

Abstract
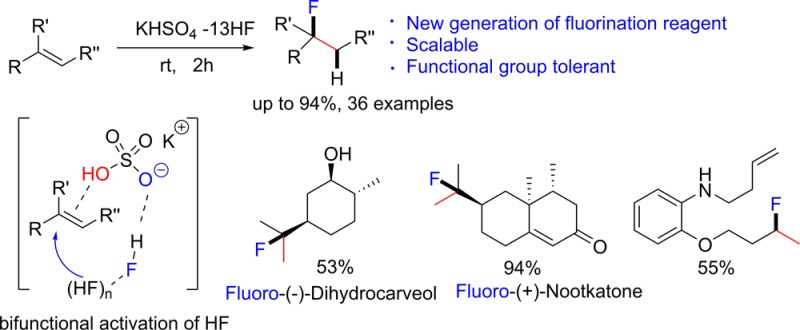
Expanding the use of fluorine in pharmaceuticals, agrochemicals and materials requires a widely applicable and more efficient protocol for the preparation of fluorinated compounds. We have developed a new generation nucleophilic fluorination reagent, KHSO4-13HF, HF 68 wt/wt %, that is not only easily handled and inexpensive but also capable of hydrofluorinating diverse, highly functionalized alkenes, including natural products. The high efficiency observed in this reaction hinges on the activation of HF using a highly “acidic” hydrogen bond acceptor.
The growing application of fluorine in pharmaceuticals, agrochemicals and materials1 has stimulated wide interest in fluorination methodologies.2 Because alkenes are one of the most important functionalities, finding a broadly applicable alkene hydrofluorination protocol is of fundamental importance in the preparation of fluorinated compounds. The direct hydrofluorination of alkenes using hydrogen fluoride (HF) reagents is the most straightforward and atom-economical protocol. Pioneering work by Olah and co-workers accomplished the direct hydrofluorination of alkenes using pyridine-HF (Scheme 1a),3 but this method only worked for limited functional-free alkenes, such as isobutene and cyclohexene, and it required using a large excess of pyridine-HF (as solvent). Later, Thibaudeau and co-workers reported an HF-based superacid system (HF/SbF5) to prepare β-fluoroamines (Scheme 1b) from allylic amines.2f Despite its great importance, the direct and widely applicable hydrofluorination of alkenes remains a synthetic challenge. To overcome the narrow scope and other limitations of direct hydrofluorination, several formal hydrofluorinationprotocols were developed using combinations of electrophilic fluorination reagents/reductants mediated by transition metals (Scheme 1c–e). For example, Boger and co-workers developed a Fe(III)/NaBH4/Selectfluor fluorination system via a free-radical mechanism.2h Hiroya and co-workers reported a cobalt/silane/N-fluoropyridinium system for mono- and α,α′-disubstituted alkenes (Scheme 1d).4 Gouverneur and co-workers developed a palladium-catalyzed hydrofluorination of aryl alkenes through sequential H– and F+ additions (Scheme 1e).2g
Scheme 1. Strategies for the Hydrofluorination of Alkenes.
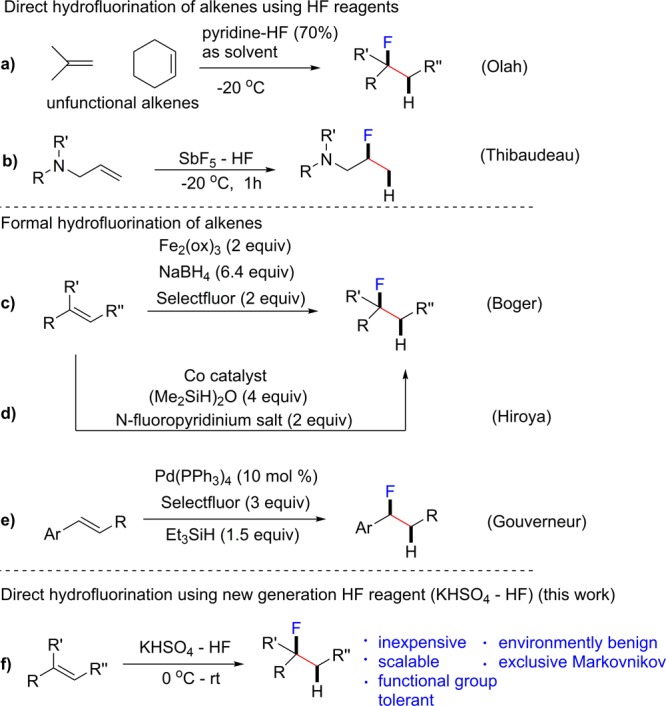
The aforementioned methods led to new reactivity pathways and solved many problems associated direct hydrofluorination; however, these methods have limitations such as low atom-economy and low functional group tolerance caused by using of strong reductants (such as NaBH4) and/or strong oxidants (such as Selectfluor). We herein report a metal-free, alkene hydrofluorination protocol using an easily handled, liquid HF complex that relies on an unprecedented bifunctional HF activation mode and shows unmatched scope and functional group tolerance (Scheme 1f).
Whereas a widely applicable direct alkene hydrofluorination has been hitherto elusive, the hydrochlorination or hydrobromination of alkenes are classic textbook reactions5 with wide applications.6 We conjectured that the shortcomings observed in hydrofluorination were caused by the acidity of HF (pKa = 3.2), less than that of HCl (pKa = −8.0) or HBr (pKa = −9.0), which is not strong enough to activate functionalized alkenes. Furthermore, HF is a toxic and corrosive gas at room temperature, and that is the reason why organic bases (or hydrogen bond acceptors) like Et3N,7 pyridine3,8 or DMPU9 are used to complex with HF, condensing it into a liquid solution and easing its handling. However, these bases or hydrogen bond acceptors reduce the acidity of HF even further. A good hydrogen bond acceptor (HBA) is bound to complex with HF and form a liquid solution but common organic bases, or neutral H-bond acceptors, will reduce the acidity of the system. To address this acidity/HF condensation conundrum, we proposed a multifunctional activation strategy capable of generating a liquid phase HF reagent that features both, high acidity and high fluoride nucleophilicity. Toward this end, we looked for a highly “acidic” hydrogen bond acceptor and chose an inexpensive and readily available inorganic salt, potassium bisulfate (KHSO4) (Scheme 2a).
Scheme 2. Bifunctional Activation Strategy for Hydrofluorination of Alkenes.
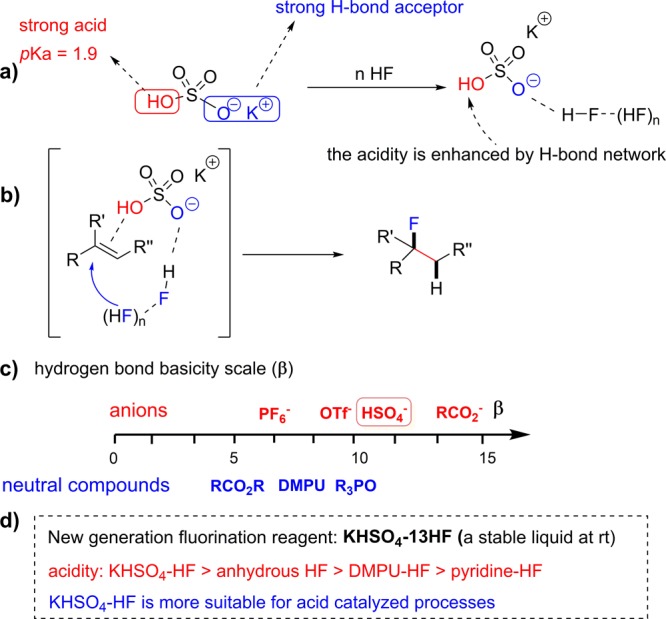
The bisulfate anion exhibits a bifunctional behavior: its −OH terminus is strongly acidic (pKa = 1.9) and its ionic −O– K+ terminus is a strong hydrogen bond acceptor. Thus, KHSO4 can form a hydrogen bond network with multiple molecules of HF. This hydrogen bonding interaction achieves two aims: (i) it condenses gaseous HF and forms a stable liquid at room temperature; (ii) it enhances the nucleophilicity of HF.10 Specifically, in the hydrofluorination of alkenes (Scheme 2b), we postulate that the acid terminus activates the alkene substrate while the H-bond acceptor terminus directs the nucleophilic attack of HF toward the acid-activated alkene, thereby accomplishing high acidity/high nucleophilicity and HF condensation simultaneously.
We found that KHSO4 formed a stable liquid at room temperature with unexpectedly large amounts of HF (up to 13 molecules of HF per molecule of KHSO4). This HF mole content is higher than that of any commercial reagents, including Olah’s reagent (pyridine-9HF) and our previously reported DMPU-12HF.9 The high HF affinity of KHSO4 could be rationalized using Hunter and co-workers’ recently reported H-bond scale (parameter β).11 In this scale, the hydrogen bonding basicities of anionic species were found to be significantly higher than those of neutral organic HBAs (Scheme 2c). Hydrogen bond basicity (measured by β) of HSO4– is comparable to the best neutral HBA, such as R3PO, and is higher than that of DMPU.9,12 An additional advantage of KHSO4 is its low cost, ready availability and easy removal in aqueous workup. So, we consider MHSO4-HF (M = alkali metals or ammonium) as the ideal next generation nucleophilic fluorination reagent (Scheme 2d). Indeed, KHSO4-HF is stable and can be easily handled at room temperature (see SI for more information). KHSO4 itself is a strong acid, and the acidity can be further enhanced by HF hydrogen network (Scheme 2a).
We used the hydrofluorination of alkene 1a as our model reaction (Table 1). As expected, Olah’s reagent (pyridine-9HF, HF 70 wt/wt %) failed to give a product (Table 1, entry 1), and the more acidic DMPU-12HF (HF 65 wt/wt %) reagent gave only trace amounts of hydrofluorination product 2a (Table 1, entry 2). To our great satisfaction, our new nucleophilic fluorination reagent, KHSO4-13HF (HF 68 wt/wt %), produced 2a, albeit in moderate yield (Table 1, entry 3). The K2SO4-14HF complex (Table 1, entry 4) was less effective: a larger excess of HF was present and an extended reaction time was needed. This result underscored the importance of our bifunctional activation strategy. The yield of the product was improved using 1,2–dichloroethane (DCE) as solvent (Table 1, entry 5). We also prepared an HF complex with lower HF content (KHSO4-8HF), but its reaction was slower compared with KHSO4-13HF (Table 1, entry 6). The HF complex of another bifunctional salt, KH2PO4, was also investigated but it gave no conversion (Table 1, entry 7). This result demonstrated that the acidic terminus of KHSO4 plays a crucial role in the addition of HF. Of all the solvents screened (Table 1, entries 8–13), toluene and DCE showed similar satisfactory results, but we selected DCE because of improved substrate solubility. The reaction in other solvents did not give a good conversion even after extended reaction times. Extending the reaction time to 2 h in DCE further improved the yield to 83% (Table 1, entry 15).
Table 1. Reaction Condition Optimization of Hydrofluorination of Alkenes*.
| entry | solvent | HF complex (A-xHF) | conditions | 1a/2a (%)a |
|---|---|---|---|---|
| 1 | DCM | Pyridine-9HF | 0 °C–rt, 0.5 h | 100/0 |
| 2 | DCM | DMPU-12HF | 0 °C–rt, 0.5 h | 96/4 |
| 3 | DCM | KHSO4-13HF | 0 °C–rt, 0.5 h | 57/43 |
| 4 | DCM | K2SO4-14HF | 0 °C–rt, 18 h | 84/16 |
| 5 | DCE | KHSO4-13HF | 0 °C–rt, 0.5 h | 29/71 |
| 6 | DCE | KHSO4-8HF | 0 °C–rt, 0.5 h | 58/32 |
| 7 | DCE | KH2PO4-9HF | 0 °C–rt, 0.5 h | 100/0 |
| 8 | dioxane | KHSO4-13HF | 0 °C–rt, 0.5 h | 100/0 |
| 9 | Et2O | KHSO4-13HF | 0 °C–rt, 0.5 h | 100/0 |
| 10 | CH3CN | KHSO4-13HF | 0 °C–rt, 0.5 h | 100/0 |
| 11 | EtOAc | KHSO4-13HF | 0 °C–rt, 0.5 h | 100/0 |
| 12 | DMSO | KHSO4-13HF | 0 °C–rt, 0.5 h | 100/0 |
| 13 | DMF | KHSO4-13HF | 0 °C–rt, 0.5 h | 100/0 |
| 14 | toluene | KHSO4-13HF | 0 °C–rt, 0.5 h | 26/74 |
| 15 | DCE | KHSO4-13HF | 0 °C–rt, 2 h | 3/83 |
Reaction conditions: 1 (0.2 mmol), HF complex (1 equiv based on the complex A-xHF, equivalents of HF is x), solvent (0.2 mL), 0 °C to rt.
GC–MS yield.
After establishing the optimal conditions for the hydrofluorination of alkenes, we explored the scope of this protocol (Table 2). First, we investigated the fluorination of monosubstituted alkenes (Table 2a). As shown in (Table 2a), a wide range of functional groups such as esters (2a, 2b), sulfonate (2c), amides (2d, 2e, 2f), ethers (2g, 2h, 2i, 2j), nitro (2g), nitrile (2i), aldehyde (2j), amine (2h), and alkyne (2r, 2s) were well tolerated. Also, alkenes with various heterocycles such as quinoline (2k), furan (2l), thiophene (2m), pyrrole (2n) and thiazole (2o) also gave good to excellent yields.
Table 2. Scope of Hydrofluorination of Alkenes*.
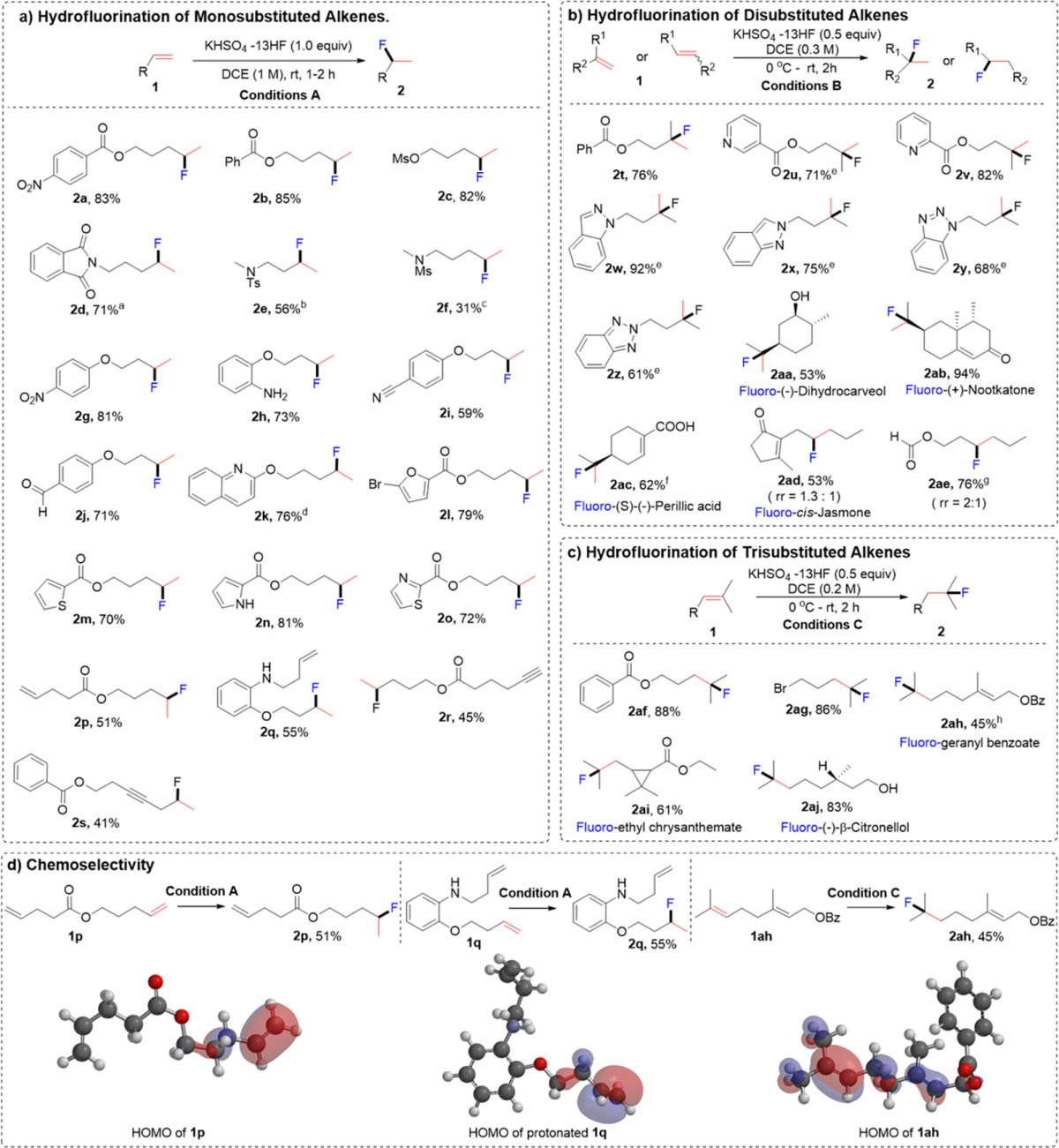
Conditions A: alkene 1 (0.2 mmol), KHSO4-13HF (1 equiv), DCE (0.2 mL), 0 °C to rt, 2 h. Conditions B: alkene 1 (0.2 mmol), KHSO4-13HF (0.5 equiv), DCE (0.6 mL), 0 °C to rt, 2 h. Conditions C: alkene 1 (0.2 mmol), KHSO4-13HF (0.5 equiv), DCE (1 mL), 0 °C to rt, 2 h.
rt, 15h.
50 °C, 2 h.
rt, 15h.
2 equiv of KHSO4-13HF was added.
1.5 equiv of KHSO4-13HF was added.
Isomer ratio = 6:1.
Neat reaction, 30 min, NMR yields.
Reverse addition: substrate solution was added to KHSO4-13HF reagent.
When this protocol was applied to more reactive disubstituted alkenes, we needed milder conditions, that is, more dilution (0.3 M) and less equivalents of KHSO4-13HF (conditions B) (Table 2b). Acceptable to good yields were observed withheterocyclic substrates like pyridines (2u, 2v), indazoles (2w, 2x) and benzotriazoles (2y, 2z). A 1.5 equiv of KHSO4-13HF was needed for full conversion, possibly because the basicity of these substrates neutralizes the acidity of the system, and therefore, more KHSO4-HF is needed to compensate it. Because disubstituted alkenes are commonly found in natural products, we screened natural products featuring various functionalities. We found that a natural product with a secondary alcohol, such as (−)-dihydrocarveol, tolerated the acidic reaction conditions and gave a moderate yield of the product (Table 2b, 2aa). Nootkatone, possessing an α,β unsaturated ketone moiety, and perillic acid, exhibiting a carboxylic acid moiety, also gave products 2ab and 2ac, respectively, in good yields. Similarly, 1,2-disubstituted substrates gave the regioisomeric products 2ad, 2ae in good yields.
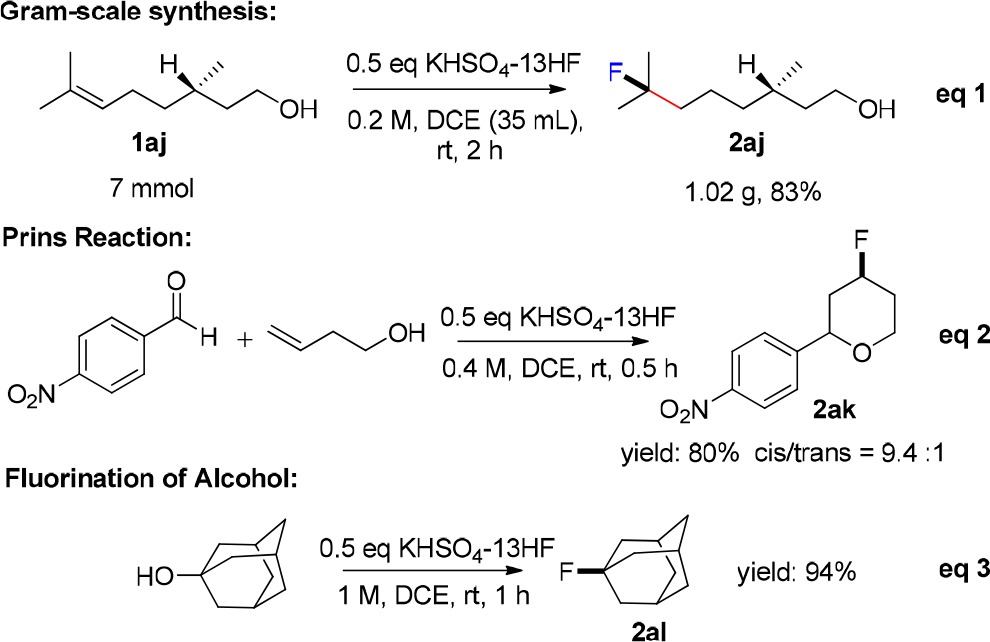
The reaction of KHSO4-13HF reagent was also investigated with the more reactive trisubstituted alkenes (Table 2c). The reaction concentration needed to be diluted further to 0.2 M to avoid the decomposition of the products (conditions C). The reaction of geranyl benzoate showed good chemoselectivity, with the more electron-rich double bond participatingin the hydrofluorination (2ah). We found that the cyclopropane motif remained intact after the reaction with ethyl chrysanthemate(2ai). Reaction with (−)-β-citronellol, which bears a primary alcohol functionality, also gave a good yield of the product 2aj. It is worth noting that our reagent exhibited very high and predictable chemoselectivity. In general, electron rich double bonds were more reactive (see 2ab, 2ac, 2ad in Table 2b). Dienes 1p, 1q and 1ah, whose two double bonds are located in very similar environments, underwent hydrofluorination selectively (Table 2d). We calculated the HOMO of the starting alkenes and found that it correctly predicted the chemoselectivity in each case. The hydrofluorination always occurred at the double bond bearing higher HOMO orbital density (Table 2d). This reagent was employed in a gram-scale hydrofluorination of (−)-β-citronellol. As shown in eq 1, 1.02 g of the fluorinated product 2aj was obtained (83% yield). It also showed good reactivity in the Prins reaction12g (eq 2) and the fluorination of alcohol (eq 3).
 |
1 |
In summary, we have developed a new generation HF reagent that is not only inexpensive and easily handled but is also highly efficient for the hydrofluorination of various highly functionalized alkenes. The excellent functional group tolerance, exclusive Markovnikov addition regioselectivity and high atom economy may facilitate the preparation of other fluorinated products at both the lab and industrial scale. For example, the reagent is a high activity fluorinating agent that could be recycled after regeneration by HF itself or HF generated at the time of use by other means, thereby minimizing HF storage. The reagent is an inherently safer means of storing HF and the bifunctional salts can be used as HF sequestering agents in an emergency. The reagent may also be a safer and easier-to-handle alkylation catalyst and etchant. Work on further applications for this new HF reagent is currently underway in our laboratory.
Acknowledgments
We are grateful to the National Institutes of Health for financial support (R01GM121660). B.X. is grateful to the National Science Foundation of China for financial support (NSFC-21672035).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b12704.
KHSO4-HF complex, starting materials and product preparation procedures, characterization data, NMR spectra (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Muller K.; Faeh C.; Diederich F. Science 2007, 317, 1881–1886. 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- a Fier P. S.; Hartwig J. F. J. Am. Chem. Soc. 2012, 134, 10795–10798. 10.1021/ja304410x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Grushin V. V. Acc. Chem. Res. 2010, 43, 160–171. 10.1021/ar9001763. [DOI] [PubMed] [Google Scholar]; c Chan K. S. L.; Wasa M.; Wang X.; Yu J.-Q. Angew. Chem., Int. Ed. 2011, 50, 9081–9084. 10.1002/anie.201102985. [DOI] [PubMed] [Google Scholar]; d Katcher M. H.; Doyle A. G. J. Am. Chem. Soc. 2010, 132, 17402–17404. 10.1021/ja109120n. [DOI] [PubMed] [Google Scholar]; e Furuya T.; Kamlet A. S.; Ritter T. Nature 2011, 473, 470–477. 10.1038/nature10108. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Thibaudeau S.; Martin-Mingot A.; Jouannetaud M.-P.; Karam O.; Zunino F. Chem. Commun. 2007, 3198–3200. 10.1039/b703629a. [DOI] [PubMed] [Google Scholar]; g Emer E.; Pfeifer L.; Brown J. M.; Gouverneur V. Angew. Chem., Int. Ed. 2014, 53, 4181–4185. 10.1002/anie.201310056. [DOI] [PubMed] [Google Scholar]; h Barker T. J.; Boger D. L. J. Am. Chem. Soc. 2012, 134, 13588–13591. 10.1021/ja3063716. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Champagne P. A.; Desroches J.; Hamel J.-D.; Vandamme M.; Paquin J.-F. Chem. Rev. 2015, 115, 9073–9174. 10.1021/cr500706a. [DOI] [PubMed] [Google Scholar]
- a Olah G. A.; Nojima M.; Kerekes I. Synthesis 1973, 1973, 779–780. 10.1055/s-1973-22297. [DOI] [Google Scholar]; b Olah G. A.; Watkins M. Org. Synth. 1978, 58, 75–9. 10.15227/orgsyn.058.0075. [DOI] [Google Scholar]
- Shigehisa H.; Nishi E.; Fujisawa M.; Hiroya K. Org. Lett. 2013, 15, 5158–5161. 10.1021/ol402696h. [DOI] [PubMed] [Google Scholar]
- a Carey F. A.; Sundberg R. J.. Advanced Organic Chemistry, Part A: Structure and Mechanisms, 5th ed.; Springer, 2007; Vol. A, pp 536–540. [Google Scholar]; b Ashtekar K. D.; Vetticatt M.; Yousefi R.; Jackson J. E.; Borhan B. J. Am. Chem. Soc. 2016, 138, 8114–8119. 10.1021/jacs.6b02877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schevenels F. T.; Shen M.; Snyder S. A. J. Am. Chem. Soc. 2017, 139, 6329–6337. 10.1021/jacs.6b12653. [DOI] [PubMed] [Google Scholar]
- Haufe G. J. Prakt. Chem./Chem.-Ztg. 1996, 338, 99–113. 10.1002/prac.19963380122. [DOI] [Google Scholar]
- a Olah G. A.; Welch J. T.; Vankar Y. D.; Nojima M.; Kerekes I.; Olah J. A. J. Org. Chem. 1979, 44, 3872–3881. 10.1021/jo01336a027. [DOI] [Google Scholar]; b Yoneda N. Tetrahedron 1991, 47, 5329–5365. 10.1016/S0040-4020(01)80970-4. [DOI] [Google Scholar]; c Bucsi I.; Török B.; Marco A. I.; Rasul G.; Prakash G. K. S.; Olah G. A. J. Am. Chem. Soc. 2002, 124, 7728–7736. 10.1021/ja0124109. [DOI] [PubMed] [Google Scholar]
- Okoromoba O. E.; Han J.; Hammond G. B.; Xu B. J. Am. Chem. Soc. 2014, 136, 14381–14384. 10.1021/ja508369z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond G.; Xu B.; Liang S. Chem. - Eur. J. 2017, 10.1002/chem.201702664. [DOI] [Google Scholar]
- a Pike S. J.; Hutchinson J. J.; Hunter C. A. J. Am. Chem. Soc. 2017, 139, 6700–6706. 10.1021/jacs.7b02008. [DOI] [PubMed] [Google Scholar]; b Smith D. A.; Beweries T.; Blasius C.; Jasim N.; Nazir R.; Nazir S.; Robertson C. C.; Whitwood A. C.; Hunter C. A.; Brammer L.; Perutz R. N. J. Am. Chem. Soc. 2015, 137, 11820–11831. 10.1021/jacs.5b07509. [DOI] [PubMed] [Google Scholar]
- a Abraham M. H. Chem. Soc. Rev. 1993, 22, 73–83. 10.1039/cs9932200073. [DOI] [Google Scholar]; b Abraham M. H.; Grellier P. L.; Prior D. V.; Morris J. J.; Taylor P. J. J. Chem. Soc., Perkin Trans. 2 1990, 521–529. 10.1039/p29900000521. [DOI] [Google Scholar]; c Abraham M. H.; Zhao Y. H. J. Org. Chem. 2004, 69, 4677–4685. 10.1021/jo049766y. [DOI] [PubMed] [Google Scholar]; d Abraham M. H.; Grellier P. L.; Prior D. V.; Duce P. P.; Morris J. J.; Taylor P. J. J. Chem. Soc., Perkin Trans. 2 1989, 699–711. 10.1039/p29890000699. [DOI] [Google Scholar]; e Abraham M. H.; Platts J. A. J. Org. Chem. 2001, 66, 3484–3491. 10.1021/jo001765s. [DOI] [PubMed] [Google Scholar]; f Laurence C.; Brameld K. A.; Graton J.; Le Questel J.-Y.; Renault E. J. Med. Chem. 2009, 52, 4073–4086. 10.1021/jm801331y. [DOI] [PubMed] [Google Scholar]; g Okoromoba O. E.; Hammond G. B.; Xu B. Org. Lett. 2015, 17, 3975–3977. 10.1021/acs.orglett.5b01919. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Okoromoba O. E.; Li Z.; Robertson N.; Mashuta M. S.; Couto U. R.; Tormena C. F.; Xu B.; Hammond G. B. Chem. Commun. 2016, 52, 13353–13356. 10.1039/C6CC07855A. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Xu P.; Wang F.; Fan G.; Xu X.; Tang P. Angew. Chem., Int. Ed. 2017, 56, 1101–1104. 10.1002/anie.201609741. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.