

Abstract
Like all herpesvirus, the ability of Epstein-Barr virus (EBV) to establish life-long persistent infections is related to a biphasic viral lifecycle that involves latency and reactivation/lytic replication. Memory B cells serve as the EBV latency compartment where silencing of viral gene expression allows maintenance of the viral genome, avoidance of immune surveillance, and life-long carriage. Upon viral reactivation, viral gene expression is induced for replication, progeny virion production, and viral spread. EBV uses the host epigenetic machinery to regulate its distinct viral gene expression states. However, epigenetic manipulation by EBV affects the host epigenome by reprogramming cells in ways that leave long-lasting, oncogenic phenotypes. Such virally-induced epigenetic alterations are evident in EBV-associated cancers.
Graphical abstract
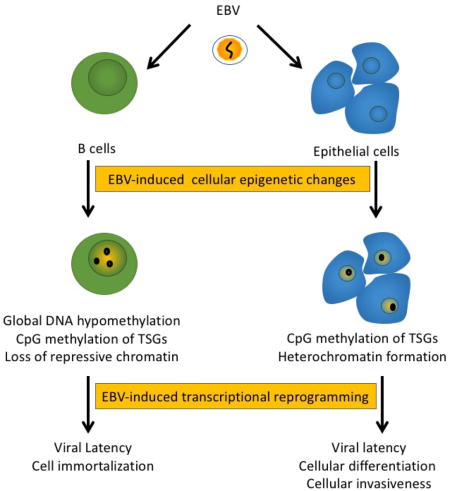
Consequences of EBV-induced epigenetic reprogramming. EBV infection of B cells and epithelial cells leads to cellular epigenetic changes that involve DNA methylation and histone modifications. Such epigenetic changes also affect the host epigenome altering cellular gene expression to states that are conducive for viral latency and replication, having effects on cell growth and differentiation. However, epigenetic changes to the host persist from one generation to the next, increasing the cellular heterogeneity of the population. In the context of cancer, virally-induced epigenetic changes may act as preneoplastic lesions that are retained in virally silent states or after loss of the viral genome as a mechanism for “hit-and-run” oncogenesis.
Introduction
Epstein-Barr virus (EBV) is a herpesvirus that infects greater than 90% of adults worldwide [1,2]. EBV is shed and transmitted in saliva infecting B cells and epithelial cells in the oral cavity [3–7]. Memory B cells provide the lifelong site for EBV latency and persistence. EBV can infect memory B cells directly or can navigate naïve B cells through their differentiation program to become memory B cells [8,9]. Viral latency is guided by promoter silencing and promoter switching, which results in an increasingly restricted viral gene expression program as B cells differentiate [10]. In naïve B cells, EBV latency III (the growth program) is observed with the expression of EBV nuclear antigens (EBNA) 1, EBNA2, EBNA3A, EBNA3B, EBNA3C, EBNA-LP, latent membrane protein (LMP) 1, LMP2A, LMP2B, and viral noncoding RNAs: EBV encoded RNAs (EBER) 1, EBER2 and the BamHI A rightward transcripts (BARTs). Germinal B cells show expression of EBNA1, LMP1, LMP2A, and noncoding RNAs, known as latency II (the default program). In memory B cells, latent genes are restricted to EBNA1, LMP2A, and noncoding RNAs, referred as latency I. Terminal differentiation of memory B cells into antibody producing plasma cells signals EBV reactivation into the productive phase of the viral lifecycle [11,12]. Epithelial cells support the productive phase of the viral lifecycle, with EBV replicating in the uppermost differentiated layers of the stratified epithelium [4,5,13–15]. Latent epithelial cell infections have been detected in basal tonsillar epithelial cells, but the nature of such latent infections is not well understood [16].
The distinct viral gene expression states adopted by EBV throughout the viral lifecycle are epigenetically regulated [17]. Epigenetic modifications stably propagate gene expression patterns in a heritable manner without affecting the DNA sequence. Epigenetic modifications involve DNA methylation where a methyl group is added to the C-5 position of cytosine residues, usually in the context of a CpG dinucleotide and post-translational modifications (acetylation, phosphorylation, ubiquitination, ADP-ribosylation) on lysine or arginine residues in the tail region of the core histones [18,19]. Such epigenetic marks alter the accessibility and recruitment of transcription factors and transcription machinery to chromatin. The regulatory proteins (chromatin readers, writers, and erasers) involved in epigenetic maintenance and reprogramming include DNA methyltransferases (DNMTs), methyl-CpG-binding proteins, histone modifying enzymes, and chromatin remodeling factors. Formation of higher order chromatin structures and gene looping also contribute to the regulation of epigenetic gene expression states. Such epigenetic states are reversible and can be altered by environmental factors (including viruses) in ways that have long-lasting detrimental effects on phenotype.
Epigenetic alterations acquired on the EBV genome
The EBV genome is encapsidated as a linear, double-stranded DNA that is free of nucleosomes and as well as methylated CpG residues [20,21]. Following nuclear entry, the viral genome circularizes by recombination of its terminal repeats present at each end of the linear genome [22,23]. An epigenetic switch occurs where the naked viral DNA assembles into nucleosomes and CpG methylation progressively increases over time. The circular viral genome is maintained as an extrachromosomal element having a similar chromatin structure as the host genome.
The acquired epigenetic modifications on the viral genome are essential for latency and regulation of the productive phase of the viral lifecycle [24]. Formation of heterochromatin and DNA methylation restrict viral gene expression to latency genes. Epigenetic modifications, including DNA methylation, are also required for the productive phase of the viral lifecycle. Despite expression of the EBV immediate early transcription factors BZLF1 and BRLF1 required for initiation of the lytic replication cascade at early times post infection of B cells, a block in late viral gene expression and progeny virus production was shown to occur up to a week following infection [25]. Transfection of unmethylated viral DNA genomes showed a similar block, yet transfection of methylated viral genomes fully activated the lytic cascade producing progeny virus within 2 days [25–27]. These results suggested that DNA methylation is a necessary epigenetic modification that protects viral genomes from abruptly entering the lytic cycle being required for reactivation and long term persistence of the virus.
The pattern of histone modifications and chromatin conformations are complex, with variations noted between latency and productive phases of the viral lifecycle. In general, active latency promoters associate with euchromatin marks, such as acetylated histone 3 lysine 9 (H3K9ac), H3K27ac, and H3K4ac, while heterochromatin marks such as tri-methylated H3K9 (H3K9me3) and H3K27me3, associate with transcriptionally silent promoters [17]. In latency, the BZLF1 and BRLF1 promoters are associated with the repressive H3K27me3 chromatin mark deposited by EZH2, the catalytic subunit of the polycomb repressive complex 2 (PRC2) [28].The EBV genome also folds into distinct loops that separate transcriptionally active and repressed regions. The cellular CCCCTC-binding factor (CTCF), which regulates chromatin organization of the host genome, binds the EBV genome at least 19 mapped sites organizing the EBV genome into loops that differ between latency III and latency I. CTCF association promotes long-range interactions between enhancers and promoters on the EBV genome [29]. Long-range interactions between the viral and host genomes likely occur, but have yet to be mapped. CTCF also acts as a boundary factor insulating euchromatin/transcriptionally-active regions from heterochromatin/transcriptionally-inactive regions. In sum, EBV gene expression is complex with layers of epigenetic control that involve chromatin composition and chromatin organization, and EBV is well adapted to use the host epigenetic machinery for completion of its lifecycle.
EBV manipulation of the host epigenetic machinery
EBV encodes a number of viral factors that interact with chromatin and chromatin remodelers. EBNA1 is an essential multi-functional viral protein that binds the latent origin of replication, OriP, is required for EBV episome replication, and tethers the viral episome to mitotic chromosomes for genome segregation and partitioning in dividing cells [30–33]. EBNA1 is also a viral transactivator with known effects on both viral and cellular promoters [34,35]. As a chromatin modifier, EBNA1 can change nucleosomal positioning and recruit chromatin remodeling factors [36]. EBNA1 participates in gene looping at OriP, and EBNA1 binding keeps OriP in an unmethylated state [30,37]. EBNA2 is a viral transactivator that does not directly bind DNA, but interacts with cellular DNA binding proteins, such as RBPJκ and EBF1, that target EBNA2 to viral and cellular promoters [38]. EBNA2 also interacts with transcriptional activators such as histone acetyltransferases and SWI/SNF chromatin remodeling factors. EBNA2 localizes to over 5000 sites on the cellular genome including super-enhancer regions, which are richly bound with transcription factors involved in the regulation of cell growth [39]. The EBNA3 proteins are transcriptional activators or repressors of viral and cellular genes [40]. The EBNA3 proteins also do not bind DNA directly, but interact with cellular DNA binding proteins and chromatin remodelers to regulate transcription [41]. The EBNA3 proteins interact with RBPJκ at distinct sites from EBNA2, and repress EBNA2/RBPJκ activation of gene expression [42,43]. As epigenetic modifiers, EBNA3A and EBNA3C were shown to recruit PRC2 to cellular promoters, such as proapoptotic BCL2L11 and p16ink4 tumor suppressor genes (TSGs), catalyzing H3K27 methylation and formation of heterochromatin [44,45].
LMP1 and LMP2A are integral membrane proteins that mimic CD40 and B cell receptor signaling, respectively. As epigenetic modifiers, LMP1 and LMP2A signaling can induce the expression and activity of DNMT1, 3A and 3B. LMP1 activation of DNMT1 involved the JNK-AP-1 pathway, while LMP1 activation of DNMT3B involved NF-κB signaling. LMP2A activated DNMT1 through the STAT3 signaling pathway [46–48]. In germinal center B cells, LMP1 expression induced a single DNMT (DNMT3A) and the H3K27 demethylase, KDM6B, [49,50]. In addition, reduced global levels of the repressive H3K27me3 mark were observed in LMP1-expressing B cells, where LMP1 activation of PARP1 was shown to inhibit EZH2, blocking the accumulation of H3K27me3 [51,52].
EBV encodes noncoding viral RNAs (EBER1, EBER2 and viral miRNAs) which regulate viral and host gene expression. Noncoding RNAs have been implicated as epigenetic regulators by recruiting transcriptional factors and chromatin regulators to target sites or by modulating the expression of chromatin remodelers [53]. EBER2 was shown to target the B cell specific transcription factor PAX5 to the viral genome through an RNA:RNA interaction, establishing the possibility that EBV noncoding RNAs may guide the recruitment of transcription factors to not only the viral genome, but also the cellular genome [54].
The lytic cycle transactivators BZLF1 and BRLF1 are influenced by the DNA methylation state of the viral genome. BZLF1 preferentially binds to methylated DNA and interacts with histone acetyltransferases to reverse the repressive chromatin marks at lytic promoters [55,56]. BRLF1 can bind methylated and unmethylated DNA, but DNA methylation reduces the ability of BRLF1 to acetylate H3K9 when bound to the promoters of lytic genes [27]. Overall, EBV encodes the capacity to manipulate the host epigenetic machinery through a number of viral effectors. The activities of these viral factors have the potential to affect the host epigenome as well.
Virally-induced epigenetic alterations to the host genome
EBV is a tumor virus associated with various lymphoid and epithelial malignancies. The oncogenic process is a result of not only genetic mutations, but also epigenetic changes involving DNA methylation and chromatin structure that in turn alter the expression of growth promoting or suppressing genes. Given such capacity to regulate its own transcriptional programs, and with latency as its default setting, EBV may induce epigenetic reprogramming of host chromatin with potential oncogenic consequences to infected cells.
EBV’s oncogenic activity is illustrated by its ability to immortalize B cells in culture into lymphoblastoid cells (LCLs) [57–59]. B cell immortalization requires the expression of several EBV latent proteins (EBNA1, EBNA2, EBNA3A, EBNA3C, LMP1, and LMP2), which interact with components of the host epigenetic machinery and have the potential to alter the host cell as well [60]. DNA methylation analysis of LCLs using whole genome bisulfite sequencing showed a global DNA hypomethylation affecting two-thirds of the B cell genome, which was not observed in activated or quiescent EBV-negative B cells [61]. At the gene level, DNA hypomethylation affected one-third of genes, including a subset of genes associated with the conversion of resting B cells to LCLs [61,62]. Localized CpG island DNA hypermethylation of TSG promoters was observed within 15 days of infection consistent with viral activation of DNMTs [63]. LCLs also showed global decreases in repressive histone modifications (H3K9me3, H3K27me3, and H3K20me3) and increased chromatin accessibility [64]. Such global changes in DNA methylation and chromatin structure indicate a virally-induced reprogramming of B cells that supports long term cell renewal through changes in differentiation, survival, and senescence required for the establishment of immortal growth.
Several EBV-associated lymphomas display distinct epigenetic changes not evident in EBV-negative counterparts. In Hodgkin’s lymphoma (HL), latent EBV (latency II) is present in ~50% of tumors. The malignant Reed-Sternberg (RS) cell of HL comprises less than 2% of the overall tumor in a background of non-neoplastic cells. The scarcity of the RS cells in HL and the mixed cellularity have limited the analysis of EBV-induced epigenetic effects in tumor tissue. Thus, in vitro infection of germinal center B cells, thought to be the cell of origin of the RS cell, and HL cell lines have been used as surrogates for EBV-positive RS cells. In HL cell lines and EBV-infected germinal center B cells, only DNMT3A levels increased while DNMT1 and DNMT3B levels were decreased, consistent with a possible DNA hypomethylation phenotype as seen in LCLs [50]. The EBV DNMT modulators, LMP1 and LMP2, were shown to reprogram germinal center B cells with characteristics of the malignant RS cells of HL [65,66].
A different EBV-induced epigenetic pattern is observed in Burkitt’s lymphoma (BL), a rapidly proliferating tumor characterized by a hallmark c-myc translocation and elevated c-myc expression. In BL, EBV displays the restricted, type I latency program. Methylome analysis of an EBV-positive BL cell line showed up to 60% of CpGs were methylated [67]. EBV-positive BL tumors show frequent DNA hypermethylation of the PRDM1/BLIMP1 gene, expressed during terminal B cell differentiation, when compared to EBV-negative BL [68]. In addition, EBV-positive BL carry fewer mutations than EBV-negative BL, suggesting that EBV-induced epigenetic changes could replace mutation [69].
EBV is also well associated with undifferentiated nasopharyngeal carcinoma (NPC) and a subset of gastric carcinomas (GC). In these tumors, EBV displays a latency type II infection with variable LMP1 expression. Methylome analysis has shown that EBV-positive GC and NPC are among the most highly DNA methylated tumors, with fewer mutations than other carcinomas [70–73]. EBV-positive NPC and GC display a CpG island DNA hypermethylator phenotype (CIMP) at promoters of TSGs not evident in the EBV-negative counterparts [74–76]. EBV-positive NPC and GC tumors show a unique profile of frequently DNA hypermethylated genes, sharing some common gene targets [77]. Chromosome 6p21.3 was DNA methylated in both EBV-positive NPC and EBV-positive GC, potentially serving as an epigenetic signature of EBV infection [71,77]. Chromatin modifications are also altered in NPC, with high levels of H3K27me3 and EZH2 observed [78,79]. An overlap between DNA hypermethylation and the bivalent marks H3K27me3 and H3K4me3 was also observed on chromosome 6, indicating epigenetic deregulation in NPC involves both DNA methylation and histone modifications [71].
EBV epigenetic reprogramming of epithelial cells is also evident in vitro. Expression of LMP1 has been shown to induce DNMTs resulting in DNA hypermethylation and silencing of the CDH1 and PTEN promoters, TSGs also shown to be DNA methylated in NPC and GC [46,47,80]. To define the epigenetic consequence of EBV epithelial infection, we used a transient infection model based on the observation that EBV is not stably maintained in cultured epithelial cells and requires selection pressure for episomal maintenance. As epigenetic changes are heritable, cellular phenotypes retained following loss of the viral genome would be epigenetic in nature. In a lung carcinoma cell line and telomerase-immortalized normal oral keratinocytes (NOK), EBV infection resulted in a delayed responsiveness to differentiation and increased invasiveness that was maintained upon loss of the virus [81,82]. Methylome analysis showed that EBV infection of NOK induced CIMP at 28 genes, with RARRES1 being a common methylated target also found in NPC tumors [81,83]. Epigenetic reprogramming of gene expression was also evident with 260 differentially expressed genes being common to EBV-infected NOK and clones that lost EBV. Ontological classification revealed gene associations with cancer, cell-to-cell signaling, cell movement, and development [81]. The members of the WNT signaling pathway, LEF1 and WNT5A, were among the top upregulated genes. Aberrant expression and activation of the Wnt signaling pathway has also been reported in NPC tumors [84]. Thus, these studies support that transit of EBV through a cell can leave an epigenetic imprint on gene expression that also permanently alters the biological properties of the cells, conferring features found in EBV-associated carcinomas that includes poor differentiation, metastasis, oncogenic gene expression alterations, and CIMP.
Conclusion
EBV carries out its lifecycle in the context of epigenetics, being well versed in using the host epigenetic machinery to establish latency and overcoming an epigenetically repressed state during viral reactivation. Virally-encoded epigenetic modulators can also affect the host epigenome resulting in cellular changes that likely benefit viral persistence. Such epigenetic changes typically occur without any appreciable effects to the host, yet can have detrimental effects in the context of cancer. Epigenetic silencing of TSGs through DNA methylation and repressive chromatin are frequently observed in EBV-positive cancers providing a selective advantage during tumor evolution. EBV-associated cancers display a high frequency of epigenetic changes and lower mutation frequencies, suggesting that epigenetic changes may substitute for mutations. Whether these epigenetic changes arise from stochastic events or are targeted to specific gene loci is still unclear. However, virally-induced epigenetic changes would be maintained in the absence of continued viral gene expression as is the case in latency or after loss of the viral genome. Such epigenetic changes would be lasting contributions to the oncogenic process and act as a mechanism for viral “hit-and run” oncogenesis.
Highlights.
EBV encodes viral epigenetic effectors that alter viral and host epigenomes.
EBV-induced epigenetic changes to the host epigenome have oncogenic consequences.
Epigenetic changes can maintain phenotypes in latency or after viral genome loss.
EBV epigenetic changes provide a framework for viral “hit-and-run” oncogenesis.
Acknowledgments
The author thanks Tawsha Munroe, Julia Myers, J. Tod Guidry and Kanchanjunga Prasai for proof reading the manuscript.
Funding: This work was supported by the National Institutes of Health grants R01DE025565 and 5P30GM11070.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest:
There are no conflicts of interests.
References
- 1.de-The G, Geser A, Day NE, Tukei PM, Williams EH, Beri DP, Smith PG, Dean AG, Bronkamm GW, Feorino P, et al. Epidemiological evidence for causal relationship between Epstein-Barr virus and Burkitt’s lymphoma from Ugandan prospective study. Nature. 1978;274:756–761. doi: 10.1038/274756a0. [DOI] [PubMed] [Google Scholar]
- 2.Henle G, Henle W, Clifford P, Diehl V, Kafuko GW, Kirya BG, Klein G, Morrow RH, Munube GM, Pike P, et al. Antibodies to Epstein-Barr virus in Burkitt’s lymphoma and control groups. J Natl Cancer Inst. 1969;43:1147–1157. [PubMed] [Google Scholar]
- 3.Pegtel DM, Middeldorp J, Thorley-Lawson DA. Epstein-Barr virus infection in ex vivo tonsil epithelial cell cultures of asymptomatic carriers. J Virol. 2004;78:12613–12624. doi: 10.1128/JVI.78.22.12613-12624.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sixbey JW, Nedrud JG, Raab-Traub N, Hanes RA, Pagano JS. Epstein-Barr virus replication in oropharyngeal epithelial cells. N Engl J Med. 1984;310:1225–1230. doi: 10.1056/NEJM198405103101905. [DOI] [PubMed] [Google Scholar]
- 5.Walling DM, Flaitz CM, Nichols CM, Hudnall SD, Adler-Storthz K. Persistent productive Epstein-Barr virus replication in normal epithelial cells in vivo. J Infect Dis. 2001;184:1499–1507. doi: 10.1086/323992. [DOI] [PubMed] [Google Scholar]
- 6.Ling PD, Lednicky JA, Keitel WA, Poston DG, White ZS, Peng R, Liu Z, Mehta SK, Pierson DL, Rooney CM, et al. The dynamics of herpesvirus and polyomavirus reactivation and shedding in healthy adults: a 14-month longitudinal study. J Infect Dis. 2003;187:1571–1580. doi: 10.1086/374739. [DOI] [PubMed] [Google Scholar]
- 7.Yao QY, Rickinson AB, Epstein MA. A re-examination of the Epstein-Barr virus carrier state in healthy seropositive individuals. Int J Cancer. 1985;35:35–42. doi: 10.1002/ijc.2910350107. [DOI] [PubMed] [Google Scholar]
- 8.Kurth J, Hansmann ML, Rajewsky K, Kuppers R. Epstein-Barr virus-infected B cells expanding in germinal centers of infectious mononucleosis patients do not participate in the germinal center reaction. Proc Natl Acad Sci U S A. 2003;100:4730–4735. doi: 10.1073/pnas.2627966100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thorley-Lawson DA, Gross A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N Engl J Med. 2004;350:1328–1337. doi: 10.1056/NEJMra032015. [DOI] [PubMed] [Google Scholar]
- 10.Young LS, Murray PG. Epstein-Barr virus and oncogenesis: from latent genes to tumours. Oncogene. 2003;22:5108–5121. doi: 10.1038/sj.onc.1206556. [DOI] [PubMed] [Google Scholar]
- 11.Crawford DH, Ando I. EB virus induction is associated with B-cell maturation. Immunology. 1986;59:405–409. [PMC free article] [PubMed] [Google Scholar]
- 12.Laichalk LL, Thorley-Lawson DA. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J Virol. 2005;79:1296–1307. doi: 10.1128/JVI.79.2.1296-1307.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greenspan JS, Greenspan D, Lennette ET, Abrams DI, Conant MA, Petersen V, Freese UK. Replication of Epstein-Barr virus within the epithelial cells of oral “hairy” leukoplakia, an AIDS-associated lesion. N Engl J Med. 1985;313:1564–1571. doi: 10.1056/NEJM198512193132502. [DOI] [PubMed] [Google Scholar]
- 14.Tugizov SM, Berline JW, Palefsky JM. Epstein-Barr virus infection of polarized tongue and nasopharyngeal epithelial cells. Nat Med. 2003;9:307–314. doi: 10.1038/nm830. [DOI] [PubMed] [Google Scholar]
- 15.Temple RM, Zhu J, Budgeon L, Christensen ND, Meyers C, Sample CE. Efficient replication of Epstein-Barr virus in stratified epithelium in vitro. Proc Natl Acad Sci U S A. 2014;111:16544–16549. doi: 10.1073/pnas.1400818111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **16.Nawandar DM, Wang A, Makielski K, Lee D, Ma S, Barlow E, Reusch J, Jiang R, Wille CK, Greenspan D, et al. Differentiation-Dependent KLF4 Expression Promotes Lytic Epstein-Barr Virus Infection in Epithelial Cells. PLoS Pathog. 2015;11:e1005195. doi: 10.1371/journal.ppat.1005195. Using laser capture microdissection, the authors show that the immediate early BZLF1 and BRLF1 transcriptis were not detected in undeifferentiated basal tonsil epithelial cells whereas BZLF1 and BRLF1 were deted in differentiated tonsil epithelial cells. These data suggest that EBV may establish latent infection in basal epithelial cell, restricting replication to more differentiated epithelial cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tempera I, Lieberman PM. Epigenetic regulation of EBV persistence and oncogenesis. Semin Cancer Biol. 2014;26:22–29. doi: 10.1016/j.semcancer.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17:487–500. doi: 10.1038/nrg.2016.59. [DOI] [PubMed] [Google Scholar]
- 19.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 20.Shaw JE, Levinger LF, Carter CW., Jr Nucleosomal structure of Epstein-Barr virus DNA in transformed cell lines. J Virol. 1979;29:657–665. doi: 10.1128/jvi.29.2.657-665.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szyf M, Eliasson L, Mann V, Klein G, Razin A. Cellular and viral DNA hypomethylation associated with induction of Epstein-Barr virus lytic cycle. Proc Natl Acad Sci U S A. 1985;82:8090–8094. doi: 10.1073/pnas.82.23.8090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lindahl T, Adams A, Bjursell G, Bornkamm GW, Kaschka-Dierich C, Jehn U. Covalently closed circular duplex DNA of Epstein-Barr virus in a human lymphoid cell line. J Mol Biol. 1976;102:511–530. doi: 10.1016/0022-2836(76)90331-4. [DOI] [PubMed] [Google Scholar]
- 23.Raab-Traub N, Flynn K. The structure of the termini of the Epstein-Barr virus as a marker of clonal cellular proliferation. Cell. 1986;47:883–889. doi: 10.1016/0092-8674(86)90803-2. [DOI] [PubMed] [Google Scholar]
- *24.Hammerschmidt W. The Epigenetic Life Cycle of Epstein-Barr Virus. Curr Top Microbiol Immunol. 2015;390:103–117. doi: 10.1007/978-3-319-22822-8_6. An excellent review on the epigenetic switches that occur to EBV throught its lifecycle. [DOI] [PubMed] [Google Scholar]
- 25.Kalla M, Schmeinck A, Bergbauer M, Pich D, Hammerschmidt W. AP-1 homolog BZLF1 of Epstein-Barr virus has two essential functions dependent on the epigenetic state of the viral genome. Proc Natl Acad Sci U S A. 2010;107:850–855. doi: 10.1073/pnas.0911948107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalla M, Gobel C, Hammerschmidt W. The lytic phase of epstein-barr virus requires a viral genome with 5-methylcytosine residues in CpG sites. J Virol. 2012;86:447–458. doi: 10.1128/JVI.06314-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wille CK, Nawandar DM, Panfil AR, Ko MM, Hagemeier SR, Kenney SC. Viral genome methylation differentially affects the ability of BZLF1 versus BRLF1 to activate Epstein-Barr virus lytic gene expression and viral replication. J Virol. 2013;87:935–950. doi: 10.1128/JVI.01790-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Comet I, Riising EM, Leblanc B, Helin K. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat Rev Cancer. 2016;16:803–810. doi: 10.1038/nrc.2016.83. [DOI] [PubMed] [Google Scholar]
- 29.Tempera I, Klichinsky M, Lieberman PM. EBV latency types adopt alternative chromatin conformations. PLoS Pathog. 2011;7:e1002180. doi: 10.1371/journal.ppat.1002180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frappier L, O’Donnell M. Overproduction, purification, and characterization of EBNA1, the origin binding protein of Epstein-Barr virus. J Biol Chem. 1991;266:7819–7826. [PubMed] [Google Scholar]
- 31.Grogan EA, Summers WP, Dowling S, Shedd D, Gradoville L, Miller G. Two Epstein-Barr viral nuclear neoantigens distinguished by gene transfer, serology, and chromosome binding. Proc Natl Acad Sci U S A. 1983;80:7650–7653. doi: 10.1073/pnas.80.24.7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rawlins DR, Milman G, Hayward SD, Hayward GS. Sequence-specific DNA binding of the Epstein-Barr virus nuclear antigen (EBNA-1) to clustered sites in the plasmid maintenance region. Cell. 1985;42:859–868. doi: 10.1016/0092-8674(85)90282-x. [DOI] [PubMed] [Google Scholar]
- 33.Reisman D, Yates J, Sugden B. A putative origin of replication of plasmids derived from Epstein-Barr virus is composed of two cis-acting components. Mol Cell Biol. 1985;5:1822–1832. doi: 10.1128/mcb.5.8.1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Canaan A, Haviv I, Urban AE, Schulz VP, Hartman S, Zhang Z, Palejev D, Deisseroth AB, Lacy J, Snyder M, et al. EBNA1 regulates cellular gene expression by binding cellular promoters. Proc Natl Acad Sci U S A. 2009;106:22421–22426. doi: 10.1073/pnas.0911676106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dresang LR, Vereide DT, Sugden B. Identifying sites bound by Epstein-Barr virus nuclear antigen 1 (EBNA1) in the human genome: defining a position-weighted matrix to predict sites bound by EBNA1 in viral genomes. J Virol. 2009;83:2930–2940. doi: 10.1128/JVI.01974-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang S, Frappier L. Nucleosome assembly proteins bind to Epstein-Barr virus nuclear antigen 1 and affect its functions in DNA replication and transcriptional activation. J Virol. 2009;83:11704–11714. doi: 10.1128/JVI.00931-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hsieh CL. Evidence that protein binding specifies sites of DNA demethylation. Mol Cell Biol. 1999;19:46–56. doi: 10.1128/mcb.19.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *38.Lu F, Chen HS, Kossenkov AV, DeWispeleare K, Won KJ, Lieberman PM. EBNA2 Drives Formation of New Chromosome Binding Sites and Target Genes for B-Cell Master Regulatory Transcription Factors RBP-jkappa and EBF1. PLoS Pathog. 2016;12:e1005339. doi: 10.1371/journal.ppat.1005339. The authors provide evidence that EBNA2 redirects its binding partners to new chromosomal sites to drive B cell growth. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou H, Schmidt SC, Jiang S, Willox B, Bernhardt K, Liang J, Johannsen EC, Kharchenko P, Gewurz BE, Kieff E, et al. Epstein-Barr virus oncoprotein super-enhancers control B cell growth. Cell Host Microbe. 2015;17:205–216. doi: 10.1016/j.chom.2014.12.013. The authors provide an explanation for the paradox for how EBNA2 affects the transcription of B cell growth factors in the absence of binding to gene proximal sites. The authors show that EBNA2 binds to super enhancers where long-range interactions influence the activity of a subset of genes involved in B cell growth. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Allday MJ, Bazot Q, White RE. The EBNA3 Family: Two Oncoproteins and a Tumour Suppressor that Are Central to the Biology of EBV in B Cells. Curr Top Microbiol Immunol. 2015;391:61–117. doi: 10.1007/978-3-319-22834-1_3. [DOI] [PubMed] [Google Scholar]
- 41.Paschos K, Bazot Q, Ho G, Parker GA, Lees J, Barton G, Allday MJ. Core binding factor (CBF) is required for Epstein-Barr virus EBNA3 proteins to regulate target gene expression. Nucleic Acids Res. 2016 doi: 10.1093/nar/gkw1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robertson ES, Lin J, Kieff E. The amino-terminal domains of Epstein-Barr virus nuclear proteins 3A, 3B, and 3C interact with RBPJ(kappa) J Virol. 1996;70:3068–3074. doi: 10.1128/jvi.70.5.3068-3074.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Waltzer L, Perricaudet M, Sergeant A, Manet E. Epstein-Barr virus EBNA3A and EBNA3C proteins both repress RBP-J kappa-EBNA2-activated transcription by inhibiting the binding of RBP-J kappa to DNA. J Virol. 1996;70:5909–5915. doi: 10.1128/jvi.70.9.5909-5915.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paschos K, Smith P, Anderton E, Middeldorp JM, White RE, Allday MJ. Epstein-barr virus latency in B cells leads to epigenetic repression and CpG methylation of the tumour suppressor gene Bim. PLoS Pathog. 2009;5:e1000492. doi: 10.1371/journal.ppat.1000492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Skalska L, White RE, Franz M, Ruhmann M, Allday MJ. Epigenetic repression of p16(INK4A) by latent Epstein-Barr virus requires the interaction of EBNA3A and EBNA3C with CtBP. PLoS Pathog. 2010;6:e1000951. doi: 10.1371/journal.ppat.1000951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hino R, Uozaki H, Murakami N, Ushiku T, Shinozaki A, Ishikawa S, Morikawa T, Nakaya T, Sakatani T, Takada K, et al. Activation of DNA methyltransferase 1 by EBV latent membrane protein 2A leads to promoter hypermethylation of PTEN gene in gastric carcinoma. Cancer Res. 2009;69:2766–2774. doi: 10.1158/0008-5472.CAN-08-3070. [DOI] [PubMed] [Google Scholar]
- 47.Peng H, Chen Y, Gong P, Cai L, Lyu X, Jiang Q, Wang J, Lu J, Yao K, Liu K, et al. Higher methylation intensity induced by EBV LMP1 via NF-kappaB/DNMT3b signaling contributes to silencing of PTEN gene. Oncotarget. 2016;7:40025–40037. doi: 10.18632/oncotarget.9474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tsai CL, Li HP, Lu YJ, Hsueh C, Liang Y, Chen CL, Tsao SW, Tse KP, Yu JS, Chang YS. Activation of DNA methyltransferase 1 by EBV LMP1 Involves c-Jun NH(2)-terminal kinase signaling. Cancer Res. 2006;66:11668–11676. doi: 10.1158/0008-5472.CAN-06-2194. [DOI] [PubMed] [Google Scholar]
- 49.Anderton JA, Bose S, Vockerodt M, Vrzalikova K, Wei W, Kuo M, Helin K, Christensen J, Rowe M, Murray PG, et al. The H3K27me3 demethylase, KDM6B, is induced by Epstein-Barr virus and over-expressed in Hodgkin’s Lymphoma. Oncogene. 2011;30:2037–2043. doi: 10.1038/onc.2010.579. [DOI] [PubMed] [Google Scholar]
- 50.Leonard S, Wei W, Anderton J, Vockerodt M, Rowe M, Murray PG, Woodman CB. Epigenetic and transcriptional changes which follow Epstein-Barr virus infection of germinal center B cells and their relevance to the pathogenesis of Hodgkin’s lymphoma. J Virol. 2011;85:9568–9577. doi: 10.1128/JVI.00468-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **51.Martin KA, Cesaroni M, Denny MF, Lupey LN, Tempera I. Global Transcriptome Analysis Reveals That Poly(ADP-Ribose) Polymerase 1 Regulates Gene Expression through EZH2. Mol Cell Biol. 2015;35:3934–3944. doi: 10.1128/MCB.00635-15. The authors are the first to demonstrate that PARP1 is an epigenetic regulator, inhibiting the activity of the catalytic subunit of the polycomb repressive complex 2 (PRC2), EZH2. Knockdown of PARP1 increased EZH2 and its associated chromatin mark, H3K27me3, at EZH2 target genes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *52.Martin KA, Lupey LN, Tempera I. Epstein-Barr Virus Oncoprotein LMP1 Mediates Epigenetic Changes in Host Gene Expression through PARP1. J Virol. 2016;90:8520–8530. doi: 10.1128/JVI.01180-16. The authors show that LMP1 activation of gene expression and its transformation potential is mediated by PARP1 repression of EZH2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee N, Steitz JA. Noncoding RNA-guided recruitment of transcription factors: A prevalent but undocumented mechanism? Bioessays. 2015;37:936–941. doi: 10.1002/bies.201500060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **54.Lee N, Moss WN, Yario TA, Steitz JA. EBV noncoding RNA binds nascent RNA to drive host PAX5 to viral DNA. Cell. 2015;160:607–618. doi: 10.1016/j.cell.2015.01.015. The authors are the first to show that the EBV noncoding RNA, EBER2, can recuit a host transcription factor to the viral genome being guided by interactions with nascent viral transcripts. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bhende PM, Seaman WT, Delecluse HJ, Kenney SC. The EBV lytic switch protein, Z, preferentially binds to and activates the methylated viral genome. Nat Genet. 2004;36:1099–1104. doi: 10.1038/ng1424. [DOI] [PubMed] [Google Scholar]
- 56.Dickerson SJ, Xing Y, Robinson AR, Seaman WT, Gruffat H, Kenney SC. Methylation-dependent binding of the epstein-barr virus BZLF1 protein to viral promoters. PLoS Pathog. 2009;5:e1000356. doi: 10.1371/journal.ppat.1000356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gerber P, Whang-Peng J, Monroe JH. Transformation and chromosome changes induced by Epstein-Barr virus in normal human leukocyte cultures. Proc Natl Acad Sci U S A. 1969;63:740–747. doi: 10.1073/pnas.63.3.740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Henle W, Diehl V, Kohn G, Zur Hausen H, Henle G. Herpes-type virus and chromosome marker in normal leukocytes after growth with irradiated Burkitt cells. Science. 1967;157:1064–1065. doi: 10.1126/science.157.3792.1064. [DOI] [PubMed] [Google Scholar]
- 59.Miller G, Lisco H, Kohn HI, Stitt D, Enders JF. Establishment of cell lines from normal adult human blood leukocytes by exposure to Epstein-Barr virus and neutralization by human sera with Epstein-Barr virus antibody. Proc Soc Exp Biol Med. 1971;137:1459–1465. doi: 10.3181/00379727-137-35810. [DOI] [PubMed] [Google Scholar]
- 60.Kang MS, Kieff E. Epstein-Barr virus latent genes. Exp Mol Med. 2015;47:e131. doi: 10.1038/emm.2014.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hansen KD, Sabunciyan S, Langmead B, Nagy N, Curley R, Klein G, Klein E, Salamon D, Feinberg AP. Large-scale hypomethylated blocks associated with Epstein-Barr virus-induced B-cell immortalization. Genome Res. 2014;24:177–184. doi: 10.1101/gr.157743.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hernando H, Shannon-Lowe C, Islam AB, Al-Shahrour F, Rodriguez-Ubreva J, Rodriguez-Cortez VC, Javierre BM, Mangas C, Fernandez AF, Parra M, et al. The B cell transcription program mediates hypomethylation and overexpression of key genes in Epstein-Barr virus-associated proliferative conversion. Genome Biol. 2013;14:R3. doi: 10.1186/gb-2013-14-1-r3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **63.Saha A, Jha HC, Upadhyay SK, Robertson ES. Epigenetic silencing of tumor suppressor genes during in vitro Epstein-Barr virus infection. Proc Natl Acad Sci U S A. 2015;112:E5199–5207. doi: 10.1073/pnas.1503806112. The authors demonstrate that DNA hypermethylation at a number of tumor suppressor genes occurs within 15 days of B cell infection. The epigenetic alteration was associated with increased DNMT3B levles during the course of infection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hernando H, Islam AB, Rodriguez-Ubreva J, Forne I, Ciudad L, Imhof A, Shannon-Lowe C, Ballestar E. Epstein-Barr virus-mediated transformation of B cells induces global chromatin changes independent to the acquisition of proliferation. Nucleic Acids Res. 2014;42:249–263. doi: 10.1093/nar/gkt886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Portis T, Dyck P, Longnecker R. Epstein-Barr Virus (EBV) LMP2A induces alterations in gene transcription similar to those observed in Reed-Sternberg cells of Hodgkin lymphoma. Blood. 2003;102:4166–4178. doi: 10.1182/blood-2003-04-1018. [DOI] [PubMed] [Google Scholar]
- 66.Vockerodt M, Morgan SL, Kuo M, Wei W, Chukwuma MB, Arrand JR, Kube D, Gordon J, Young LS, Woodman CB, et al. The Epstein-Barr virus oncoprotein, latent membrane protein-1, reprograms germinal centre B cells towards a Hodgkin’s Reed-Sternberg-like phenotype. J Pathol. 2008;216:83–92. doi: 10.1002/path.2384. [DOI] [PubMed] [Google Scholar]
- 67.Kreck B, Richter J, Ammerpohl O, Barann M, Esser D, Petersen BS, Vater I, Murga Penas EM, Bormann Chung CA, Seisenberger S, et al. Base-pair resolution DNA methylome of the EBV-positive Endemic Burkitt lymphoma cell line DAUDI determined by SOLiD bisulfite-sequencing. Leukemia. 2013;27:1751–1753. doi: 10.1038/leu.2013.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang T, Ma J, Nie K, Yan J, Liu Y, Bacchi CE, Queiroga EM, Gualco G, Sample JT, Orazi A, et al. Hypermethylation of the tumor suppressor gene PRDM1/Blimp-1 supports a pathogenetic role in EBV-positive Burkitt lymphoma. Blood Cancer J. 2014;4:e261. doi: 10.1038/bcj.2014.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Giulino-Roth L, Wang K, MacDonald TY, Mathew S, Tam Y, Cronin MT, Palmer G, Lucena-Silva N, Pedrosa F, Pedrosa M, et al. Targeted genomic sequencing of pediatric Burkitt lymphoma identifies recurrent alterations in antiapoptotic and chromatin-remodeling genes. Blood. 2012;120:5181–5184. doi: 10.1182/blood-2012-06-437624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–209. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *71.Dai W, Cheung AK, Ko JM, Cheng Y, Zheng H, Ngan RK, Ng WT, Lee AW, Yau CC, Lee VH, et al. Comparative methylome analysis in solid tumors reveals aberrant methylation at chromosome 6p in nasopharyngeal carcinoma. Cancer Med. 2015;4:1079–1090. doi: 10.1002/cam4.451. The author analyzed the DNA methylation profile of nasopharyngeal carcinoma compared to solid tumors. Nasopharyngeal carcinoma is a top DNA hypermethylated tumors displaying a CpG island DNA methylator phentoype. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li L, Zhang Y, Fan Y, Sun K, Su X, Du Z, Tsao SW, Loh TK, Sun H, Chan AT, et al. Characterization of the nasopharyngeal carcinoma methylome identifies aberrant disruption of key signaling pathways and methylated tumor suppressor genes. Epigenomics. 2015;7:155–173. doi: 10.2217/epi.14.79. [DOI] [PubMed] [Google Scholar]
- 73.Dai W, Zheng H, Cheung AK, Lung ML. Genetic and epigenetic landscape of nasopharyngeal carcinoma. Chin Clin Oncol. 2016;5:16. doi: 10.21037/cco.2016.03.06. [DOI] [PubMed] [Google Scholar]
- 74.Kang GH, Lee S, Kim WH, Lee HW, Kim JC, Rhyu MG, Ro JY. Epstein-barr virus-positive gastric carcinoma demonstrates frequent aberrant methylation of multiple genes and constitutes CpG island methylator phenotype-positive gastric carcinoma. Am J Pathol. 2002;160:787–794. doi: 10.1016/S0002-9440(10)64901-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Matsusaka K, Kaneda A, Nagae G, Ushiku T, Kikuchi Y, Hino R, Uozaki H, Seto Y, Takada K, Aburatani H, et al. Classification of Epstein-Barr virus-positive gastric cancers by definition of DNA methylation epigenotypes. Cancer Res. 2011;71:7187–7197. doi: 10.1158/0008-5472.CAN-11-1349. [DOI] [PubMed] [Google Scholar]
- 76.Zhou L, Jiang W, Ren C, Yin Z, Feng X, Liu W, Tao Q, Yao K. Frequent hypermethylation of RASSF1A and TSLC1, and high viral load of Epstein-Barr Virus DNA in nasopharyngeal carcinoma and matched tumor-adjacent tissues. Neoplasia. 2005;7:809–815. doi: 10.1593/neo.05217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *77.Niller HH, Banati F, Salamon D, Minarovits J. Epigenetic Alterations in Epstein-Barr Virus-Associated Diseases. Adv Exp Med Biol. 2016;879:39–69. doi: 10.1007/978-3-319-24738-0_3. A recent comprehensive review detailing the epigenetic alterations associated with EBV infection and disease. [DOI] [PubMed] [Google Scholar]
- 78.Cai MY, Tong ZT, Zhu W, Wen ZZ, Rao HL, Kong LL, Guan XY, Kung HF, Zeng YX, Xie D. H3K27me3 protein is a promising predictive biomarker of patients’ survival and chemoradioresistance in human nasopharyngeal carcinoma. Mol Med. 2011;17:1137–1145. doi: 10.2119/molmed.2011.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hwang CF, Huang HY, Chen CH, Chien CY, Hsu YC, Li CF, Fang FM. Enhancer of zeste homolog 2 overexpression in nasopharyngeal carcinoma: an independent poor prognosticator that enhances cell growth. Int J Radiat Oncol Biol Phys. 2012;82:597–604. doi: 10.1016/j.ijrobp.2010.11.062. [DOI] [PubMed] [Google Scholar]
- 80.Tsai CN, Tsai CL, Tse KP, Chang HY, Chang YS. The Epstein-Barr virus oncogene product, latent membrane protein 1, induces the downregulation of E-cadherin gene expression via activation of DNA methyltransferases. Proc Natl Acad Sci U S A. 2002;99:10084–10089. doi: 10.1073/pnas.152059399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Birdwell CE, Queen KJ, Kilgore PC, Rollyson P, Trutschl M, Cvek U, Scott RS. Genome-wide DNA methylation as an epigenetic consequence of Epstein-Barr virus infection of immortalized keratinocytes. J Virol. 2014;88:11442–11458. doi: 10.1128/JVI.00972-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Queen KJ, Shi M, Zhang F, Cvek U, Scott RS. Epstein-Barr virus-induced epigenetic alterations following transient infection. Int J Cancer. 2013;132:2076–2086. doi: 10.1002/ijc.27893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yanatatsaneejit P, Chalermchai T, Kerekhanjanarong V, Shotelersuk K, Supiyaphun P, Mutirangura A, Sriuranpong V. Promoter hypermethylation of CCNA1, RARRES1, and HRASLS3 in nasopharyngeal carcinoma. Oral Oncol. 2008;44:400–406. doi: 10.1016/j.oraloncology.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 84.Zeng ZY, Zhou YH, Zhang WL, Xiong W, Fan SQ, Li XL, Luo XM, Wu MH, Yang YX, Huang C, et al. Gene expression profiling of nasopharyngeal carcinoma reveals the abnormally regulated Wnt signaling pathway. Hum Pathol. 2007;38:120–133. doi: 10.1016/j.humpath.2006.06.023. [DOI] [PubMed] [Google Scholar]