

Abstract
Introduction
Pathogenesis of Alzheimer’s disease (AD) in apolipoprotein E ε4 (APOE ε4) carriers remains unclear. We hypothesize that APOE isoforms have differential effects on synaptic function.
Methods
We compared levels of CSF neurogranin (Ng) between APOE ε4 carriers and noncarriers in 399 subjects with normal cognition, mild cognitive impairment (MCI), and AD. We examined associations between Ng levels and age, education, gender, CSF-Aβ42, and tau protein.
Results
Neurogranin levels were significantly higher in APOE ε4 carriers compared to APOE ε4 noncarriers with MCI. Levels of Ng between the APOE ε4 carriers and APOE ε4 noncarriers with AD did not differ. Ng levels were correlated with MMSE and levels of tau and Aβ42.
Discussion
Significantly higher CSF Ng levels in APOE ε4 carriers with MCI may reflect synaptic injury underlying early cognitive impairment. Neurogranin may be an early biomarker of AD and important for disease diagnosis and timing of intervention in APOE ε4 carriers.
Keywords: Synaptic function, Neurogranin, APOE ε4, Alzheimer’s disease, APOE, Mild cognitive impairment
1. Introduction
Alzheimer’s disease (AD) is the most common form of dementia. Pathologically, it is characterized by extracellular amyloid deposition and intracellular accumulation of hyperphosphorylated tau protein in a patient’s brain. In late-onset AD, over half of all AD cases are associated with the APOE ε4 genotype, highlighting the important role of APOE ε4 in AD pathogenesis [1].
APOE ε4 carriers with AD or amnestic mild cognitive impairment (MCI) have lower β-amyloid 42 (Aβ42), elevated total tau (t-tau), and phospho-tau (p-tau) in cerebrospinal fluid (CSF), compared to APOE ε4 noncarriers [2]. Structural magnetic resonance imaging (MRI) studies have shown that APOE ε4 carriers with AD and MCI have greater medial temporal lobe atrophy, particularly in the hippocampal area, compared to APOE ε4 noncarriers [3]. The accumulated evidence strongly supports the hypothesis that the APOE ε4 genotype exerts multiple effects on brain metabolism and structure.
Currently, the mechanism by which the APOE ε4 genotype contributes to the earlier onset and rapid progression of AD remains unclear. Although the APOE ε4 genotype affects amyloid metabolism, the association between amyloid deposition and cognitive impairment in AD is inconsistent. This suggests that other pathophysiological factors may be involved in cognitive decline seen among APOE ε4 carriers.
Synaptic dysfunction has been postulated as a central mechanism underlying cognitive impairment in AD [4]. Additionally, neuropathologic and biochemical studies demonstrate that postsynaptic components are damaged in AD [5]. The postsynaptic protein, drebrin, is remarkably reduced in the frontotemporal region of the AD brain, including in the hippocampus, compared to normal controls [6,7]. Neurogranin (Ng) is a postsynaptic protein which is highly expressed in hippocampus and involved in memory consolidation [8]. Kvartsberg’s and Maartje’s groups reported that Ng was markedly elevated in the CSF of patients with AD and MCI, indicating that Ng may be a biological marker reflecting synaptic integrity [9,10]. We thus hypothesized that APOE ε4 has detrimental effects on synaptic function, leading to elevated central nervous system Ng levels, which may in turn contribute to cognitive impairment in those APOE ε4 carriers who develop MCI and AD.
To investigate the effect of APOE ε4 on Ng, CSF Ng levels were examined in participants with normal cognition, MCI, and AD from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) dataset. We compared CSF Ng levels between APOE ε4 carriers and APOE ε4 noncarriers and examined the gene dose-effect of APOE ε4 on CSF Ng levels. We examined the correlation between Ng and mini-mental state examination (MMSE) score and levels of CSF Aβ42, t-tau protein, and p-tau protein. Finally, we analyzed the association of APOE ε4 with CSF Ng by controlling for age, education, gender, clinical diagnosis, and CSF levels of Aβ42, t-tau, and p-tau.
2. Materials and methods
2.1. ADNI study
Data used in the preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database. The ADNI was launched in 2003 as a public-private partnership, led by Principal Investigator Michael W. Weiner, MD. The primary goal has been to test whether serial MRI, positron emission tomography, other biological markers, and clinical and neuropsychological assessments can be combined to measure the progression of MCI and early AD. In our study, participants with an initial analysis of CSF Ng were included. Institutional review board approval was obtained at each ADNI site, and informed consent was obtained from each participant or authorized representative. Demographic information was extracted from the ADNI database. In this study, there were 111 participants with normal cognition, 193 participants with MCI, and 95 participants with AD.
2.2. CSF analyses
2.2.1. Quantification of Ng in CSF
The levels of CSF neurogranin (Ng) were determined at the Clinical Neurochemistry Laboratory, University of Gothenburg, Sweden, and are available in the ADNI database. A validation study of the Ng assay was performed [9]. CSF Ng levels of the ADNI samples were measured in duplicate. CSF Ng was analyzed by electrochemiluminescence technology (Meso Scale Discovery, Gaithersburg, Maryland, USA) using Ng 7, a monoclonal antibody specific for Ng as a coating antibody, and polyclonal Ng anti-rabbit (ab 23,570, Upstate) as a detector antibody [9]. Values are given in pg/mL.
2.2.2. Quantification of CSF Aβ42, t-tau, and p-tau
The CSF levels of Aβ42, t-tau, and p-tau were analysed by Leslie M. Shaw and John Q. Trojanowski’s group, Department of Pathology & Laboratory Medicine and Center for Neurodegenerative Diseases Research, Perelman School of Medicine, University of Pennsylvania. The xMAP Luminex platform and Innogenetics/Fujirebio Alz-Bio3 immunoassay kits were used [11]. Linear regression analyses were performed for Aβ1–42 and t-tau to compare the CSF concentration results obtained. Values are given as pg/mL for both tau and Aβ42.
2.3. APOE allele genotyping
APOE (gene map locus 19q13.2) genotypes of the study subjects were obtained from the ADNI database (adni.loni.usc.edu).
2.4. Statistical analysis
The F-test was used to examine the differences in continuous variables, and the χ2 test was used to compare the frequencies of categorical variables among the control, MCI, and AD groups. For multiple comparisons of means, statistically significant results for individual variables were followed by post hoc pairwise comparisons with the Tukey’s HSD (honest significant difference) test and the Tukey-Kramer adjustment (unequal group sizes) [12]. The Pearson correlation test was used to analyze the correlation between CSF Ng and other variables. To examine the potential association between the APOE ε4 genotype and CSF Ng levels, several general linear regression models were constructed: model 1 was unadjusted; model 2 was adjusted for age, sex, and education attainment; model 3 was additionally adjusted for diagnostic status; and model 4 was additionally adjusted for tau and Aβ42 levels. All data analyses were performed with SAS statistical software version 9.3 package for Windows (SAS Institute, Cary, NC) [13]. The level of statistical significance was set at P < .05. In Table 1, values are expressed as mean ± standard deviation. In Fig. 1, the error bars represent standard deviation. In Fig. 2, the error bars represent 95% confidence intervals adjusted by age, education, and gender.
Table 1.
Demographic information on normal cognition, MCI and AD subjects
| Control (N = 111) |
MCI (N = 193) |
AD (N = 95) |
P value | |
|---|---|---|---|---|
| Age (y) | 76 ± 5 | 74 ± 8 | 75 ± 8 | .317 |
| Education (y) | 15.8 ± 2.8 | 15.7 ± 3.0 | 15.1 ± 3.2 | .221 |
| Female (%) | 50a | 33b | 44 | .01 |
| APOE ε4 (%) | 24a | 53b | 71c | <.001 |
| MMSE | 29a ± 1.0 | 27b ± 2.0 | 24c ± 2 | <.001 |
| CDGLOB | 0a ± 0 | 0.5b ± 0 | 0.7c ± 0.3 | <.001 |
| Aβ42 (pg/mL) | 206.0a ± 55.4 | 165.6b ± 54.5 | 143.2c ± 39.7 | <.001 |
| t-tau (pg/mL) | 69.1a ± 29.90 | 102.2b ± 60.2 | 122.9c ± 57.4 | <.001 |
| p-tau (pg/mL) | 24.9a ± 14.8 | 35.5b ± 18.1 | 41.4c ± 19.7 | <.001 |
Abbreviations: MCI, mild cognitive impairment; AD, Alzheimer’s disease; MMSE, mini-mental state examination; CDGLOB, global clinical dementia rating; t-tau, total tau protein; p-tau, phospho-tau protein; Aβ42, β-amyloid 42.
NOTE. Values are expressed as mean ± standard deviation. Alphabetic “a”, “b”, and “c” superscripts indicate that the pairwise groups have statistical significance with the Tukey HSD procedure after the Tukey-Kramer adjustment for multiple comparison of unequal sample sizes [12].
Fig. 1.
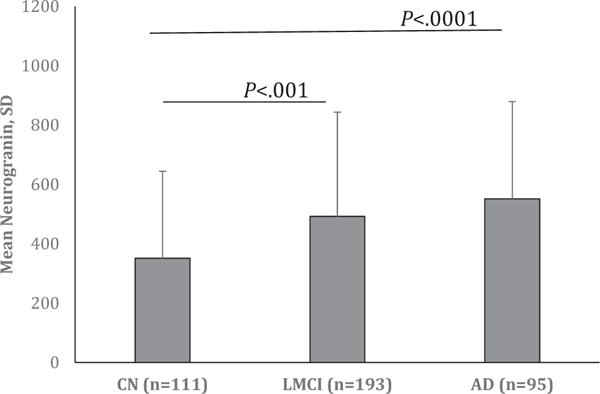
Significantly higher CSF neurogranin levels in the patients with MCI and AD compared to normal controls. Fig. 1 shows that CSF neurogranin levels are significantly higher in the subjects with MCI (P < .001) and AD (P < .0001) compared to that in subjects with normal cognition. The mean levels of CSF neurogranin with standard deviation are expressed on the Y-axis. Abbreviations: CSF, cerebrospinal fluid; MCI, mild cognitive impairment; AD, Alzheimer’s disease. NOTE. The error bars represent standard deviation.
Fig. 2.
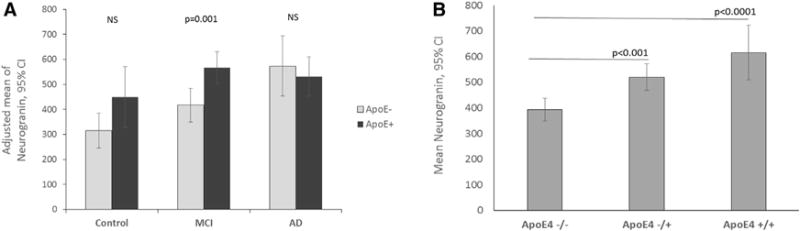
Comparison of CSF neurogranin levels in APOE ε4 carriers and APOE ε4 noncarriers with normal cognition, MCI and AD. (A) CSF neurogranin levels are significantly higher in APOE ε4 carriers with MCI compared to APOE ε4 noncarriers with MCI (P =.001). The Y-axis expresses the mean of CSF Ng levels adjusted for age, education, and gender with 95% confidence intervals. (B) The CSF neurogranin levels are significantly increased in a gene dose-dependent manner of APOE ε4 across the entire sample. The Y-axis expresses the mean of CSF Ng levels with 95% confidence intervals. Abbreviations: MCI, mild cognitive impairment; AD, Alzheimer’s disease. NOTE. The error bars represent 95% confidence intervals.
3. Results
3.1. Demographic information on study subjects
Table 1 depicts the demographic information: 111 subjects with normal cognition, 193 subjects with MCI, and 95 subjects with AD. There were no significant differences in age and education across the three groups. As expected, there was a significant difference in MMSE scores across the three groups (P < .001). The mean MMSE score in the patients with AD was 24 ± 2 (mean ± SD), consistent with mild AD dementia (Table 1). In addition, the global clinical dementia rating scores (CDR) among the three groups were compared. There were significant differences in global CDR scores across the three groups (P <.001). In agreement with the previous findings, >50% of the subjects with MCI and AD were APOE ε4 carriers [14]. There were significant differences in CSF Aβ42, t-tau, and p-tau across the three groups. Consistent with previous findings, the subjects with AD had the lowest CSF Aβ42 and the highest CSF t-tau and p-tau protein [15]. The demographic information on the subjects by gender is reported in the Supplementary data.
3.2. Significantly greater levels of CSF Ng in the APOE ε4 carriers
To study Ng in AD, CSF Ng levels were analyzed among the three groups: control, MCI, and AD. As shown in Fig. 1, the results showed that CSF Ng levels were significantly higher in the subjects with AD, followed by MCI, then normal controls (control vs. MCI, P < .001; controls vs. AD, P < .0001). The Y-axis of Fig. 1 expresses the mean of CSF Ng levels with their standard deviations.
To examine the association of the APOE ε4 genotype with CSF Ng, CSF Ng levels were compared between the APOE ε4 carriers and APOE ε4 noncarriers in the three groups. In the normal control group, the APOE ε4 carriers had a tendency to have higher CSF Ng compared to APOE ε4 noncarriers, although there was no statistical significance (P =.06; Fig. 2A). In the MCI group, the APOE ε4 carriers had significantly higher levels of CSF Ng compared to APOE ε4 noncarriers (P =.001). In the AD group, there was no statistical difference in CSF Ng levels between the APOE ε4 carriers and APOE ε4 noncarriers (P = .57). The Y-axis of Fig. 2A expresses the mean CSF Ng levels adjusted by age, education, and gender with 95% confidence intervals.
To confirm the effect of APOE ε4 on CSF Ng, the gene dose-effect of APOE ε4 on CSF Ng was analyzed. Across the entire sample, the CSF Ng levels were increased in a gene dose-dependent manner (heterozygous APOE ε4 vs. homozygous APOE ε4). This result confirmed that APOE ε4 is directly associated with CSF Ng levels (Fig. 2B). The Y-axis of Fig. 2B expresses the mean of CSF Ng levels with 95% confidence intervals.
We further explored the APOE-by-sex interaction on CSF Ng levels with a general linear model. Across the entire sample, the APOE-by-sex interaction on Ng levels was significant after adjusting for education, age, and diagnosis (P = .007). Female APOE ε4 carriers had significantly higher levels of CSF Ng compared to female noncarriers, whereas male carriers showed modestly higher levels of CSF Ng compared to male noncarriers (difference between means, 218; 2 in females, P <.001; 35.9 in males, P =.401).
3.3. Correlation of Ng with MMSE score, Aβ42, t-tau, and p-tau
To understand the mechanism underlying elevated Ng levels in APOE ε4 carriers, the correlations between CSF Ng and different variables were analyzed in this cohort using Pearson correlation test (Table 2). An inverse correlation between CSF Ng and MMSE scores was found (r = −0.16, P = .001). In addition, an inverse relationship between CSF Ng and Aβ42 levels was observed (r = −0.34, P < .0001). We further analyzed the relationship between Ng and tau protein. Strong positive correlations between Ng and t-tau protein, and also p-tau protein were observed (r = 0.71, P < .0001; r = 0.67, P < .0001, respectively).
Table 2.
Correlation between CSF Ng and MMSE, t-tau, p-tau, and Aβ42
| t-tau | Aβ42 | p-tau | MMSE | |
|---|---|---|---|---|
| CSF Ng | ||||
| R | 0.71 | −0.34 | 0.67 | −0.16 |
| P | <.0001 | <.0001 | <.0001 | 0.001 |
Abbreviations: CSF Ng, cerebrospinal fluid neurogranin; t-tau, total tau protein; Aβ42, β-amyloid 42; p-tau, phospho-tau protein; MMSE, mini-mental state.
NOTE. Pearson’s correlation test was applied to analyze the correlation between CSF Ng and other variables. The correlation coefficient is expressed as r.
3.4. Association of Ng with APOE ε4
To explore the relationship between APOE ε4 and CSF Ng, the potential association of CSF Ng with APOE genotype was analyzed using a general linear regression model, both with and without adjustment of other factors (Table 3). CSF Ng was significantly associated with APOE ε4 (standardized β = 0.22 (0.05); P < .0001) without controlling for other factors (model 1). A significant association of CSF Ng with APOE ε4 was found after adjusting for age, education, and gender (β = 0.21 (0.05); P < .0001; model 2). We further confirmed that Ng is significantly associated with APOE ε4 after adjusting for age, education, gender, and diagnosis (β = 0.15 (0.05); P =.0029; model 3). Finally, we examined the potential association of Ng with two typically assessed biomarkers of AD: CSF Aβ42 and tau. We found that the association of CSF Ng with APOE ε4 was no longer present after controlling for CSF amyloid and tau protein (β = 0.01 (0.04); P = .84; model 4).
Table 3.
Modeling of potential association of neurogranin with APOE ε4 adjusted for age, education, gender, clinical diagnosis, CSF tau, and Aβ 42
| Model
|
||||||||
|---|---|---|---|---|---|---|---|---|
| 1
|
2
|
3
|
4
|
|||||
| Beta (se) | P | Beta (se) | P | Beta (se) | P | Beta (se) | P | |
| APOE ε4 (+) vs. (−) | 0.22 (0.05) | <.0001 | 0.21 (0.05) | <.0001 | 0.15 (0.05) | .0029 | 0.01 (0.04) | .8405 |
| Age, y | −0.06 (0.05) | .2268 | −0.05 (0.05) | .2901 | −0.09 (0.04) | .0152 | ||
| Female vs. male | 0.09 (0.05) | .0622 | 0.11 (0.05) | .0264 | 0.02 (0.04) | .6258 | ||
| Education, y | −0.08 (0.05) | .1208 | −0.07 (0.05) | .1545 | −0.08 (0.04) | .0300 | ||
| MCI vs. control | 0.18 (0.06) | .0032 | −0.03 (0.05) | .5614 | ||||
| AD vs. control | 0.19 (0.06) | .0022 | −0.08 (0.05) | .0751 | ||||
| t-tau | 0.70 (0.04) | <.0001 | ||||||
| Aβ42 | −0.07 (0.04) | .1336 | ||||||
Abbreviations: MCI, mild cognitive impairment; AD, Alzheimer’s disease; t-tau, total tau protein; CSF Ng, cerebrospinal fluid neurogranin; Aβ42, β-amyloid 42. Beta is standardized beta.
NOTE. General linear regression models were constructed to explore the association of neurogranin with other variables. Model 1 was unadjusted; model 2 was adjusted for age, sex, and education attainment; model 3 was additionally adjusted for diagnostic status; and model 4 was additionally adjusted for tau and Aβ42 levels. Beta is standardized beta.
4. Discussion
In this study, CSF neurogranin (Ng) levels were significantly greater in APOE ε4 carriers compared to APOE ε4 noncarriers with MCI. Comparison of CSF Ng levels in APOE ε4 carriers versus APOE ε4 noncarriers with normal cognition showed a similar trend. CSF Ng levels were increased in an APOE ε4 gene dose-dependent manner. In contrast, there was no difference in CSF Ng levels between APOE ε4 carriers and noncarriers with AD. CSF Ng levels were correlated with MMSE score, CSF t-tau, p-tau, and Aβ42 levels. CSF Ng levels were significantly associated with APOE ε4 independent of age, education, and gender.
Although APOE ε4 has been reported as a strong risk factor for AD for decades, the mechanism underlying the pathogenesis of AD in APOE ε4 carriers remains unclear. Our findings provide evidence that APOE ε4 carriers may undergo synaptic damage to confer risk of AD. To our knowledge, this is the first report of significantly increased CSF Ng in APOE ε4 carriers compared to noncarriers with MCI. Higher CSF Ng levels were also observed among control APOE ε4 carriers compared to noncarriers, although there was no statistical significance between the two groups. Given the modest number of cognitively normal participants who were APOE ε4-positive, a larger number may have yielded statistically significant findings. Regarding the subjects with AD, there was no significant difference in CSF Ng levels between APOE ε4 carriers and noncarriers. Collectively, these data suggest that significantly increased CSF Ng in APOE ε4 carriers with MCI reflects an early event in the pathogenesis of AD. This finding is consistent with previous studies, which showed that high CSF Ng levels in subjects with MCI-predicted progression to dementia [9,10].
In this study, the effect of APOE ε4 on Ng was shown to be gene dose-dependent. This result is congruent with the previous findings of Reiman et al., who reported that APOE ε4 gene dose was correlated with lower regional cerebral metabolic rate of glucose [16].
With regard to gender, we observed that the APOE ε4 effect on CSF Ng levels was significantly stronger in females than males. This result is consistent with Sampedro’s findings that cerebral hypometabolism and atrophy were greater in female APOE ε4 carriers than male carriers [17]. These data suggest that increased CSF Ng levels, together with other reported metabolic and structural alterations in female APOE ε4 carriers, may contribute to a higher risk of AD in women [14,18,19].
Regarding the biology of Ng, it is localized in neuronal cell bodies and dendrites of cerebral cortex, and hippocampus [20]. Ng is implicated in long-term potentiation and visual-spatial learning [21,22]. Davidsson and Blennow found that the expression of Ng was reduced in AD brains [23]. In AD neocortical tissue, Ng mRNA translocation to dendrites was reduced, whereas in frontotemporal dementia, Ng mRNA translocation to dendrites was preserved [24,25]. Taken together, these findings suggest that Ng may play an integral role in the cascade of neural events leading to AD. Regarding the nature of CSF Ng, Kvartsberg’s group reported that CSF Ng was the C-terminally truncated species [9]. It is speculated that enzymatic activities may generate C-terminal fragments. It is possible that multiple synaptic injuries may upregulate enzymatic activities to generate C-terminal fragments of Ng, which are released into the lymphatic system in close proximity to the synaptically injured dendritic trees of injured cells. Another explanation is that dying neurons may contribute to the elevated Ng level in APOE carriers with MCI.
Regarding APOE genotype and synaptic function, animal studies have shown that APOE isoforms encoded by different APOE alleles differentially regulate synaptic plasticity and repair [3]. White et al. demonstrated that after entorhinal cortex lesioning, compensatory sprouting in association with synaptophysin and GAP-43 was impaired in transgenic mice expressing human APOE4 compared with APOE3 transgenic mice [26]. Their results are consistent with the association of impaired synaptic plasticity with APOE ε4 genotype. APOE ε4-targeted replacement mice showed reduced excitatory synaptic transmission and dendritic arborization, compared to APOE ε3-targeted replacement mice [27]. This finding indicates that APOE ε4 genotype may modulate postsynaptic function. Although mounting evidence shows an association of APOE genotype with synaptic function in animal models, the data regarding the APOE effect on synaptic function in human are limited. Our study provides in vivo evidence that the APOE isoforms may differentially regulate synaptic function in patients.
Although the correlations presented above do not imply any type of causation, a number of new investigative directions are suggested. In our study, the strongest correlation was found between Ng and tau protein. Yet, the pathway between synaptic injury and diffuse neuronal degeneration in AD remains to be determined, as tau pathology is well known to contribute to synapse degeneration and resulting dementia [28,29]. Although tau protein is considered to be primarily an axonal protein, recent studies show that tau is present in postsynaptic terminals of nondemented patients and AD patients, as well as in the somatodendritic compartment of cultured primary hippocampal neurons which were treated by Aβ oligomers [30,31]. Increased tau in CSF may be attributed to either a presynaptic-postsynaptic dysfunction of significant magnitude to be detectable, or that there is a primary postsynaptic injury. Aberrant function of tau and Ng protein mediated by APOE ε4 may represent a critical event in the pathogenesis of AD.
In this study, we note a second correlation between CSF Ng and Aβ42. This observation warrants further investigation to determine the extent to which amyloid pathology is associated with synaptic injury in APOE ε4 carriers. Emerging evidence suggests that Aβ-induced synaptic dysfunction is dependent on the elevation of cytoplasmic Ca2+ and N-methyl-D-aspartate (NMDA) receptor-mediated activity, and this process results in dendritic spine loss [32–34]. A recent study showed that Ng rescues Aβ-mediated depression in synaptic transmission in organotypic hippocampal slices [35]. Because Ng is localized in dendritic spines and involved in calcium-mediated NMDA receptor activity, the association of Ng with amyloid-induced pathology should be further investigated. Another possible explanation is that a correlation between CSF Ng and Aβ42 merely reflects the association of Aβ42 and tau. Indeed, when we controlled for CSF tau levels, the association between amyloid and Ng was no longer present. Of note, Kvartsberg et al. did not find a relationship between Aβ42 and Ng [9]. This discrepancy might be attributable to differences in the level of cognitive impairment as measured by MMSE between the two populations. Another interpretation of the lack of reproducibility compared to the referenced study may be due to the sample processing methodology and variability of polyclonal antibodies among lots used in the assay.
In summary, we found that CSF Ng levels were significantly elevated in APOE ε4 carriers with MCI compared to noncarriers. This finding indicates that postsynaptic injury may be an early pathologic event in APOE ε4 carriers who develop AD. CSF Ng levels were correlated with MMSE score, CSF tau, and Aβ42 levels. CSF Ng levels were likely associated with APOE ε4 and clinical diagnosis and independent of age, gender, and education. An association of Ng with APOE ε4 was not found after adjusting for CSF amyloid and tau protein. We believe that the interaction among Ng, tau protein, and Aβ42 may contribute to an important mechanism underlying synaptic damage in APOE ε4 carriers who develop AD.
Our study has limitations: (1) The cross-sectional design used in our study does not permit us to address the sequence of the events that may lead to cognitive impairment as a result of elevated Ng, decreased Aβ42, and increased tau protein in CSF. A longitudinal study, with multiple repeated measures, to monitor the levels of CSF Ng, Aβ42, and tau protein in APOE ε4 carriers is needed to provide such information pertaining to possible mechanisms underlying synaptic damage in the APOE ε4 carriers who develop AD. (2) Because the ADNI cohort is a selected convenience sample of volunteers, sample selection bias should be taken into consideration for interpretating the data. (3) Our study finding is based on ADNI samples. The result needs to be confirmed in other samples, ideally in a longitudinal population. (4) With a cross-sectional design, correlation and association analyses are performed. There may be other unknown additional mediating factors underlying observed correlations between Ng and other variables. Our finding is interesting, in that it demonstrates a key pathway which presages further in-depth investigation of in vivo synaptic function in AD.
Supplementary Material
RESEARCH IN CONTEXT.
Systematic review: The authors reviewed the literature using PubMed and AAIC meeting abstracts. CSF neurogranin (Ng) was significantly elevated in the subjects with Alzheimer’s disease (AD) and mild cognitive impairment (MCI). The effect of APOE ε4 on Ng has not been reported. Relevant citations are appropriately cited.
Interpretation: Significantly higher CSF Ng levels in APOE ε4 carriers with MCI may reflect synaptic injury underlying early cognitive impairment. Neurogranin may be an early biomarker of AD and important for disease diagnosis and timing of intervention in APOE ε4 carriers.
Future directions: The study suggests that APOE ε4 carriers may undergo synaptic damage conferring risk of Alzheimer’s disease. A longitudinal study should be designed to compare cognitive change between normal APOE ε4 carriers with high Ng and low Ng. A clinical outcome of the elevated Ng in APOE ε4 carriers should be investigated including brain structure and medication response.
Acknowledgments
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Eisai; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd. and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
We thank Drs. Brant Watson and Kristen Upchurch for critical reading of this article.
Dr. Blennow has served as a consultant for Eli Lilly, Novartis, Roche Diagnostics, and Sanofi-Aventis, on Advisory Boards for Amgen and IBL International, and given lectures for Fujirebio Europe and Lundbeck. Drs. Blennow and Zetterberg are co-founders of Brain Biomarker Solutions in Gothenburg AB, a GU Holding-based platform company at the University of Gothenburg.
Footnotes
Drs Sun, Dong, Levin, Crocco, Loewenstein, and Wright report no disclosures.
Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wpcontent/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.jalz.2016.05.003.
References
- 1.Mahley RW, Huang Y. Apolipoprotein E sets the stage: response to injury triggers neuropathology. Neuron. 2012;76:871–85. doi: 10.1016/j.neuron.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holtzman DM, Herz J, Bu G. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006312. doi: 10.1101/cshperspect.a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–18. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–91. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 5.Gong Y, Lippa CF. Review: disruption of the postsynaptic density in Alzheimer’s disease and other neurodegenerative dementias. Am J Alzheimers Dis Other Demen. 2010;25:547–55. doi: 10.1177/1533317510382893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harigaya Y, Shoji M, Shirao T, Hirai S. Disappearance of actin-binding protein, drebrin, from hippocampal synapses in Alzheimer’s disease. J Neurosci Res. 1996;43:87–92. doi: 10.1002/jnr.490430111. [DOI] [PubMed] [Google Scholar]
- 7.Shim KS, Lubec G. Drebrin, a dendritic spine protein, is manifold decreased in brains of patients with Alzheimer’s disease and Down syndrome. Neurosci Lett. 2002;324:209–12. doi: 10.1016/s0304-3940(02)00210-0. [DOI] [PubMed] [Google Scholar]
- 8.Diez-Guerra FJ. Neurogranin, a link between calcium/calmodulin and protein kinase C signaling in synaptic plasticity. IUBMB Life. 2010;62:597–606. doi: 10.1002/iub.357. [DOI] [PubMed] [Google Scholar]
- 9.Kvartsberg H, Duits FH, Ingelsson M, Andreasen N, Ohrfelt A, Andersson K, et al. Cerebrospinal fluid levels of the synaptic protein neurogranin correlates with cognitive decline in prodromal Alzheimer’s disease. Alzheimers Dement. 2015;11:1180–90. doi: 10.1016/j.jalz.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 10.Kester MI, Teunissen CE, Crimmins DL, Herries EM, Ladenson JH, Scheltens P, et al. Neurogranin as a Cerebrospinal Fluid Biomarker for Synaptic Loss in Symptomatic Alzheimer Disease. JAMA Neurol. 2015;72:1275–80. doi: 10.1001/jamaneurol.2015.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403–13. doi: 10.1002/ana.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gabriel KR. A simple method of multiple comparisons of means. J Am Stat Assoc. 1978;73:724–9. [Google Scholar]
- 13.Freund RJ, Littell RC, Spector PC. SAS System for Linear Models. 1986 [Google Scholar]
- 14.Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278:1349–56. [PubMed] [Google Scholar]
- 15.Rosenmann H. CSF biomarkers for amyloid and tau pathology in Alzheimer’s disease. J Mol Neurosci. 2012;47:1–14. doi: 10.1007/s12031-011-9665-5. [DOI] [PubMed] [Google Scholar]
- 16.Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, et al. Correlations between apolipoprotein E epsilon4 gene dose and brain-imaging measurements of regional hypometabolism. Proc Natl Acad Sci U S A. 2005;102:8299–302. doi: 10.1073/pnas.0500579102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sampedro F, Vilaplana E, de Leon MJ, Alcolea D, Pegueroles J, Montal V, et al. APOE-by-sex interactions on brain structure and metabolism in healthy elderly controls. Oncotarget. 2015;6:26663–74. doi: 10.18632/oncotarget.5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Payami H, Montee KR, Kaye JA, Bird TD, Yu CE, Wijsman EM, et al. Alzheimer’s disease, apolipoprotein E4, and gender. JAMA. 1994;271:1316–7. [PubMed] [Google Scholar]
- 19.Poirier J, Davignon J, Bouthillier D, Kogan S, Bertrand P, Gauthier S. Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet. 1993;342:697–9. doi: 10.1016/0140-6736(93)91705-q. [DOI] [PubMed] [Google Scholar]
- 20.Gerendasy DD, Sutcliffe JG. RC3/neurogranin, a postsynaptic calpacitin for setting the response threshold to calcium influxes. Mol Neurobiol. 1997;15:131–63. doi: 10.1007/BF02740632. [DOI] [PubMed] [Google Scholar]
- 21.Pak JH, Huang FL, Li J, Balschun D, Reymann KG, Chiang C, et al. Involvement of neurogranin in the modulation of calcium/ calmodulin-dependent protein kinase II, synaptic plasticity, and spatial learning: A study with knockout mice. Proc Natl Acad Sci U S A. 2000;97:11232–7. doi: 10.1073/pnas.210184697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miyakawa T, Yared E, Pak JH, Huang FL, Huang KP, Crawley JN. Neurogranin null mutant mice display performance deficits on spatial learning tasks with anxiety related components. Hippocampus. 2001;11:763–75. doi: 10.1002/hipo.1092. [DOI] [PubMed] [Google Scholar]
- 23.Davidsson P, Blennow K. Neurochemical dissection of synaptic pathology in Alzheimer’s disease. Int Psychogeriatr. 1998;10:11–23. doi: 10.1017/s1041610298005110. [DOI] [PubMed] [Google Scholar]
- 24.Chang JW, Schumacher E, Coulter PM, 2nd, Vinters HV, Watson JB. Dendritic translocation of RC3/neurogranin mRNA in normal aging, Alzheimer disease and fronto-temporal dementia. J Neuropathol Exp Neurol. 1997;56:1105–18. doi: 10.1097/00005072-199710000-00004. [DOI] [PubMed] [Google Scholar]
- 25.Landry CF, Watson JB, Kashima T, Campagnoni AT. Cellular influences on RNA sorting in neurons and glia: an in situ hybridization histochemical study. Brain Res Mol Brain Res. 1994;27:1–11. doi: 10.1016/0169-328x(94)90178-3. [DOI] [PubMed] [Google Scholar]
- 26.White F, Nicoll JA, Roses AD, Horsburgh K. Impaired neuronal plasticity in transgenic mice expressing human apolipoprotein E4 compared to E3 in a model of entorhinal cortex lesion. Neurobiol Dis. 2001;8:611–25. doi: 10.1006/nbdi.2001.0401. [DOI] [PubMed] [Google Scholar]
- 27.Wang C, Wilson WA, Moore SD, Mace BE, Maeda N, Schmechel DE, et al. Human apoE4-targeted replacement mice display synaptic deficits in the absence of neuropathology. Neurobiol Dis. 2005;18:390–8. doi: 10.1016/j.nbd.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 28.Spires-Jones TL, Hyman BT. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron. 2014;82:756–71. doi: 10.1016/j.neuron.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hyman BT. Amyloid-dependent and amyloid-independent stages of Alzheimer disease. Arch Neurol. 2011;68:1062–4. doi: 10.1001/archneurol.2011.70. [DOI] [PubMed] [Google Scholar]
- 30.Tai HC, Serrano-Pozo A, Hashimoto T, Frosch MP, Spires-Jones TL, Hyman BT. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am J Pathol. 2012;181:1426–35. doi: 10.1016/j.ajpath.2012.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zempel H, Thies E, Mandelkow E, Mandelkow EM. Abeta oligomers cause localized Ca(21) elevation, missorting of endogenous tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010;30:11938–50. doi: 10.1523/JNEUROSCI.2357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat Neurosci. 2010;13:812–8. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–13. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tu S, Okamoto S, Lipton SA, Xu H. Oligomeric Abeta-induced synaptic dysfunction in Alzheimer’s disease. Mol Neurodegener. 2014;9:48. doi: 10.1186/1750-1326-9-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaleka KS, Gerges NZ. Neurogranin restores amyloid beta-mediated synaptic transmission and long-term potentiation deficits. Exp Neurol. 2015;277:115–23. doi: 10.1016/j.expneurol.2015.12.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.