

Abstract
Since the 1990s, the International Union of Immunological Societies (IUIS) PID expert committee (EC), now called Inborn Errors of Immunity Committee, has published every other year a classification of the inborn errors of immunity. This complete catalog serves as a reference for immunologists and researchers worldwide. However, it was unadapted for clinicians at the bedside. For those, the IUIS PID EC is now publishing a phenotypical classification since 2013, which proved to be more user-friendly. There are now 320 single-gene inborn errors of immunity underlying phenotypes as diverse as infection, malignancy, allergy, auto-immunity, and auto-inflammation. We herein propose the revised 2017 phenotypic classification, based on the accompanying 2017 IUIS Inborn Errors of Immunity Committee classification.
Keywords: Primary immunodeficiencies, Classification, Phenotypic, IUIS, Inborn errors of immunity
Human primary immunodeficiency diseases (PID) comprise 330 distinct disorders with 320 different gene defects listed [1]. Long considered as rare diseases, recent studies tend to show that they are more common than generally thought, if only by their rapidly increasing number [2, 3].The International Union of Immunological Societies (IUIS) PID expert committee proposed a PID classification since 1999 [1], which facilitates clinical research and comparative studies worldwide; it is updated every other year to include new disorders or disease-causing genes. This classification is organized in tables, each of which groups PIDs that share a given pathogenesis. As this catalog is not adapted for use by the clinician at the bedside, the now called Inborn Errors of Immunity Committee proposed since 2013 a phenotypic complement to its classification [4]. Moreover, a smartphone application has been published, based on the 2015 phenotypic classification [5]. As the number of inborn errors of immunity is quickly increasing, and at an even faster pace since the advent of next-generation sequencing, this phenotypic classification requires revision at the same pace as the classical IUIS classification.
Here, we present an update of these figures (Figs. 1, 2, 3, 4, 5, 6, 7, 8, and 9), based on the accompanying 2017 report in inborn errors of immunity. We included all diseases included in the 2017 update of the IUIS classification [1] and split some categories in two parts to ease the lecture. An algorithm was assigned to each of the nine main groups of the classification and the same color was used for each group of similar conditions. Disease names are presented in red and genes in bold and italics. Mode of inheritance is expressed when adequate; if not expressed, the default mode of transmission is autosomal recessive. Clinical features that point to several diseases are presented in italics before the disease names.
Fig. 1.
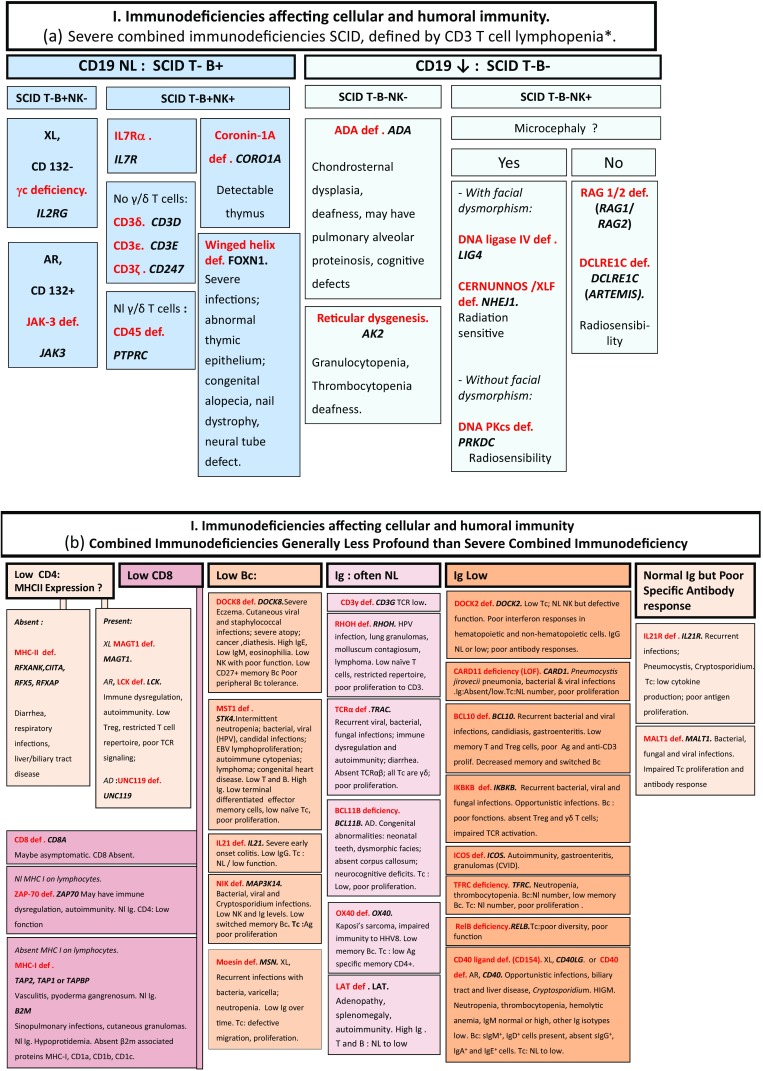
Immunodeficiencies affecting cellular and humoral immunity. a Severe combined immunodeficiencies defined by T cell lymphopenia. b Combined immunodeficiencies. * T cell lymphopenia in SCID is defined by CD3+ T cells < 300/µL. AD: autosomal dominant transmission; ADA: adenosine deaminase; Ag: antigen; AR: autosomal recessive transmission; β2m: bêta-2 microglobulin; Bc: B cells; CBC: complete blood count; CD: cluster of differentiation; CVID: common variable immunodeficiency; def: deficiency; EBV: Epstein Barr virus; HHV8: human herpes virus 8; HIGM: hyper IgM syndrome; HPV: human papillomavirus; Ig: immunoglobulins; MHC: major histocompatibility complex; Nl: normal; NK: natural killer; SCID: severe combined immunodeficiency; Tc: T cells; TCR: T cell receptor; Treg: regulatory T cells; XL: X-linked transmission
Fig. 2.

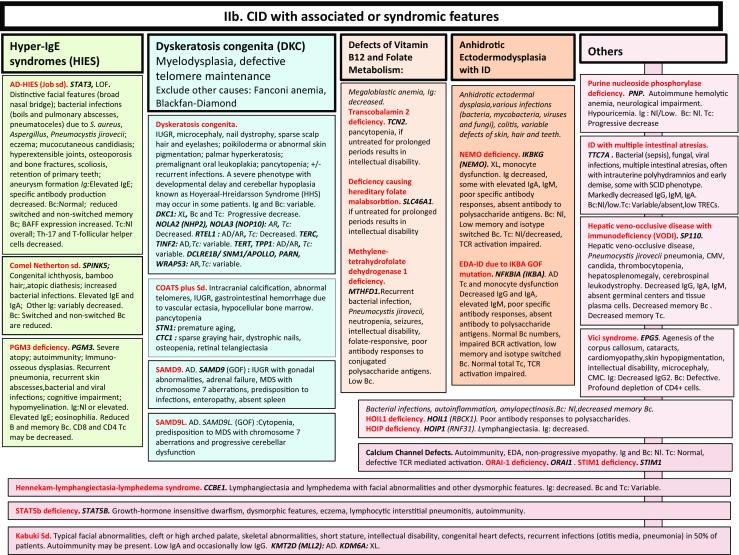
a, b CID with associated or syndromic features. Ab: antibody; AD: autosomal dominant transmission; ANA: anti-nuclear antibodies; ANCA: anti-neutrophil cytoplasm antibodies; AR: autosomal recessive transmission; Bc: B cells; BCG: Bacillus Calmette-Guerin; BCR: B cell receptor; CD: cluster of differentiation; CMV: cytomegalovirus; CNS: central nervous system; def: deficiency; DNA: desoxyribonucleic acid; DKC: dyskeratosis congenita; EDA: anhidrotic ectodermal dysplasia; GOF: gain-of-function; HIES: hyper IgE syndrome; FILS: facial dysmorphism, immunodeficiency, livedo and short stature; ID: immunodeficiency; Ig: immunoglobulins; IUGR: intrauterine growth retardation; LOF: loss-of-function; MDS: myelodysplasia; Nl: normal; NK: natural killer; PHA: phytohemagglutinin; PPS: polysaccharides; SCID: severe combined immunodeficiency; sd: syndrome; Tc: T cells; TCR: T cell receptor; TREC: T cell receptor excision circle; XL: X-linked transmission
Fig. 3.
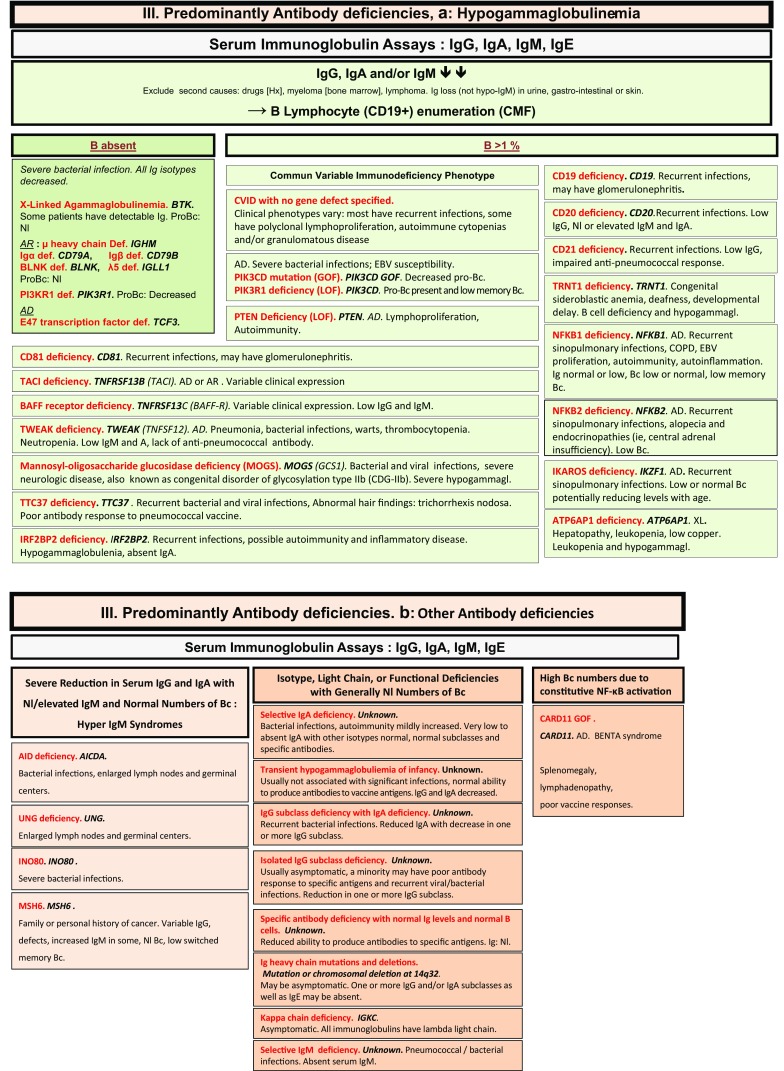
Predominantly antibody deficiencies. a Hypogammaglobulinemias. b Other antibody deficiencies. AD: autosomal dominant transmission; AR: autosomal recessive transmission; Bc: B cells; BENTA: B cell expansion with NF-κB and T cell anergy; CD: cluster of differentiation; CMF: flow cytometry; COPD: chronic obstructive pulmonary disease; def: deficiency; EBV: Epstein Barr virus; GOF: gain-of-function; Hx: patient history; Ig: immunoglobulins; Nl: normal; XL: X-linked transmission
Fig. 4.

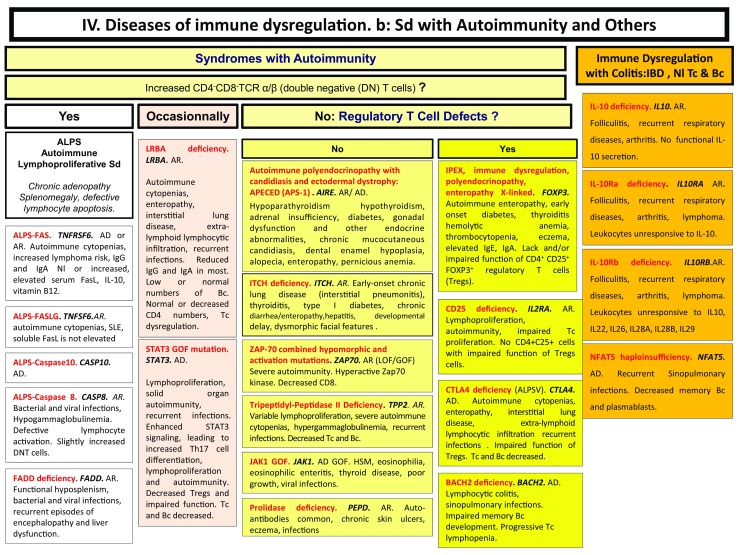
Diseases of immune dysregulation. a Hemophagocytic lymphohistiocytosis. b Other diseases of immune dysregulation. Ab: antibody; AD: autosomal dominant transmission; Ag: antigen; ALPS: autoimmune lymphoproliferative syndrome; APS: autoimmune polyendocrinopathy syndrome; AR: autosomal recessive transmission; Bc: B cells; CD: cluster of differentiation; CMF: flow cytometry; CTL: cytotoxic T lymphocytes; def: deficiency; DNT: double negative T cells; EBV: Epstein Barr virus; FHL: familial hemophagocytic lymphohistiocytosis; GOF: gain-of-function; HLH: hemophagocytic lymphohistiocytosis; (H)SM: (hepato)splenomegalia; IBD: inflammatory bowel disease; Ig: immunoglobulin; IL-10: interleukin-10; LOF: loss-of-function; iNKT: invariant NKT cells; NK: natural killer cells; Nl: normal; sd: syndrome; SLE: systemic lupus erythematous disease; Tc: T cells; TCR: T cell receptor; XL: X-linked transmission
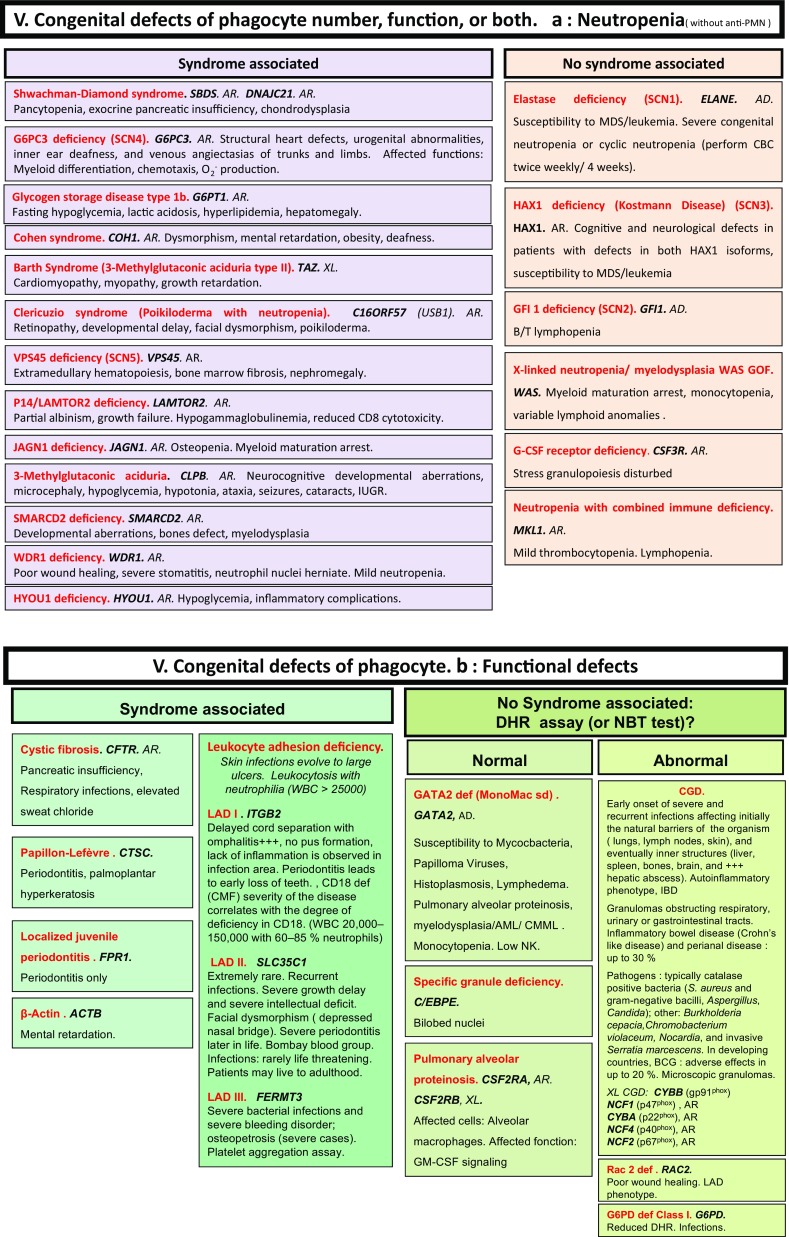
Congenital defects of phagocyte number, function, or both. a Neutropenia. b Functional defects of phagocytes. AD: autosomal dominant transmission; AML: acute myeloid leukemia; AR: autosomal recessive transmission; BCG: Bacillus Calmette-Guerin; CD: cluster of differentiation; CGD: chronic granulomatous disease; CMF: flow cytometry; CMML: chronic myelomonocytic leukemia; def: deficiency; DHR: dihydrorhodamine-1,2,3; GOF: gain-of-function; IUGR: intrauterine growth retardation; MDS: myelodysplasia; NBT: nitroblue of tetrazolium; NK: natural killer cells; WBC: white blood cells; XL: X-linked transmission
Fig. 6.
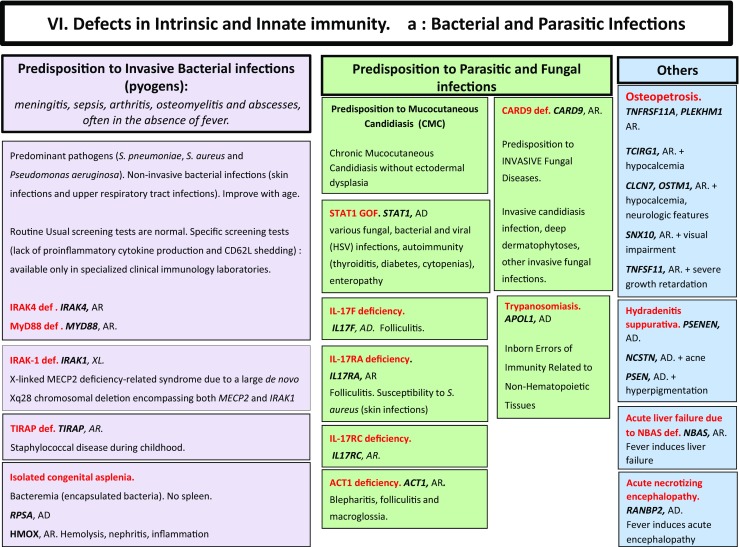
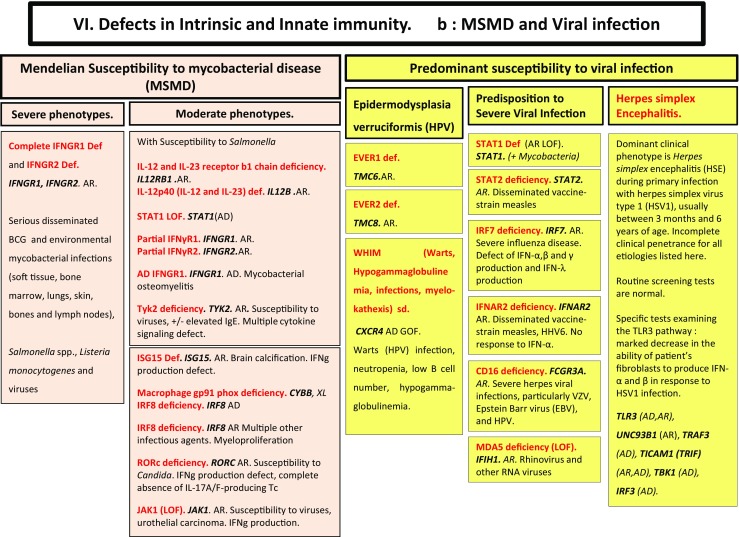
Defects in intrinsic and innate immunity. a Bacterial and parasitic infections. b MSMD and viral infection. AD: autosomal dominant transmission; AR: autosomal recessive transmission; BCG: Bacillus Calmette-Guerin; CD: cluster of differentiation; CMC: chronic mucocutaneous candidiasis; GOF: gain-of-function; IFNg: interferon-gamma; HHV6: human herpes virus type 6; HPV: human papilloma virus; HSV: herpes simplex virus; LOF: loss-of-function; MSMD: Mendelian susceptibility to mycobacterial disease; NK: natural killer cells; RNA: ribonucleic acid; sd: syndrome; Tc: T cells; TLR3: Toll-like receptor type 3; VZV: varicella zoster virus; XL: X-linked transmission
Fig. 7.
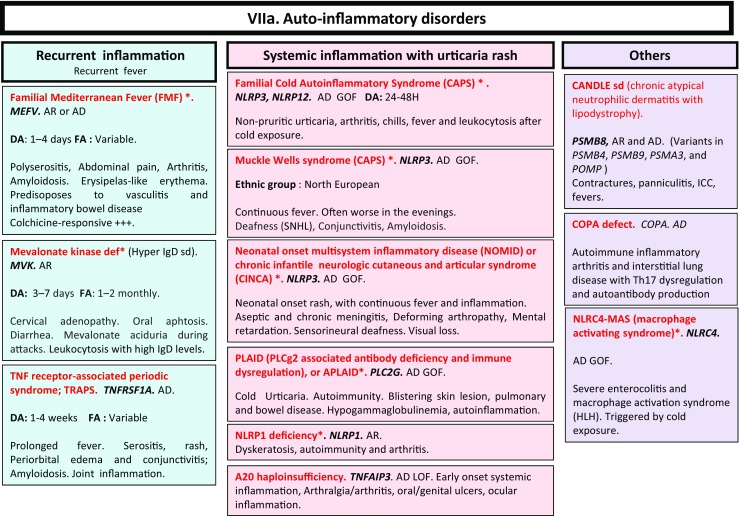
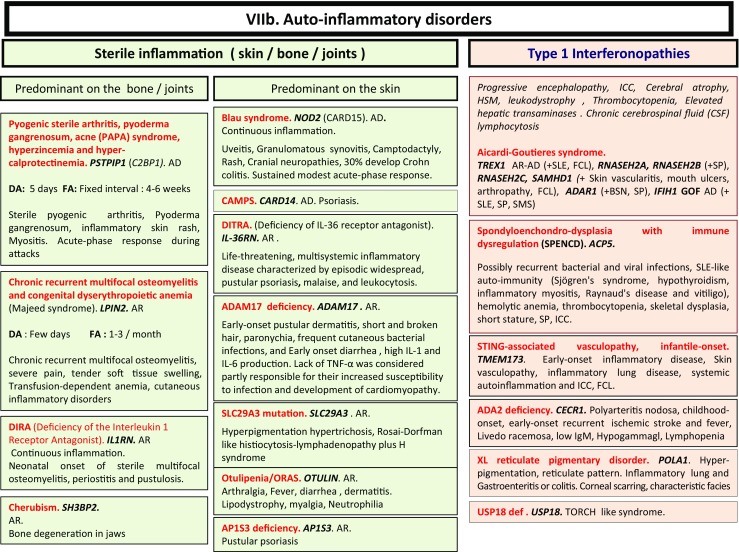
a, b Autoinflammatory disorders. *Diseases affecting the inflammasome. AD: autosomal dominant transmission; AR: autosomal recessive transmission; BSN: bilateral striatal necrosis; CAPS: cryopirin-associated periodic syndrome; DA: duration of inflammation episode; FA: frequency of inflammation episode; FCL: familial chilblain lupus; GOF: gain-of-function; HLH: hemophagocytic lymphohistiocytosis; HSM: hepatosplenomegalia; ICC: intracranial calcifications; IL: interleukin; LOF: loss-of-function; sd: syndrome; SLE: systemic lupus erythematosus; SMS: Singleton-Merten syndrome; SNHL: sensorineural hearing loss; SP: spastic paraparesis; TORCH: toxoplasmosis, other, rubella, cytomegalovirus, and herpes infections
Fig. 8.

Complement deficiencies. AD: autosomal dominant transmission; AH50: alternate pathway hemolytic activity; AR: autosomal recessive transmission; CH50: complement hemolytic activity; def: deficiency; LOF: loss-of-function; sd: syndrome; SLE: systemic lupus erythematosus; XL: X-linked transmission
Fig. 9.

Phenocopies of PID. ALPS: autoimmune lymphoproliferative syndrome; AutoAb: auto-antibodies; CID: combined immunodeficiency; CMC: chronic mucocutaneous candidiasis; GOF: gain-of-function; MSMD: Mendelian susceptibility to mycobacterial disease; PRCA: pure red cell aplasia
Compliance with Ethical Standards
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- 1.Picard C, Gaspar HB, Al-Herz W, Bousfiha A, Chatila T, Crow YJ, et al. International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee report on inborn errors of immunity. J Clin Immunol 2017(in Press). [DOI] [PMC free article] [PubMed]
- 2.Bousfiha AA, Jeddane L, Ailal F, Benhsaien I, Mahlaoui N, Casanova JL, Abel L. Primary immunodeficiency diseases worldwide: more common than generally thought. J Clin Immunol. 2013;33(1):1–7. doi: 10.1007/s10875-012-9751-7. [DOI] [PubMed] [Google Scholar]
- 3.Kobrynski L, Powell RW, Bowen S. Prevalence and morbidity of primary immunodeficiency diseases, United States 2001-2007. J Clin Immunol. 2014;34(8):954–961. doi: 10.1007/s10875-014-0102-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bousfiha AA, Jeddane L, Ailal F, Al Herz W, Conley ME, Cunningham-Rundles C, et al. A phenotypic approach for IUIS PID classification and diagnosis: guidelines for clinicians at the bedside. J Clin Immunol. 2013;33(6):1078–1087. doi: 10.1007/s10875-013-9901-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jeddane L, Ouair H, Benhsaien I, El Bakkouri J, Bousfiha AA. Primary immunodeficiency classification on smartphone. J Clin Immunol. 2017;37(1):1–2. doi: 10.1007/s10875-016-0354-6. [DOI] [PubMed] [Google Scholar]