

Abstract
Ectodysplasin A (Eda), a member of the tumour necrosis factor superfamily, plays an important role in ectodermal organ development. An EDA mutation underlies the most common of ectodermal dysplasias, that is X‐linked hypohidrotic ectodermal dysplasia (XLHED) in humans. Even though it lacks a developmental function, the role of Eda during the postnatal stage remains elusive. In this study, we found tight junctional proteins ZO‐1 and claudin‐1 expression is largely reduced in epidermal, corneal and lung epithelia in Eda mutant Tabby mice at different postnatal ages. These declines are associated with tail ulceration, corneal pannus formation and lung infection. Furthermore, topical application of recombinant Eda protein markedly mitigated corneal barrier dysfunction. Using cultures of a human corneal epithelial cell line and Tabby mouse skin tissue explants, Eda up‐regulated expression of ZO‐1 and claudin‐1 through activation of the sonic hedgehog signalling pathway. We conclude that EDA gene expression contributes to the maintenance of epithelial barrier function. Such insight may help efforts to identify novel strategies for improving management of XLHED disease manifestations in a clinical setting.
Keywords: Ectodysplasin A, epithelial barrier dysfunction, X‐linked hypohidrotic ectodermal dysplasia, tight junction
Introduction
Eda is a member of the tumour necrosis factor (TNF) superfamily regulating ectodermal development in various organs. In humans, germline EDA mutations cause a syndrome designated as XLHED characterized by the absence or malformation of multiple ectodermal appendages resulting in hypotrichosis, hypodontia, anhidrosis or hypohidrosis 1, 2, 3, 4. Spontaneous EDA mutant penetrance appears in other species including dog 5, cattle 6 and mouse 7.
XLHED‐afflicted individuals in their youth are at increased risk for developing severe, acute pneumonia and respiratory infections 2, 8. Respiratory infections, wheezing and recurrent sinus infections can last throughout adulthood in most cases 9, 10. XLHED patients also experience an increased prevalence of allergic diseases 11, atopic diathesis as well as chronic dermatitis throughout their lifetime 12. It was presumed that exocrine gland insufficiencies of the respiratory tract 8, 13, 14 and skin surface 10 could lead to chronic inflammatory and recurrent infection. However, the pathogenic mechanism underlying these changes remains unclear.
Ocular surface anomalies are another major category of XLHED‐related diseases. All adult XLHED patients have corneal pathology such as pannus formation 15. Eda mutant Tabby mice also experience dramatic increases in ocular surface inflammation and corneal pannus in their adult stage 16. Previous studies favour the hypothesis that keratopathy in XLHED patients results from an altered tear film lipid layer caused by a meibomian gland developmental defect 17. However, this possibility is not supported by the finding that transfection of the Eda‐A1 gene to Tabby mice prevented corneal defects without restoring meibomian gland formation. This result instead indicates that Eda is required for maintenance of ocular surface integrity rather than meibomian gland morphogenesis 16.
The clinical manifestations of XLHED indicate that not all of the symptoms result from EDA control of mechanisms underlying morphogenesis. Instead this realization prompted suggestions that EDA may play a role in maintaining the homeostasis of organs such as skin, cornea and pulmonary tissues after the developmental stage. Clarifying the functional roles of EDA in maintaining normal tissue function still warrants further study.
We show here that epithelial tight junctional integrity is compromised in the cornea, skin and lung of Tabby mice, which was accompanied by abnormal ZO‐1 and claudin‐1 protein expression leading to epithelial barrier dysfunction. This defect markedly increases tissue susceptibility to surface pathogenic infiltration, which leads to chronic inflammation in these mice. Moreover, we found that the Eda protein regulates epithelial tight junction formation through activation of the sonic hedgehog (Shh) signalling pathway. Our findings provide new insights into pathophysiological mechanism of XLHED disease and identify a new Eda protein function unrelated to morphogenesis during the postnatal stage.
Materials and methods
Animals
The EDA mutant Tabby mice (C57BL/6J‐AW‐J‐EdaTa−6J/J) and wild‐type C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). The mice were housed at the Experimental Animal Center of Xiamen University, and some were kept in a specific pathogen‐free (SPF) barrier facility, while others were kept in a conventional facility (CF) with free access to water and food. All animal protocols were performed in accordance with the Association for Research in Vision and Ophthalmology (ARVO) statement for the use of Animals in Ophthalmic and Vision Research and approved by the Animal Ethical Committee of Xiamen University. All experiments were performed in accordance with the tenets set forth in the Declaration of Helsinki.
Haematoxylin & eosin staining
After the animals were sacrificed, the eye, skin and lung tissues were dissected and fixed in 4% paraformaldehyde overnight and embedded in paraffin. The tissue sections (5 μm) were deparaffinized and stained with haematoxylin for 1 min. and eosin for 2 min. After that, the sections were mounted using mounting medium and examined under a light microscope (Eclipse 50i, Nikon, Japan).
Immunostaining
Cryostat sections (6 μm) of mice eyes or skin tissues or cultured cells were fixed in 4% paraformaldehyde for 20 min. The samples were washed 3 times with PBS, followed by incubation in 0.2% Triton X‐100 for 10 min. After rinsing three times with PBS for 5 min. each and pre‐incubation with 2% bovine serum albumin (BSA) for 1 hr at room temperature, sections were incubated with anti‐CD45 (sc‐52491,1:50,Santa Cruz, Dallas, TX, USA), ZO‐1 (61‐7300, 1:100, Invitrogen, Carlsbad, CA, USA), Claudin‐1 (519,000, 1:100, Invitrogen), Gli‐1 (sc‐20687, 1:50,Santa Cruz) primary antibodies at 4°C overnight. After three washes with PBS for 10 min. each, they were incubated with AlexaFluor 488‐conjugated secondary antibody (donkey anti‐rabbit, goat, rat or mouse IgG, 1:300, Life Technologies, Carlsbad, CA, USA) for 1 hr at room temperature. After three additional PBS washes for 5 min., the samples were counterstained with DAPI and then mounted for analysis under the confocal laser scanning microscope (Fluoview 1000, Olympus, Tokyo, Japan).
Western blotting
The skin, corneal epithelium, lung tissue or cultured cells were extracted with cold lysis buffer comprising 50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1% Nonidet P‐40, 0.5% sodium deoxycholate, 0.1% SDS, and protease and phosphatase inhibitor cocktails. Equal amounts of protein extracts were subjected to electrophoresis on 8% or 10% SDS‐PAGE and then electrophoretically transferred to PVDF membrane. After blocking in 1% BSA for 1 hr, the membranes were incubated with primary antibodies ZO‐1 (61‐7300, 1:1000, Invitrogen), claudin‐1 (519,000, 1:1000, Invitrogen), Gli‐1 (sc‐20687, 1:500,Santa Cruz) and β‐actin (A3854, 1:10,000, Sigma‐Aldrich) overnight at 4°C. After three washes with Tris‐buffered saline with 0.05% Tween‐20 for 10 min. each, the membranes were incubated with HRP‐conjugated goat or rabbit anti‐mouse or rabbit IgG (1:10,000) for 1 hr. The results were visualized by enhanced chemiluminescence reagents and recorded with an imaging system (ChemiDoc XRS, Bio‐Rad, Hercules, CA, USA).
RNA isolation and quantitative real‐time RT‐PCR analysis
Total RNA was isolated from the samples using Trizol Reagent (Invitrogen) and extracted following the manufacturer's protocol. RNA was reverse transcribed to cDNA by the ExScript RT Reagent kit (Takara Bio, Shiga, Japan). Quantitative real‐time RT‐PCR (qRT‐PCR) was performed with a StepOne Real‐Time detection system (Applied Biosystems, Foster City, CA, USA) using an SYBR Premix Ex Taq Kit (Takara Bio, Shiga, Japan). The amplification program included an initial denaturation step at 95°C for 10 min., followed by 40 cycles of 95°C for 10 sec., 57°C for 30 sec., and 75°C for 10 sec.; then, the melting curve analysis was conducted at once from 65 to 95°C. SYBR Green fluorescence was measured after each extension step, and the specificity of amplification was evaluated by melting curve analysis. The primers were designed using Primer 3 system, and they are listed in Table S1. The results of the relative quantitative real‐time PCR were analysed by the comparative threshold cycle (CT) method and normalized to β‐actin as the reference gene.
Evaluation of sodium fluorescein staining
One microlitre of 0.1% liquid sodium fluorescein was dropped into the conjunctival sac of mice. Ninety seconds later, corneal epithelial damage was photographed and graded with a cobalt blue filter under the slit‐lamp microscope. The cornea was divided into four quadrants, which were scored, respectively, as previously described 18. Briefly, as follows: absent, 0; slightly punctate staining less than 30 spots, 1; punctate staining more than 30 spots, but not diffuse, 2; severe diffuse staining but no positive plaque, 3; positive fluorescein plaque, 4. The scores of each quadrant were added to arrive at a final grade.
Measurement of epithelial permeability to carboxy fluorescein
Corneal epithelial barrier function was evaluated based on measurements of time dependent carboxy fluorescein penetrance. Briefly, 1 μl of 0.3% carboxy fluorescein was administered to the ocular surface; 10 min. later, the eyes were washed with 1 ml PBS to remove the residual carboxy fluorescein. After that, the animals were sacrificed with an overdose of chloral hydrate anaesthesia. The corneas were excised, and each cornea was placed in a 1.5‐ml tube containing 500 μl of PBS. The tubes were wrapped in aluminium foil to protect the solution from light and placed on an orbital shaker for 90 min. After 1 min. centrifugation, the supernatants were measured with a Gilford Fluoro IV fluorometer (Corning, Oberlin, OH, USA) to detect the concentration of carboxy fluorescein.
Bacterial infection examination
Tabby mice and the wild‐type mice were sacrificed, and the eyeballs and lung tissues were harvested and washed three times with sterile PBS. After that, the corneas and lung tissues were dissected and homogenized with 500 μl PBS. After low‐speed centrifugation, the supernatant was coated on LB medium (1% tryptone, 0.5% yeast extract, 1% NaCl, 1.5% agar, pH7.0) plates in triplicate and incubated at 37°C overnight, the number of bacterial colonies was then counted.
Skin tissue explants culture
The postnatal five‐day‐old mice were sacrificed and soaked in 1% iodine for 5 min., followed by three washes with PBS for 15 min. After that, the skin tissue was separated under the dissecting microscope and cut into small pieces about 5 × 5 mm size. The skin tissue explants were then placed and cultured in six‐well plates containing Defined Keratinocyte‐SFM basal medium (10744‐019, GIBCO, Carlsbad, CA, USA). Mouse recombinant Eda protein (191‐ED, R&D, Minneapolis, MN, USA), Shh protein(S0191, Sigma‐Aldrich) and Shh inhibitor cyclopamine (C4116, Sigma‐Aldrich) at different concentrations were added in different cultures. Cultures were incubated at 37°C under 5% CO2 and 95% humidity for 24 hrs.
Human corneal epithelial cell culture
Human corneal epithelial (HCE) cell line was obtained from RIKEN Biosource Center (Tokyo, Japan). The basic medium for the HCE cells is DMEM/F12, containing 6% FBS, 25 mM insulin, 10 ng/ml epidermal growth factor (EGF). The conditioned medium, which contained 20 ng/ml human recombinant Eda (3944‐ED, R&D, Minneapolis, MN) or 10 μM cyclopamine was replaced when HCE cells were grown to 70% confluence. After 6‐hrs or 24‐hrs culture, the culture medium was removed and washed immediately with PBS. The RIPA buffer or Trizol buffer was added to harvest the protein or mRNA.
pcDNA‐EDA plasmid transfection
The EDA cDNA was cloned into SV40‐driven pcDNA3.1 vector. The resulting plasmid was transfected into HCE cells by Lipofectamine® 2000 (11668‐019, Invitrogen, Carlsbad, CA, USA) when the cells reached 70% confluence in six‐well plates. Simultaneously, the empty pcDNA3.1 vector was transfected as the negative control.
Statistical analysis
Statistical analysis was performed with SPSS 16.0.0 (SPSS, Chicago, IL, USA). All summary data are reported as mean ± S.E.M. Comparisons between groups were performed by an unpaired, two‐tailed Student's t‐test. P ≤ 0.05 was considered statistically significant.
Results
Reduction of epithelial tight junctional protein expression in Tabby mice
As the morphological changes in Eda‐deficient Tabby mice mirror those occurring in humans, this strain is frequently used to study the underlying pathophysiological mechanisms of the XLHED syndrome 19, 20. In mice, eyelid opening occurs around 2 weeks after birth. We harvested corneal epithelia, dorsal skin epithelia as well as their lung tissue from four‐ and eight‐week‐old Tabby mice and wild‐type littermates. Western blot results show that the corneal epithelial tight junctional ZO‐1 and claudin‐1 protein expression both dramatically decreased (Fig. 1A). Similarly such declines also occurred in their skin epithelium (Fig. 1B) and lung tissue (Fig. S1) at all time points when compared to wild‐type littermates. Immunofluorescent staining also showed discontinuous and weaker expression patterns of ZO‐1 and claudin‐1 in the corneal (Fig. 1C) and skin (Fig. 1D) epithelia of Tabby mice. These results suggest that there is epithelial barrier dysfunction in both neonatal and adult Tabby mice.
Figure 1.
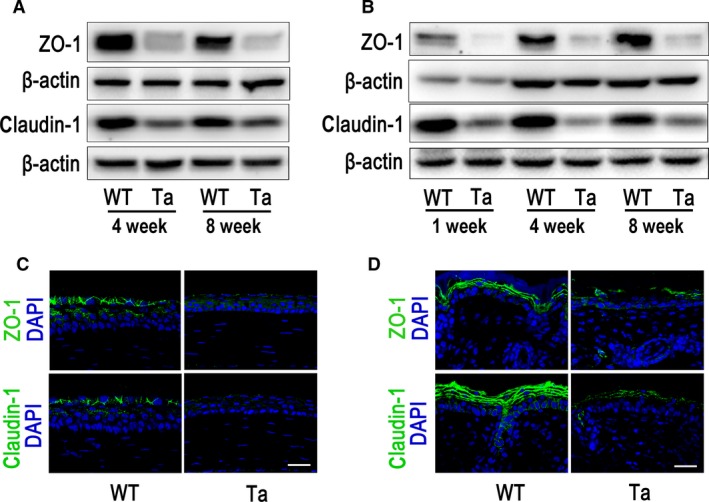
Expression of tight junction proteins, ZO‐1 and claudin‐1, in the corneal and skin epithelia of Tabby mice (Ta) and wild‐type mice (WT). (A) Western blot results show that ZO‐1 and claudin‐1 decreased in the corneal epithelia of four‐ and eight‐week‐old Tabby mice when compared with wild‐type control. (B) ZO‐1 and claudin‐1 expression was lower in the skin tissue of one‐, four‐ and eight‐week‐old Tabby mice. (C) ZO‐1 and claudin‐1 was strongly expressed in the surface layer corneal epithelium of eight‐week‐old wild‐type mice, while they were weakly expressed in Tabby mice. (D) ZO‐1 and claudin‐1 was expressed in the superficial layers of the skin epithelium of eight‐week‐old wild‐type mice, while they were weakly and discontinuously expressed in Tabby mice. Scale bars: 30 μm.
Inflammation in corneas, skin and lung tissues of Tabby mice
In Tabby mice, ocular surface diseases occur postnatally as early as 4 weeks followed by age dependent increases in pathological severity 16. Slit‐lamp microscopy observation revealed a normal ocular surface in Tabby mice at 4 weeks. However, the corneal surface was scabrous and mildly oedematous in the central or peri‐central corneal stroma in eight‐week‐old Tabby mice (Fig. S2A). H&E staining showed that the corneal epithelial surface was irregular in Tabby mice at 4 weeks and 8 weeks. In contrast, the epithelial surface was smooth in wild‐type mice (Fig. S2B). Sporadic CD45‐positive macrophages were present in the anterior part of the central corneal stroma of Tabby mice at 4 weeks. Their numbers increased and spread into the corneal epithelium at 8 weeks, while they were absent in both layers of wild‐type littermates (Fig. 2A).
Figure 2.
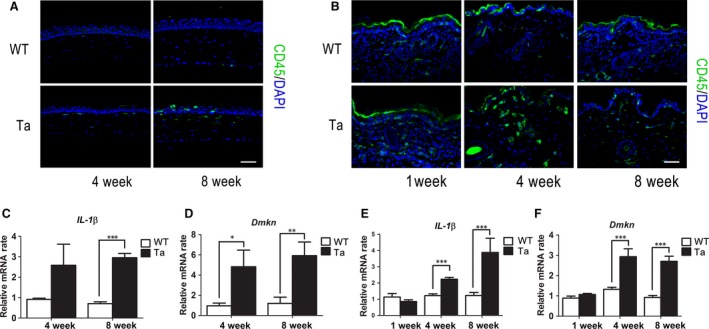
The Corneal and skin epithelial inflammation augmentation in Tabby mice. (A) Immunofluorescent staining of CD45 was negative in the wild‐type mice, while there were sporadic positive cells in the superficial corneal stroma at 4 weeks, which spread into the epithelial layer in eight‐week‐old Tabby mice. (B) CD45‐positive cells were dramatically increased in the dermis of Tabby mice at 4 and 8 weeks of age. The mRNA levels of IL‐1β (C) and Dmkn (D) significantly increased in four‐ and eight‐week‐old Tabby mice corneal epithelia when compared to the wild‐type littermates(n = 6 mice for each group). In skin epithelia, IL‐1β (E) and Dmkn (F) expression was also up‐regulated in four‐ and eight‐week‐old Tabby mice (n = 5 mice for each group). Data represent the mean ± S.E.M. Significance was analysed using an unpaired, two‐tailed Student's t‐test (*P < 0.05, **P < 0.01, ***P < 0.001). Scale bars: 60 μm.
The Tabby mice exhibited pink skin 1 week after birth, which was maintained throughout their life (Fig. S2C). H&E staining showed mildly increased cellularity of their inter‐follicular connective tissue compared with wild‐type littermates (Fig. S2D). CD45‐positive cells dramatically increased in the skin of four‐ and eight‐week‐old Tabby mice (Fig. 2B), indicating chronic inflammation of the skin. We also detected in them IL‐1β and Dmkn chemokine gene expression. qRT‐PCR results showed that both chemokines very markedly increased in the corneal (Fig. 2C and D) and skin (Fig 2E and F) epithelial layers of four‐week‐old Tabby mice with larger rises at 8 weeks.
There was sporadic hyperaemia in one‐week‐old Tabby mice lung tissue along with dramatic increases in cell infiltration within the pulmonary alveoli. Immunostaining revealed numerous CD45‐positive macrophages in the lung tissue of Tabby mice, while they were not present in the wild‐type littermates (Fig. S3), indicating pulmonary inflammation in the neonatal Tabby mice.
Corneal and pulmonary bacterial infection accompany epithelial barrier dysfunction in Tabby mice
Intact corneal tight junctions are crucial for maintaining barrier function against pathogenic infiltration and preventing dysregulated immune response activation. As corneal epithelial tight junctional ZO‐1 and claudin‐1 protein expression declined in Tabby mice, we hypothesized that compromise of its protective barrier function would increase the possibility of bacterial infection. Accordingly, we then harvested corneal tissues from eight‐week‐old Tabby mice, homogenized and suspended them in PBS. Bacterial culture of the suspension from Tabby mice resulted in more bacterial colonies than on plates containing cultures obtained from wild‐type mice (Fig. 3A). This difference was statistically significant (Fig. 3B). Furthermore, corneal epithelial gene expression of lysozyme 1 (Fig. 3C) and lysozyme 2 (Fig. 3D) both dramatically increased in four‐ and eight‐week‐old Tabby mice compared with the wild‐type littermates, indicating more bacterial infection in Tabby mice. Similarly, bacterial culture of lung tissue suspensions from one‐week‐old Tabby mice was also positive while there was no bacterial colony formation from wild‐type littermate mice (Fig. S4). These differences indicate that the corneal and pulmonary inflammation was at least partly because of bacterial infection in Tabby mice.
Figure 3.
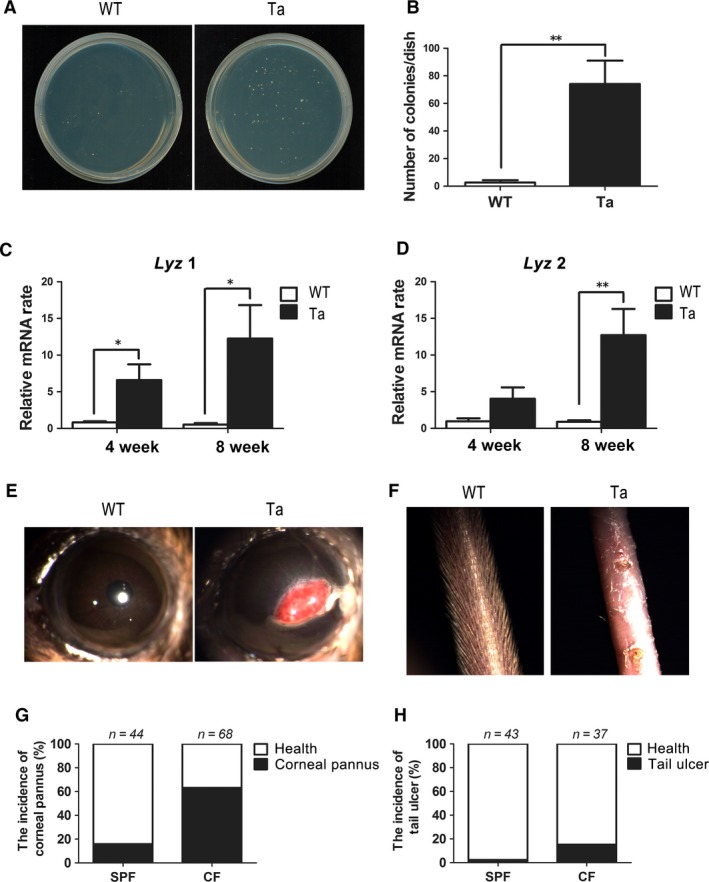
Corneal bacterial infection, corneal pannus and tail ulcer in Tabby mice. (A) Bacterial culture of the corneal tissue suspension from eight‐week‐old Tabby mice and wild‐type littermates. (B) The number of bacterial colonies was significantly larger in Tabby mice compared with that in wild‐type mice (n = 5 mice for each group). Lysozyme 1 (Lyz 1) (C) and Lysozyme 2 (Lyz 2) (D) mRNA expression was both up‐regulated in the corneal epithelia of four‐ and eight‐week‐old Tabby mice (n = 6 mice for each group). (E) Corneal pannus presented in the central cornea of 16‐week‐old Tabby mouse, while it was not found in wild‐type mice. (F) The hair of tail skin was absent in the 16‐week‐old Tabby mouse, and there were small ulcers present in the tail skin surface. No tail ulcer was found in wild‐type mice. (G) The morbidity of the corneal pannus was 15.9% in those housed in the SPF environment, while it was 63.2% in Tabby mice housed in the CF environment. (H) The morbidity of tail ulcer in Tabby mice was 2.4% in SPF, while it was 13.5% in CF. Data represent the mean ± S.E.M. Significance was analysed using an unpaired, two‐tailed Student's t‐test (*P < 0.05, **P < 0.01).
To further investigate the susceptibility of Tabby mice to bacterial infection, we bred different groups of 16‐week‐old Tabby mice in either CF or SPF environments and compared the incidence of corneal pannus (Fig. 3E) and tail ulcer formation (Fig. 3F) maintained in these two different environments. The results show that the morbidity of the corneal pannus was about 63.2% for those housed in the CF while it was only 15.9% in the SPF environment (Fig. 3G). Similarly, the morbidity of tail ulcer was 13.5% in the CF environment, while it was only 2.4% in SPF environment (Fig. 3H). These differences in morbidity strongly suggest that epithelial barrier dysfunction is associated with increases in corneal bacterial infection and skin ulcer formation in Tabby mice.
Recombinant Eda protein rescues corneal epithelial barrier dysfunction in Tabby mice
The Eda protein is essential for meibomian gland development 20. Interestingly, we found that the Eda protein is highly expressed in the meibomian gland and is present in human tear samples. However, its cognate receptor (Edar) is highly expressed in the corneal epithelium, but at a lower density in the meibomian gland (unpublished observation). As Eda is a secretory protein, we proposed that it could be secreted from the meibomian gland and targeted for controlling ocular surface epithelial function. One possibility is that corneal epithelial tight junctional compromise may result from an Eda deficiency because of the absence of meibomian glands in Tabby mice. To test whether exogenous Eda application could rescue corneal barrier dysfunction, 5 μl of a 20 ng/ml mouse recombinant Eda protein solution was administered 3 times per day to the ocular surface of four‐week‐old wild‐type and Tabby mice. After 3 weeks, the corneal surface fluorescein dye staining pattern clearly decreased more in Tabby mice relative to what occurred in Tabby mice treated instead with the PBS vehicle (Fig. 4A), which was confirmed by fluorescein score determination (Fig. 4B). Additional validation of this difference was obtained based on measurements in corneal epithelial carboxy fluorescein (CF) permeation. There was greater infiltration of CF in the Tabby mice, which declined after Eda treatment (Fig. 4C and D). On the other hand, tight junctional ZO‐1 and claudin‐1 protein expression was significantly up‐regulated by Eda protein application in the Tabby mice (Fig. 4E). Whole mount staining of corneal epithelia also documented ZO‐1 and claudin‐1 expression rescue in Eda treated Tabby mice (Fig. 4F). These results support the notion that corneal epithelial barrier dysfunction stems from the absence of Eda in Tabby mice.
Figure 4.
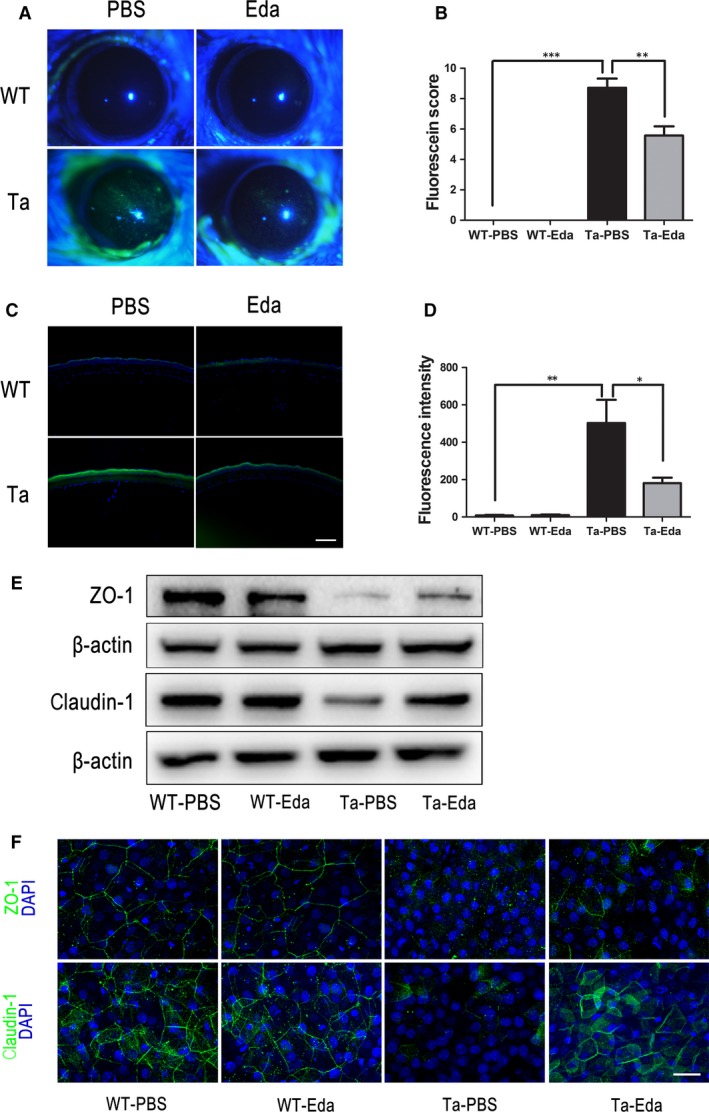
Topical Eda application partially rescues corneal epithelial barrier dysfunction in Tabby mice. (A) Wild‐type mice lack fluorescein dye staining in the cornea. Tabby mice cornea strongly stain in the PBS treatment group, whereas it is markedly less prominent in the Eda treatment group. (B) Fluorescein score dramatically decreased after Eda treatment. (C) Carboxy fluorescein (CF) permeation was very evident across the corneal epithelium of Tabby mice, while it was reduced after Eda treatment. (D) Fluorescence intensity significantly decreased in Tabby mice after Eda treatment. (E) Corneal epithelial ZO‐1 and claudin‐1 expression was up‐regulated after application of Eda protein in Tabby mice. (F) Whole mount staining of ZO‐1 and claudin‐1 showed both proteins were up‐regulated in the corneal epithelia of Tabby mice after Eda treatment. Data represent the mean ± S.E.M. Significance was analysed using an unpaired, two‐tailed Student's t‐test (*P < 0.05, **P < 0.01, ***P < 0.001), n = 4 for each group. Scale bars: 30 μm.
Eda regulates epithelial barrier function through the sonic hedgehog signalling pathway
Eda mediates its function through binding Edar and activates downstream signal cascades 21. Different studies indicate that Eda‐Edar signalling is mediated for the most part, if not totally, by the I‐κB kinase‐dependent canonical NF‐κB signalling pathway 22, 23. It was also shown that bone morphogenetic protein (BMP) activity could be suppressed while the sonic hedgehog (Shh) signalling pathway was instead up‐regulated by the Eda‐linked pathway during ectodermal organogenesis 24, 25, 26, 27, 28. Previous reports suggested that the Shh pathway regulates tight junctional protein expression 29, 30. This was validated by showing that the stomach epithelial tight junction protein expression pattern was disrupted in Shh knockout mice 31. Based on those observations, we hypothesized that Eda regulates the tight junction protein expression through the Shh signalling pathway. To test this hypothesis, skin explants from one‐week‐old wild‐type and Tabby mice were cultured in the absence or presence of Eda (0.1 μg/ml), Shh (0.5 μg/ml) and a Shh inhibitor cyclopamine (20 μM). After a 24‐hrs or 48‐hrs culture, total mRNA and protein harvested from the explants were analysed by qRT‐PCR and Western blot, respectively. The results show that the baseline of Shh expression was significantly lower in Tabby mice compared with that in wild‐type mice. Addition of Eda significantly increased the expression of Shh in Tabby mice skin explants, and this rise could be inhibited by cyclopamine (Fig. 5A). However, both Eda and Shh medium supplementation did not up‐regulate Shh gene expression on wild‐type mice skin explants (Fig. 5A). We also monitored GLI‐1 gene expression, a downstream mediator in the Shh signalling pathway. GLI‐1 was also significantly down‐regulated in Tabby mice. Eda and Shh application did not increase GLI‐1 expression in wild‐type mice; however, both Eda and Shh supplementation up‐regulated GLI‐1 mRNA expression in Tabby mice (Fig. 5B). Western blot results further confirmed that Eda up‐regulated Gli‐1 protein expression as well as tight junctional proteins ZO‐1 and claudin‐1, and this up‐regulation was blocked by cyclopamine (Fig. 5C).
Figure 5.
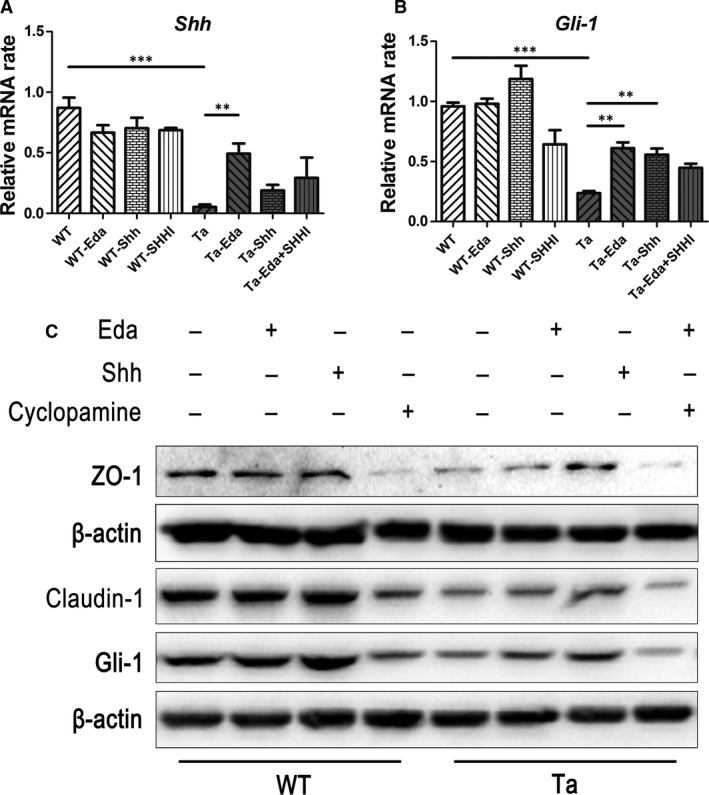
Eda activates sonic hedgehog signalling pathway in Tabby mice skin. One‐week‐old wild‐type and Tabby mice skin explants were cultured in the absence or presence of Eda (0.1 μg/ml), Shh (0.5 μg/ml), and Shh inhibitor (SHHI) cyclopamine (20 μM). Total mRNA and protein were, respectively, harvested after 24‐ and 48‐hrs culture. (A) The Shh mRNA expression in Tabby mice skin was much lower than that in the wild‐type mice. Addition of Eda increased the expression of Shh. (B) Gli‐1 mRNA expression in Tabby mice skin was also much lower compared with wild‐type mice. Addition of Eda or Shh up‐regulated Gli‐1 expression. (C) Western blot showed Shh inhibitor cyclopamine reduced expression of ZO‐1 and claudin‐1 in wild‐type skin tissue. Application of Eda or Shh protein in Tabby mice skin tissue culture up‐regulated ZO‐1, claudin‐1, and Gli‐1 expression. Addition of cyclopamine attenuated the effect of Eda. Data represent the mean ± S.E.M. Significance was analyzed using an unpaired, two‐tailed Student's t‐test (**P < 0.01, ***P < 0.001), n = 3 for each group.
To further validate the contribution made by Eda in regulating epithelial tight junctional protein expression and integrity, we constructed a pcDNA3.1‐EDA plasmid and transfected it into a human corneal epithelial (HCE) cell line whose EDA mRNA expression level was very low. One week after transfection, qRT‐PCR results showed that the EDA plasmid was successfully transfected as EDA expression dramatically increased in HCE cells (Fig. S5). As expected, ZO‐1 and claudin‐1 tight junctional protein expression also significantly increased in EDA transfected HCE cells (Fig. 6A). Moreover, both SHH and GLI‐1 gene expression were up‐regulated in these EDA transfected cells (Fig. 6B).
Figure 6.
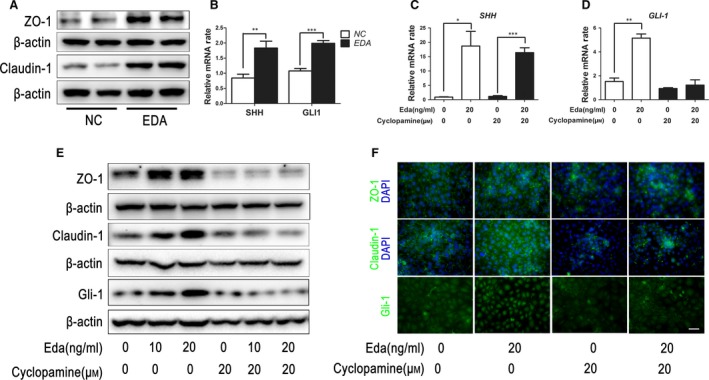
Eda regulates epithelial barrier function through the sonic hedgehog signalling pathway. (A) The pcDNA3.1‐EDA plasmid was transfected into human corneal epithelial (HCE) cell line for 1 week. The expression of ZO‐1 and claudin‐1 increased in EDA transfected HCE cells. (B) qRT‐PCR results showed SHH and GLI‐1 mRNA was significantly up‐regulated in EDA transfected cells. (C) SHH gene expression in HCE cells was significantly up‐regulated by Eda protein and could not be blocked by cyclopamine. (D) GLI‐1 gene expression in HCE cells was also up‐regulated by Eda protein and could be blocked by cyclopamine. (E) Western blot results show that Eda protein up‐regulated the expression of ZO‐1, claudin‐1 and Gli‐1 in HCE cells in a dose‐dependent manner, and this effect could be blocked by cyclopamine. (F) Immunofluorescent staining showed ZO‐1 and claudin‐1 expression in HCE cells was remarkably inhibited by cyclopamine. Nuclear expression of Gli‐1 was enhanced in HCE cells after Eda treatment and was down‐regulated with additional cyclopamine. Data represent the mean ± S.E.M. Significance was analysed using an unpaired, two‐tailed Student's t‐test (*P < 0.05, **P < 0.01, ***P < 0.001), n = 3 for each group. Scale bar: 60 μm.
To further document the functional role of Eda, we also applied human Eda recombinant protein to the HCE cells. Eda protein application dramatically up‐regulated SHH gene expression, whereas cyclopamine had no effect on this response (Fig. 6C). Similarly, GLI‐1 gene expression was also up‐regulated by Eda, while cyclopamine markedly decreased this rise (Fig. 6D). Western blot results showed that Eda dose dependently up‐regulated ZO‐1, claudin‐1 and Gli‐1 expression, and such an effect could be blocked by cyclopamine (Fig. 6E). Immunofluorescent staining further confirmed that ZO‐1 and claudin‐1 was strongly enhanced in HCE cells after Eda treatment, while cyclopamine remarkably inhibited the expression of these proteins. Meanwhile, Gli‐1 nuclear expression was dramatically induced by Eda and could be blocked by cyclopamine (Fig. 6F).
Discussion
Eda mRNA is expressed in various organs and tissues, including the heart, kidney, pancreas, brain, lung, liver, skeletal muscle, teeth, as well as the skin during both embryonic development and adulthood 1, 32. Even though it is known that the Eda gene has a developmental function in controlling the morphogenesis of various ectodermal structures such as hair, teeth, nails and exocrine glands, little is known about its postnatal function. In the current study, for the first time, we found tight junction proteins such as ZO‐1 and claudin‐1 were dramatically down‐regulated in Eda mutant Tabby mice, resulting in epithelial barrier dysfunction in various tissues such as skin, cornea and lung. We further proved that it is Eda that makes an essential contribution to maintaining surface epithelial homeostasis through regulation of tight junction formation.
Epithelial apical tight junctional integrity plays a critical role in providing an epithelial barrier function against pathogenic infiltration. Its intactness is also needed for cell–cell communication and establishing cell polarity as well as regulating paracellular movement of ions and macromolecules 33. Disruption of the tight junction is characteristic of a number of diseases including dermatitis, pathogenic infection and cancer 34. It was proposed long ago that genetically impaired epidermal barrier formation may be the primary cause of the rapid increase in the prevalence of atopic dermatitis and respiratory atopy 35. Recently, defective expression of epidermal ZO‐1 and claudin‐1 was observed in the skin of patients with atopic dermatitis 36, where such pathology is a risk factor for viral infection and allergen sensitization 37, 38. Our results clearly demonstrate that compromise of tight junctional integrity is associated with marked increases in vulnerability to bacterial infection and chronic inflammation in Tabby mice. Losses in junctional compaction may help explain why XLHED patients are at a greater of risk of suffering from pneumonia, respiratory infections and are also more frequently afflicted with allergic diseases, chronic dermatitis and ocular surface inflammation than the general population.
Although the Eda‐Edar signalling pathway was intensively studied, most studies focused on the effect of Eda linked signalling pathway activation on morphogenesis 3. In our study, we found that Eda regulates ZO‐1 and claudin‐1 tight junctional protein expression through activation of Shh signalling pathway using in vivo and in vitro methodology. It was reported that the stomach epithelial cell–cell junction was disrupted in Shh conditional knockout mice 31, which also supports the notion that Shh is situated downstream from Eda in this signalling pathway.
Recombinant Eda protein injection into pregnant Tabby mouse could reverse the XLHED phenotype in the offspring 39. Similarly administration of Fc‐Eda to dog models of XLHED during their first 2 weeks of life also rescued developmental abnormalities 40, 41. Animals treated with recombinant Eda protein in prenatal or early postnatal stage showed dramatically decreased, if not all, clinical signs such as eye and airway infections with improved lacrimation, meibomain gland and bronchial glands formation 40, 41. Therefore, the ocular surface defect and pulmonary infection in EDA mutant animals may synergistically resulted from both secretory glands defect and the absence of biological function of EDA. In other words, normal secretory glands in the ocular surface and pulmonary tissues may compensate the deficiency of EDA. However, this interpretation does not fit previous study which showed transfection of the Eda‐A1 gene to Tabby mice prevented corneal defects without restoring meibomian gland formation 16. To further address this contradiction, inducible Eda knockout mice should be generated to knock down EDA gene in the adult stage, allowing us to investigate the function of Eda during the adult stage while eliminating any involvement of morphogenesis.
Currently, phase 2 neonate clinical trials are being conducted using humanized Fc‐Eda‐A1 protein, EDI200 to treat XLHED (www.clinicaltrials.gov, NCT01775462). There is optimism that EDI200 treatment will correct the developmental disorders of XLHED in humans. We tested if recombinant Eda protein application to the ocular surface of postnatal Tabby mice reverses declines in epithelial barrier tightness. This procedure largely rescued ZO‐1 and claudin‐1 tight junction protein expression. Our results point to the possibility that repeated exogenous Eda treatment may be necessary to sustain this protein at high enough levels to maintain normal tight junction barrier function from childhood into adulthood in XLHED patients. In summary, EDA gene expression provides essential support for the maintenance of epithelial tight junctional integrity during postnatal stage. This effect is of importance in hindering pathogenic infiltration and protecting tissues from incurring declines in their normal function. As such control by EDA gene expression of barrier function is mediated through the Shh signalling pathway, this finding pinpoints another potential drug target for treating XLHED‐related diseases.
Conflict of interests
The authors have declared that no competing interests exist.
Supporting information
Figure S1 ZO‐1 and claudin‐1 expression in lung tissue of Tabby mice.
Figure S2 Inflammation of skin tissue in Tabby mice.
Figure S3 Inflammation of lung tissue in Tabby mice.
Figure S4 Pulmonary bacterial infection in Tabby mice.
Figure S5 EDA plasmid transfection in HCE cells.
Table S1 Primer sequence pairs used for quantitative real‐time PCR.
Acknowledgements
We are especially grateful to Prof. Ting Xie from Stowers Institute for his valuable suggestions on the study and reviewing the manuscript. This study was supported in part by the grants from Chinese National Key Scientific Research Project (No.2013CB967003 [to WL]), the National Natural Science Foundation of China (NSFC, No.81270977, No.81470602 [to WL], and 81330022 [to ZL]), the Fundamental Research Funds for the Central Universities of China (No.2012121049, and No.20720150171 [to WL]).
S.L., J.Z. and W.L. designed research, S.L., J.Z., L.Z., J.L., J.Y., K.N., Y.Q. and H.H. performed research, S.L., J.Z., Y.C. and W.L. analysed the experimental data, S.L., Z.L, P.S.R and W.L. wrote the manuscript.
Contributor Information
Zuguo Liu, Email: zuguoliu@xmu.edu.cn.
Wei Li, Email: wei1018@xmu.edu.cn.
References
- 1. Kere J, Srivastava AK, Montonen O, et al X‐linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat Genet. 1996; 13: 409–16. [DOI] [PubMed] [Google Scholar]
- 2. Clarke A, Phillips DI, Brown R, et al Clinical aspects of X‐linked hypohidrotic ectodermal dysplasia. Arch Dis Child. 1987; 62: 989–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lefebvre S, Mikkola ML. Ectodysplasin research–where to next? Semin Immunol. 2014; 26: 220–8. [DOI] [PubMed] [Google Scholar]
- 4. Nguyen‐Nielsen M, Skovbo S, Svaneby D, et al The prevalence of X‐linked hypohidrotic ectodermal dysplasia (XLHED) in Denmark, 1995–2010. Eur J Med Genet. 2013; 56: 236–42. [DOI] [PubMed] [Google Scholar]
- 5. Casal ML, Mauldin EA, Ryan S, et al Frequent respiratory tract infections in the canine model of X‐linked ectodermal dysplasia are not caused by an immune deficiency. Vet Immunol Immunopathol. 2005; 15: 95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Drögemüller C, Distl O, Leeb T. Partial deletion of the bovine ED1 gene causes anhidrotic ectodermal dysplasia in cattle. Genome Res. 2001; 11: 1699–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Srivastava AK, Pispa J, Hartung AJ, et al The Tabby phenotype is caused by mutation in a mouse homologue of the EDA gene that reveals novel mouse and human exons and encodes a protein (ectodysplasin‐A) with collagenous domains. Proc Natl Acad Sci U S A. 1997; 94: 13069–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beahrs JO, Lillington GA, Rosan RC, et al Anhidrotic ectodermal dysplasia: predisposition to bronchial disease. Ann Intern Med. 1971; 74: 92–6. [DOI] [PubMed] [Google Scholar]
- 9. Fete M, Hermann J, Behrens J, et al X‐linked hypohidrotic ectodermal dysplasia (XLHED): clinical and diagnostic insights from an international patient registry. Am J Med Genet A. 2014; 164A: 2437–42. [DOI] [PubMed] [Google Scholar]
- 10. Dietz J, Kaercher T, Schneider AT, et al Early respiratory and ocular involvement in X‐linked hypohidrotic ectodermal dysplasia. Eur J Pediatr. 2013; 172: 1023–31. [DOI] [PubMed] [Google Scholar]
- 11. Vanselow NA, Yamate M, Adams MS, et al The increased prevalence of allergic disease in anhidrotic congenital ectodermal dysplasia. J Allergy. 1970; 45: 302–9. [DOI] [PubMed] [Google Scholar]
- 12. Koguchi‐Yoshioka H, Wataya‐Kaneda M, Yutani M, et al Atopic diathesis in hypohidrotic/anhidrotic ectodermal dysplasia. Acta Derm Venereol. 2015; 95: 476–9. [DOI] [PubMed] [Google Scholar]
- 13. Capitanio MA, Chen JT, Arey JB, et al Congenital anhidrotic ectodermal dysplasia. Am J Roentgenol Radium Ther Nucl Med. 1968; 103: 168–72. [DOI] [PubMed] [Google Scholar]
- 14. Siegel MB, Potsic WP. Ectodermal dysplasia: the otolaryngologic manifestations and management. Int J Pediatr Otorhinolaryngol. 1990; 19: 265–71. [DOI] [PubMed] [Google Scholar]
- 15. Kaercher T. Ocular symptoms and signs in patients with ectodermal dysplasia syndromes. Graefes Arch Clin Exp Ophthalmol. 2004; 242: 495–500. [DOI] [PubMed] [Google Scholar]
- 16. Cui CY, Smith JA, Schlessinger D, et al X‐linked anhidrotic ectodermal dysplasia disruption yields a mouse model for ocular surface disease and resultant blindness. Am J Pathol. 2005; 167: 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wilson FM 2nd, Grayson M, Pieroni D. Corneal changes in ectodermal dysplasia. Case report, histopathology, and differential diagnosis. Am J Ophthalmol. 1973; 75: 17–27. [DOI] [PubMed] [Google Scholar]
- 18. Pauly A, Brignole‐Baudouin F, Labbé A, et al New tools for the evaluation of toxic ocular surface changes in the rat. Invest Ophthalmol Vis Sci. 2007; 48: 5473–83. [DOI] [PubMed] [Google Scholar]
- 19. Gruneberg H. The tabby syndrome in the mouse. Proc R Soc Lond B Biol Sci. 1971; 179: 139–56. [DOI] [PubMed] [Google Scholar]
- 20. Gruneberg H. The glandular aspects of the tabby syndrome in the mouse. J Embryol Exp Morphol. 1971; 25: 1–19. [PubMed] [Google Scholar]
- 21. Mikkola ML, Thesleff I. Ectodysplasin signaling in development. Cytokine Growth Factor Rev. 2003; 14: 211–24. [DOI] [PubMed] [Google Scholar]
- 22. Schmidt‐Ullrich R, Aebischer T, Hulsken J, et al Requirement of NF‐kappaB/Rel for the development of hair follicles and other epidermal appendices. Development. 2001; 128: 3843–53. [DOI] [PubMed] [Google Scholar]
- 23. Puel A, Picard C, Ku CL, et al Inherited disorders of NF‐kappaB‐mediated immunity in man. Curr Opin Immunol. 2004; 16: 34–41. [DOI] [PubMed] [Google Scholar]
- 24. Pummila M, Fliniaux I, Jaatinen R, et al Ectodysplasin has a dual role in ectodermal organogenesis: inhibition of Bmp activity and induction of Shh expression. Development. 2007; 134: 117–25. [DOI] [PubMed] [Google Scholar]
- 25. Schmidt‐Ullrich R, Tobin DJ, Lenhard D, et al NF‐kappaB transmits Eda A1/EdaR signalling to activate Shh and cyclin D1 expression, and controls post‐initiation hair placode down growth. Development. 2006; 133: 1045–57. [DOI] [PubMed] [Google Scholar]
- 26. Laurikkala J, Mikkola M, Mustonen T, et al TNF signaling via the ligand‐receptor pair ectodysplasin and edar controls the function of epithelial signaling centers and is regulated by Wnt and activin during tooth organogenesis. Dev Biol. 2001; 229: 443–55. [DOI] [PubMed] [Google Scholar]
- 27. Laurikkala J, Pispa J, Jung HS, et al Regulation of hair follicle development by the TNF signal ectodysplasin and its receptor Edar. Development. 2002; 129: 2541–53. [DOI] [PubMed] [Google Scholar]
- 28. Cui CY, Yin M, Sima J, et al Involvement of Wnt, Eda and Shh at defined stages of sweat gland development. Development. Oct 2014; 141: 3752–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xia YP, He QW, Li YN, et al Recombinant human sonic hedgehog protein regulates the expression of ZO‐1 and occludin by activating angiopoietin‐1 in stroke damage. PLoS One. 2013; 8: e68891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang Y, Jin S, Sonobe Y, et al Interleukin‐1beta induces blood‐brain barrier disruption by downregulating Sonic hedgehog in astrocytes. PLoS One. 2014; 9: e110024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xiao C, Ogle SA, Schumacher MA, et al Hedgehog signaling regulates E‐cadherin expression for the maintenance of the actin cytoskeleton and tight junctions. Am J Physiol Gastrointest Liver Physiol. 2010; 299: G1252–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Elomaa O, Pulkkinen K, Hannelius U, et al Ectodysplasin is released by proteolytic shedding and binds to the EDAR protein. Hum Mol Genet. 2001; 10: 953–62. [DOI] [PubMed] [Google Scholar]
- 33. Anderson JM, Van Itallie CM. Tight junctions and the molecular basis for regulation of paracellular permeability. Am J Physiol. 1995; 269: G467–75. [DOI] [PubMed] [Google Scholar]
- 34. Sawada N, Murata M, Kikuchi K, et al Tight junctions and human diseases. Med Electron Microsc. 2003; 36: 147–56. [DOI] [PubMed] [Google Scholar]
- 35. Taieb A. Hypothesis: from epidermal barrier dysfunction to atopic disorders. Contact Dermatitis. 1999; 41: 177–80. [DOI] [PubMed] [Google Scholar]
- 36. De Benedetto A, Rafaels NM, McGirt LY, et al Tight junction defects in patients with atopic dermatitis. J Allergy Clin Immunol. 2011; 127: e771–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. De Benedetto A, Slifka MK, Rafaels NM, et al Reductions in claudin‐1 may enhance susceptibility to herpes simplex virus 1 infections in atopic dermatitis. J Allergy Clin Immunol. 2011; 128: 242–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. De Benedetto A, Kubo A, Beck LA. Skin barrier disruption: a requirement for allergen sensitization? J Invest Dermatol. 2012; 132: 949–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gaide O, Schneider P. Permanent correction of an inherited ectodermal dysplasia with recombinant EDA. Nat Med. 2003; 9: 614–18. [DOI] [PubMed] [Google Scholar]
- 40. Casal ML, Lewis JR, Mauldin EA, et al Significant correction of disease after postnatal administration of recombinant ectodysplasin A in canine X‐linked ectodermal dysplasia. Am J Hum Genet. 2007; 81: 1050–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mauldin EA, Gaide O, Schneider P, et al Neonatal treatment with recombinant ectodysplasin prevents respiratory disease in dogs with X‐linked ectodermal dysplasia. Am J Med Genet A. 2009; 149A: 2045–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 ZO‐1 and claudin‐1 expression in lung tissue of Tabby mice.
Figure S2 Inflammation of skin tissue in Tabby mice.
Figure S3 Inflammation of lung tissue in Tabby mice.
Figure S4 Pulmonary bacterial infection in Tabby mice.
Figure S5 EDA plasmid transfection in HCE cells.
Table S1 Primer sequence pairs used for quantitative real‐time PCR.