

Abstract
Background
Oxidative stress plays a crucial role in the development of alcoholic liver disease (ALD), however effective pharmacological treatments for oxidative injury is still lacking. The objective of this study was to determine whether inhibition of NADPH oxidase activity could reverse alcohol-induced liver injury via protecting mitochondrial functions.
Methods
C57BL/6J mice were pair-fed with Lieber-DeCarli control or ethanol diet for four week with or without administration with 30mg/kg/d GKT137831, a NOX4 inhibitor for the last two weeks. H4IIEC3 cells were transfected with scrambled or NOX4 shRNA. Cells were then treated with 200mM ethanol for 48h.
Results
Alcohol exposure induced NOX4 expression in the liver and mitochondrial fraction. GKT137831 partially reversed alcohol-induced liver injury and elevation of serum H2O2. The levels of mitochondrial ROS, mitochondrial DNA, respiratory chain complex IV, and hepatic ATP were partially reversed by GKT137831 after alcohol exposure. Furthermore GKT137831 ameliorated alcohol-induced lipid accumulation and increased HNF-4α and β-oxidation enzymes. GKT137831 also decreased alcohol-induced apoptosis coupled with decreased insertion of Bax into mitochondria and decreased activation of cleaved caspase-9 and cleaved PARP. Mechanistic study shows that ethanol induced expression of NOX4 in H4IIEC3 cells. Knockdown of NOX4 caused an increased mitochondrial membrane potential, decreased mitochondrial superoxide levels, reduced number of apoptotic cells, decreased lipid accumulation, and improved ATP levels and NAD+/NADH ratio after ethanol treatment.
Conclusion
Pharmacological inhibition of NOX4 activity protects against alcohol-induced fat accumulation and activation of intrinsic apoptosis via improving mitochondrial function. General significance: Pharmacological inhibition of NOX4 could be a promising treatment for ALD.
Keywords: Alcoholic liver disease, mitochondria, NADPH oxidase, NOX4, GKT137831, ROS
Graphical abstract
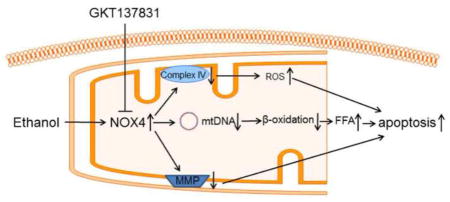
1. Introduction
Excessive alcohol consumption results in alcoholic liver disease (ALD). ALD evolves from alcoholic fatty liver to alcoholic steatohepatitis and the last stage is alcoholic cirrhosis, which is irreversible1. The generation of toxic metabolites and byproducts by ethanol oxidation, for instance acetaldehyde and lipid peroxidation products contribute to the pathogenesis of alcoholic liver injury. It has been known that prolonged alcohol consumption induces hepatic cytochrome P450 2E1 (CYP2E1), which not only accelerates oxidation of ethanol to acetaldehyde but also generates of reactive oxygen species (ROS)2. Previous study reported that knockdown of CYP2E1 protected mice from chronic alcohol consumption-induced liver injury via reduction of ROS3, 4. Therefore, removal of hepatic ROS could be an effective strategy in preventing the development of ALD.
Mitochondrion is a vital organelle that maintains biological functions of a cell by providing energy. Defective mitochondria result in shortage in ATP, leakage of ROS, accumulation of fat, and activation of intrinsic apoptotic cell death pathway5–7. It has been shown that approximately 1–2% of the oxygen consumed in the mitochondria is converted to ROS7. Within the mitochondrial matrix, SOD2 catalyzes the conversion of superoxide to hydrogen peroxide that can be readily oxidized by glutathione peroxidase (Gpx)8. Ethanol and oxidative stress exacerbate ROS formation in mitochondria6. The uncontrolled level of ROS in the mitochondria could damage the mitochondrial membrane and lead to increased diffusion of hydrogen peroxide to cytoplasm. Dysfunctional mitochondria also contributes to defective lipid metabolism manifested by decreased carnitine palmitoyltransferase I (CPT1) activity and β-oxidation8. In addition, dysfunctional mitochondria after alcohol exposure release cytochrome C to cytosol, leading to activation of apoptosis9. Thus, it is of interest to investigate whether decreases of ROS levels could rescue the function of mitochondria after alcohol exposure.
In addition to CYP2E1 and mitochondrial electron transport chain (ETC), NADPH oxidase (NOX) is another well-known source of ROS generation10, 11. NOX generates superoxide from oxygen using NAD(P)H as an electron donor. It is known that NOX family consists of seven members, NOX1-5 and DUOX1-211. NOX1, NOX2, and NOX4 have been found in the liver12. Different from NOX2 that is expressed in phagocytes such as Kupffer cell13, NOX1 and NOX4 are expresses in nonphagocytes12. In comparison with NOX1, NOX4 is able to generate ROS constitutively due to not requiring recruitment of structural subunits to form active enzyme14. It has been shown that NOX1 promotes hepatic stellate cell proliferation and aggravates fibrosis15–17. In addition, NOX4 have been found to play a critical role in lung, kidney and liver fibrosis18–20. Recently, a NOX4/NOX1 dual inhibitor (GKT137831) has been shown a promising effect in treating pulmonary fibrosis and bile duct ligation-induced hepatic fibrosis21.
Previous studies have shown that NOX4 predominantly exists in the mitochondria in cardiac myocytes and kidney cortex19, 22 Our previous reports demonstrated that chronic alcohol exposure impairs hepatic mitochondrial function23, 24; however, it is unclear if NOX4 locates in hepatic mitochondria and if NOX4 contributes to alcohol-induced oxidative stress. Therefore, in the present study, we first determined the subcellular distribution of NOX4 in the liver and effect of alcohol exposure on NOX4 expression, and then tested the protective effects of NOX4 inhibitor, GKT137831, on alcohol-induced ROS generation and mitochondrial dysfunction. We also performed cell culture study to define the link between NOX4 expression and mitochondrial ROS levels and function.
2. Materials and Methods
2.1. Animal and alcohol feeding experiments
Male Wistar rats were obtained from Harlan (Indianapolis, IN). The animal protocol was approved by the Institutional Animal Care and Use Committee of the North Carolina Research Campus. Eight-week-old male rats were pair-fed a modified Lieber-DeCarli alcohol (Dyets, Bethlehem) or isocaloric maltose dextrin control liquid diet (Dyets) for 5 months (n= 6 for each group) with a stepwised feeding procedure, as described previously24. Briefly, on the day of feeding, the ethanol content (w/v%) in the diet was 5.0 (36% of total calories) and gradually increased to 6.3 (44% of total calories). At the end of 5-month feeding, rats were anesthetized with inhalational isoflurane.
Male C57BL/6J mice were obtained from the Jackson Laboratory (Bar Harbor, ME). All mice were treated according to the experimental procedures approved by Institutional Animal Care and Use Committee. The mice were pair-fed a stepwise and modified Lieber-DeCarli ethanol or control liquid diet for four weeks (n=6) with or without 30mg /kg/d GKT137831 (Selleck Chemicals, Houston, TX) supplements in food for the last two weeks. On the day of feeding, ethanol content (%, w/v) in the diet was 3.2 (22.4% of total calories) and gradually increased to 4.2 (29.4% of total calories). The mice were gavage with ethanol (3g /kg) or isocaloric dextrin (5.2g/kg) once a week. At the end of 4-week of feeding, mice were anesthetized with inhalational isoflurane.
Male C57BL/6J mice were treated with recombinant TNF-alpha (R&D systems, Minneapolis, MN) via intravenous injection at 20 μg/kg body weight 30 minutes after intraperitoneal injection of D-galactosamine (D-gal) (800 mg/kg body weight) (Sigma-Aldrich, St. Louis, MO). Tissues were collected 4 hours after TNF-alpha treatment.
2.2. Histopathology analysis of liver
Liver tissues were fixed in 10% formalin, and processed for paraffin embedding. Paraffin sections were cut at 5 μm and stained with hematoxylin and eosin (H&E) to assess the histological features of steatosis and inflammation.
2.3. Alanine aminotransferase (ALT) and aspartate aminotransferase (AST)
The ALT and AST activity in the serum were measured with an Infinity kit (Thermo Scientific, Waltham, MA).
2.4. Immunoblot staining
T-PER tissue extraction reagent (Thermo scientific, Waltham, MA) and M-PER mammalian extraction reagent (Thermo scientific) containing protease inhibitors (Sigma-Aldrich, St. Louis, MO) were used for extraction of proteins from liver tissue and cell culture, respectively. Aliquots containing 60μg protein from liver tissue or 20μg protein from cell lysate were loaded onto an 8%–15% sodium dodecyl sulfate-polyacrylamide gel (SDS-PAGE). After electrophoresis, proteins were transferred to a polyvinylidene fluoride (PVDF) membrane. The membrane was probed with polyclonal antibodies against NADPH oxidase 4 (NOX4, Cat. # ab133303), CYP2E1, aldehyde dehydrogenase 2 (ALDH2), oxphos (Abcam, Cambridge, MA), 4HNE (Northwest Life Science Specialties, Vancouver, WA), superoxide dismutase 1 (SOD1), glutathione S-transferase Mu 1 (GSTM1), Bax (Santa Cruz Biotechnologies, Santa Cruz, CA), calnexin, anti-heat shock protein 60 (HSP60) (BD, Franklin Lake, NJ), alcohol dehydrogenase (ADH), peroxisome proliferator-activated receptor alpha (PPAR-α), hepatocyte nuclear factor 4 (HNF-4α), glutathione peroxidase1 (Gpx1), (Novus biological, Littleton, CO), catalase, superoxide dismutase 2 (SOD2), voltage-dependent anion channel (VDAC) (Millipore, Billerica, MA), caspase-9, cleaved caspase-9, cleaved-PARP (Cell Signaling Technology, Danvers, MA) ACADVL, CPT-1 (Proteintech, Chicago, IL), and β-actin (Sigma-Aldrich) respectively. The membrane was then incubated with HRP-conjugated goat anti-rabbit IgG, or goat anti-mouse IgG, or rabbit anti-goat IgG antibody. The protein bands were visualized by an Enhanced Chemiluminescence detection system (GE Healthcare, Piscataway, NJ) and quantified by densitometry analysis.
2.5. Oil red O staining
Liver tissue were frozen in Tissue-Tek OCT (Optimum Cutting Temperature) Compound (VWR, Batavia, IL). Cryostat liver tissue section were cut at 7 μm, fixed and processed with Oil red O solution to stain neutral lipid.
2.6. Quantitative assay of triglycerides, and free fatty acid (FFA)
The levels of triglycerides and FFA in liver tissue and hepatoma cells were measured with assay kits (BioVision, Milpitas, CA).
2.7. Measurement of ATP
ATP levels in both liver and hepatocytes were measured by commercial ATP Assay Kit (Abcam). The method was described previously23.
2.8. qPCR
The DNA was extracted by QIAamp DNA Mini Kit (Qiagen, Venlo, Limburg). The forward and reverse primers were purchased from Integrated DNA Technologies (Coralville, IA). Primer sequences of mice NADH dehydrogenase for the qPCRs are (Forwarding: CACACAAACATAACCACTTTAAC, Reverse: GTAGGTCAATGAATGAGTGGTT). The18S rRNA served as a nuclear DNA reference (Forwarding: CGC GGT TCT ATT TTG TTG GT, Reverse: AGT CGG CAT CGT TTA TGG TC). qPCR analysis with SYBR green PCR Master Mix (Qiagen, Valencia, CA) was performed on the Applied Biosystems 7500 Real Time PCR System (Applied Biosystems). The result was calculated as mitochondrial DNA:nuclear DNA ratio.
2.9. Hydrogen peroxide
Serum hydrogen peroxide levels were measured by Amplex Red Hydrogen Peroxide/Peroxidase Assay kit (Invitrogen, Waltham, MA). Amplex Red reagent react with hydrogen peroxide in a 1:1 stoichiometry to produce the red oxidation product, resorufin. Resorufin can be measured fluoremetrically or spectrophotometrically (OD=560nm).
2.10. Terminal Deoxynucleotidyl transferase dUTP Nick End Labeling (TUNEL) assay
Apoptotic cell death in the liver and hepatoma cells was assessed by detection of DNA fragmentation using an ApopTag peroxidase in situ Apoptosis Detection Kit (Millipore, Billerica, MA). Briefly, cell slides were fixed with 1% paraformaldehyde for 5min. Then liver tissue slides or cell slides were pretreated with proteinase K and H2O2 and incubated with the reaction mixture containing terminal deoxynucleotidyl transferase (TdT) and digoxigenin-conjugated dUTP for 1h at 37 °C. The labeled DNA was visualized with HRP-conjugated anti-digoxigenin antibody with DAB as the chromagen.
2.11. NAD+/NADH
The levels of NAD+ and NADH were measured using NAD+/NADH Quantitation Colorimetric Kit (BioVisio).
2.12. Cell culture and treatment
Rat H4IIEC3 hepatoma cells obtained from the American Type Culture Collection (Manassas, VA) were grown in Dulbecco’s modified Eagle medium (DMEM; Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) and penicillin (100 U/ml) streptomycin sulfate (100 μg/mL) (Invitrogen, Carlsbad, CA). H4IIEC3 hepatoma cells were seeded at 2×105 cells per 6-well plate, or 3×104 cells per well for 8-well chamber slide overnight. Then cells were treated with 200mM ethanol for 48 hours (Sigma-Aldrich) in absence of FBS.
2.13. Silence of NOX4 in H4IIEC3 cells
H4IIEC3 cells were transfected with rat NOX4 shRNA or scrambed shRNA (Santa Cruz Biotechnologies, Santa Cruz, CA). Stable clones were generated by deliver shRNA lentivirus into cells. The selection of stable silenced clones was started 48 h later with 2μM puromycin.
2.14. Mitochondrial membrane potential
The mitochondrial membrane potential of hepatoma cells is assessed by TMRE mitochondrial kit (Abcam), as described previously23.
2.15. Mitochondrial ROS
The production of superoxide in the mitochondrial was visualized in fluorescence microscopy by MitoSOX Red reagent (ThermoFisher, Rockville, MD). MitoSOX Red reagent permeates live cells and selectively targets mitochondrial superoxide. The oxidized product generates red fluorescent upon binding to nuclei acid.
2.16. Liver Caspase 9 activity
The activity of Caspase 9 was measured with Caspase-9 colorimetic assay kit (BioVision, Milpitas, CA) according to manufacturer’s instruction.
2.17. Statistical analysis
Results are expressed as mean ± standard deviation. Differences among multiple groups were analyzed by analysis of variance followed by Tukey’s test. Differences between two groups were analyzed by two-tail Student’s t-test. The significance between groups was defined as P < 0.05
3. Results
3.1. The effect of alcohol exposure on the expression and subcellular distribution of NOX4 in the liver
To determine the role of NOX4 in the pathogenesis of ALD, the protein levels of NOX4 was measured. NOX4 proteins were significantly increased in the liver of rats or mice after alcohol feeding (Figures 1A–1B). The protein levels of NOX4 were further measured in the mitochondrial and microsomal fractions. As shown in Figure 1C, NOX4 proteins were higher in the mitochondrial fraction in comparison with microsomal fraction, and alcohol feeding significantly increased the protein levels of NOX4 in the mitochondria of the rat liver (Figure 1C). The distribution of NOX4 was then determined in the liver of mice. The result showed that the mitochondria have a higher levels of NOX4 compared to that of the microsome and alcohol feeding significantly increased the expression of NOX4 in the mitochondria of mouse liver (Figure 1D).
Figure 1.

Expression of NOX4 in the liver and subcellular compartments after alcohol feeding. (A) The immunoblot bands of NOX4 in the liver of rats fed alcohol for 5 months (B) The immunoblot bands of NOX4 in the liver of mice fed alcohol for 1 month. (C) The immunobands of NOX4 in mitochondrial and microsomal fraction in the liver of rats fed alcohol for 5 months. (D) The immunoblot bands of NOX4 in the mitochondrial and microsomal fraction in the liver of mice fed alcohol for 1 month. The bands density was quantified by densitometry analysis. The ratio to β-actin was calculated by setting the value of control (ctrl) as one. Results are mean ± SD (n=3). Significant differences (*P < 0.05) between control-and ethanol-fed rats and mice are determined by Student’s t-test. EtOH: ethanol.
3.2. GKT137831 ameliorates chronic alcohol-induced liver injury
The hematoxylin and eosin (H&E) staining revealed that four weeks of alcohol feeding caused formation of lipid droplets (arrowheads) and necrosis (arrows) in the hepatocytes, which was partially reversed by GKT137831 treatment for the last two weeks (Figure 2A). In addition, as shown in Figure 2B, GKT137831 treatment attenuated alcohol feeding-induced elevation of serum ALT and AST (Figures 2B and 2C).
Figure 2.

Serum markers of liver injury and liver pathology in mice fed control (Ctrl) or ethanol (EtOH) for four weeks with or without GKT137831 for the last two weeks. (A) Light microscopy with hematoxylin & eosin staining shows accumulation of lipid droplets (arrows) and necrosis (arrowheads) in the liver of ethanol-fed mice, which was attenuated by GKT137831 treatment. (B) Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activities. Serum ALT and AST activities were measured by using Infinity ALT and AST Reagents. Scale bar: 20μM. Results are mean ± SD (n=6). Results for bars that do not share a letter differed significantly among groups (P <0.05). Significant differences among groups are determined by ANOVA followed by Tukey’s test.
3.3. GKT137831 decreases alcohol-induced elevation of serum hydrogen peroxide but does not affect alcohol-induced alteration of alcohol metabolism enzymes and ROS scavenger enzymes
Four weeks of alcohol feeding remarkably increased the expression of NOX4 in the liver and mitochondrial fraction, and GKT137831 treatment for the last two weeks did not affect the protein levels of NOX4 after alcohol exposure (Figures 3A and 3B). Alcohol feeding increased the hepatic protein levels of CYP2E1 but did not affect alcohol metabolism enzymes, including alcohol dehydrogenase (ADH), catalase, and aldehyde dehydrogenase 2 (ALDH2) ) as shown in Figure 3C. GKT137831 treatment did not have additional effect on the alcohol metabolism enzymes after alcohol exposure. The antioxidant enzymes were then measured. Alcohol feeding increased the expression of GSTM1 but not SOD1, SOD2 and Gpx1 (Figure 3D). Alcohol feeding along with GKT137831 treatment did not affect the expression of antioxidant enzymes in the liver of mice compared with mice fed with alcohol alone (Figure 3D). Moreover, the levels of serum H2O2 was measured, as shown in figure 3E, GKT137831 treatment reduced alcohol feeding-induced elevation of H2O2 levels in the serum.
Figure 3.

Protein levels of NOX4, alcohol oxidation enzymes, antioxidant enzymes and serum H2O2 in the liver of mice fed control (Ctrl) or ethanol (EtOH) for four weeks with or without GKT137831 for the last two weeks. (A) The immunoblot bands of NOX4 in the liver. (B) The immunoblot bands of NOX4 in the mitochondria. (C) The immunoblot bands of ADH, CYP2E1, Catalase, and ALDH2 in the liver. (D) The immunoblot bands of GSTM1, SOD1, SOD2, and Gpx1 in the liver. (E) The levels of H2O2 in the serum. Serum H2O2 was measured by Amplex Red Peroxide/Peroxidase Assay kit. The bands density was quantified by densitometry analysis. The ratio to HSP60 or β-actin was calculated by setting the value of ctrl as one. Results are mean ± SD (n=3). Results for bars that do not share a letter differed significantly among groups (P <0.05). Significant differences among groups are determined by ANOVA followed by Tukey’s test.
3.4. GKT137831 reduces alcohol-induced the production of ROS in the mitochondria and protects against reduction of mitochondrial respiratory chain complex IV, mitochondrial DNA, and ATP levels in the liver of mice
As shown in Figure 4A, alcohol exposure significantly increased the levels of 4HNE and Gpx1 in the mitochondria, which were partially reversed by GKT137831 treatment. The expression of mitochondrial respiratory chain proteins was measured by western blot. Alcohol exposure significantly decreased the levels of complex IV, and GKT137831 treatment partially reversed alcohol-induced reduction of complex IV (Figure 4B). Additionally, the levels of mitochondrial DNA (mtDNA) and hepatic ATP levels were determined. As shown in Figures 4C and 4D, GKT137831 treatment reduced alcohol-induced reduction of mitochondrial DNA and ameliorated alcohol-induced decrease of ATP in the liver.
Figure 4.

The protein levels of mitochondrial 4HNE and Gpx1, mitochondrial respiratory complexes, the number of mtDNA, and the concentration of ATP in the liver of mice fed control (Ctrl) or ethanol (EtOH) for four weeks with or without GKT137831 for the last two weeks. (A) The immunoblot bands of mitochondrial 4HNE and Gpx1. (B) The immunoblot bands of mitochondrial respiratory complex I-V. (C) The mtDNA levels were measured with NADH dehydrogenase subunit 6 by qPCR. (D) ATP levels in the liver of mice were detected by commercial ATP assay kit. The bands density was quantified by densitometry analysis. The ratio to β-actin or mitochondrial HSP60 was calculated by setting the value of ctrl as one. Results are mean ± SD (n=3). Results for bars that do not share a letter differed significantly among groups (P <0.05). Significant differences among groups are determined by ANOVA followed by Tukey’s test.
3.5. GKT137831 reverses alcohol-induced lipid accumulation and increases the expression of HNF-4α and β-oxidation enzymes in the liver of mice
Oil red O staining shows that alcohol exposure increased fat accumulation in the liver manifested by the increased size of lipid droplets (arrows) (Figure 5A). GKT137831 treatment reduced the size of lipid droplets in the liver of mice (Figure 5A). Quantitative measurement of hepatic lipid contents showed that alcohol exposure significantly increased the hepatic triglyceride (P < 0.05) and free fatty acid (FFA) (P < 0.05) levels, which were reversed by GKT137831 treatment (Figures 5B and 5C).
Figure 5.

Hepatic lipid accumulation, protein levels of lipid oxidation enzymes, and NAD+/NADH ratio in the liver of mice fed control (Ctrl) or ethanol (EtOH) for four weeks with or without GKT137831 for the last two weeks. (A) Oil red O staining of neutral lipids (arrows) in the liver. (B) Quantitative measurements of hepatic concentration of triglycerides and (C) free fatty acids. (D) The immunoblot bands of PPAR-α, HNF-4α, ACADVL and CPT1. The bands density was quantified by densitometry analysis. The ratio to β-actin was calculated by setting the value of ctrl as one. (E) The ratio of NAD+/NADH in the liver was measured by commercial kit. Scale bar: 50μM. Results are mean ± SD (n=3). Results for bars that do not share a letter differed significantly among groups (P <0.05). Significant differences among groups are determined by ANOVA followed by Tukey’s test.
To determine the potential mechanism, by which GKT137831 reduces lipid accumulation in the liver after alcohol exposure, the protein levels of PPAR-α, HNF-4α, and important β-oxidation enzymes were measured. As shown in Figure 5D, alcohol feeding significantly decreased the expression of HNF-4α, but not PPAR-α, ACADVL and CPT1. GKT137831 treatment reversed alcohol feeding-induced reduction of HNF-4α. In addition, the expression of ACADVL and CPT1 were significantly increased by GKT137831 treatment in comparison with control-and alcohol-feeding groups, respectively (Figure 5D). The ratio of NAD+ to NADH was measured as well, and GKT137831 treatment did not reverse alcohol-induced reduction of NAD+ to NADH ratio (Figure 5E).
3.6. GKT137831 ameliorates alcohol-induced mitochondrial apoptotic signaling
Apoptotic cells were measured by TUNEL assay. As shown in Figure 6A, GKT137831 treatment decreased the number of TUNEL positive cells (dark brown color indicated by arrows) in the liver after alcohol exposure.
Figure 6.

TUNEL assay, caspase activity assay, and mitochondrial apoptotic markers in the liver of mice fed control (Ctrl) or ethanol (EtOH) for four weeks with or without GKT137831 for the last two weeks. (A) Representative image of TUNEL assay and quantification of TUNEL assay (right panel). (B) Immunoblot bands of mitochondrial Bax and mitochondrial VDAC. (C) Immunoblot bands of cleaved caspase-9, caspase-9, cleaved PARP, and PARP. The bands density was quantified by densitometry analysis. The ratio to β-actin or mitochondrial HSP60 was calculated by setting the value of ctrl as one. (D). Liver caspase-9 activity assay. Results are mean ± SD (n=3). Results for bars that do not share a letter differed significantly among groups (P <0.05). Significant differences among groups are determined by ANOVA followed by Tukey’s test.
To determine the potential mechanism of GKT137831 in protecting against alcohol feeding induced apoptosis, mitochondrial apoptotic cell death markers were measured. GKT137831 treatment reversed alcohol feeding-induced insertion of Bax into mitochondria and loss of VDAC from mitochondria (Figure 6B). Moreover, GKT137831 treatment reduced the level of cleaved-caspase 9 and cleaved PARP in the liver after alcohol exposure (Figure 6C). Hepatic caspase-9 activity was increased by alcohol exposure, which was reversed by GKT137831 treatment (Figure 6D).
3.7. Knockdown of NOX4 in H4IIEC3 cells reverses ethanol-increased loss of mitochondrial membrane potential, decreased levels of mitochondrial ROS, promoted lipid accumulation and apoptosis, reduced levels of ATP and NAD+/NADH
NOX4 was successfully knocked down in H4IIEC3 cells (Figure 7A, left panel). H4IIEC3 cells were then treated with 200 mM ethanol for 48 hours. As shown in the right panel of Figure 7A, ethanol treatment significantly increased the levels of NOX4 in H4IIEC3 cells transfected with scrambled shRNA. The ethanol induced up-regulation of NOX4 was significantly alleviated in H4IIEC3 cells transfected with NOX4 shRNA.
Figure 7.

Effect of NOX4 on mitochondrial membrane potential (MMP), mitochondrial ROS generation, apoptosis, ATP production, lipid accumulation, and NAD+/NADH levels. (A) The immunoblot bands of NOX4 in H4IIEC3 cells transfected with scrambled or NOX4 shRNA (left panel) and in H4IIEC3 treated with 200mM ethanol for 48 hours (right panel). H4IIEC3 cells transfected with scrambled or NOX4 shRNA were treated with 200mM of ethanol for 48 hours. TMRE labeled active mitochondria were shown in (B), ROS production was shown in (C), apoptosis was shown in (D), levels of triglyceride were shown in (H), levels of NAD+/NADH were shown in (I), levels of ATP were shown in (J). (E) Quantification of MMP. (F) Quantification of Mitosox staining. (G) Quantification of TUNEL staining. The ratio to β-actin or mitochondrial HSP60 was calculated by setting the value of ctrl as one. Results are mean ± SD (n=3). Statistical difference (P < 0.05) was analyzed by Student’s t-test.
To determine the effect of NOX4 on the integrity and function of mitochondria, mitochondrial membrane potential (MMP) and ROS levels were measured. As shown in Figure 7B, cells transfected with NOX4 shRNA had higher MMP as indicated by stronger red fluorescence, compared to cells transfected with scrambled shRNA after ethanol treatment. Quantitative analysis shows that ethanol treatment significantly decreased MMP in cells transfected with scrambled shRNA, but not in cells transfected with NOX4 shRNA (Figure 7E). Moreover, cells were stained with Mitosox to determine the levels of ROS in the mitochondria and cells were counterstained with HOECHST to indicate nuclei. The result shows that cells transfected with NOX4 shRNA had weaker red fluorescence staining after ethanol treatment in comparison with cells transfected with scrambled shRNA (Figure 7C). Quantitative analysis illustrates that ethanol treatment remarkably increased intensity of positive staining in cells transfected with scrambled shRNA, which was attenuated in cells transfected with NOX4 shRNA (Figure 7F). TUNEL staining showed that ethanol treatment significantly increased number of apoptotic cells indicated by red fluorescence in H4IIEC3 cells transfected with scrambled shRNA, which was significantly alleviated in H4IIEC3 cells transfected with NOX4 shRNA (Figures 7D–7G). Moreover, ethanol treatment significantly increased levels of triglyceride in H4IIEC3 cells transfected with scrambled shRNA and silence of NOX4 significantly inhibited this ethanol-induced up-regulation of triglyceride accumulation (Figure 7H). Ethanol treatment also significantly decreased levels of ATP and NAD+/NADH in control H4IIEC3 cells which were attenuated by silence of NOX4 (Figures 7I–7J).
4. Discussion
The present study demonstrated that inhibition of NOX4 activity protects against ALD. First, we found that chronic alcohol feeding induced the protein levels of NOX4 in the liver and mitochondria, and mitochondrial fraction had higher levels of NOX4 than microsomal fraction. Then, we used GKT137831, a NADPH oxidase inhibitor, to treat alcohol-fed mice, and we observed that GKT137831 ameliorated alcohol-induced liver injury along with decreased ROS levels in mitochondria. Improved mitochondrial functions are coupled with increased expression of β-oxidation enzymes and decreased apoptotic cells after alcohol exposure. Last, mechanistic study shows that ethanol challenging-induced loss of MMP, decrease of ATP, elevation of mitochondrial ROS levels, increase of apoptosis, enhance of TG accumulation, and decrease of NAD+/NADH ratio were restored in H4IIEC3 cells transfected with NOX4 shRNA. This study revealed a novel mechanism of NOX4 in the pathogenesis of ALD.
NOXs overexpression is associated with increased oxidative stress and pathogenesis of diseases. It has been shown that NOXs are the major enzyme responsible for superoxide generation through transferring electrons from NADPH or NADH to molecular oxygen22. Activation of NOXs was observed in heart failure25, lung fibrosis20, diabetic kidney disease26, and liver disease17, 27. Previous study demonstrated that NOX-deficient (p47 phox−/− ) mice had a decrease of oxidative stress and were resistant to alcohol-induced hepatic steatosis and hepatitis13. The p47phox is necessary for NOX1 and NOX2 to form active complexes, therefore decreased NOXs activity may have protective effect on the development of ALD. Different from other NOXs, NOX4 is constitutively active. Active NOX4 directly produces H2O2. During chronic liver injury, the balance between the amount of H2O2 and antioxidant enzymes is perturbed. Human study showed that NOX4 was significantly upregulated at mRNA levels in the liver of alcoholic hepatitis patients compared to patients with normal liver28. In the present study, we found that alcohol exposure significantly increased protein of NOX4 in the liver and hepatic mitochondria. NOX4 inhibitor ameliorated alcohol-induced elevation of H2O2. Knockdown of NOX4 in H4IIEC3 cells attenuated loss of MMP and mitochondrial oxidative stress after ethanol challenge. Increased hepatic inflammation has been reported in chronic alcohol fed mice and rats. The p47phox knockout mice, which lack a critical subunit of NADPH oxidase protected against alcohol-induced hepatitis by inactivating NF-kappa B and reducing production of cytotoxic TNF-alpha13. In the present study, immunohistochemical staining showed that NOX4 inhibitor attenuated alcohol-induced inflammatory cell infiltration. Thus, the current study demonstrated that NOX4 levels were upregulated in the liver by alcohol exposure, and increased NOX4 levels are associated with the development of ALD.
GKT137831 is a drug used to determine the role of NOX-mediated tissue injury and fibrosis. Previous studies showed that GKT137831 treatment effectively decreased hydrogen peroxide levels, attenuated apoptosis, and reversed fibrosis in the liver21, 29 and heart 30. Consistent with previous studies, we found that GKT137831 treatment decreased ROS level in the mitochondria after alcohol exposure. In addition, mitochondrial glutathione peroxidase-1 levels were significantly decreased in mice fed GKT137831along with alcohol compared with mice fed alcohol alone. This result suggests that decrease of mitochondrial ROS by GKT137831 is achieved by reducing the production instead of increasing clearance. Previous studies demonstrated that mtDNA and mitochondrial respiratory chain complex IV are sensitive to oxidative stress23, 31–34. In addition, it is known that increased mitochondrial oxidative stress activates intrinsic apoptotic cell death pathway via releasing cytochrome C and pro-apoptotic proteins35. In the current study, we found that alcohol-induced reduction of mitochondrial respiratory chain complex IV, mtDNA, and ATP was reversed by inhibition of NOX4 activity. Intrinsic apoptosis induced by chronic alcohol feeding was also rescued by inhibition of NOX4 activity. Thus, the present study demonstrated that alcohol-induced mitochondria oxidative stress is attenuated by inhibition of NOX4 activity, which results in improved mitochondria functions and decreased apoptosis. In addition, it will be of interest to feed alcohol to mice with hepatocyte-specific deletion of NOX4. Previous study showed that hepatocyte-specific deletion of NOX4 attenuated oxidative stress, lipid peroxidation, and liver fibrosis in mice with dietary induced steatohepatitis 36. Thus, mice with hepatocyte-specific deletion of NOX4 may protect from alcohol-induced liver injury via decrease of oxidative stress and apoptosis.
PPARα and HNF-4α are both transcription factors, and play critical roles in regulation of hepatic lipid homeostasis. HNF-4α is a master regulator of hepatic gene expression, and liver-specific knockout of HNF-4α led to severe steatosis in mice37. In addition activation of HNF-4α by zinc attenuated alcohol-induced hepatic steatosis along with upregulated genes involves in β-oxidation (Cpt-1 and Acadl)38. In the present study, we found an increase of HNF-4α, ACVDAL, and CPT1but not PPARα at proteins levels by GKT137831 treatment after alcohol exposure. These data suggests that upregulation of HNF-4α and important β-oxidation enzymes may account for the mechanism, by which inhibition of NOX4 activity ameliorates alcohol-induced hepatic steatosis. Moreover, we measured the ratio of NAD+/NADH after alcohol exposure with or without GKT137831 treatment for the last two weeks. It is well known that decrease NAD+/NADH ratio favoring lipid accumulation39. In the current study, GKT137831 did not restore the ratio of NAD+/NADH. It is possibly due to that inhibition of NOX4 did not affect the expression of alcohol oxidation enzymes, in particular ALDH2, or accelerate the clearance of acetaldehyde and the conversion of NAD+ to NADH. Thus, the data suggest that attenuated lipid accumulation in the liver by inhibition of NOX4 activity is not resulted from increased ratio of NAD+ to NADH but enhanced levels of HNF-4α and β-oxidation.
Collectively, the present study demonstrated that NOX4 predominantly locates in the mitochondria of the liver and is upregulated by alcohol exposure, and pharmacological inhibition of NOX4 reversed alcohol-induced hepatic steatosis and apoptosis. Alcohol exposure increased oxidative stress in mitochondria, which impaired levels of mitochondrial respiratory chain complex IV, mtDNA, and ATP. Attenuation of alcohol-induced mitochondrial oxidative stress is likely the most important mechanism underlying the protective action of NOX4 inhibitor on mitochondrial function. These results suggest that mitochondrial NOX4 is a promising therapeutic target for treating ALD.
Highlights.
NOX4 predominantly localizes in the mitochondria, but not in the ER, in the liver of mice, and chronic alcohol exposure increase hepatic NOX4 in the mitochondria.
Inhibition of NOX4 activity decreases alcohol-elevated serum hydrogen peroxide levels.
Inhibition of NOX4 activity decreases fat accumulation coupled with increased HNF-4α and CPT-1 in the liver after alcohol exposure.
Inhibition of NOX4 activity decreases alcohol-induced mitochondrial apoptotic cell death pathway.
Knockdown of NOX4 in hepatoma cells protects against ethanol-induced loss of mitochondrial membrane potential and generation of ROS.
Acknowledgments
This research was supported by the National Institutes of Health (R01AA020212 and 2R01AA018844).
Sources of support: This research was supported by the National Institutes of Health (R01AA020212 and 2R01AA018844).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mathews S, Xu M, Wang H, Bertola A, Gao B. Animals models of gastrointestinal and liver diseases. Animal models of alcohol-induced liver disease: pathophysiology, translational relevance, and challenges. American journal of physiology Gastrointestinal and liver physiology. 2014;306:G819–23. doi: 10.1152/ajpgi.00041.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neuman MG, Malnick S, Maor Y, Nanau RM, Melzer E, Ferenci P, Seitz HK, Mueller S, Mell H, Samuel D, Cohen LB, Kharbanda KK, Osna NA, Ganesan M, Thompson KJ, McKillop IH, Bautista A, Bataller R, French SW. Alcoholic liver disease: Clinical and translational research. Experimental and molecular pathology. 2015;99:596–610. doi: 10.1016/j.yexmp.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 3.Lu Y, Wu D, Wang X, Ward SC, Cederbaum AI. Chronic alcohol-induced liver injury and oxidant stress are decreased in cytochrome P4502E1 knockout mice and restored in humanized cytochrome P4502E1 knock-in mice. Free radical biology & medicine. 2010;49:1406–16. doi: 10.1016/j.freeradbiomed.2010.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang X, Wu D, Yang L, Gan L, Cederbaum AI. Cytochrome P450 2E1 potentiates ethanol induction of hypoxia and HIF-1alpha in vivo. Free radical biology & medicine. 2013;63:175–86. doi: 10.1016/j.freeradbiomed.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lemire J, Mailloux R, Puiseux-Dao S, Appanna VD. Aluminum-induced defective mitochondrial metabolism perturbs cytoskeletal dynamics in human astrocytoma cells. Journal of neuroscience research. 2009;87:1474–83. doi: 10.1002/jnr.21965. [DOI] [PubMed] [Google Scholar]
- 6.Auger C, Alhasawi A, Contavadoo M, Appanna VD. Dysfunctional mitochondrial bioenergetics and the pathogenesis of hepatic disorders. Frontiers in cell and developmental biology. 2015;3:40. doi: 10.3389/fcell.2015.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murphy MP. Modulating mitochondrial intracellular location as a redox signal. Science signaling. 2012;5:pe39. doi: 10.1126/scisignal.2003386. [DOI] [PubMed] [Google Scholar]
- 8.Willems PH, Rossignol R, Dieteren CE, Murphy MP, Koopman WJ. Redox Homeostasis and Mitochondrial Dynamics. Cell metabolism. 2015;22:207–18. doi: 10.1016/j.cmet.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 9.Adachi M, Ishii H. Role of mitochondria in alcoholic liver injury. Free radical biology & medicine. 2002;32:487–91. doi: 10.1016/s0891-5849(02)00740-2. [DOI] [PubMed] [Google Scholar]
- 10.Altenhofer S, Kleikers PW, Radermacher KA, Scheurer P, Rob Hermans JJ, Schiffers P, Ho H, Wingler K, Schmidt HH. The NOX toolbox: validating the role of NADPH oxidases in physiology and disease. Cellular and molecular life sciences : CMLS. 2012;69:2327–43. doi: 10.1007/s00018-012-1010-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nature reviews Drug discovery. 2011;10:453–71. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paik YH, Kim J, Aoyama T, De Minicis S, Bataller R, Brenner DA. Role of NADPH oxidases in liver fibrosis. Antioxidants & redox signaling. 2014;20:2854–72. doi: 10.1089/ars.2013.5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kono H, Rusyn I, Yin M, Gabele E, Yamashina S, Dikalova A, Kadiiska MB, Connor HD, Mason RP, Segal BH, Bradford BU, Holland SM, Thurman RG. NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. The Journal of clinical investigation. 2000;106:867–72. doi: 10.1172/JCI9020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiological reviews. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 15.Bataller R, Schwabe RF, Choi YH, Yang L, Paik YH, Lindquist J, Qian T, Schoonhoven R, Hagedorn CH, Lemasters JJ, Brenner DA. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. The Journal of clinical investigation. 2003;112:1383–94. doi: 10.1172/JCI18212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Minicis S, Seki E, Oesterreicher C, Schnabl B, Schwabe RF, Brenner DA. Reduced nicotinamide adenine dinucleotide phosphate oxidase mediates fibrotic and inflammatory effects of leptin on hepatic stellate cells. Hepatology. 2008;48:2016–26. doi: 10.1002/hep.22560. [DOI] [PubMed] [Google Scholar]
- 17.Adachi T, Togashi H, Suzuki A, Kasai S, Ito J, Sugahara K, Kawata S. NAD(P)H oxidase plays a crucial role in PDGF-induced proliferation of hepatic stellate cells. Hepatology. 2005;41:1272–81. doi: 10.1002/hep.20719. [DOI] [PubMed] [Google Scholar]
- 18.Tanaka M, Tanaka K, Masaki Y, Miyazaki M, Kato M, Kotoh K, Enjoji M, Nakamuta M, Takayanagi R. Intrahepatic microcirculatory disorder, parenchymal hypoxia and NOX4 upregulation result in zonal differences in hepatocyte apoptosis following lipopolysaccharide- and D-galactosamine-induced acute liver failure in rats. International journal of molecular medicine. 2014;33:254–62. doi: 10.3892/ijmm.2013.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Block K, Gorin Y, Abboud HE. Subcellular localization of Nox4 and regulation in diabetes. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:14385–90. doi: 10.1073/pnas.0906805106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carnesecchi S, Deffert C, Donati Y, Basset O, Hinz B, Preynat-Seauve O, Guichard C, Arbiser JL, Banfi B, Pache JC, Barazzone-Argiroffo C, Krause KH. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxidants & redox signaling. 2011;15:607–19. doi: 10.1089/ars.2010.3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang JX, Chen X, Serizawa N, Szyndralewiez C, Page P, Schroder K, Brandes RP, Devaraj S, Torok NJ. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free radical biology & medicine. 2012;53:289–96. doi: 10.1016/j.freeradbiomed.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:15565–70. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun Q, Zhong W, Zhang W, Zhou Z. Defect of mitochondrial respiratory chain is a mechanism of ROS overproduction in a rat model of alcoholic liver disease: role of zinc deficiency. American journal of physiology Gastrointestinal and liver physiology. 2016;310:G205–14. doi: 10.1152/ajpgi.00270.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun Q, Zhong W, Zhang W, Li Q, Sun X, Tan X, Sun X, Dong D, Zhou Z. Zinc deficiency mediates alcohol-induced apoptotic cell death in the liver of rats through activating ER and mitochondrial cell death pathways. American journal of physiology Gastrointestinal and liver physiology. 2015;308:G757–66. doi: 10.1152/ajpgi.00442.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li JM, Gall NP, Grieve DJ, Chen M, Shah AM. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension. 2002;40:477–84. doi: 10.1161/01.hyp.0000032031.30374.32. [DOI] [PubMed] [Google Scholar]
- 26.Gorin Y, Block K, Hernandez J, Bhandari B, Wagner B, Barnes JL, Abboud HE. Nox4 NAD(P)H oxidase mediates hypertrophy and fibronectin expression in the diabetic kidney. The Journal of biological chemistry. 2005;280:39616–26. doi: 10.1074/jbc.M502412200. [DOI] [PubMed] [Google Scholar]
- 27.Thakur V, Pritchard MT, McMullen MR, Wang Q, Nagy LE. Chronic ethanol feeding increases activation of NADPH oxidase by lipopolysaccharide in rat Kupffer cells: role of increased reactive oxygen in LPS-stimulated ERK1/2 activation and TNF-alpha production. Journal of leukocyte biology. 2006;79:1348–56. doi: 10.1189/jlb.1005613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colmenero J, Bataller R, Sancho-Bru P, Bellot P, Miquel R, Moreno M, Jares P, Bosch J, Arroyo V, Caballeria J, Gines P. Hepatic expression of candidate genes in patients with alcoholic hepatitis: correlation with disease severity. Gastroenterology. 2007;132:687–97. doi: 10.1053/j.gastro.2006.12.036. [DOI] [PubMed] [Google Scholar]
- 29.Aoyama T, Paik YH, Watanabe S, Laleu B, Gaggini F, Fioraso-Cartier L, Molango S, Heitz F, Merlot C, Szyndralewiez C, Page P, Brenner DA. Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology. 2012;56:2316–27. doi: 10.1002/hep.25938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Somanna NK, Valente AJ, Krenz M, Fay WP, Delafontaine P, Chandrasekar B. The Nox1/4 Dual Inhibitor GKT137831 or Nox4 Knockdown Inhibits Angiotensin-II-Induced Adult Mouse Cardiac Fibroblast Proliferation and Migration. AT1 Physically Associates With Nox4 Journal of cellular physiology. 2016;231:1130–41. doi: 10.1002/jcp.25210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coleman WB, Cunningham CC. Effect of chronic ethanol consumption on hepatic mitochondrial transcription and translation. Biochimica et biophysica acta. 1991;1058:178–86. doi: 10.1016/s0005-2728(05)80235-x. [DOI] [PubMed] [Google Scholar]
- 32.Thayer WS, Rubin E. Molecular alterations in the respiratory chain of rat liver after chronic ethanol consumption. The Journal of biological chemistry. 1981;256:6090–7. [PubMed] [Google Scholar]
- 33.Venkatraman A, Landar A, Davis AJ, Chamlee L, Sanderson T, Kim H, Page G, Pompilius M, Ballinger S, Darley-Usmar V, Bailey SM. Modification of the mitochondrial proteome in response to the stress of ethanol-dependent hepatotoxicity. The Journal of biological chemistry. 2004;279:22092–101. doi: 10.1074/jbc.M402245200. [DOI] [PubMed] [Google Scholar]
- 34.Caro AA, Bell M, Ejiofor S, Zurcher G, Petersen DR, Ronis MJ. N-acetylcysteine inhibits the up-regulation of mitochondrial biogenesis genes in livers from rats fed ethanol chronically. Alcoholism, clinical and experimental research. 2014;38:2896–906. doi: 10.1111/acer.12576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis : an international journal on programmed cell death. 2007;12:913–22. doi: 10.1007/s10495-007-0756-2. [DOI] [PubMed] [Google Scholar]
- 36.Bettaieb A, Jiang JX, Sasaki Y, Chao TI, Kiss Z, Chen X, Tian J, Katsuyama M, Yabe-Nishimura C, Xi Y, Szyndralewiez C, Schroder K, Shah A, Brandes RP, Haj FG, Torok NJ. Hepatocyte Nicotinamide Adenine Dinucleotide Phosphate Reduced Oxidase 4 Regulates Stress Signaling, Fibrosis, and Insulin Sensitivity During Development of Steatohepatitis in Mice. Gastroenterology. 2015;149:468–80. e10. doi: 10.1053/j.gastro.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hayhurst GP, Lee YH, Lambert G, Ward JM, Gonzalez FJ. Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Molecular and cellular biology. 2001;21:1393–403. doi: 10.1128/MCB.21.4.1393-1403.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kang X, Zhong W, Liu J, Song Z, McClain CJ, Kang YJ, Zhou Z. Zinc supplementation reverses alcohol-induced steatosis in mice through reactivating hepatocyte nuclear factor-4alpha and peroxisome proliferator-activated receptor-alpha. Hepatology. 2009;50:1241–50. doi: 10.1002/hep.23090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Q, Xie G, Zhang W, Zhong W, Sun X, Tan X, Sun X, Jia W, Zhou Z. Dietary nicotinic acid supplementation ameliorates chronic alcohol-induced fatty liver in rats. Alcoholism, clinical and experimental research. 2014;38:1982–92. doi: 10.1111/acer.12396. [DOI] [PMC free article] [PubMed] [Google Scholar]