57], excessive ROS production can lead to the oxidation of proteins, lipids, and DNA [58]. The relative contribution of ROS to induce oxidative stress and damage is dependent on both ROS production and the level of cellular antioxidants [55]. For example, glycolytic fibers have increased susceptibility to oxidative stress, which involves enhanced ROS generation and decreased scavenging capabilities, compared to oxidative fibers [59]. Therefore, the cycle of aberrant ROS production and damage accumulation has been attributed to muscle atrophy associated with aging and inflammatory pathologies.
2.3.2. ER Stress and UPR
Pathological stressors such as dysregulated protein synthesis, accumulation of misfolded proteins, calcium imbalance, and energy deprivation can all induce ER stress [60]. Excessive ROS production can induce ER stress and activate the UPR [61]. Additionally, the UPR can be activated when the balance between newly synthesized peptides and degrading misfolded proteins occurs, and an exacerbated UPR can disrupt muscle homeostasis. Under normal conditions, the UPR functions to return homeostasis through three major pathways: inositol-requiring protein (IRE1), RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK), and activating transcription factor-6 (ATF6) [61–63]. PERK promotes protein synthesis inhibition through the inactivation of eIF2α [64]. ATF6 in conjunction with ATF4 and XBP1 can decrease ROS and ER stress [65]. IRE1 has both kinase and endoribonuclease activities to alter gene expression favoring apoptosis signaling or metabolic adaptations that promote survival [61, 64]. Overall, ER stress and UPR activation inhibit translation at the level of initiation through the phosphorylation of eIF2α, thereby preventing further unfolded protein accumulation, while preserving nutrients and energy [60]. Thus, while ER stress and UPR are adaptive responses to maintain homeostasis, the aberrant activation of these cellular processes can compromise muscle mass and metabolic function.
2.3.3. Insulin Resistance
Insulin resistance accompanies many wasting conditions [66], and disrupted oxidative metabolism has been implicated in decreased skeletal muscle insulin sensitivity [67]. Altered mitochondrial function and metabolic flexibility can disrupt glucose and lipid homeostasis, which are also associated with insulin resistance [68, 69]. Disrupted muscle glycolytic flux is associated with altered amino acid utilization as tricarboxylic acid (TCA) intermediates [70], which could negatively affect protein metabolism. Indeed, the TCA cycle has emerged as a critical point in metabolic regulation linking bioenergetics with anabolic and catabolic pathways. Lipid accumulation and bioactive lipid intermediates, such as diacylglycerols (DAG) and ceramides, can inhibit insulin signaling and contribute to anabolic resistance. Increased ceramide can inhibit Akt through the activation protein kinase C (PKC) isoforms or protein phosphatase 2A (PP2A) [71]. Moreover, ceramide can impair insulin receptor function through glycosphingolipid GM3 synthesis [72, 73]. DAG can also inhibit insulin signaling transduction through PKC activation and serine phosphorylation of insulin receptor substrate 1 (IRS-1) [74, 75]. Lastly, ceramide can modulate nutrient uptake through the regulation of the sodium-dependent neutral amino acid transporter 2 (SNAT2) [76, 77], which could impair amino acid activation of mTORC1. Collectively, impaired insulin sensitivity through altered oxidative metabolism could have significant ramifications on metabolic flexibility during pathological conditions.
2.4. Reciprocal Regulation through mTORC1
Several lines of evidence suggest that mTORC1 signaling can regulate energy production and oxidative metabolism across various cell types [78–80]. mTORC1 activity and mTOR-Raptor complex formation has been tightly correlated with mitochondrial metabolism [80]. Specifically, lower mTORC1 activity is associated with decreased mitochondrial oxygen consumption, whereas increased mTORC1 activity (e.g., TSC2 knockdown) is accompanied by enhanced mitochondrial oxygen consumption [80]. Moreover, the inhibition of mTORC1 signaling reduces mitochondrial respiration (coupled and uncoupled), impairs TCA cycle activity, and lowers ATP production capacity [81]. In line with a potential regulatory role of mTORC1 activity on oxidative metabolism, reduced mitochondrial content and function has been observed in muscle-specific mTOR or raptor knockout (RAmKO) mice [82, 83]. Interestingly, mTORC1 inactivation in these mice is associated with altered systemic metabolic homeostasis and muscle atrophy/dystrophic phenotype [82, 83]. mTORC1 signaling also has the potential to perturb mitochondrial function through the regulation of translation and transcription to modulate expression of genes involved in oxidative metabolism [78, 81, 84]. The activation of mTORC1/4E-BP pathway has been implicated in the control of energy homeostasis via the translation of nuclear-encoded mitochondria-related genes such as mitochondrial transcription factor A (TFAM), mitochondrial ribosomal proteins, and complex I and IV components [81]. mTORC1 can also regulate mitochondrial content and function by modulating transcription factors that regulate energy metabolism [78, 85]. Indeed, mTORC1 has been implicated in the regulation of mitochondrial gene expression through the direct modulation of Yin Yang 1 (YY1) and PGC-1α transcriptional complex activity [78]. In skeletal muscle, YY1 physically interacts with and recruits PGC-1α to the promoters of nuclear mitochondrial genes, and the inhibition of mTORC1 by rapamycin prevents the coactivation of YY1 by PGC-1α [78]. In addition to its role in translation and transcription, mTORC1 has also been implicated in the regulation of autophagy, a process associated with catabolism and the regulation of mitochondrial quality control. mTORC1 inhibits autophagy by phosphorylating the proautophagic kinase ULK1, which prevents its association and subsequent activation by AMPK [46, 86]. Thus, chronic mTORC1 suppression could accelerate autophagy and perturb mitochondrial homeostasis. Collectively, these studies highlight a critical role for mTORC1 signaling in energy homeostasis through the regulation of protein synthesis and oxidative metabolism. Future research is warranted to determine the complex relationship between suppressed mTORC1 signaling and disrupted oxidative metabolism during pathological conditions.
2.5. Cancer-Induced Changes in Protein Synthesis and Oxidative Metabolism
Disrupted protein synthesis and oxidative metabolism have established roles in many wasting conditions and are intricately linked to wasting processes during cancer cachexia progression. Several studies have demonstrated suppressed basal protein synthesis in preclinical models of cancer cachexia [10, 87–89]. Interestingly, muscle protein synthesis is suppressed during the initial stages of weight loss (<5% body weight loss) and is further reduced throughout cachexia progression in tumor bearing mice [11]. Cancer cachexia has the potential to disrupt protein translation at several regulatory steps (e.g., initiation, elongation, and termination), and the regulation of translation initiation during cancer cachexia has been an active area of investigation. The phosphorylation of eIF2α, which attenuates translation initiation and overall protein synthesis, is elevated in cancer patients and preclinical models [90–92]. Moreover, mTORC1 signaling is suppressed during late stage cachexia [11, 93, 94], which corresponds to disrupted S6K1 and 4EBP-1 regulation [11, 91]. Lewis lung carcinoma (LLC) conditioned media or recombinant IL-6 can suppress mTORC1 signaling in C2C12 myotubes [94–96], which highlights a direct role of circulating factors on muscle. Elevated eEF2 phosphorylation has also been observed in cachectic mice bearing Murine adenocarcinoma 16 (MAC16) tumors [91]. Collectively, these studies highlight a role for translation initiation and elongation in disrupted protein synthesis regulation, which may serve as a potential therapeutic target to treat or prevent cancer cachexia.
Disrupted muscle oxidative metabolism coincides with suppressed protein synthesis and mTORC1 signaling [97, 98]. Recent evidence also suggests that disrupted mitochondrial function may also precede muscle atrophy during cancer [99], which highlights a potential role in the regulation of anabolic processes. Cancer cachexia can disrupt several processes regulating mitochondrial quality, which encompasses function, content, dynamics, and mitophagy [100]. Mitochondrial morphology and function related to ATP production and coupling efficiency are disrupted during cachexia progression [98, 101–103]. Moreover, ATP synthesis rates and electron transport chain (ETC) complex activities are reduced in cachectic muscle [101, 104–106], and tumor-derived factors can impair basal and ATP-related oxygen consumption in C2C12 myotubes [107]. Furthermore, recent genomic and metabolomic approaches have demonstrated altered carbohydrate and lipid metabolic flux coincide with mitochondrial dysfunction [108, 109]. Indices of mitochondrial quality control related to content (e.g., mitochondrial DNA expression), dynamics (e.g., Mfn1/2 and Fis1), and autophagy are disrupted throughout cachexia progression [11, 97, 98]. While these studies collectively demonstrate suppressed oxidative metabolism regulation during cancer cachexia progression, whether these alterations interact with disrupted protein turnover has not been firmly established.
2.6. Cancer-Induced Metabolic and Energy Stress
2.6.1. AMPK Activation
AMPK activity is disrupted during cancer cachexia progression, which can alter skeletal muscle metabolism and gene expression [43]. Several preclinical models of cancer cachexia demonstrate chronically elevated muscle AMPK activity during late-stage cachexia [11, 94, 96, 110, 111], which coincides with the suppression of mTORC1 signaling and disrupted mitochondrial quality control mechanisms. While AMPK activation by systemic IL-6 also corresponds with mTORC1 suppression in tumor-bearing mice [94], AMPK inhibition could rescue IL-6-induced suppression of mTORC1 signaling in C2C12 myotubes [94]. Moreover, inhibition of myotube AMPK activity during LLC treatment improved mTORC1 signaling and protein synthesis [95]. In contrast to the acute effects of exercise on AMPK activity in healthy skeletal muscle, the chronic activation of AMPK by cachexia or IL-6 overexpression is uncoupled from mitochondrial biogenesis [11, 97]. While these studies highlight a potential role for disrupted mitochondrial function in the suppression of mTORC1 signaling during cancer cachexia, further work is needed to determine the relationship between AMPK activation in the suppression of anabolic signaling and disrupted oxidative metabolism. Given the potential role of AMPK in muscle protein synthesis and metabolic homeostasis, further work is required to determine if restoring dysregulated AMPK activity may be a potential therapeutic target for muscle wasting syndrome.
2.6.2. ER Stress and UPR Activation
While muscle ER stress and UPR pathways are activated in several wasting conditions [112], their role in cancer-induced atrophy has not been widely investigated. Nonetheless, in two preclinical models of cancer cachexia, the activation of several markers of ER stress (e.g., IRE1a, XBP-1, and ATF6) was accompanied by the suppression of mTORC1 signaling, activation of protein breakdown, and dysregulated AMPK activation [113]. Moreover, tumor-derived factors within LLC conditioned media were sufficient to induce the expression of ER stress molecules and phosphorylate eIF2α in cultured C2C12 myotubes [113]. Interestingly, global inhibition of ER stress and UPR pathways induced muscle wasting in wild-type mice and resulted in a more pronounced cachectic muscle phenotype in tumor-bearing mice [113]. Overall, these findings demonstrate initial evidence that tumor-derived factors can induce ER stress and UPR activation in muscle; however, these pathways may also be important regulatory mechanisms to preserve muscle mass during cancer cachexia progression.
2.6.3. Insulin Resistance
Related to cancer cachexia, there is evidence that mitochondrial dysfunction and excess lipid accumulation may coincide with insulin resistance [114]. Intramyocellular lipid accumulation has been observed in cachectic cancer patients [115, 116]. Moreover, the regulation of lipolysis through adipose triglyceride lipase (ATGL) and HSL activity has been negatively associated with the BMI of cachectic cancer patients [117]. The relationship between mitochondrial dysfunction and insulin resistance in many chronic disease states provides a strong rationale for impaired insulin signaling in the etiology of cancer-associated anabolic resistance in muscle.
3. Cancer-Induced Anabolic Resistance
Many cancer patients experience weight loss at the time of diagnosis [3], and skeletal muscle depletion is indicative of poor prognoses in many cancers [4, 118, 119]. Therefore, whether cachectic muscle retains the anabolic plasticity to treatment therapies is clinically relevant and could greatly impact a cancer patient's survival. While circadian fluctuations in protein turnover have established roles in muscle mass regulation, it is evident that protein synthesis is dynamically responsive to environmental cues. Nutrients and exercise are potent stimulators of protein synthesis in healthy skeletal muscle; however, several wasting conditions demonstrate reduced sensitivity to these anabolic stimuli [14, 120]. Given that small decrements in daily protein synthesis could significantly impact long-term muscle maintenance in wasting conditions [121], anabolic resistance could also contribute to muscle during cachexia progression. Therefore, determining the molecular mechanisms that contribute to decreased anabolic plasticity could significantly impact treatment of the cachectic cancer patient.
3.1. Regulation of Anabolic Resistance
3.1.1. Nutrition
It has long been recognized that feeding can stimulate whole-body and skeletal muscle protein synthesis compared to the fasted state in healthy individuals [122, 123]. Similarly, consumption or infusion of essential amino acids (EAA), particularly leucine, can stimulate protein synthesis in young individuals [124–126]. The protein synthesis induction by feeding and/or EAA is associated with the activation of mTORC1 signaling [127–129]. In the presence of amino acids, the Ragulator-Rag complex targets mTORC1 to the lysosomal surface, where it can interact with and become activated by the small GTPase Rheb upon amino stimulation [130]. While the maintenance of muscle mass is critically important during aging and wasting conditions, significant progress has been made in our understanding of the anabolic response to feeding in elderly individuals [13]. Several studies have demonstrated a blunted response to lower doses of EAA in elderly individuals [131, 132], with this decreased sensitivity and responsiveness to nutrients associated with impaired mTORC1 signaling [127, 133]. However, the ability to stimulate protein synthesis remains intact when higher doses are ingested [124, 125, 134]. These findings indicate that while a threshold is needed to stimulate protein synthesis, the synthetic machinery required for protein synthesis remains intact. Related to pathological wasting conditions, muscle resistance to leucine stimulation has also been observed in severe inflammatory disorders such as sepsis and endotoxemia [135, 136]. Collectively, these studies highlight that reduced sensitivity of skeletal muscle to feeding contributes to muscle mass loss observed during aging and disease state.
3.1.2. Exercise
Muscle contraction is a potent stimulator of protein synthesis in healthy skeletal muscle [14, 137] and has been shown to synergistically improve nutrient-induced stimulation of protein synthesis [138–140]. The activation of protein synthesis by resistance exercise can be observed as early as 1 h postexercise [141, 142] and can remain elevated for up to 24–48 h, dependent on the manipulation of training variables [137, 143, 144]. The cellular mechanisms associated with loading-induced protein synthesis include phosphatidic acid (PA), extracellular-related kinase 1 and 2 (ERK1/2), and mTORC1 signaling [13, 145], which can occur independent of upstream IGF-1 receptor signaling [146, 147]. Indeed, the downstream activation of P70S6K by high-force contractions has been associated with protein synthesis and muscle growth in both humans and rodents [142, 148, 149]. However, disrupted mTORC1 signaling has been implicated in the impaired anabolic response to contraction in aging and pathological conditions [142, 150, 151]. Therefore, anabolic resistance to exercise may be present in many wasting conditions.
3.2. Cancer-Induced Anabolic Resistance
3.2.1. Nutrition
An improved understanding of the cachectic muscle response to anabolic stimuli is of great clinical importance, given that cachexia cannot be fully reversed by conventional nutritional support [2]. While emerging evidence suggests that basal muscle protein synthesis is disrupted throughout cachexia progression, few studies have examined whether nutrition and exercise can stimulate anabolic signaling in cachectic muscle. While cancer patients can still induce protein synthesis in response to protein ingestion [152–157], this anabolic response to nutrients is strongly impaired [152, 155–157]. However, a specifically formulated medical food (high protein and leucine) could overcome anabolic resistance in cancer patients with involuntary weight loss [152]. Additionally, resistance exercise could enhance the myofibrillar protein synthesis response to feeding response in men with prostate cancer on androgen deprivation [155]. These studies demonstrate that the translational machinery may be responsive to feeding, but highlight that individualized, multimodal approaches may be needed to maximally stimulate protein synthesis. Interestingly, few preclinical studies have examined the acute feeding response during cancer cachexia. However, we have demonstrated suppressed mTORC1 responsiveness to glucose administration in cachectic skeletal muscle [94]. Overall, there is clear evidence that future studies are needed to improve our understanding of the mechanisms regulating anabolic resistance to feeding, which could have significant clinical and physical ramifications in the cachectic cancer patient.
3.2.2. Exercise
While the mechanisms associated with feeding-induced protein synthesis during cancer are starting to emerge, much less is known related to the acute anabolic response to contraction in cachectic muscle. We have reported that mechanoactivation of protein synthesis in stretched myotubes is disrupted by conditioned media from Lewis lung carcinoma (LLC) cells [95], suggesting that tumor-derived cachectic factors can interfere with mechanical signaling inducing protein synthesis in vitro. In addition, we found that severe cachexia could disrupt the metabolic and anabolic signaling response to a single bout of stimulated concentric muscle contractions [110]. However, these contractions were associated with the sustained activation of AMPK, which may be related to exacerbated metabolic stress given that cachectic muscle develops mitochondrial dysfunction [97, 102, 106]. Thus, it is imperative that future research determines the cachectic muscle's anabolic and metabolic sensitivity to different types of muscle contractions. In addition, futures studies should also examine cachectic muscle response in fasted and fed states throughout cachexia progression. Given that exercise can induce protein synthesis for many hours, the potential benefits of exercise for cachectic cancer patients will likely occur in the hours postexercise and coincide with enhanced nutritional responsiveness.
4. Role for Exercise Training to Impede Anabolic Resistance during Cancer Cachexia
While exercise training has been discussed as a potential therapy to mitigate muscle atrophy, current understanding of the acute response and training adaptation to exercise during cancer cachexia is limited. Given that exercise involves muscle contractions that can vary in intensity and metabolic demand, the molecular responses related to growth and metabolism can differ between contraction types. In general, a single exercise bout can induce metabolic signaling pathways linked to energy production and proteostasis, whereas repeated bouts can stimulate adaptations related to oxidative metabolism and growth [158]. However, the extent to which these adaptations are related to remodeling or growth can be impacted by variables such as the exercise type, intensity and workload, and the nutritional status during the postexercise recovery [159, 160]. The following section will highlight our current understanding of exercise training to impede suppressed anabolic and metabolic plasticity during cachexia progression (Figure 3).
Figure 3.

Mechanisms associated with suppressed anabolic potential during cancer cachexia. The systemic cachectic environment induces several muscle disruptions that can impede the protein synthesis induction by anabolic stimuli. Exercise training has the potential to improve muscle oxidative metabolism, which can have direct effects on protein metabolism, insulin sensitivity, and the unfolded protein response. Abbreviations: ROS, reactive oxygen species; UPR, unfolded protein response; mTORC1, mammalian target of rapamycin complex 1.
While the field of exercise oncology is currently limited in the number of studies that have been completed in the cachectic cancer patient, initial progress has been made in our understanding of exercise adaptations during the development and progression of cachexia in preclinical models. Treadmill exercise training alone or in combination with nutritional support can reduce tumor growth and improve muscle mass [161–163]. Moreover, we have demonstrated that treadmill training during systemic IL-6 overexpression enhanced mTORC1 signaling and mitochondrial quality control in tumor-bearing mice [94, 98], which was associated with improved insulin sensitivity [164]. Related to resistance exercise, rodent models of overload hypertrophy and stimulated eccentric contractions have demonstrated preserved muscle mass whether it was initiated prior to or at tumor-implantation [165–168]. In addition, we found that repeated bouts of stimulated eccentric contractions after the initiation of cachexia attenuated myofiber atrophy that coincided with improved oxidative capacity in tumor-bearing mice [111]. However, whether these changes were associated with improved basal protein synthesis regulation requires further investigation. Collectively, these studies demonstrate that exercise training can improve indices of muscle mass, protein turnover regulation, and oxidative metabolism throughout the progression of cancer cachexia. Additional studies are needed to determine the potential interactions between nutrients and exercise during the treatment of the cancer patient.
5. Conclusion and Future Research Perspectives
Muscle protein synthesis and oxidative metabolism have established roles in physical function and metabolic health. Given the significant energy requirement for protein synthesis to maintain muscle homeostasis, it is not surprising that mitochondria can regulate cellular processes related to remodeling and growth. While there is considerable evidence that protein synthesis and oxidative metabolism are disrupted throughout cachexia progression, the potential interaction between these two regulatory pathways has not been fully appreciated. Therefore, the primary objective of this review was to highlight the relationship of dysfunctional oxidative metabolism to anabolic resistance during cancer-induced muscle wasting. First, we described oxidative metabolism regulation of basal protein synthesis in skeletal muscle. Mitochondrial dysfunction has clear implications for the regulation of protein synthesis through the induction of metabolic and energetic stress pathways. While evidence exists that these pathways are altered in preclinical models of cancer cachexia, it is unclear whether this occurs in cachectic cancer patients. However, many of these studies have limitations related to sample size, patient population, and the degree and duration of the cachectic phenotype. Further research is required to establish if the mechanisms disrupting protein turnover are differentially regulated by a muscle's oxidative phenotype. Second, we assessed cancer-induced alterations to an anabolic stimulus. While not observed in all cancer patients or preclinical models, evidence exists that cachexia can disrupt the anabolic response to nutrients and exercise. However, further research is needed to clearly describe anabolic resistance throughout cachexia progression. Finally, we reviewed a role for resistance exercise to impede cancer-induced anabolic suppression and mitochondrial dysfunction. While evidence suggests that exercise maintains muscle mass in tumor-bearing mice, further investigation is warranted to determine if these improvements are related to enhanced anabolic and metabolic plasticity of cachectic skeletal muscle. Clearly defining the interactions between muscle protein synthesis, mTORC1 signaling, and oxidative metabolism will provide greater insight into the regulation of skeletal muscle wasting and systemic metabolic dysfunction during cancer cachexia progression. Additionally, understanding whether exercise training enhances skeletal muscle's sensitivity to nutrients will improve our efforts for treating the cachectic cancer patient.
Acknowledgments
The authors thank Gaye Christmus for the editorial review of the manuscript. This work was supported by National Institutes of Health Grants R01 CA-121249 (National Cancer Institute) and P20 RR-017698 (National Institute of General Medical Science) to James A. Carson, SPARC Graduate Research Grant from the Office of the Vice President for Research at the University of South Carolina to Justin P. Hardee, and an ACSM Foundation Research Grant from the American College of Sports Medicine Foundation to Justin P. Hardee.
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper. In addition, the received funding did not lead to any conflict of interests regarding the publication of this manuscript.
References
- 1.Evans W. J., Morley J. E., Argiles J., et al. Cachexia: a new definition. Clinical Nutrition. 2008;27(6):793–799. doi: 10.1016/j.clnu.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 2.Fearon K., Strasser F., Anker S. D., et al. Definition and classification of cancer cachexia: an international consensus. The Lancet Oncology. 2011;12(5):489–495. doi: 10.1016/S1470-2045(10)70218-7. [DOI] [PubMed] [Google Scholar]
- 3.Tisdale M. J. Mechanisms of cancer cachexia. Physiological Reviews. 2009;89(2):381–410. doi: 10.1152/physrev.00016.2008. [DOI] [PubMed] [Google Scholar]
- 4.Prado C. M., Sawyer M. B., Ghosh S., et al. Central tenet of cancer cachexia therapy: do patients with advanced cancer have exploitable anabolic potential? The American Journal of Clinical Nutrition. 2013;98(4):1012–1019. doi: 10.3945/ajcn.113.060228. [DOI] [PubMed] [Google Scholar]
- 5.Jung H. W., Kim J. W., Kim J. Y., et al. Effect of muscle mass on toxicity and survival in patients with colon cancer undergoing adjuvant chemotherapy. Support Care Cancer. 2015;23(3):687–694. doi: 10.1007/s00520-014-2418-6. [DOI] [PubMed] [Google Scholar]
- 6.Barret M., Antoun S., Dalban C., et al. Sarcopenia is linked to treatment toxicity in patients with metastatic colorectal cancer. Nutrition and Cancer. 2014;66(4):583–589. doi: 10.1080/01635581.2014.894103. [DOI] [PubMed] [Google Scholar]
- 7.Cespedes Feliciano E. M., Lee V. S., Prado C. M., et al. Muscle mass at the time of diagnosis of nonmetastatic colon cancer and early discontinuation of chemotherapy, delays, and dose reductions on adjuvant FOLFOX: the C-SCANS study. Cancer. 2017 doi: 10.1002/cncr.30950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carson J. A., Hardee J. P., VanderVeen B. N. The emerging role of skeletal muscle oxidative metabolism as a biological target and cellular regulator of cancer-induced muscle wasting. Seminars in Cell & Developmental Biology. 2016;54:53–67. doi: 10.1016/j.semcdb.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.White J. P., Puppa M. J., Narsale A., Carson J. A. Characterization of the male ApcMin/+ mouse as a hypogonadism model related to cancer cachexia. Biology Open. 2013;2(12):1346–1353. doi: 10.1242/bio.20136544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dworzak F., Ferrari P., Gavazzi C., Maiorana C., Bozzetti F. Effects of cachexia due to cancer on whole body and skeletal muscle protein turnover. Cancer. 1998;82(1):42–48. doi: 10.1002/(SICI)1097-0142(19980101)82:1<42::AID-CNCR5>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 11.White J. P., Baynes J. W., Welle S. L., et al. The regulation of skeletal muscle protein turnover during the progression of cancer cachexia in the Apc(Min/+) mouse. PLoS One. 2011;6(9, article e24650) doi: 10.1371/journal.pone.0024650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Emery P. W., Lovell L., Rennie M. J. Protein synthesis measured in vivo in muscle and liver of cachectic tumor-bearing mice. Cancer Research. 1984;44(7):2779–2784. [PubMed] [Google Scholar]
- 13.Fry C. S., Rasmussen B. B. Skeletal muscle protein balance and metabolism in the elderly. Current Aging Science. 2011;4(3):260–268. doi: 10.2174/1874609811104030260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dickinson J. M., Volpi E., Rasmussen B. B. Exercise and nutrition to target protein synthesis impairments in aging skeletal muscle. Exercise and Sport Sciences Reviews. 2013;41(4):216–223. doi: 10.1097/JES.0b013e3182a4e699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.VanderVeen B. N., Fix D. K., Carson J. A. Disrupted skeletal muscle mitochondrial dynamics, mitophagy, and biogenesis during cancer cachexia: a role for inflammation. Oxidative Medicine and Cellular Longevity. 2017;2017:p. 3292087. doi: 10.1155/2017/3292087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fearon K. C., Glass D. J., Guttridge D. C. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metabolism. 2012;16(2):153–166. doi: 10.1016/j.cmet.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 17.Powers S. K., Smuder A. J., Judge A. R. Oxidative stress and disuse muscle atrophy: cause or consequence? Current Opinion in Clinical Nutrition and Metabolic Care. 2012;15(3):240–245. doi: 10.1097/MCO.0b013e328352b4c2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schiaffino S., Reggiani C. Fiber types in mammalian skeletal muscles. Physiological Reviews. 2011;91(4):1447–1531. doi: 10.1152/physrev.00031.2010. [DOI] [PubMed] [Google Scholar]
- 19.Ciciliot S., Rossi A. C., Dyar K. A., Blaauw B., Schiaffino S. Muscle type and fiber type specificity in muscle wasting. The International Journal of Biochemistry & Cell Biology. 2013;45(10):2191–2199. doi: 10.1016/j.biocel.2013.05.016. [DOI] [PubMed] [Google Scholar]
- 20.Habets P. E., Franco D., Ruijter J. M., Sargeant A. J., Pereira J. A., Moorman A. F. RNA content differs in slow and fast muscle fibers: implications for interpretation of changes in muscle gene expression. The Journal of Histochemistry and Cytochemistry. 1999;47(8):995–1004. doi: 10.1177/002215549904700803. [DOI] [PubMed] [Google Scholar]
- 21.Dickinson J. M., Lee J. D., Sullivan B. E., Harber M. P., Trappe S. W., Trappe T. A. A new method to study in vivo protein synthesis in slow- and fast-twitch muscle fibers and initial measurements in humans. Journal of Applied Physiology. 2010;108(5):1410–1416. doi: 10.1152/japplphysiol.00905.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goodman C. A., Kotecki J. A., Jacobs B. L., Hornberger T. A. Muscle fiber type-dependent differences in the regulation of protein synthesis. PLoS One. 2012;7(5):p. e37890. doi: 10.1371/journal.pone.0037890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sonenberg N., Hinnebusch A. G. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136(4):731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hershey J. W., Sonenberg N., Mathews M. B. Principles of translational control: an overview. Cold Spring Harbor Perspectives in Biology. 2012;4(12) doi: 10.1101/cshperspect.a011528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hinnebusch A. G., Lorsch J. R. The mechanism of eukaryotic translation initiation: new insights and challenges. Cold Spring Harbor Perspectives in Biology. 2012;4(10) doi: 10.1101/cshperspect.a011544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kimball S. R. Eukaryotic initiation factor eIF2. The International Journal of Biochemistry & Cell Biology. 1999;31(1):25–29. doi: 10.1016/S1357-2725(98)00128-9. [DOI] [PubMed] [Google Scholar]
- 27.Wek R. C., Jiang H. Y., Anthony T. G. Coping with stress: eIF2 kinases and translational control. Biochemical Society Transactions. 2006;34(1):7–11. doi: 10.1042/BST0340007. [DOI] [PubMed] [Google Scholar]
- 28.Laplante M., Sabatini D. M. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laplante M., Sabatini D. M. mTOR signaling at a glance. Journal of Cell Science. 2009;122(20):3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma X. M., Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nature Reviews. Molecular Cell Biology. 2009;10(5):307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 31.Proud C. G. mTORC1 signalling and mRNA translation. Biochemical Society Transactions. 2009;37(1):227–231. doi: 10.1042/BST0370227. [DOI] [PubMed] [Google Scholar]
- 32.Redpath N. T., Foulstone E. J., Proud C. G. Regulation of translation elongation factor-2 by insulin via a rapamycin-sensitive signalling pathway. The EMBO Journal. 1996;15(9):2291–2297. [PMC free article] [PubMed] [Google Scholar]
- 33.Laplante M., Sabatini D. M. Regulation of mTORC1 and its impact on gene expression at a glance. Journal of Cell Science. 2013;126(8):1713–1719. doi: 10.1242/jcs.125773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buttgereit F., Brand M. D. A hierarchy of ATP-consuming processes in mammalian cells. The Biochemical Journal. 1995;312(1):163–167. doi: 10.1042/bj3120163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Romanello V., Sandri M. Mitochondrial biogenesis and fragmentation as regulators of muscle protein degradation. Current Hypertension Reports. 2010;12(6):433–439. doi: 10.1007/s11906-010-0157-8. [DOI] [PubMed] [Google Scholar]
- 36.Rolfe D. F., Brown G. C. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiological Reviews. 1997;77(3):731–758. doi: 10.1152/physrev.1997.77.3.731. [DOI] [PubMed] [Google Scholar]
- 37.Guillet C., Boirie Y., Walrand S. An integrative approach to in-vivo protein synthesis measurement: from whole tissue to specific proteins. Current Opinion in Clinical Nutrition and Metabolic Care. 2004;7(5):531–538. doi: 10.1097/00075197-200409000-00005. [DOI] [PubMed] [Google Scholar]
- 38.Kressler D., Hurt E., Bassler J. Driving ribosome assembly. Biochimica et Biophysica Acta. 2010;1803(6):673–683. doi: 10.1016/j.bbamcr.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 39.Thomson E., Ferreira-Cerca S., Hurt E. Eukaryotic ribosome biogenesis at a glance. Journal of Cell Science. 2013;126(21):4815–4821. doi: 10.1242/jcs.111948. [DOI] [PubMed] [Google Scholar]
- 40.Hardie D. G. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nature Reviews. Molecular Cell Biology. 2007;8(10):774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 41.Ruderman N. B., Xu X. J., Nelson L., et al. AMPK and SIRT1: a long-standing partnership? American Journal of Physiology. Endocrinology and Metabolism. 2010;298(4):E751–E760. doi: 10.1152/ajpendo.00745.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kemp B. E., Oakhill J. S., Scott J. W. AMPK structure and regulation from three angles. Structure. 2007;15(10):1161–1163. doi: 10.1016/j.str.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 43.Hardie D. G., Sakamoto K. AMPK: a key sensor of fuel and energy status in skeletal muscle. Physiology. 2006;21(1):48–60. doi: 10.1152/physiol.00044.2005. [DOI] [PubMed] [Google Scholar]
- 44.Bolster D. R., Crozier S. J., Kimball S. R., Jefferson L. S. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. The Journal of Biological Chemistry. 2002;277(27):23977–23980. doi: 10.1074/jbc.C200171200. [DOI] [PubMed] [Google Scholar]
- 45.Tong J. F., Yan X., Zhu M. J., Du M. AMP-activated protein kinase enhances the expression of muscle-specific ubiquitin ligases despite its activation of IGF-1/Akt signaling in C2C12 myotubes. Journal of Cellular Biochemistry. 2009;108(2):458–468. doi: 10.1002/jcb.22272. [DOI] [PubMed] [Google Scholar]
- 46.Kim J., Kundu M., Viollet B., Guan K. L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature Cell Biology. 2011;13(2):132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laker R. C., Drake J. C., Wilson R. J., et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nature Communications. 2017;8(1):p. 548. doi: 10.1038/s41467-017-00520-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jager S., Handschin C., St-Pierre J., Spiegelman B. M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(29):12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shaw R. J. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiologica (Oxford, England) 2009;196(1):65–80. doi: 10.1111/j.1748-1716.2009.01972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Inoki K., Zhu T., Guan K. L. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–590. doi: 10.1016/S0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 51.Gwinn D. M., Shackelford D. B., Egan D. F., et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Molecular Cell. 2008;30(2):214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Inoki K., Li Y., Zhu T., Wu J., Guan K. L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nature Cell Biology. 2002;4(9):648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 53.Powers S. K., Smuder A. J., Criswell D. S. Mechanistic links between oxidative stress and disuse muscle atrophy. Antioxidants & Redox Signaling. 2011;15(9):2519–2528. doi: 10.1089/ars.2011.3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoppeler H. Exercise-induced ultrastructural changes in skeletal muscle. International Journal of Sports Medicine. 1986;07(04):187–204. doi: 10.1055/s-2008-1025758. [DOI] [PubMed] [Google Scholar]
- 55.Powers S. K., Jackson M. J. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiological Reviews. 2008;88(4):1243–1276. doi: 10.1152/physrev.00031.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barbieri E., Sestili P. Reactive oxygen species in skeletal muscle signaling. Journal of Signal Transduction. 2012;2012:p. 982794. doi: 10.1155/2012/982794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Powers S. K., Duarte J., Kavazis A. N., Talbert E. E. Reactive oxygen species are signalling molecules for skeletal muscle adaptation. Experimental Physiology. 2010;95(1):1–9. doi: 10.1113/expphysiol.2009.050526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ott M., Gogvadze V., Orrenius S., Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis. 2007;12(5):913–922. doi: 10.1007/s10495-007-0756-2. [DOI] [PubMed] [Google Scholar]
- 59.Anderson E. J., Neufer P. D. Type II skeletal myofibers possess unique properties that potentiate mitochondrial H(2)O(2) generation. American Journal of Physiology. Cell Physiology. 2006;290(3):C844–C851. doi: 10.1152/ajpcell.00402.2005. [DOI] [PubMed] [Google Scholar]
- 60.Kaufman R. J. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes & Development. 1999;13(10):1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- 61.Santos C. X., Tanaka L. Y., Wosniak J., Laurindo F. R. Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxidants & Redox Signaling. 2009;11(10):2409–2427. doi: 10.1089/ars.2009.2625. [DOI] [PubMed] [Google Scholar]
- 62.Wang M., Kaufman R. J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nature Reviews. Cancer. 2014;14(9):581–597. doi: 10.1038/nrc3800. [DOI] [PubMed] [Google Scholar]
- 63.Oslowski C. M., Urano F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods in Enzymology. 2011;490:71–92. doi: 10.1016/B978-0-12-385114-7.00004-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schroder M., Kaufman R. J. The mammalian unfolded protein response. Annual Review of Biochemistry. 2005;74(1):739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 65.Wu J., Kaufman R. J. From acute ER stress to physiological roles of the unfolded protein response. Cell Death and Differentiation. 2006;13(3):374–384. doi: 10.1038/sj.cdd.4401840. [DOI] [PubMed] [Google Scholar]
- 66.Honors M. A., Kinzig K. P. The role of insulin resistance in the development of muscle wasting during cancer cachexia. Journal of Cachexia, Sarcopenia and Muscle. 2012;3(1):5–11. doi: 10.1007/s13539-011-0051-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Montgomery M. K., Turner N. Mitochondrial dysfunction and insulin resistance: an update. Endocrine Connections. 2015;4(1):R1–R15. doi: 10.1530/EC-14-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Unger R. H. Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology. 2003;144(12):5159–5165. doi: 10.1210/en.2003-0870. [DOI] [PubMed] [Google Scholar]
- 69.Koopman R., van Loon L. J. Aging, exercise, and muscle protein metabolism. Journal of Applied Physiology. 2009;106(6):2040–2048. doi: 10.1152/japplphysiol.91551.2008. [DOI] [PubMed] [Google Scholar]
- 70.Harfmann B. D., Schroder E. A., Kachman M. T., Hodge B. A., Zhang X., Esser K. A. Muscle-specific loss of Bmal1 leads to disrupted tissue glucose metabolism and systemic glucose homeostasis. Skeletal Muscle. 2016;6(1):p. 12. doi: 10.1186/s13395-016-0082-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mahfouz R., Khoury R., Blachnio-Zabielska A., et al. Characterising the inhibitory actions of ceramide upon insulin signaling in different skeletal muscle cell models: a mechanistic insight. PLoS One. 2014;9(7, article e101865) doi: 10.1371/journal.pone.0101865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Leskawa K. C., Erwin R. E., Buse P. E., Hogan E. L. Glycosphingolipid biosynthesis during myogenesis of rat L6 cells in vitro. Molecular and Cellular Biochemistry. 1988;83(1):47–54. doi: 10.1007/BF00223197. [DOI] [PubMed] [Google Scholar]
- 73.Anastasia L., Papini N., Colazzo F., et al. NEU3 sialidase strictly modulates GM3 levels in skeletal myoblasts C2C12 thus favoring their differentiation and protecting them from apoptosis. The Journal of Biological Chemistry. 2008;283(52):36265–36271. doi: 10.1074/jbc.M805755200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yu C., Chen Y., Cline G. W., et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. The Journal of Biological Chemistry. 2002;277(52):50230–50236. doi: 10.1074/jbc.M200958200. [DOI] [PubMed] [Google Scholar]
- 75.Li Y., Soos T. J., Li X., et al. Protein kinase C theta inhibits insulin signaling by phosphorylating IRS1 at Ser(1101) The Journal of Biological Chemistry. 2004;279(44):45304–45307. doi: 10.1074/jbc.C400186200. [DOI] [PubMed] [Google Scholar]
- 76.Hyde R., Hajduch E., Powell D. J., Taylor P. M., Hundal H. S. Ceramide down-regulates system a amino acid transport and protein synthesis in rat skeletal muscle cells. The FASEB Journal. 2005;19(3):461–463. doi: 10.1096/fj.04-2284fje. [DOI] [PubMed] [Google Scholar]
- 77.Guenther G. G., Peralta E. R., Rosales K. R., Wong S. Y., Siskind L. J., Edinger A. L. Ceramide starves cells to death by downregulating nutrient transporter proteins. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(45):17402–17407. doi: 10.1073/pnas.0802781105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cunningham J. T., Rodgers J. T., Arlow D. H., Vazquez F., Mootha V. K., Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450(7170):736–740. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- 79.Polak P., Cybulski N., Feige J. N., Auwerx J., Ruegg M. A., Hall M. N. Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metabolism. 2008;8(5):399–410. doi: 10.1016/j.cmet.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 80.Schieke S. M., Phillips D., JP M. C., Jr, et al. The mammalian target of rapamycin (mTOR) pathway regulates mitochondrial oxygen consumption and oxidative capacity. The Journal of Biological Chemistry. 2006;281(37):27643–27652. doi: 10.1074/jbc.M603536200. [DOI] [PubMed] [Google Scholar]
- 81.Morita M., Gravel S. P., Chenard V., et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metabolism. 2013;18(5):698–711. doi: 10.1016/j.cmet.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 82.Bentzinger C. F., Romanino K., Cloetta D., et al. Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metabolism. 2008;8(5):411–424. doi: 10.1016/j.cmet.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 83.Risson V., Mazelin L., Roceri M., et al. Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy. The Journal of Cell Biology. 2009;187(6):859–874. doi: 10.1083/jcb.200903131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Morita M., Gravel S. P., Hulea L., et al. mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle. 2015;14(4):473–480. doi: 10.4161/15384101.2014.991572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Duvel K., Yecies J. L., Menon S., et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Molecular Cell. 2010;39(2):171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Egan D., Kim J., Shaw R. J., Guan K. L. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy. 2011;7(6):643–644. doi: 10.4161/auto.7.6.15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Narsale A. A., Puppa M. J., Hardee J. P., et al. Short-term pyrrolidine dithiocarbamate administration attenuates cachexia-induced alterations to muscle and liver in ApcMin/+ mice. Oncotarget. 2016;7(37):59482–59502. doi: 10.18632/oncotarget.10699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lopes M. N., Black P., Ashford A. J., Pain V. M. Protein metabolism in the tumour-bearing mouse. Rates of protein synthesis in host tissues and in an Ehrlich ascites tumour at different stages in tumour growth. The Biochemical Journal. 1989;264(3):713–719. doi: 10.1042/bj2640713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Emery P. W., Edwards R. H., Rennie M. J., Souhami R. L., Halliday D. Protein synthesis in muscle measured in vivo in cachectic patients with cancer. British Medical Journal (Clinical Research Ed.) 1984;289(6445):584–586. doi: 10.1136/bmj.289.6445.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Eley H. L., Russell S. T., Baxter J. H., Mukerji P., Tisdale M. J. Signaling pathways initiated by beta-hydroxy-beta-methylbutyrate to attenuate the depression of protein synthesis in skeletal muscle in response to cachectic stimuli. American Journal of Physiology. Endocrinology and Metabolism. 2007;293(4):E923–E931. doi: 10.1152/ajpendo.00314.2007. [DOI] [PubMed] [Google Scholar]
- 91.Eley H. L., Russell S. T., Tisdale M. J. Effect of branched-chain amino acids on muscle atrophy in cancer cachexia. The Biochemical Journal. 2007;407(1):113–120. doi: 10.1042/BJ20070651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Eley H. L., Skipworth R. J., Deans D. A., Fearon K. C., Tisdale M. J. Increased expression of phosphorylated forms of RNA-dependent protein kinase and eukaryotic initiation factor 2alpha may signal skeletal muscle atrophy in weight-losing cancer patients. British Journal of Cancer. 2008;98(2):443–449. doi: 10.1038/sj.bjc.6604150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Smith K. L., Tisdale M. J. Increased protein degradation and decreased protein synthesis in skeletal muscle during cancer cachexia. British Journal of Cancer. 1993;67(4):680–685. doi: 10.1038/bjc.1993.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.White J. P., Puppa M. J., Gao S., Sato S., Welle S. L., Carson J. A. Muscle mTORC1 suppression by IL-6 during cancer cachexia: a role for AMPK. American Journal of Physiology. Endocrinology and Metabolism. 2013;304(10):E1042–E1052. doi: 10.1152/ajpendo.00410.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gao S., Carson J. A. Lewis lung carcinoma regulation of mechanical stretch-induced protein synthesis in cultured myotubes. American Journal of Physiology. Cell Physiology. 2016;310(1):C66–C79. doi: 10.1152/ajpcell.00052.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Puppa M. J., Gao S., Narsale A. A., Carson J. A. Skeletal muscle glycoprotein 130’s role in Lewis lung carcinoma-induced cachexia. The FASEB Journal. 2014;28(2):998–1009. doi: 10.1096/fj.13-240580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.White J. P., Baltgalvis K. A., Puppa M. J., Sato S., Baynes J. W., Carson J. A. Muscle oxidative capacity during IL-6-dependent cancer cachexia. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology. 2011;300(2):R201–R211. doi: 10.1152/ajpregu.00300.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.White J. P., Puppa M. J., Sato S., et al. IL-6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the ApcMin/+ mouse. Skeletal Muscle. 2012;2(1):p. 14. doi: 10.1186/2044-5040-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Brown J. L., Rosa-Caldwell M. E., Lee D. E., et al. Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour-bearing mice. Journal of Cachexia, Sarcopenia and Muscle. 2017 doi: 10.1002/jcsm.12232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yan Z., Lira V. A., Greene N. P. Exercise training-induced regulation of mitochondrial quality. Exercise and Sport Sciences Reviews. 2012;40(3):159–164. doi: 10.1097/JES.0b013e3182575599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Constantinou C., Fontes de Oliveira C. C., Mintzopoulos D., et al. Nuclear magnetic resonance in conjunction with functional genomics suggests mitochondrial dysfunction in a murine model of cancer cachexia. International Journal of Molecular Medicine. 2011;27(1):15–24. doi: 10.3892/ijmm.2010.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tzika A. A., Fontes-Oliveira C. C., Shestov A. A., et al. Skeletal muscle mitochondrial uncoupling in a murine cancer cachexia model. International Journal of Oncology. 2013;43(3):886–894. doi: 10.3892/ijo.2013.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shum A. M., Mahendradatta T., Taylor R. J., et al. Disruption of MEF2C signaling and loss of sarcomeric and mitochondrial integrity in cancer-induced skeletal muscle wasting. Aging. 2012;4(2):133–143. doi: 10.18632/aging.100436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Julienne C. M., Dumas J. F., Goupille C., et al. Cancer cachexia is associated with a decrease in skeletal muscle mitochondrial oxidative capacities without alteration of ATP production efficiency. Journal of Cachexia, Sarcopenia and Muscle. 2012;3(4):265–275. doi: 10.1007/s13539-012-0071-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Padrao A. I., Oliveira P., Vitorino R., et al. Bladder cancer-induced skeletal muscle wasting: disclosing the role of mitochondria plasticity. The International Journal of Biochemistry & Cell Biology. 2013;45(7):1399–1409. doi: 10.1016/j.biocel.2013.04.014. [DOI] [PubMed] [Google Scholar]
- 106.Antunes D., Padrao A. I., Maciel E., et al. Molecular insights into mitochondrial dysfunction in cancer-related muscle wasting. Biochimica et Biophysica Acta. 2014;1841(6):896–905. doi: 10.1016/j.bbalip.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 107.McLean J. B., Moylan J. S., Andrade F. H. Mitochondria dysfunction in lung cancer-induced muscle wasting in C2C12 myotubes. Frontiers in Physiology. 2014;5:p. 503. doi: 10.3389/fphys.2014.00503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Der-Torossian H., Wysong A., Shadfar S., Willis M. S., McDunn J., Couch M. E. Metabolic derangements in the gastrocnemius and the effect of compound a therapy in a murine model of cancer cachexia. Journal of Cachexia, Sarcopenia and Muscle. 2013;4(2):145–155. doi: 10.1007/s13539-012-0101-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fukawa T., Yan-Jiang B. C., Min-Wen J. C., et al. Excessive fatty acid oxidation induces muscle atrophy in cancer cachexia. Nature Medicine. 2016;22(6):666–671. doi: 10.1038/nm.4093. [DOI] [PubMed] [Google Scholar]
- 110.Puppa M. J., Murphy E. A., Fayad R., Hand G. A., Carson J. A. Cachectic skeletal muscle response to a novel bout of low-frequency stimulation. Journal of Applied Physiology. 2014;116(8):1078–1087. doi: 10.1152/japplphysiol.01270.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hardee J. P., Mangum J. E., Gao S., et al. Eccentric contraction-induced myofiber growth in tumor-bearing mice. Journal of Applied Physiology. 2016;120(1):29–37. doi: 10.1152/japplphysiol.00416.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bohnert K. R., McMillan J. D., Kumar A. Emerging roles of ER stress and unfolded protein response pathways in skeletal muscle health and disease. Journal of Cellular Physiology. 2017;233(1):67–78. doi: 10.1002/jcp.25852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bohnert K. R., Gallot Y. S., Sato S., Xiong G., Hindi S. M., Kumar A. Inhibition of ER stress and unfolding protein response pathways causes skeletal muscle wasting during cancer cachexia. The FASEB Journal. 2016;30(9):3053–3068. doi: 10.1096/fj.201600250RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Das S. K., Hoefler G. The role of triglyceride lipases in cancer associated cachexia. Trends in Molecular Medicine. 2013;19(5):292–301. doi: 10.1016/j.molmed.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Stephens N. A., Skipworth R. J., Macdonald A. J., Greig C. A., Ross J. A., Fearon K. C. Intramyocellular lipid droplets increase with progression of cachexia in cancer patients. Journal of Cachexia, Sarcopenia and Muscle. 2011;2(2):111–117. doi: 10.1007/s13539-011-0030-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Weber M. A., Krakowski-Roosen H., Schroder L., et al. Morphology, metabolism, microcirculation, and strength of skeletal muscles in cancer-related cachexia. Acta Oncologica. 2009;48(1):116–124. doi: 10.1080/02841860802130001. [DOI] [PubMed] [Google Scholar]
- 117.Das S. K., Eder S., Schauer S., et al. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science. 2011;333(6039):233–238. doi: 10.1126/science.1198973. [DOI] [PubMed] [Google Scholar]
- 118.Iritani S., Imai K., Takai K., et al. Skeletal muscle depletion is an independent prognostic factor for hepatocellular carcinoma. Journal of Gastroenterology. 2015;50(3):323–332. doi: 10.1007/s00535-014-0964-9. [DOI] [PubMed] [Google Scholar]
- 119.Prado C. M., Lieffers J. R., McCargar L. J., et al. Prevalence and clinical implications of sarcopenic obesity in patients with solid tumours of the respiratory and gastrointestinal tracts: a population-based study. The Lancet Oncology. 2008;9(7):629–635. doi: 10.1016/S1470-2045(08)70153-0. [DOI] [PubMed] [Google Scholar]
- 120.Burd N. A., Gorissen S. H., van Loon L. J. Anabolic resistance of muscle protein synthesis with aging. Exercise and Sport Sciences Reviews. 2013;41(3):169–173. doi: 10.1097/JES.0b013e318292f3d5. [DOI] [PubMed] [Google Scholar]
- 121.Horstman A. M., Olde Damink S. W., Schols A. M., van Loon L. J. Is cancer cachexia attributed to impairments in basal or postprandial muscle protein metabolism? Nutrients. 2016;8(8) doi: 10.3390/nu8080499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rennie M. J., Edwards R. H., Halliday D., Matthews D. E., Wolman S. L., Millward D. J. Muscle protein synthesis measured by stable isotope techniques in man: the effects of feeding and fasting. Clinical Science. 1982;63(6):519–523. doi: 10.1042/cs0630519. [DOI] [PubMed] [Google Scholar]
- 123.Garlick P. J., Clugston G. A., Swick R. W., Waterlow J. C. Diurnal pattern of protein and energy metabolism in man. The American Journal of Clinical Nutrition. 1980;33(9):1983–1986. doi: 10.1093/ajcn/33.9.1983. [DOI] [PubMed] [Google Scholar]
- 124.Paddon-Jones D., Sheffield-Moore M., Zhang X. J., et al. Amino acid ingestion improves muscle protein synthesis in the young and elderly. American Journal of Physiology. Endocrinology and Metabolism. 2004;286(3):E321–E328. doi: 10.1152/ajpendo.00368.2003. [DOI] [PubMed] [Google Scholar]
- 125.Volpi E., Mittendorfer B., Wolf S. E., Wolfe R. R. Oral amino acids stimulate muscle protein anabolism in the elderly despite higher first-pass splanchnic extraction. The American Journal of Physiology. 1999;277(3, Part 1):E513–E520. doi: 10.1152/ajpendo.1999.277.3.E513. [DOI] [PubMed] [Google Scholar]
- 126.Liu Z., Jahn L. A., Wei L., Long W., Barrett E. J. Amino acids stimulate translation initiation and protein synthesis through an Akt-independent pathway in human skeletal muscle. The Journal of Clinical Endocrinology and Metabolism. 2002;87(12):5553–5558. doi: 10.1210/jc.2002-020424. [DOI] [PubMed] [Google Scholar]
- 127.Cuthbertson D., Smith K., Babraj J., et al. Anabolic signaling deficits underlie amino acid resistance of wasting, aging muscle. The FASEB Journal. 2005;19(3):422–424. doi: 10.1096/fj.04-2640fje. [DOI] [PubMed] [Google Scholar]
- 128.Fujita S., Dreyer H. C., Drummond M. J., et al. Nutrient signalling in the regulation of human muscle protein synthesis. The Journal of Physiology. 2007;582(2):813–823. doi: 10.1113/jphysiol.2007.134593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Anthony J. C., Yoshizawa F., Anthony T. G., Vary T. C., Jefferson L. S., Kimball S. R. Leucine stimulates translation initiation in skeletal muscle of postabsorptive rats via a rapamycin-sensitive pathway. The Journal of Nutrition. 2000;130(10):2413–2419. doi: 10.1093/jn/130.10.2413. [DOI] [PubMed] [Google Scholar]
- 130.Sancak Y., Bar-Peled L., Zoncu R., Markhard A. L., Nada S., Sabatini D. M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141(2):290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Katsanos C. S., Kobayashi H., Sheffield-Moore M., Aarsland A., Wolfe R. R. Aging is associated with diminished accretion of muscle proteins after the ingestion of a small bolus of essential amino acids. The American Journal of Clinical Nutrition. 2005;82(5):1065–1073. doi: 10.1093/ajcn/82.5.1065. [DOI] [PubMed] [Google Scholar]
- 132.Katsanos C. S., Kobayashi H., Sheffield-Moore M., Aarsland A., Wolfe R. R. A high proportion of leucine is required for optimal stimulation of the rate of muscle protein synthesis by essential amino acids in the elderly. American Journal of Physiology. Endocrinology and Metabolism. 2006;291(2):E381–E387. doi: 10.1152/ajpendo.00488.2005. [DOI] [PubMed] [Google Scholar]
- 133.Guillet C., Prod'homme M., Balage M., et al. Impaired anabolic response of muscle protein synthesis is associated with S6K1 dysregulation in elderly humans. The FASEB Journal. 2004;18(13):1586–1587. doi: 10.1096/fj.03-1341fje. [DOI] [PubMed] [Google Scholar]
- 134.Rasmussen B. B., Wolfe R. R., Volpi E. Oral and intravenously administered amino acids produce similar effects on muscle protein synthesis in the elderly. The Journal of Nutrition, Health & Aging. 2002;6(6):358–362. [PMC free article] [PubMed] [Google Scholar]
- 135.Lang C. H., Frost R. A. Differential effect of sepsis on ability of leucine and IGF-I to stimulate muscle translation initiation. American Journal of Physiology. Endocrinology and Metabolism. 2004;287(4):E721–E730. doi: 10.1152/ajpendo.00132.2004. [DOI] [PubMed] [Google Scholar]
- 136.Lang C. H., Frost R. A. Endotoxin disrupts the leucine-signaling pathway involving phosphorylation of mTOR, 4E-BP1, and S6K1 in skeletal muscle. Journal of Cellular Physiology. 2005;203(1):144–155. doi: 10.1002/jcp.20207. [DOI] [PubMed] [Google Scholar]
- 137.Chesley A., MacDougall J. D., Tarnopolsky M. A., Atkinson S. A., Smith K. Changes in human muscle protein synthesis after resistance exercise. Journal of Applied Physiology. 1992;73(4):1383–1388. doi: 10.1152/jappl.1992.73.4.1383. [DOI] [PubMed] [Google Scholar]
- 138.Tipton K. D., Ferrando A. A., Phillips S. M., Doyle D., Jr., Wolfe R. R. Postexercise net protein synthesis in human muscle from orally administered amino acids. The American Journal of Physiology. 1999;276(4, Part 1):E628–E634. doi: 10.1152/ajpendo.1999.276.4.E628. [DOI] [PubMed] [Google Scholar]
- 139.Dreyer H. C., Drummond M. J., Pennings B., et al. Leucine-enriched essential amino acid and carbohydrate ingestion following resistance exercise enhances mTOR signaling and protein synthesis in human muscle. American Journal of Physiology. Endocrinology and Metabolism. 2008;294(2):E392–E400. doi: 10.1152/ajpendo.00582.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rasmussen B. B., Tipton K. D., Miller S. L., Wolf S. E., Wolfe R. R. An oral essential amino acid-carbohydrate supplement enhances muscle protein anabolism after resistance exercise. Journal of Applied Physiology. 2000;88(2):386–392. doi: 10.1152/jappl.2000.88.2.386. [DOI] [PubMed] [Google Scholar]
- 141.Dreyer H. C., Fujita S., Cadenas J. G., Chinkes D. L., Volpi E., Rasmussen B. B. Resistance exercise increases AMPK activity and reduces 4E-BP1 phosphorylation and protein synthesis in human skeletal muscle. The Journal of Physiology. 2006;576(2):613–624. doi: 10.1113/jphysiol.2006.113175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kumar V., Selby A., Rankin D., et al. Age-related differences in the dose-response relationship of muscle protein synthesis to resistance exercise in young and old men. The Journal of Physiology. 2009;587(1):211–217. doi: 10.1113/jphysiol.2008.164483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.MacDougall J. D., Gibala M. J., Tarnopolsky M. A., MacDonald J. R., Interisano S. A., Yarasheski K. E. The time course for elevated muscle protein synthesis following heavy resistance exercise. Canadian Journal of Applied Physiology. 1995;20(4):480–486. doi: 10.1139/h95-038. [DOI] [PubMed] [Google Scholar]
- 144.Phillips S. M., Tipton K. D., Aarsland A., Wolf S. E., Wolfe R. R. Mixed muscle protein synthesis and breakdown after resistance exercise in humans. The American Journal of Physiology. 1997;273(1, Part 1):E99–107. doi: 10.1152/ajpendo.1997.273.1.E99. [DOI] [PubMed] [Google Scholar]
- 145.O'Neil T. K., Duffy L. R., Frey J. W., Hornberger T. A. The role of phosphoinositide 3-kinase and phosphatidic acid in the regulation of mammalian target of rapamycin following eccentric contractions. The Journal of Physiology. 2009;587(14):3691–3701. doi: 10.1113/jphysiol.2009.173609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Spangenburg E. E., Le Roith D., Ward C. W., Bodine S. C. A functional insulin-like growth factor receptor is not necessary for load-induced skeletal muscle hypertrophy. The Journal of Physiology. 2008;586(1):283–291. doi: 10.1113/jphysiol.2007.141507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Witkowski S., Lovering R. M., Spangenburg E. E. High-frequency electrically stimulated skeletal muscle contractions increase p70s6k phosphorylation independent of known IGF-I sensitive signaling pathways. FEBS Letters. 2010;584(13):2891–2895. doi: 10.1016/j.febslet.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Terzis G., Georgiadis G., Stratakos G., et al. Resistance exercise-induced increase in muscle mass correlates with p70S6 kinase phosphorylation in human subjects. European Journal of Applied Physiology. 2008;102(2):145–152. doi: 10.1007/s00421-007-0564-y. [DOI] [PubMed] [Google Scholar]
- 149.Baar K., Esser K. Phosphorylation of p70(S6k) correlates with increased skeletal muscle mass following resistance exercise. The American Journal of Physiology. 1999;276(1, Part 1):C120–C127. doi: 10.1152/ajpcell.1999.276.1.C120. [DOI] [PubMed] [Google Scholar]
- 150.Steiner J. L., Lang C. H. Sepsis attenuates the anabolic response to skeletal muscle contraction. Shock. 2015;43(4):344–351. doi: 10.1097/SHK.0000000000000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Fry C. S., Drummond M. J., Glynn E. L., et al. Aging impairs contraction-induced human skeletal muscle mTORC1 signaling and protein synthesis. Skeletal Muscle. 2011;1(1):p. 11. doi: 10.1186/2044-5040-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Deutz N. E., Safar A., Schutzler S., et al. Muscle protein synthesis in cancer patients can be stimulated with a specially formulated medical food. Clinical Nutrition. 2011;30(6):759–768. doi: 10.1016/j.clnu.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Dillon E. L., Basra G., Horstman A. M., et al. Cancer cachexia and anabolic interventions: a case report. Journal of Cachexia, Sarcopenia and Muscle. 2012;3(4):253–263. doi: 10.1007/s13539-012-0066-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Dillon E. L., Volpi E., Wolfe R. R., et al. Amino acid metabolism and inflammatory burden in ovarian cancer patients undergoing intense oncological therapy. Clinical Nutrition. 2007;26(6):736–743. doi: 10.1016/j.clnu.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hanson E. D., Nelson A. R., West D. W., et al. Attenuation of resting but not load-mediated protein synthesis in prostate cancer patients on androgen deprivation. The Journal of Clinical Endocrinology and Metabolism. 2017;102(3):1076–1083. doi: 10.1210/jc.2016-3383. [DOI] [PubMed] [Google Scholar]
- 156.Horstman A. M., Sheffield-Moore M. Nutritional/metabolic response in older cancer patients. Nutrition. 2015;31(4):605–607. doi: 10.1016/j.nut.2014.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Williams J. P., Phillips B. E., Smith K., et al. Effect of tumor burden and subsequent surgical resection on skeletal muscle mass and protein turnover in colorectal cancer patients. The American Journal of Clinical Nutrition. 2012;96(5):1064–1070. doi: 10.3945/ajcn.112.045708. [DOI] [PubMed] [Google Scholar]
- 158.Egan B., Zierath J. R. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metabolism. 2013;17(2):162–184. doi: 10.1016/j.cmet.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 159.Atherton P. J., Smith K. Muscle protein synthesis in response to nutrition and exercise. The Journal of Physiology. 2012;590(5):1049–1057. doi: 10.1113/jphysiol.2011.225003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wilkinson S. B., Phillips S. M., Atherton P. J., et al. Differential effects of resistance and endurance exercise in the fed state on signalling molecule phosphorylation and protein synthesis in human muscle. The Journal of Physiology. 2008;586(15):3701–3717. doi: 10.1113/jphysiol.2008.153916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Deuster P. A., Morrison S. D., Ahrens R. A. Endurance exercise modifies cachexia of tumor growth in rats. Medicine and Science in Sports and Exercise. 1985;17(3):385–392. [PubMed] [Google Scholar]
- 162.Penna F., Busquets S., Pin F., et al. Combined approach to counteract experimental cancer cachexia: eicosapentaenoic acid and training exercise. Journal of Cachexia, Sarcopenia and Muscle. 2011;2(2):95–104. doi: 10.1007/s13539-011-0028-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Salomao E. M., Toneto A. T., Silva G. O., Gomes-Marcondes M. C. Physical exercise and a leucine-rich diet modulate the muscle protein metabolism in Walker tumor-bearing rats. Nutrition and Cancer. 2010;62(8):1095–1104. doi: 10.1080/01635581.2010.492082. [DOI] [PubMed] [Google Scholar]
- 164.Puppa M. J., White J. P., Velazquez K. T., et al. The effect of exercise on IL-6-induced cachexia in the Apc (Min/+) mouse. Journal of Cachexia, Sarcopenia and Muscle. 2012;3(2):117–137. doi: 10.1007/s13539-011-0047-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.al-Majid S., McCarthy D. O. Resistance exercise training attenuates wasting of the extensor digitorum longus muscle in mice bearing the colon-26 adenocarcinoma. Biological Research for Nursing. 2001;2(3):155–166. doi: 10.1177/109980040100200301. [DOI] [PubMed] [Google Scholar]
- 166.Jaweed M. M., Herbison G. J., Miller E. E., Ditunno J. F. Compensatory hypertrophy of the soleus in tumor-bearing rats. Journal of the Neurological Sciences. 1983;61(2):171–179. doi: 10.1016/0022-510X(83)90003-5. [DOI] [PubMed] [Google Scholar]
- 167.Norton J. A., Lowry S. F., Brennan M. F. Effect of work-induced hypertrophy on skeletal muscle of tumor- and nontumor-bearing rats. Journal of Applied Physiology: Respiratory, Environmental and Exercise Physiology. 1979;46(4):654–657. doi: 10.1152/jappl.1979.46.4.654. [DOI] [PubMed] [Google Scholar]
- 168.Otis J. S., Lees S. J., Williams J. H. Functional overload attenuates plantaris atrophy in tumor-bearing rats. BMC Cancer. 2007;7(1):p. 146. doi: 10.1186/1471-2407-7-146. [DOI] [PMC free article] [PubMed] [Google Scholar]