

Abstract
Congenital scoliosis (CS) is a three‐dimensional deformity of the spine affecting quality of life. We have demonstrated TBX6 haploinsufficiency is the most important contributor to CS. However, the pathophysiology at the protein level remains unclear. Therefore, this study was to explore the differential proteome in serum of CS patients with TBX6 haploinsufficiency. Sera from nine CS patients with TBX6 haploinsufficiency and nine age‐ and gender‐matched healthy controls were collected and analysed by isobaric tagged relative and absolute quantification (iTRAQ) labelling coupled with mass spectrometry (MS). In total, 277 proteins were detected and 20 proteins were designated as differentially expressed proteins, which were submitted to subsequent bioinformatics analysis. Gene Ontology classification analysis showed the biological process was primarily related to ‘cellular process’, molecular function ‘structural molecule activity’ and cellular component ‘extracellular region’. IPA analysis revealed ‘LXR/RXR activation’ was the top pathway, which is a crucial pathway in lipid metabolism. Hierarchical clustering analysis generated two clusters. In summary, this study is the first proteomic research to delineate the total and differential serum proteins in TBX6 haploinsufficiency‐caused CS. The proteins discovered in this experiment may serve as potential biomarkers for CS, and lipid metabolism might play important roles in the pathogenesis of CS.
Keywords: congenital scoliosis, TBX6, proteomics, isobaric tagged relative and absolute quantification, lipid metabolism
Introduction
CS is a complex three‐dimensional deformity of the spine with a lateral curvature of more than ten degrees 1. The overall incidence of CS has been estimated at approximately one of 2000 live births 2. CS originates from defects of the axial skeletal development during embryogenesis. Disruption of normal somitogenesis in the embryonic period which is regulated by a multitude of genes can result in vertebral malformations 3. If left untreated, the scoliotic deformity of the CS, specifically in the thoracic region, can progress to serious conditions devastating the pulmonary function, hence augmenting the mortality 4, 5.
The T‐box genes are an evolutionarily conserved family of genes encoding transcription factors that play a key role in regulating the development processes 6. The T‐box gene TBX6, located at the 16p11.2 region, with 6092 bp in size, is accountable for normal somitogenesis by interacting with the Notch pathway in the clock‐wavefront model 7, 8. More recently, our team reported that TBX6 haploinsufficiency is associated with sporadic cases of CS 1. Involvement of TBX6 haploinsufficiency in CS cases is endorsed by the identification of TBX6 null mutation in conjunction with a hypomorphic allele to account for 8‐11% of CS cases 1. To date, the TBX6 compound inheritance model has shed light on the maximum ratio of CS 9, suggesting TBX6 one of the most important genes in the occurrence and pathogenesis of CS.
Although the involvement of TBX6 gene in CS has been well identified, further biological researches are needed to clarify the exact mechanisms of TBX6 in CS from more aspects. While gene is the carrier for genetic information, protein is the final output of gene and the executor of exact biological functions 10, 11. In this regard, it is quite compelling to elucidate the role of TBX6 in CS from the protein perspective. One desirable way is to utilize the proteomics technique to detect the total proteins, namely the global proteome profiles, thus complementing our previous findings on TBX6. However, to the best of our knowledge, there are no publications using proteomics techniques to study the CS disease. Therefore, this study is conceived to establish the serum proteome profiles of CS cases with TBX6 haploinsufficiency by iTRAQ technique.
Materials and methods
Patients and sample obtainment
In the previous genetic experiment, genome‐wide analyses of copy number variants were performed by Agilent oligonucleotide comparative genomic hybridization (CGH) microarrays. Additionally, a custom 4 × 180k CGH microarray with approximately 180,000 oligonucleotide probes was utilized. DNA processing and microarray experiment were following the Agilent oligonucleotide CGH instruction (Protocol version 6.0). Quantitative PCR analysis was conducted through ∆Ct method using SYBR Green Realtime PCR Master Mix (TOYOBO, Osaka, Japan) 1.
As for the proteomic experiment, a case–control study was adopted. The sera of the nine patients and nine healthy controls were drawn from the previous cohort. The exclusion criteria were exactly the same as previously mentioned 1, in which known syndromes (including Alagille syndrome, Goldenhar's syndrome, hemifacial microsomia, Klippel–Feil syndrome, spondylocostal dysostosis, spondylothoracic dysostosis and VACTERL syndrome) were excluded clinically. Herein, the inclusion criteria for case group were as follows: (i) clinical diagnosis of CS; and (ii) carrying a genotype of 16p11.2 deletion and a common TBX6 haplotype of TCA (defined by the non‐reference alleles of three common single‐nucleotide polymorphisms [SNPs]‐rs2289292, rs3809624 and rs3809627, respectively). Age and gender were matched between the case and control groups to guarantee there was no statistical significance. This work had been approved by the ethical committee of Peking Union Medical College Hospital, and written informed consent had been obtained from the participants or their guardians.
Depletion of high‐abundance serum proteins
Serum IgG and albumin were depleted using ProteoPrep Blue Albumin and IgG Depletion kit (PROTBA, Sigma‐Aldrich Company, Darmstadt, Germany), according to the manufacturer's instruction. A total of 600 μl serum was used for the depletion, and finally, 400 μl was eluted at the end of the procedure. The protein concentration after depletion was evaluated by Bradford method.
Trypsin digestion and iTRAQ labelling
Peptides from 100 μg of each depleted serum were labelled with 8‐plex iTRAQ reagents (Applied Biosystems, Foster City, CA, USA), according to the manufacturer's protocol. In short, the proteins were denatured, alkylated and trypsin‐digested at 37°C overnight. Then the peptides were labelled with one unit of iTRAQ reagent that was reconstituted in 150 μl isopropanol. The labelling arrangement was shown in Table 1. After the labelling, the samples were combined into one tube and dried in vacuo. Dried peptides were resuspended in 100 μl of mobile phase A and were centrifuged at 14,000 g for 20 min. The supernatants were loaded on the column (Durashell‐C18,4.6 × 250 mm, 5 μm, 100 Å, Agela, DC952505‐0) and eluted stepwise by injecting mobile B in the RIGOL L‐3000 system (RIGOL, Beijing, China). Mobile phase A consisted of 2% (v/v) acetonitrile, 98% (v/v) ddH2O and pH 10, and phase B consisted of 98% (v/v) acetonitrile, 2% (v/v) ddH2O and pH 10. The 60‐min. gradients comprised 5% mobile B for 5 min., 5–30% mobile B for 35 min., 30–95% mobile B for 10 min. and equilibrated with 5% mobile B for 10 min. at a 300 nl/min. flow rate. The fractions were eluted at 1.5‐min. intervals and collected using step gradients of mobile B.
Table 1.
Labelling design for samples from CS and control individuals. The digested peptides were labelled with 113–121 iTRAQ isotopes. A total of 100 μg proteins were labelled with 1 unit of label. Three replicates were performed for the pooled sample (121 isotopes). CS is the acronym of congenital scoliosis
| 113 | 114 | 115 | 116 | 117 | 118 | 119 | 121 | ||
|---|---|---|---|---|---|---|---|---|---|
| 1 | CS 1 | CS 2 | CS 3 | CS 4 | Control 1 | Control 2 | Control 3 | Pool | 8‐plex |
| 2 | Control 4 | Control 5 | Control 6 | Control 7 | CS 5 | CS 6 | CS 7 | Pool | 8‐plex |
| 3 | CS 8 | CS 9 | Control 8 | Control 9 | Pool | 8‐plex |
Mass spectrometer analysis
LC‐MS/MS analysis was performed on Q‐Exactive mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) equipped with a nano‐liquid chromatography system (Thermo Scientific EASY‐nLC 1000 System, Waltham, MA, USA). The dried iTRAQ‐labelled peptides were dissolved in 20 μl of liquid containing 2% methanol and 0.1% formic acid. After centrifugation at 12,000 g for 10 min., the supernatant was collected. Then, peptide mixtures were separated using a binary solvent system with 99.9% H2O, 0.1% formic acid (phase A) and 99.9% acetonitrile, 0.1% formic acid (phase B). Linear gradients of 8–30% B for 77 min., 30–50% B for 10 min., 50–95% B for 15 min. and finally 5% B for 11 min., with a flow rate of 350 nl/min., were employed. The eluent was submitted to Q‐Exactive mass spectrometer through EASY‐Spray ion source with parameters as follows: spray voltage, 2.3 kv; capillary temperature, 320°C; and declustering potential, 100 V. The mass spectrometer was adjusted automatically between MS and MS/MS mode. The full‐scan MS mode was operated with the following parameters: automatic gain control (AGC) target, 3e6; maximum ion transfer, 20 ms; resolution, 70,000 FWHM; and scan range, 300–1800 m/z. The MS/MS mode was set as follows: AGC target, 1e5; maximum ion transfer, 120 ms; intensity threshold, 8.30E+03; fragmentation methods, high‐energy collisional dissociation (HCD); normalized collision energy, 29%; and top number, 20.
Database searching and protein identification
We used ProteoWizard (version 3.0.8789) to extract the raw data files from MS. Charge state deconvolution and deisotoping were not performed. The MS/MS samples were searched using the Mascot search engine against the SwissProt database (2016_03, selected for Homo sapiens, unknown version, 20200 entries). The search was governed by the following parameters: trypsin was assumed as the digestion enzyme; two missed cleavages were allowed at maximum; fragment ion mass tolerance of 20 mmu; parent ion tolerance of 15 ppm; carbamidomethylation of cysteine was selected as fixed modification; oxidation of methionine, iTRAQ 8 plex of lysine and the N‐terminus were set as variable modifications. Protein quantification was performed by Scaffold Q+ (4.6.1, Proteome Software Inc., Portland, OR, USA) to calculate the relative abundance of iTRAQ‐labelled peptides and the corresponding proteins. Peptide identification was accepted at greater than 95% probability to achieve FDR < 1.0%. Proteins identifications were accepted if more than 96% probability to achieve FDR < 1.0%. Only the proteins with at least two identified peptides were considered for quantification. Protein probabilities were calculated by the Protein Prophet algorithm 12. Principle of parsimony was applied for grouping of proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone. Normalization calculation was performed with median protein ratio iteratively on intensities. Spectral data were log‐transformed, pruned of those that can match to multiple proteins and with a missing value.
Bioinformatics analysis
The distribution description of the data in each group is generally presented as mean ± standard deviation (S.D.) The total proteins were submitted to Student's t‐tests to determine the significance between two groups by Perseus software (version 1.5.0.31, Berlin, Germany). The q values were calculated on the basis of permutation‐based FDR default setting in Perseus. Ratio is defined as 2difference, while difference equals the mean of CS log‐transformed spectral intensities minus the mean of control log‐transformed spectral intensities. The significance threshold of q values was 0.05. Ratio cut‐off values were set as 1.42 or 0.70. Differentially expressed proteins were designated when meeting the two requirements simultaneously: (i) q values < 0.05; and (ii) ratios ≥ 1.42 (up‐regulated) or ≤0.70 (down‐regulated). Gene Ontology (GO) analysis was performed on the PANTHER platform (http://www.pantherdb.org/), in which the biological process, molecular function and cellular component were analysed. The Ingenuity Pathway Analysis software (IPA@, v01‐04, QIANGEN Redwood City, CA, USA) was utilized to conduct the network and pathway analysis. In the IPA parameters, no expression cut‐off value was selected, and both up‐regulated and down‐regulated proteins were focused on. Hierarchical clustering analysis with Euclidean distance was carried out after Z‐score transformation, and K means pre‐processing by Perseus.
Results
Detection and identification of total proteins and differentially expressed proteins
In total, we identified 277 proteins that were expressing in both CS and control groups. Among the identified total proteins, 65 proteins showed a q value < 0.05. There are 108 up‐regulated (ratio > 1) and 169 down‐regulated proteins (ratio < 1) in the CS group in comparison with the control group, with ratio (CS/control) ranging from 0.573 to 2.052 (Table S1). Taking into account both the q value and the ratio, we designated 20 differentially expressed proteins (q < 0.05 and ratio ≥ 1.42 or ≤0.70) (Table 2)(Fig. 1). The differentially expressed proteins included apolipoprotein C‐I, sex hormone‐binding globulin, complement C4‐A and transgelin‐2.
Table 2.
The 20 differentially expressed proteins in serum identified by LC‐MS/MS analysis in CS and normal control samples
| Accession number | Protein name | CS mean | CS S.D. | Control mean | Control S.D. | Ratio (CS/control) | Up‐ or down‐regulation | −Log P‐value | q value | Differentially expressed proteins |
|---|---|---|---|---|---|---|---|---|---|---|
| CO9_HUMAN | Complement component C9 OS = Homo sapiens GN = C9 PE = 1 SV = 2 | 17.432 | 0.220 | 16.912 | 0.165 | 1.434 | UP | 4.463 | 0.000 | + |
| TAGL2_HUMAN | Transgelin‐2 OS = Homo sapiens GN = TAGLN2 PE = 1 SV = 3 | 15.599 | 0.287 | 14.964 | 0.233 | 1.552 | UP | 4.014 | 0.000 | + |
| CRP_HUMAN | C‐reactive protein OS = Homo sapiens GN = CRP PE = 1 SV = 1 | 16.620 | 0.801 | 15.297 | 0.210 | 2.502 | UP | 3.702 | 0.002 | + |
| LBP_HUMAN | Lipopolysaccharide‐binding protein OS = Homo sapiens GN = LBP PE = 1 SV = 3 | 16.160 | 0.407 | 15.331 | 0.343 | 1.776 | UP | 3.593 | 0.003 | + |
| SHBG_HUMAN | Sex hormone‐binding globulin OS = Homo sapiens GN = SHBG PE = 1 SV = 2 | 16.518 | 0.254 | 17.181 | 0.335 | 0.631 | Down | 3.643 | 0.003 | + |
| VWF_HUMAN | Von Willebrand factor OS = Homo sapiens GN = VWF PE = 1 SV = 4 | 16.863 | 0.303 | 16.356 | 0.178 | 1.422 | UP | 3.293 | 0.003 | + |
| MMP9_HUMAN | Matrix metalloproteinase‐9 OS = Homo sapiens GN = MMP9 PE = 1 SV = 3 | 15.666 | 0.313 | 15.151 | 0.216 | 1.428 | UP | 3.041 | 0.005 | + |
| A1AG1_HUMAN | α‐1‐acid glycoprotein 1 OS = Homo sapiens GN = ORM1 PE = 1 SV = 1 | 17.388 | 0.269 | 16.872 | 0.289 | 1.430 | UP | 2.914 | 0.008 | + |
| SAA2_HUMAN | Serum amyloid A‐2 protein OS = Homo sapiens GN = SAA2 PE = 1 SV = 1 | 17.382 | 0.453 | 16.777 | 0.169 | 1.522 | UP | 2.766 | 0.010 | + |
| K1C10_HUMAN | Keratin, type I cytoskeletal 10 OS = Homo sapiens GN = KRT10 PE = 1 SV = 6 | 16.266 | 0.080 | 16.844 | 0.507 | 0.669 | Down | 2.422 | 0.016 | + |
| A2GL_HUMAN | Leucine‐rich α‐2‐glycoprotein OS = Homo sapiens GN = LRG1 PE = 1 SV = 2 | 16.537 | 0.528 | 15.868 | 0.266 | 1.590 | UP | 2.430 | 0.017 | + |
| CO4A_HUMAN | Complement C4‐A OS = Homo sapiens GN = C4A PE = 1 SV = 2 | 17.520 | 0.278 | 16.919 | 0.468 | 1.517 | UP | 2.357 | 0.018 | + |
| K2C1_HUMAN | Keratin, type II cytoskeletal 1 OS = Homo sapiens GN = KRT1 PE = 1 SV = 6 | 16.021 | 0.171 | 16.823 | 0.721 | 0.573 | Down | 2.296 | 0.020 | + |
| HBG2_HUMAN | Haemoglobin subunit γ‐2 OS = Homo sapiens GN = HBG2 PE = 1 SV = 2 | 15.672 | 0.503 | 16.409 | 0.482 | 0.600 | Down | 2.228 | 0.023 | + |
| KCRM_HUMAN | Creatine kinase M‐type OS = Homo sapiens GN = CKM PE = 1 SV = 2 | 15.127 | 0.469 | 14.556 | 0.279 | 1.486 | UP | 2.198 | 0.024 | + |
| APOC1_HUMAN | Apolipoprotein C‐I OS = Homo sapiens GN = APOC1 PE = 1 SV = 1 | 17.607 | 0.543 | 18.237 | 0.319 | 0.646 | Down | 2.074 | 0.029 | + |
| K22E_HUMAN | Keratin, type II cytoskeletal 2 epidermal OS = Homo sapiens GN = KRT2 PE = 1 SV = 2 | 14.629 | 0.233 | 15.213 | 0.540 | 0.667 | Down | 2.055 | 0.030 | + |
| SAA1_HUMAN | Serum amyloid A‐1 protein OS = Homo sapiens GN = SAA1 PE = 1 SV = 1 | 16.743 | 0.756 | 15.982 | 0.225 | 1.695 | UP | 1.977 | 0.032 | + |
| IGHG1_HUMAN | Ig γ‐1 chain C region OS = Homo sapiens GN = IGHG1 PE = 1 SV = 1 | 16.583 | 0.548 | 17.162 | 0.350 | 0.669 | Down | 1.777 | 0.045 | + |
| A1AT_HUMAN | α‐1‐anti‐trypsin OS = Homo sapiens GN = SERPINA1 PE = 1 SV = 3 | 17.018 | 0.559 | 16.487 | 0.235 | 1.445 | UP | 1.737 | 0.048 | + |
The spectral intensity was log‐transformed and compared by Student's t‐test with permutation‐based FDR test using Perseus. The significance cut‐off of q values was 0.05, and ratio cut‐off values were 1.42 or 0.70. Differentially expressed proteins were assigned if ratio ≥1.42 or ≤0.70 and q values < 0.05.
Figure 1.
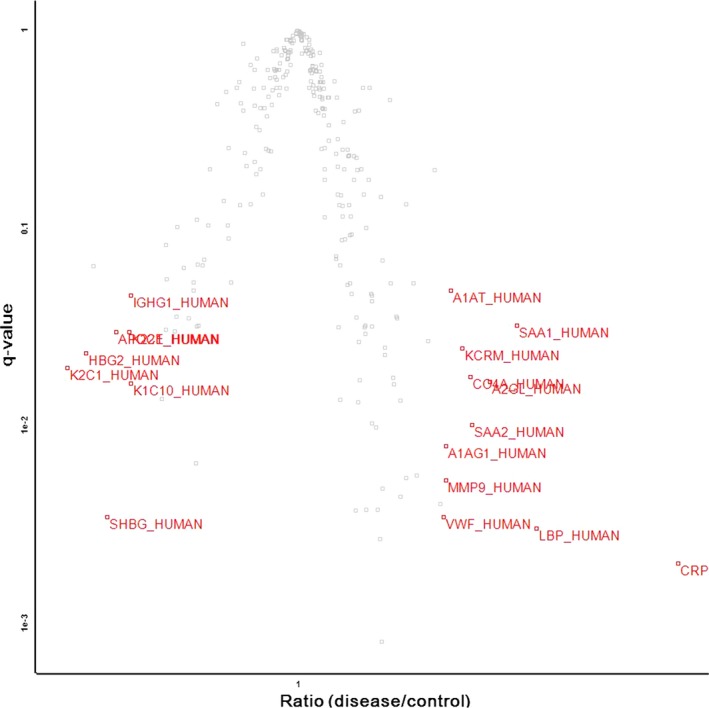
Volcano plot representation of totally identified protein results showing the ratio (x‐axis) and significance (q values, y‐axis). The volcano plot indicates that 20 proteins were significantly (q value < 0.05) differentially (ratio ≥1.42 or ≤0.70) expressed in the CS group compared with the normal control group. The accession numbers of each differentially expressed protein are shown in red.
GO classification of identified proteins
We classified the 20 differentially expressed proteins according to GO information (PANTHER platform, http://www.pantherdb.org/) concerning biological process, molecular function and cellular component. The biological processes of the 20 proteins are related to cellular process (25.6%), localization (12.8%), cellular component organization or biogenesis (10.3%), response to stimulus (10.3%), multicellular organismal process (10.3%), immune system process (10.3%), metabolic process (7.7%), biological regulation (5.1%), locomotion (5.1%) and biological adhesion (2.6%) (Fig. 2A). The top one molecular function is the structural molecule activity (36.4%); other molecular functions are involved in: binding (27.3%), catalytic activity (27.3%) and receptor activity (9.1%) (Fig. 2B). The cellular component analysis revealed that the main cellular component categories of these proteins are extracellular region (35.3%), cell part (23.5%), organelle (23.5%), membrane (5.9%), macromolecular complex (5.9%) and extracellular matrix (5.9%) (Fig. 2C).
Figure 2.
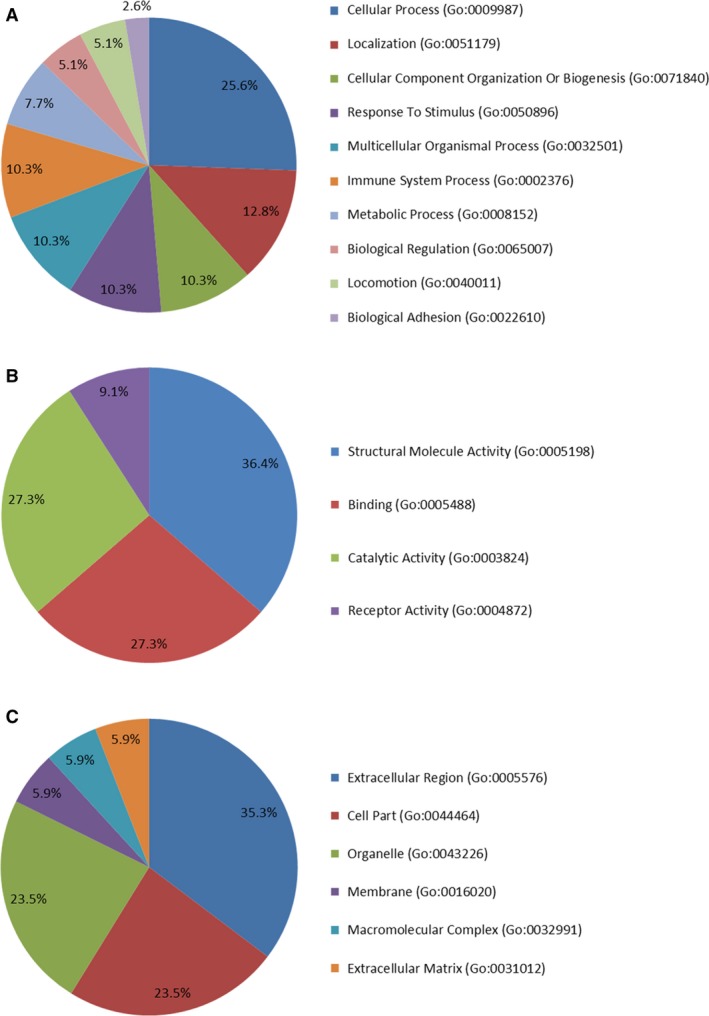
Classifications of the identified differential proteins according to GO annotation of biological process (A), molecular function (B) and cellular component (C). The GO analysis was performed on the PANTHER platform, and a total of 20 differentially expressed proteins were submitted to the analysis.
IPA network and pathway analysis
Network analyses of 20 differentially expressed proteins in the serum of CS samples versus controls were assessed through the use of IPA software. The IPA calculates the P‐values by Fisher's exact test and the p score to assess the probability of matching the submitted proteins in a protein–protein interaction by random chance. Generally, we found a total of four significant molecular networks (Table S2). The first remarkable networks are depicted in Figure 3. The first network with the highest score (37) consists of 15 ‘focus molecules’, and its related diseases and functions incorporate cellular movement, haematological system development and function and immune cell trafficking (Table S2).
Figure 3.
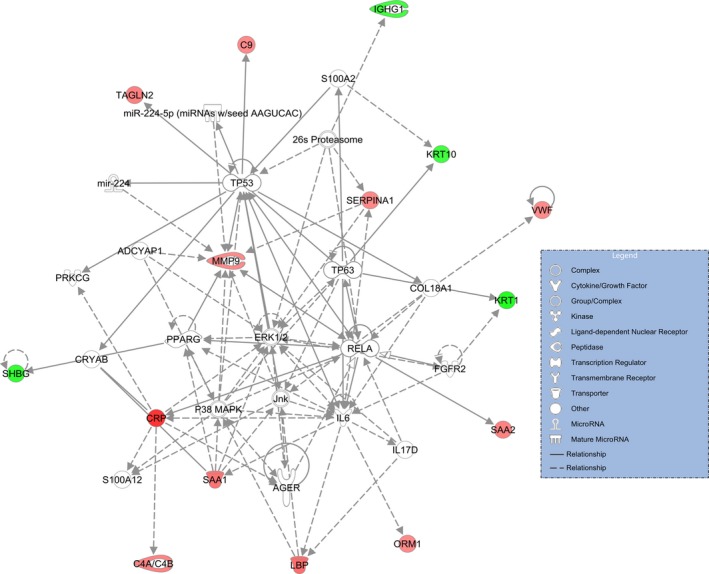
The top molecular network generated by IPA software for 20 differentially expressed proteins in serum of CS versus control. The modulated proteins are classified by established relationships into the functional analysis of a network. Solid line indicates direct interaction, and dashed line means indirect interaction.
The canonical pathways assigned by IPA which involve the 20 differentially expressed proteins indicated that the top seven pathways are LXR/RXR activation, acute phase response signalling, FXR/RXR activation, atherosclerosis signalling, IL‐12 signalling and production in macrophages, coagulation system and complement system (Table 3). The interactive relationship between proteins in the first canonical pathway is denoted in Figure 4. The whole information on the canonical pathway output is displayed in Table S3. It is noteworthy to mention that the top one network is intimately associated with lipid metabolism (Fig. 4).
Table 3.
The top seven canonical pathways enriched by IPA. The 20 differentially expressed proteins were imported to IPA software, and the generated top seven pathways are shown
| Top seven canonical pathways | −log(P‐value) | Molecules | Ratio |
|---|---|---|---|
| LXR/RXR Activation | 1.47E01 | 9 | 7.44E‐02 |
| Acute phase response signaling | 1.34E01 | 9 | 5.36E‐02 |
| FXR/RXR Activation | 1.05E01 | 7 | 5.6E‐02 |
| Atherosclerosis signaling | 5.13E00 | 4 | 3.23E‐02 |
| IL‐12 Signaling and production in macrophages | 3.37E00 | 3 | 2.08E‐02 |
| Coagulation system | 3.22E00 | 2 | 5.71E‐02 |
| Complement system | 3.2E00 | 2 | 5.56E‐02 |
Figure 4.
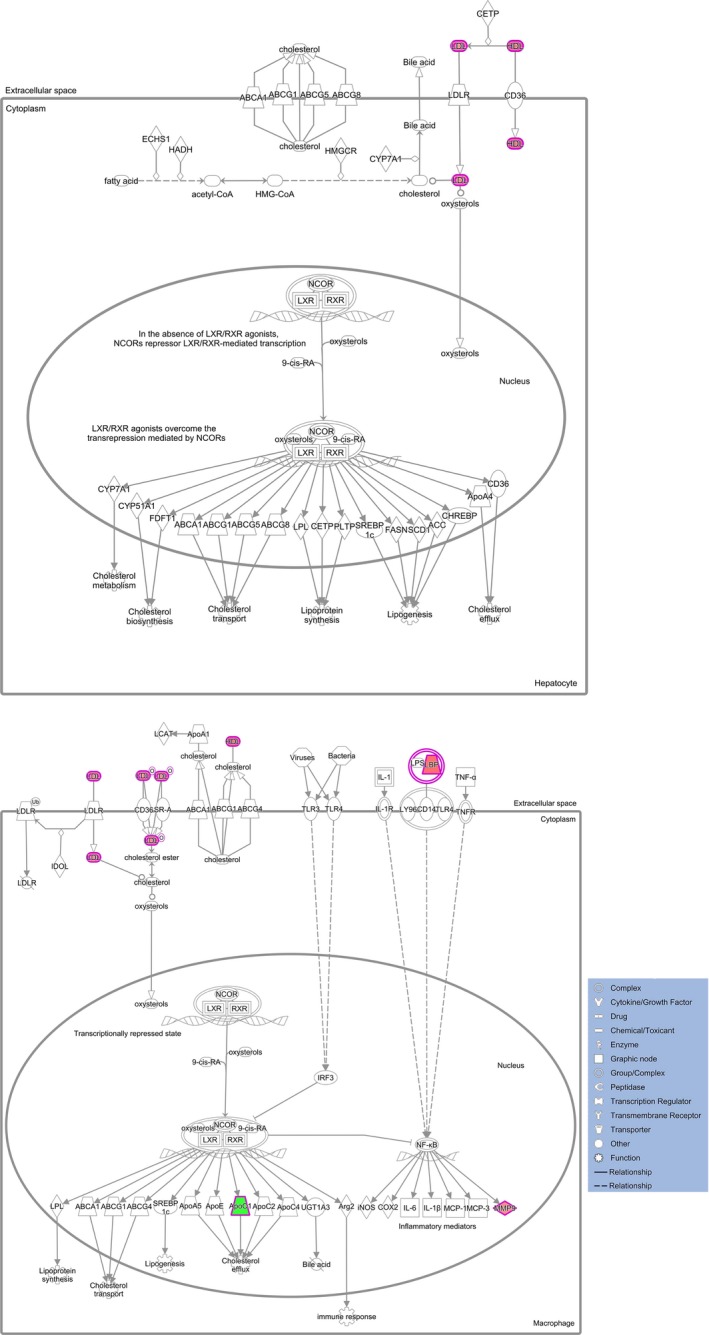
The top canonical pathway assigned by the IPA for 20 differentially expressed proteins expressed in both CS and normal control. The first canonical pathway of LXR/RXR activation. Each line and arrow suggests known functional or physical interaction. Red and green colours in each shape mean the up‐ or down‐regulation of pathway‐associated protein expression. Solid line indicates direct interaction, and dashed line means indirect interaction.
Hierarchical clustering analysis
We conducted the hierarchical clustering analysis on the basis of the expression values for the 65 proteins having q values < 0.05. The data were Z‐score transformed and were clustered in both the row and the column directions. The results showed that most of these samples were correctly classified into two groups corresponding to disease or normal control condition, apart from one sample (CS 4) that was incorrectly classified (Fig. 5A). This suggests that CS may have particular expression profiles, distinct from the normal state profiles. The 65 submitted proteins were separated into two clusters: cluster 61 (16 proteins) and cluster 62 (49 proteins) (Table S4). The corresponding expressional pattern in cluster 61 is delineated in Figure 5B.
Figure 5.
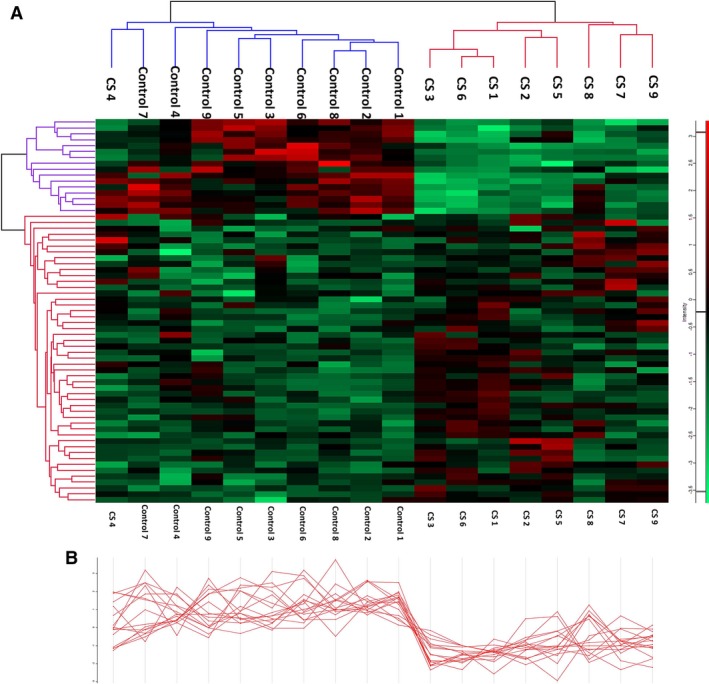
Hierarchical clustering analysis of 65 proteins with q values < 0.05. (A) Heat map representation of every protein in the congenital scoliosis (CS) and control samples. The proteins were separated into two clusters: cluster 61 (16 proteins) and cluster 62 (49 proteins). Each row represents an individual protein, and each column represents an individual sample. The dendrogram at the top suggests the similarity in protein expression profiles of the sample. (B) The expressional pattern of each protein in cluster 61 across every individual sample. Blue–red heat map values correspond to low–high protein expression levels.
Discussion
In the present study, to elucidate the mechanisms of TBX6‐induced CS at the protein level, we employed a comparative proteomics strategy of iTRAQ to explore the differential protein expression profiles, GO terms, IPA network and pathway and hierarchical clustering between normal control and CS patients with both TBX6 deletion and T‐C‐A haplotype. To the best of our knowledge, this investigation is the first proteomics research on CS, following our latest genetic revelation of TBX6 in the onset of CS 1.
Our results indicated that 277 total proteins were expressed in both CS and normal controls. Furthermore, 65 proteins showed q values < 0.05 and 20 proteins were designated as differentially expressed proteins (q < 0.05, and ratio ≥ 1.42 or ≤0.70). We then conducted bioinformatics analyses on these proteins. Generally, the GO classification suggested that annotation with highest percentage in biological process, molecular function and cellular component was cellular process, structural molecule activity and extracellular region, respectively. IPA output showed that the most significant canonical pathway was the ‘LXR/RXR activation’ pathway. Hierarchical clustering analysis separated the 65 protein (q < 0.05) into two clusters (16 versus 49 proteins). Due to the significant expression modulations as well as potential biological and functional relevance of the differentially expressed proteins in the pathogenesis and aetiology of CS, we herein choose several of the proteins to discuss in the following section.
The most appealing finding to emerge from our study is that we, for the first time, hypothesize lipid metabolism to play vital roles in the pathogenesis of CS. Supporting evidence not only arises from proteomic revelation of APOC1 and LXR/RXR pathway in this study, as illustrated in following text, but also comes from other discoveries including the doctoral thesis of Dr. Sun from our team who conducted a preliminary proteomics analysis on CS by 2‐D gel electrophoresis in 2011 (http://d.g.wanfangdata.com.cn/Thesis_Y1985714.aspx). Part of the serum proteins he found, such as APOA1 and APOA4, also point to lipid metabolism. But his results have not been submitted to any journal for publishing so far. LXR encodes highly homologous transcription factors of nuclear receptors and forms functional heterodimers with RXR via LXR‐responsive element within the targeted regulators 13. The LXR/RXR functions as sensors of cholesterol homoeostasis by regulating genes that control sterol and fatty acid metabolism 14. Another supporting evidence is that in adolescent idiopathic scoliosis, which is another type of scoliosis and may have overlapping pathogenesis with CS 15, 16, lipid metabolism was identified to be the most remarkable metabolic changes in the serum 17. Taken together, our results and other evidence strongly point to disrupted lipid metabolism in CS. More generalized studies with larger sample sizes should be conducted to investigate whether lipid metabolism is associated with CS.
Sex hormone‐binding globulin (SHBG) is a homodimeric glycoprotein, the most prominent feature of which is to bind sex steroids with equally high affinity as well as specificity. It is synthesized in the liver, and its molecular weight is approximately 90 kD 18. Each dimer of the SHBG, consisting of 373 amino acids, exhibits a tertiary structure of carboxy‐terminal laminin G‐like domain (LGLD), which is joined to an amino‐terminal LGLD by a linker region 19. SHBG has been reported to be implicated in various physiological disturbance states and diseases. In polycystic ovary syndrome (PCOS), Wassell et al. 20 found that PCOS patients exhibiting a positive response to metformin treatment showed a significantly lower pre‐treatment SHBG levels, implying the SHBG may be a predictor of response to pharmacological treatment for PCOS. In thyroid disorders, Hampl et al. 21 observed that low SHBG levels in patients treated with total thyroidectomy because of thyroid cancer were significantly elevated to physiological values after reaching normal thyroid function. In pituitary diseases, hyperprolactinemia was associated with decreased SHBG concentrations, and SHBG levels returned to normal limits following bromocriptine administration 22. In metabolic syndrome (METS) and type 2 diabetes mellitus (T2DM), SHBG levels have been demonstrated to have an inverse association with an increased risk for the development of METS and T2DM 23, 24, 25, suggesting that SHBG is likely to represent a sensitive and early biomarker for metabolic disturbances. The related molecular mechanism explaining the observational studies may be partially attributed to that monosaccharide‐induced hepatic de novo lipogenesis can reduce hepatic HNF‐4α levels and subsequently SHBG gene expression. In liver disorders, SHBG was significantly increased in patients with cirrhosis, a result and final path of various liver disorders 26. The elevation of SHBG in cirrhosis was less pronounced with progression to the decompensated state. As liver cirrhosis is inclined to develop into hepatocellular carcinoma (HCC), SHBG might constitute a potential risk marker for early detection of HCC 26. In the realm of orthopaedic researches, SHBG was found abated in rheumatoid arthritis patients than control volunteers, in both pre‐menopausal and postmenopausal status 27. SHBG was also associated with bone loss, vertebral fractures and kyphosis 28. Interestingly, Chilean scholars, Albala et al. 29, evaluated the bone mineral density and the SHBG in obese and non‐obese postmenopausal women, and concluded that obesity exerts protection against osteoporosis via decreased SHBG. Furthermore, another publication suggested that high serum SHBG levels have an increased risk of fractures in older Swedish men 30. Low bone mineral density has been observed in CS patients 31, and the distribution of bone mineral density in CS mice was highest in the curve apex 32. Our study is the first to account for SHBG protein as differentially expressed protein in CS patients. We believe the finding of SHBG may be helpful in elaborating the low bone mineral density in CS patients when taking into account the reported role of SHBG in osteoporosis.
Apolipoprotein C‐I (APOC1), with a 6.6 kD molecular mass, is one constituent of chylomicrons, high‐density lipoprotein (HDL) and triglyceride‐rich lipoproteins, which can blunt the metabolism of triglyceride‐rich lipoproteins 33. APOC1 functions as an inhibitor of lipoprotein binding to the low‐density lipoprotein (LDL) receptor and very low‐density lipoprotein (VLDL) receptor. APOC1 is also a major inhibitor of cholesterol ester transfer protein and is able to affect fatty acid uptake in a direct manner 34. High expression of apolipoproteins in embryogenesis would be helpful for transporting material, such as lipid. Therefore, it is reasonable to speculate APOC1, the smallest member of apolipoprotein family, may have an important role in embryogenesis, in addition to the well‐documented expression of other lipoproteins (APOE and APOA1) in embryonic development 35 that, potentially, may be associated with bone forming. One publication confirmed our ratiocination. Wang and his colleagues 36 observed in zebrafish that apoc1 gene is expressed in gradient along the ventral–dorsal axis with the highest expression on the ventral side, and apoc1 gene expression occurs in tail paraxial mesoderm that originates from the blastoderm margin in somitogenesis. They also revealed that apoc1 gene expression can be activated by bone morphogenetic proteins (BMPs) signalling, and this effect can be dampened by retinoic acid (RA) signalling suppression. This serves a new cynosure because RA and its receptors are demonstrated to be expressed in the paraxial mesoderm and are critical for regulating key genes in the somitogenesis clock and the HOX gene induction 37. RA inhibits the ability of the paraxial mesoderm to respond to asymmetric signalling downstream of the left‐side determinant Nodal, which could cause asymmetry of somitogenesis 38. In addition, BMP signalling has been shown to be involved in the development of the initial vertebrate body and somite 39, 40 and BMP inhibitor, dorsomorphin, produces split‐body phenotypes including bilaterally split somites 41. Considering the roles of BMP and RA in somitogenesis and their demonstrated downstream effects on APOC1 protein 36, we can infer that the down‐regulated expression of APOC1 protein in our study and the occurrence of CS are two concurrent phenotypes caused, at least in part, by dysregulation of some common upstream signallings such as RA and BMP. Furthermore, apoc1 gene expression in murine macrophages can be up‐regulated by ligands of LXR/RXR pathway 42, which is in agreement with the appearance of LXR/RXR pathway as the most remarkable canonical pathway of IPA output in our study. This further strengthens the link between APOC1 and CS. As LXR/RXR pathway is the key regulator for lipid and cholesterol metabolism 14, it is reasonable to generalize a step further from APOC1 and LXR/RXR to lipid metabolism – it is lipid metabolism that may share a common molecular background with CS under partial contributions from RA and BMPs. Given the previously demonstrated aetiological documentation of CS, we can even propose a more radical supposition that lipid metabolism may have interactions with the known mechanisms of CS, and future investigations on how the lipid metabolism is involved in the onset of CS, especially from an embryological point of view, may shed new light on the early prevention and treatment of CS.
Complement C4‐A (CO4A) is a component of the complement system and is indispensable for its classical activation pathway 43. CO4A is derived from the proteolytic degradation of complement factor 4 (C4). CO4A acts in the form of a single chain precursor, and then is cleaved into a trimer before secretion 44. Anaphylatoxin C4a, the cleaved product of alpha chain of C4, is a mediator of local inflammatory response. The gene encoding CO4A is localized at the major histocompatibility complex (MHC) class III region. Recent studies implicated that CO4A was related to neurological disorders. In schizophrenia, the level of C4 protein was observed to be significantly increased in patients' sera 45, 46, and CO4A RNA expression was significantly higher in the brain tissue 47. Besides, C4A gene structural diversity can in part serve as a bridge between MHC variation and schizophrenia 47. It is known that C4 protein lies in neuronal synapses, dendrites, axons and cell bodies. Thus, the schizophrenic phenotype can be attributed to the potential role of C4 protein in synapse remodelling and pruning during the neurodevelopmental process 48. In our study, the level of CO4A protein was significantly elevated in CS patients, all of which carry a 16p11.2 deletion. In fact, 16p11.2 deletion has demonstrated to be a likely cause for developmental delay, mental retardation and autism spectrum disorder 49. However, the detailed neurodevelopmental records of the patients in this study were not available for us to determine whether they had neurological disorders or not. Further investigations are needed to clarify this point. As CS and neurological disorder (such as schizophrenia) are two well‐demonstrated phenotypes caused by 16p11.2 deletion, one phenotype may, to some extent, be indicative of the other phenotype. Hence, we propose that CO4A, and thus neurological disorder (which has to do with CO4A) may be coincidental with some CS cases that have 16p11.2 deletion. In line with our reasoning pattern in the hypothesis, previous publications revealed that two other phenotypes of 16p11.2 deletion, obesity and low cognitive functioning, showed remarkable interrelations with each other: people who have learning disabilities have a greater chance of being obese 50, and the obese people in turn exhibit a substantially decreased cognitive ability 51. However, the altered level of CO4A in our study might be caused by 16p11.2 deletion, but not CS. Whether CO4A and CS can manifest simultaneously depends on the coverage of deletion.
Transgelin‐2 is a homolog of the protein transgelin, a 22 kD actin‐binding protein of the calponin family. Transgelin was first identified in 1987 in smooth muscle tissues of birds and therefore named as smooth muscle 22 (SM22) 52. Transgelin is widely expressed in vascular and visceral smooth muscles and is one of the earliest markers of smooth muscle differentiation 53. Transgelin‐2 has been suggested to be a tumour suppressor demonstrated by its discovery as differentially expressed protein in several proteomics studies. Through the 2‐D gel electrophoresis, transgelin‐2 was identified by MS as one of the differentially expressed proteins in lung adenocarcinoma 54. Validation by immunohistochemistry revealed the exclusive expression of transgelin and transgelin‐2 in the stromal tissue compartment and neoplastic glandular compartment, respectively, suggesting its important role in active stromal remodelling of invasive carcinomas. Similarly, in a highly metastatic variant of human breast cancer cell, transgelin‐2 was found to be the only down‐regulated protein, and verification experiments implied a significant association between lymph node metastasis and expression of transgelin‐2 55. In antineoplastic drug‐resistant breast cancer cell line, transgelin‐2 was reported to be a mediator for drug resistance by up‐regulating the adenosine triphosphate‐binding cassette transporter proteins 56. In colorectal cancer, transgelin‐2 was significantly up‐regulated, which was also discovered by quantitative proteome analysis. Further validation indicated transgelin‐2 was tissue‐specific in colorectal cancer cells, and overexpression of transgelin‐2 was associated with tumour prognostic factors 57. Apart from its identification in cancer cells, transgelin‐2 was also significantly differentially expressed in proteome of other diseases, including rheumatoid arthritis and meniscal injury. Transgelin‐2 was selected as one of the candidate markers for rheumatoid arthritis due to its elevated expression in the synovial fluid of erosive rheumatoid arthritis in comparison with non‐erosive rheumatoid arthritis 58. In injured menisci, proteomic analysis suggested transgelin‐2 was one of the interest proteins after evaluating the expressional trends between case and control groups 59. In spite of its discovery and potential biological value in many diseases, transgelin‐2 has never been reported in the threshold of scoliosis research. Our study for the first time reports the involvement of transgelin‐2 as serum differentially expressed protein in CS. As such, transgelin‐2 may have been suggested as a candidate marker for CS possibly indicating potential co‐existing smooth muscle deformity with the malformation of spine. However, more observational investigations, especially on abnormal embryogenesis, are necessary to confirm the hypothesis.
In addition to the above‐mentioned proteins, other significantly differentially expressed proteins, including serum amyloid A‐1 protein, serum amyloid A‐2 protein, leucine‐rich α‐2‐glycoprotein, creatine kinase M‐type and α‐1‐acid glycoprotein 1, are also worth our concern. Serum amyloid A proteins belong to the apolipoproteins family and are acute phase proteins during inflammatory response. Serum amyloid A‐1 and A‐2, two isoforms of serum amyloid A, are produced by hepatocyte and released into blood up to a 1000‐fold during inflammation 60. Leucine‐rich α‐2‐glycoprotein is a potential diagnostic marker for inflammation‐associated diseases for its augmented expression in ulcerative colitis 61, 62, asthma 63, acute appendicitis 64 and rheumatoid arthritis 65. Creatine kinase M‐type, a subtype of creatine kinase, reversibly catalyses the conversion of creatine into phosphocreatine and adenosine diphosphate (ADP) by facilitating the transfer of adenosine triphosphate (ATP) 66. Because it is mainly expressed in sarcomeric muscles such as skeletal and cardiac muscles, creatine kinase M‐type is a potential diagnostic marker for myocardial infarction 67, rhabdomyolysis 68 and myositis 69. α‐1‐acid glycoprotein 1 is an acute phase protein involved in immune system modulation, and elevated serum α‐1‐acid glycoprotein 1 is seen in systemic inflammatory response or infection 70. Although these proteins are firstly identified in the proteome of CS, future researches on the possible biological merits of the observed proteins are still necessary for the aetiological elucidation of CS.
In summary, we for the first time depicted the differential protein expression profiles of CS in serum. These previously unidentified proteins may offer invaluable clue for not only the aetiological mechanism but also the onset and development of CS. The application of these proteins may serve effective diagnostic tools for early detection of CS. The underlying interactions between the differential proteins and CS need further investigation.
Conflict of interest
The authors confirm that there is no conflict of interests.
Supporting information
Table S1 List of total proteins identified in sera from CS and control samples.
Table S2 Network analysis results generated by IPA software.
Table S3 Ingenuity canonical pathways assigned by IPA software.
Table S4 The separated proteins in cluster 61 and cluster 62 by HCA.
Acknowledgements
We thank Bintao Qiu and Weiliang Ye for excellent technical assistance. We thank Shugang Li, Yipeng Wang, Hong Zhao, Keyi Yu and Yu Zhao for their sample offering. This study was supported by the National Natural Science Foundation of China (81501852, 81472046, 81271942, 81130034, 81472045 and 81772301), the Distinguished Youth Foundation of Peking Union Medical College Hospital (JQ201506), Beijing Nova Program (Z161100004916123), the Central Level Public Interest Program for Scientific Research Institute (2016ZX310177), the PUMC Youth Fund & the Fundamental Research Funds for the Central Universities (3332016006) and the CAMS Initiative for Innovative Medicine (2016‐I2M‐3‐003).
Q.Z. and N.W. designed and conducted the experiment. S.L. and Y.Z. analysed the data. G.L., J.L., J.C. and Y.Z. revised the manuscript and provided critical advice. Z.L., W.C. and Y.C. assisted in manuscript writing. J.C., M.L. and Y.Z. participated in data interpretation. J.W. and X.C. provided the samples and participated in experimental conception. Y.Y., S.W., J.Z., X.Y., Y.M., J.S., Z.W. and G.Q. collected the sample and reviewed the final draft.
Contributor Information
Zhihong Wu, Email: orthoscience@126.com.
Guixing Qiu, Email: qiuguixingpumch@126.com.
References
- 1. Wu N, Ming X, Xiao J, et al TBX6 null variants and a common hypomorphic allele in congenital scoliosis. N Engl J Med. 2015; 372: 341–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wynne‐Davies R. Congenital vertebral anomalies: aetiology and relationship to spina bifida cystica. J Med Genet. 1975; 12: 280–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hubaud A, Pourquie O. Signalling dynamics in vertebrate segmentation. Nat Rev Mol Cell Biol. 2014; 15: 709–21. [DOI] [PubMed] [Google Scholar]
- 4. Day GA, Upadhyay SS, Ho EK, et al Pulmonary functions in congenital scoliosis. Spine (Phila Pa 1976). 1994; 19: 1027–31. [DOI] [PubMed] [Google Scholar]
- 5. Weiss HR, Moramarco M. Congenital scoliosis (Mini‐review). Curr Pediatr Rev. 2016; 12: 43–7. [DOI] [PubMed] [Google Scholar]
- 6. Papaioannou VE, Silver LM. The T‐box gene family. BioEssays. 1998; 20: 9–19. [DOI] [PubMed] [Google Scholar]
- 7. Baker RE, Schnell S, Maini PK. A clock and wavefront mechanism for somite formation. Dev Biol. 2006; 293: 116–26. [DOI] [PubMed] [Google Scholar]
- 8. Giudicelli F, Lewis J. The vertebrate segmentation clock. Curr Opin Genet Dev. 2004; 14: 407–14. [DOI] [PubMed] [Google Scholar]
- 9. Chen W, Liu J, Yuan D, et al Progress and perspective of TBX6 gene in congenital vertebral malformations. Oncotarget. 2016; 7: 57430–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Anderson NL, Anderson NG. Proteome and proteomics: new technologies, new concepts, and new words. Electrophoresis. 1998; 19: 1853–61. [DOI] [PubMed] [Google Scholar]
- 11. Banks RE, Dunn MJ, Hochstrasser DF, et al Proteomics: new perspectives, new biomedical opportunities. Lancet. 2000; 356: 1749–56. [DOI] [PubMed] [Google Scholar]
- 12. Nesvizhskii AI, Keller A, Kolker E, et al A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003; 75: 4646–58. [DOI] [PubMed] [Google Scholar]
- 13. Son YL, Lee YC. Molecular determinants of the interactions between SRC‐1 and LXR/RXR heterodimers. FEBS Lett. 2010; 584: 3862–6. [DOI] [PubMed] [Google Scholar]
- 14. Edwards PA, Kennedy MA, Mak PA. LXRs; oxysterol‐activated nuclear receptors that regulate genes controlling lipid homeostasis. Vascul Pharmacol. 2002; 38: 249–56. [DOI] [PubMed] [Google Scholar]
- 15. Giampietro PF. Genetic aspects of congenital and idiopathic scoliosis. Scientifica (Cairo). 2012; 2012: 152365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hayes M, Gao X, Yu LX, et al ptk7 mutant zebrafish models of congenital and idiopathic scoliosis implicate dysregulated Wnt signalling in disease. Nat Commun. 2014; 5: 4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sun ZJ, Jia HM, Qiu GX, et al Identification of candidate diagnostic biomarkers for adolescent idiopathic scoliosis using UPLC/QTOF‐MS analysis: a first report of lipid metabolism profiles. Sci Rep. 2016; 6: 22274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Walsh KA, Titani K, Takio K, et al Amino acid sequence of the sex steroid binding protein of human blood plasma. Biochemistry. 1986; 25: 7584–90. [DOI] [PubMed] [Google Scholar]
- 19. Grishkovskaya I, Avvakumov GV, Sklenar G, et al Crystal structure of human sex hormone‐binding globulin: steroid transport by a laminin G‐like domain. EMBO J. 2000; 19: 504–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wassell J, Michail M, Soliman N, et al The value of sex hormone binding globulin (SHBG) in predicting treatment response in polycystic ovary syndrome (PCOS). Clin Lab. 2011; 57: 95–8. [PubMed] [Google Scholar]
- 21. Hampl R, Kancheva R, Hill M, et al Interpretation of sex hormone‐binding globulin levels in thyroid disorders. Thyroid. 2003; 13: 755–60. [DOI] [PubMed] [Google Scholar]
- 22. Zwirska‐Korczala K, Ostrowska Z, Zych F, et al The levels of pituitary‐testicular axis hormones and SHBG in active acromegaly following bromocriptine treatment. Endocr Regul. 1991; 25: 211–6. [PubMed] [Google Scholar]
- 23. Lakshman KM, Bhasin S, Araujo AB. Sex hormone‐binding globulin as an independent predictor of incident type 2 diabetes mellitus in men. J Gerontol A Biol Sci Med Sci. 2010; 65: 503–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fenske B, Kische H, Gross S, et al Endogenous androgens and sex hormone‐binding globulin in women and risk of metabolic syndrome and type 2 diabetes. J Clin Endocrinol Metab. 2015; 100: 4595–603. [DOI] [PubMed] [Google Scholar]
- 25. Ding EL, Song Y, Manson JE, et al Sex hormone‐binding globulin and risk of type 2 diabetes in women and men. N Engl J Med. 2009; 361: 1152–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maruyama Y, Adachi Y, Aoki N, et al Mechanism of feminization in male patients with non‐alcoholic liver cirrhosis: role of sex hormone‐binding globulin. Gastroenterol Jpn. 1991; 26: 435–9. [DOI] [PubMed] [Google Scholar]
- 27. Mirone L, Altomonte L, D'Agostino P, et al A study of serum androgen and cortisol levels in female patients with rheumatoid arthritis. Correlation with disease activity. Clin Rheumatol. 1996; 15: 15–9. [DOI] [PubMed] [Google Scholar]
- 28. Woods GN, Huang MH, Cawthon PM, et al SHBG, sex steroids and kyphosis in older men: the MrOS study. J Bone Miner Res. 2016; 31: 2123–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Albala C, Yanez M, Devoto E, et al Obesity as a protective factor for postmenopausal osteoporosis. Int J Obes Relat Metab Disord. 1996; 20: 1027–32. [PubMed] [Google Scholar]
- 30. Mellstrom D, Vandenput L, Mallmin H, et al Older men with low serum estradiol and high serum SHBG have an increased risk of fractures. J Bone Miner Res. 2008; 23: 1552–60. [DOI] [PubMed] [Google Scholar]
- 31. Zhu F, Qiu Y, Yeung HY, et al Trabecular bone micro‐architecture and bone mineral density in adolescent idiopathic and congenital scoliosis. Orthop Surg. 2009; 1: 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Farley FA, Hall J, Goldstein SA. Characteristics of congenital scoliosis in a mouse model. J Pediatr Orthop. 2006; 26: 341–6. [DOI] [PubMed] [Google Scholar]
- 33. Jong MC, Hofker MH, Havekes LM. Role of ApoCs in lipoprotein metabolism: functional differences between ApoC1, ApoC2, and ApoC3. Arterioscler Thromb Vasc Biol. 1999; 19: 472–84. [DOI] [PubMed] [Google Scholar]
- 34. Westerterp M, Berbee JF, Delsing DJ, et al Apolipoprotein C‐I binds free fatty acids and reduces their intracellular esterification. J Lipid Res. 2007; 48: 1353–61. [DOI] [PubMed] [Google Scholar]
- 35. Babin PJ, Thisse C, Durliat M, et al Both apolipoprotein E and A‐I genes are present in a nonmammalian vertebrate and are highly expressed during embryonic development. Proc Natl Acad Sci USA. 1997; 94: 8622–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang Y, Li WH, Li Z, et al BMP and RA signaling cooperate to regulate Apolipoprotein C1 expression during embryonic development. Gene. 2015; 554: 196–204. [DOI] [PubMed] [Google Scholar]
- 37. Duester G. Retinoic acid regulation of the somitogenesis clock. Birth Defects Res C Embryo Today. 2007; 81: 84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vermot J, Pourquie O. Retinoic acid coordinates somitogenesis and left‐right patterning in vertebrate embryos. Nature. 2005; 435: 215–20. [DOI] [PubMed] [Google Scholar]
- 39. Danesh SM, Villasenor A, Chong D, et al BMP and BMP receptor expression during murine organogenesis. Gene Expr Patterns. 2009; 9: 255–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Miura S, Davis S, Klingensmith J, et al BMP signaling in the epiblast is required for proper recruitment of the prospective paraxial mesoderm and development of the somites. Development. 2006; 133: 3767–75. [DOI] [PubMed] [Google Scholar]
- 41. Mourabit S, Moles MW, Smith E, et al Bmp suppression in mangrove killifish embryos causes a split in the body axis. PLoS One. 2014; 9: e84786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mak PA, Laffitte BA, Desrumaux C, et al Regulated expression of the apolipoprotein E/C‐I/C‐IV/C‐II gene cluster in murine and human macrophages. A critical role for nuclear liver X receptors alpha and beta. J Biol Chem. 2002; 277: 31900–8. [DOI] [PubMed] [Google Scholar]
- 43. Fearon DT. Activation of the alternative complement pathway. CRC Crit Rev Immunol. 1979; 1: 1–32. [PubMed] [Google Scholar]
- 44. Belt KT, Carroll MC, Porter RR. The structural basis of the multiple forms of human complement component C4. Cell. 1984; 36: 907–14. [DOI] [PubMed] [Google Scholar]
- 45. Mayilyan KR, Arnold JN, Presanis JS, et al Increased complement classical and mannan‐binding lectin pathway activities in schizophrenia. Neurosci Lett. 2006; 404: 336–41. [DOI] [PubMed] [Google Scholar]
- 46. Hakobyan S, Boyajyan A, Sim RB. Classical pathway complement activity in schizophrenia. Neurosci Lett. 2005; 374: 35–7. [DOI] [PubMed] [Google Scholar]
- 47. Sekar A, Bialas AR, de Rivera H, et al Schizophrenia risk from complex variation of complement component 4. Nature. 2016; 530: 177–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mayilyan KR, Weinberger DR, Sim RB. The complement system in schizophrenia. Drug News Perspect. 2008; 21: 200–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Weiss LA, Shen Y, Korn JM, et al Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008; 358: 667–75. [DOI] [PubMed] [Google Scholar]
- 50. Chen AY, Kim SE, Houtrow AJ, et al Prevalence of obesity among children with chronic conditions. Obesity (Silver Spring). 2010; 18: 210–3. [DOI] [PubMed] [Google Scholar]
- 51. Li Y, Dai Q, Jackson JC, et al Overweight is associated with decreased cognitive functioning among school‐age children and adolescents. Obesity (Silver Spring). 2008; 16: 1809–15. [DOI] [PubMed] [Google Scholar]
- 52. Lees‐Miller JP, Heeley DH, Smillie LB. An abundant and novel protein of 22 kDa (SM22) is widely distributed in smooth muscles. Purification from bovine aorta. Biochem J. 1987; 244: 705–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Assinder SJ, Stanton JA, Prasad PD. Transgelin: an actin‐binding protein and tumour suppressor. Int J Biochem Cell Biol. 2009; 41: 482–6. [DOI] [PubMed] [Google Scholar]
- 54. Rho JH, Roehrl MH, Wang JY. Tissue proteomics reveals differential and compartment‐specific expression of the homologs transgelin and transgelin‐2 in lung adenocarcinoma and its stroma. J Proteome Res. 2009; 8: 5610–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xu SG, Yan PJ, Shao ZM. Differential proteomic analysis of a highly metastatic variant of human breast cancer cells using two‐dimensional differential gel electrophoresis. J Cancer Res Clin Oncol. 2010; 136: 1545–56. [DOI] [PubMed] [Google Scholar]
- 56. Cai J, Chen S, Zhang W, et al Paeonol reverses paclitaxel resistance in human breast cancer cells by regulating the expression of transgelin 2. Phytomedicine. 2014; 21: 984–91. [DOI] [PubMed] [Google Scholar]
- 57. Zhang Y, Ye Y, Shen D, et al Identification of transgelin‐2 as a biomarker of colorectal cancer by laser capture microdissection and quantitative proteome analysis. Cancer Sci. 2010; 101: 523–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liao H, Wu J, Kuhn E, et al Use of mass spectrometry to identify protein biomarkers of disease severity in the synovial fluid and serum of patients with rheumatoid arthritis. Arthritis Rheum. 2004; 50: 3792–803. [DOI] [PubMed] [Google Scholar]
- 59. Roller BL, Monibi F, Stoker AM, et al Characterization of meniscal pathology using molecular and proteomic analyses. J Knee Surg. 2015; 28: 496–505. [DOI] [PubMed] [Google Scholar]
- 60. Zhang N, Ahsan MH, Purchio AF, et al Serum amyloid A‐luciferase transgenic mice: response to sepsis, acute arthritis, and contact hypersensitivity and the effects of proteasome inhibition. J Immunol. 2005; 174: 8125–34. [DOI] [PubMed] [Google Scholar]
- 61. Shinzaki S, Matsuoka K, Iijima H, et al Leucine‐rich alpha‐2 glycoprotein is a serum biomarker of mucosal healing in ulcerative colitis. J Crohns Colitis. 2016; 11: 84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Serada S, Fujimoto M, Terabe F, et al Serum leucine‐rich alpha‐2 glycoprotein is a disease activity biomarker in ulcerative colitis. Inflamm Bowel Dis. 2012; 18: 2169–79. [DOI] [PubMed] [Google Scholar]
- 63. Honda H, Fujimoto M, Miyamoto S, et al Sputum leucine‐rich alpha‐2 glycoprotein as a marker of airway inflammation in asthma. PLoS One. 2016; 11: e162672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kentsis A, Ahmed S, Kurek K, et al Detection and diagnostic value of urine leucine‐rich alpha‐2‐glycoprotein in children with suspected acute appendicitis. Ann Emerg Med. 2012; 60: 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Serada S, Fujimoto M, Ogata A, et al iTRAQ‐based proteomic identification of leucine‐rich alpha‐2 glycoprotein as a novel inflammatory biomarker in autoimmune diseases. Ann Rheum Dis. 2010; 69: 770–4. [DOI] [PubMed] [Google Scholar]
- 66. Yan YB. Creatine kinase in cell cycle regulation and cancer. Amino Acids. 2016; 48: 1775–84. [DOI] [PubMed] [Google Scholar]
- 67. Morelli RL, Emilson B, Rapaport E. MM‐CK subtypes diagnose reperfusion early after myocardial infarction. Am J Med Sci. 1987; 293: 139–49. [DOI] [PubMed] [Google Scholar]
- 68. Lehmann P, Hartung W, Fleck M. Rhabdomyolysis and creatine kinase elevation. Z Rheumatol. 2013; 72: 236–41. [DOI] [PubMed] [Google Scholar]
- 69. Bhatnagar D, Carey P, Pollard A. Focal myositis and elevated creatine kinase levels in a patient with phaeochromocytoma. Postgrad Med J. 1986; 62: 197–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fournier T, Medjoubi‐N N, Porquet D. Alpha‐1‐acid glycoprotein. Biochim Biophys Acta. 2000; 1482: 157–71. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 List of total proteins identified in sera from CS and control samples.
Table S2 Network analysis results generated by IPA software.
Table S3 Ingenuity canonical pathways assigned by IPA software.
Table S4 The separated proteins in cluster 61 and cluster 62 by HCA.