

Abstract
Background:
Amyloid-β deposition and accumulation of autophagic vacuoles are pathologic features of Alzheimer's disease (AD). Dysregulation of the endosomal–autophagic–lysosomal (EAL) pathway, which impairs amyloid precursor protein processing, is one of the earliest changes in AD. However, the precise role of EAL pathway in neurodegeneration remains unclear. This study aimed to investigate the role of EAL pathway in AD and further study the mechanism of EAL dysfunction.
Methods:
We used 3-, 7-, and 12-month-old APPswe/PSEN1dE9 (APP/PS1) mice to model different stages of AD with age- and gender-matched wild-type littermates as controls (4–7 mice per group) and detected the changes of EAL markers, endosomal organizers Rab5 and Rab7, autophagosome marker LC3B, and lysosomal proteins Lamp1/2 in cortex and hippocampus by immunohistochemistry and Western blotting analysis. To further explore the mechanism of EAL dysregulation in AD, components of the class III phosphatidylinositol 3-kinase (PI3KC3) complex, activators of Rab7 (Beclin1 and UVRAG), and the negative regulator of Rab7 (Rubicon) were also measured in this two brain regions.
Results:
In 7-month-old APP/PS1 brain that amyloid beta initiated to accumulate intracellularly, EAL pathway, and related PI3KC3 members, UVRAG and Beclin1 were upregulated both in cortex and hippocampus (all P < 0.05). By the age of 12 months old, when abundant amyloid plaques formed, EAL markers, UVRAG, and Beclin1 were also upregulated in the cortex (all P < 0.05). However, Rab7 was decreased significantly (P = 0.0447), accompanied by a reduction of its activating PI3KC complex component Beclin1 (P = 0.0215) and enhancement of its inhibiting component Rubicon (P = 0.0055) in the hippocampus.
Conclusions:
Our study implies that EAL pathway, represented as Rab7 and its PI3KC3 regulators’ expressions, showed temporal and spatial variation in brains at different stages of AD. It provides new insights into the role of EAL pathway in pathogenesis and indicates potential therapeutic targets in neurodegenerative diseases.
Keywords: Autophagy, Class III Phosphatidylinositol 3-Kinase Complex, Endocytosis, Neurodegeneration, Rab7
INTRODUCTION
Alzheimer's disease (AD), a neurodegenerative disease highly prevalent in elderly people, mainly manifests as progressive memory loss and other cognitive dysfunctions. Mutations in genes encoding amyloid precursor protein (APP), Presenilin1, or Presenilin 2 can cause familial early-onset AD; however, the etiology of the more common sporadic AD remains unclear.[1]
The neuropathological hallmarks of AD include intraneuronal neurofibrillary tangles and extracellular senile plaques consisting of amyloid-β (Aβ), both of them play pivotal roles in the evolution of neurodegeneration.[2,3] Recent studies have discovered that APP, the precursor of Aβ, is processed via the endosomal–autophagic–lysosomal (EAL) pathway.[2] APP is internalized by endocytosis, then sorted in the early endosomes, and finally delivered to the late endosomes. The late endosomes then fuse with either lysosomes or autophagosomes for lysosomal degradation.[2] Mounting evidence has suggested that EAL dysfunction is one of the early neuropathological features in AD. EAL dysfunction is characterized by progressive accumulation of autophagic vacuoles (AVs) and enlargement of endosomes.[4] It has been proven that endosomes in brains of AD patients and AD mouse models are enriched in APP and APP secretases compared with healthy controls.[4,5] Therefore, trafficking, catabolism, and elimination of APP via the EAL pathway may be crucially involved in the pathogenesis of AD.
Autophagy is a highly regulated process that is responsible for degrading long-lived or aggregated proteins and damaged organelles and is characterized by sequestration of cytoplasmic cargoes into double-membrane vesicles called autophagosomes. Autophagy maintains neuronal homeostasis, and its role in the turnover of aggregated proteins is important but controversial in neurodegenerative disorders, including AD, Parkinson's disease, amyotrophic lateral sclerosis, and corticobasal degeneration.[6] On the one hand, increasing autophagy could promote the clearance of aggregated proteins. On the other hand, overactive autophagy can induce neurodegeneration and apoptosis.[7] The results of studies targeting autophagy have been conflicting each other on therapeutic effects.[8] Since a balance between the formation and clearance of AVs is required for maintaining normal autophagic flux, accumulation of AVs could be considered as an aspect of dysregulation of EAL pathway. Targeting specific markers that modulate this crucial process is a potential therapeutic strategy.
Recent studies have demonstrated that both trafficking of APP among organelles and fusion of vesicles in EAL pathway are precisely regulated by Rab proteins. Rabs, a family of small GTPases, are key players in endocytosis, recycling of cell surface molecules, identifying types of vesicles, and regulating the formation, movement, and fusion of vesicle membranes.[9] Rab proteins include the early endosome constituent Rab5 and the late endosome effector Rab7. Specifically, Rab5 regulates early endosome uptake and fusion; Rab7 is required for initiation of early-to-late endosome transition, facilitation of late endosome maturation, and crosstalk between endosomes and autophagosomes.[10] Activation of Rab5 contributes to APP-induced axonal blockage,[11] while Rab7 knockdown blocks the fusion of autophagosomes/endosomes with lysosomes.[12] Abnormal expression of Rabs has been observed in some neurodegenerative disorders, such as Charcot–Marie–Tooth type 2B, dementia with Lewy bodies, mild cognitive impairment, and AD;[13,14] however, the mechanisms by which Rabs participate in EAL pathway dysregulation in AD have yet to be elucidated.
Rab7 is known to be closely linked to the class III phosphatidylinositol 3-kinase (PI3KC3) complex, an important regulator of intercellular vesicle transport.[15] It has recently been shown that UVRAG, a component of the PI3KC3 complex, binding to Beclin1 component via its central conserved domain to stabilize the Beclin1–PI3KC3 complex, could induce autophagy.[16] In UVRAG overexpressing cells, a marked increase in autophagosomes and Rab7 activity was detected.[17] Another component of the PI3KC3 complex, Rubicon, is a negative regulator of endosomal maturation and autophagy. Rubicon competes with Rab7 for UVRAG binding.[18] However, the relationship between Rab7 and PI3KC3 in AD has not been analyzed.
To investigate the dysregulation of EAL pathway and a possible involvement of PI3KC3 component(s) in the neurodegeneration in AD, we employed a transgenic AD mouse model (the APP/PS1 mouse) that expresses mouse/human APP (Mo/Hu APP695swe) and mutant human PS1 (PS1-dE9) protein. We analyzed changes of early endosomal constituent Rab5, late endosomal marker Rab7, autophagosome marker LC3B, lysosomal marker Lamp1/2 of EAL pathway, and further detected the alterations of UVRAG, Beclin1, and Rubicon in cortex and hippocampus, the two main vulnerable regions at different aged AD mouse brain.
METHODS
Mice
B6Cg-Tg (APPswe, PSEN1dE9) 85Dbo mice were purchased from the Jackson Laboratory (Bar Harbor, USA) and crossed with C57BL/6J to maintain this line. Genotyping of tail biopsies was performed by PCR. In this study, 3-, 7-, and 12-month-old male and female transgenic mice and age- and gender-matched wild-type (Wt) littermate controls (4–7 mice per group) were used. All mice were maintained in specific pathogen-free facilities in 12-h light-dark cycle rooms and had free access to water and standard rodent chow. All animal procedures were approved by the Institutional Animal Care and Use Committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology and were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Immunohistochemistry
The mice were sacrificed by decapitation and the brains were quickly removed. The left halves of the brains were fixed in 4% paraformaldehyde and embedded in paraffin, while the right halves were quickly frozen and stored at −80°C for biochemical analysis. Immunohistochemistry was performed according to methods described previously.[19] For 6E10 staining, which detects Aβ, the blocking step was preceded by treatment with 70% formic acid for 10 min. The following primary antibodies were used: 6E10 (monoclonal mouse anti-Aβ1-16 1:1000, Biolegend, USA), polyclonal rabbit anti-Rab5 (1:300, Abcam, USA), polyclonal goat anti-Rab7 (1:100, Santa Cruz, USA), polyclonal rabbit anti-LC3B (1:200, Cell Signaling Technology, USA), polyclonal rabbit anti-Lamp1 (1:400, Abcam), polyclonal goat anti-Beclin1 (1:100, Santa Cruz), polyclonal rabbit anti-UVRAG (1:400, Millipore, USA), polyclonal rabbit anti-Rubicon (1:50, LifeSpan Biosciences, USA), Alexa Fluor-conjugated antibodies (Molecular Probes, USA), and HRP-conjugated antibodies (chromogenic). Nuclei were labeled with DAPI or hematoxylin. Images were acquired using a confocal microscope (Fluoview10, Olympus, Tokyo, Japan) or an epifluorescence microscope (Olympus).
Western blotting analysis
Western blotting analysis of the entire right hemispheres or dissected cortex/hippocampi of the right hemispheres was executed as described before.[19] Polyvinylidene difluoride membranes were incubated overnight with primary antibodies, and then HRP-conjugated secondary antibodies (Jackson ImmunoResearch, USA) were applied, and chemiluminescence (Supersignal West Pico, Thermo Fisher Scientific, USA) was used to visualize the luminescence. The following primary antibodies were used for immunoblotting: polyclonal rabbit anti-Rab5 (1:1000, Abcam), monoclonal mouse anti-Rab7 (clone Rab7-117, 1:1000, Sigma-Aldrich, USA), polyclonal rabbit anti-LC3B (1:1000, Cell Signaling), polyclonal goat anti-Lamp2 (1:200, Santa Cruz), polyclonal rabbit anti-UVRAG (1:2000, Millipore), monoclonal mouse anti-Beclin1 (1:500, BD Biosciences, USA), and β-actin (clone AC-74, 1:10,000, Sigma-Aldrich).
Statistical analysis
Data are presented as mean ± standard error. Quantification of Western blotting and immunofluorescence was carried out by Image J software (the National Institutes of Health, the US Department of Health and Human Services, USA). Normality and homoscedasticity assumptions were reached. The comparison between Wt and transgenic groups was done by two-tailed t-test with GraphPad Prism (Graphpad Software Inc., USA) and diagrams were also plotted with this software. Differences were considered statistically significant when P < 0.05.
RESULTS
Endosomal–autophagic–lysosomal pathway was dysregulated in 12-month-old APP/PS1 mice
The 12-month-old APP/PS1 mice exhibited abundant Aβ accumulation [Supplementary Figure 1 (366.5KB, tif) ] and also showed impaired spatial memory, which were similar to symptoms observed in AD patients. To confirm the EAL pathway dysfunction in AD mouse brain, we measured the expression of endosomal organizers Rab5 and Rab7, autophagosome marker LC3B, and lysosomal proteins Lamp1 and Lamp2. Western blotting revealed significantly increased expression of all markers above in hemisphere homogenates of 12-month-old APP/PS1 mouse brains compared to that of control mice (APP/PS1 vs. Wt, t = 3.718, P = 0.0040; t = 6.243, P < 0.0001; t = 7.669, P < 0.0001; and t = 2.421, P = 0.0360 for Lamp2, Rab5, Rab7, and LC3B II/I, respectively) [Figure 1a and 1b], indicating activation of EAL pathway in the AD mouse model.
Figure 1.

EAL dysregulation in 12-month-old APP/PS1 mice. (a) Western blotting analysis of Lamp2, Rab5, Rab7, and LC3B-I/II in hemisphere homogenates of Wt and APP/PS1 (Tg) mice (n = 6), using β-actin as a loading control. (b) Quantitative analysis of Lamp2, Rab5, Rab7, and LC3B-II/I levels. (c) Immunostaining of Rab5, Rab7, LC3B, and of Lamp1 in cortex and hippocampus of Wt and Tg mice (n = 4). Scale bar of immunofluorescence images = 50 μm. Scale bar of Lamp1 images = 40 μm. *P < 0.05, †P < 0.01, ‡P < 0.0001. Wt: Wildw type; EAL: Endosomal–autophagic–lysosomal; Tg: Transgenic.
Amyloid plaque deposition in 12- and 7-month-old APP/PS1 mice. (a) 12-month-old APP/PS1 mice exhibited a considerable amount of Aβ deposition, mainly in extracellular plaques in the hippocampus and cortex. Scale bar = 500 μm. (b) A significant elevation of Aβ 40/42 was confirmed by sandwich ELISA of the hemisphere homogenate from the APP/PS1 mice. (c) 7-month-old APP/PS1 mice began to develop intracellular Aβ accumulation, while 12-month-old APP/PS1 mice showed not only extracellular plaques but also intracellular Aβ deposits. Scale bar = 40 μm. Aβ: Amyloid-β.
We also analyzed the regional distributions of these markers in cortex and hippocampus via immunofluorescence staining (parietal and temporal lobes and CA1 were shown). Rab5 mainly concentrated in the neuronal soma, particularly in the perikarya, while Rab7 was located in the neuronal submembrane region and proximal neurites. LC3B was distributed both in the soma and neurites of neurons [Figure 1c]. Compared to Wt littermates, expression of Rab5, Rab7, and LC3B was significantly increased in the cortex of APP/PS1 mice [Figure 1c]. Surprisingly, we discovered markedly decreased expression of Rab7, accompanied by elevation of Rab5 and LC3B levels, in the hippocampi of AD mice [Figure 1c]. In addition, LC3B was primarily distributed in neuronal somata, rather than both in somata and neurites as found in the Wt mice [Figure 1c]. This may indicate a disturbance in autophagosome function in the AD mice. Similar to LC3B, Lamp1 expression was increased in the cortices and hippocampi of APP/PS1 mice and exhibited a plaque-like distribution which is consistent with the previous studies which revealed strong immunoreactivity of Lamp1/2 in cells and in cell process surrounding plaques in AD brains [Figure 1c].[20,21] This may be due to the accumulation of lysosomes and autophagolysosomes in neurites or glial cells around amyloid plaques. Taken together, these findings indicate that the EAL pathway was dysregulated and distinct mechanisms may be involved in cortex and hippocampus of the 12-month-old AD mice.
Rab7 was involved in amyloid-β-related pathology and relevant with class III phosphatidylinositol 3-kinase complex components in APP/PS1 mouse brain
The discrepancy of Rab7 expression between cortices and hippocampi of 12-month-old APP/PS1 mice suggests that Rab7 may play a special role in Aβ-related EAL pathway dysregulation in AD. To confirm the possible link between Rab7 and Aβ aggregation in neurons, we carried out double-labeled immunofluorescent staining of Rab7 and Aβ, and did observe the co-localization of two proteins in neurons of 12-month-old APP/PS1 mice [Figure 2a, merged, labeled in yellow]. These data implied that Rab7 and Aβ may be functionally related to AD pathogenesis.
Figure 2.

Co-localization of Rab7 with Aβ and PI3KC3 complex components in 12-month-old APP/PS1 mice. (a) Co-localization of Rab7 (green) and 6E10 (red) in 12-month-old APP/PS1 (Tg) cortex (n = 4) scale bar = 80 μm. (b) Co-localization of Rab7 (green) and Beclin1 or UVRAG (red) in 12-month-old Tg cortex (n = 4) scale bar = 40 μm. Aβ: Amyloid-β; Tg: Transgenic; PI3KC3: Class III phosphatidylinositol 3-kinase.
Previous in vitro studies have disclosed the interaction of Rab7 with components of the PI3KC3 complex, Beclin1, UVRAG, and Rubicon in mediating endosome and autophagosome maturation.[15,18] In 12-month-old APP/PS1 mouse brain, the co-localization of Rab7 with Beclin1, UVRAG [Figure 2b], and Rubicon [Figure 3a] was verified by means of immunofluorescent double labeling, which indicates that UVRAG, Beclin1, and Rubicon may also participate in regulating Rab7 in AD mice.
Figure 3.
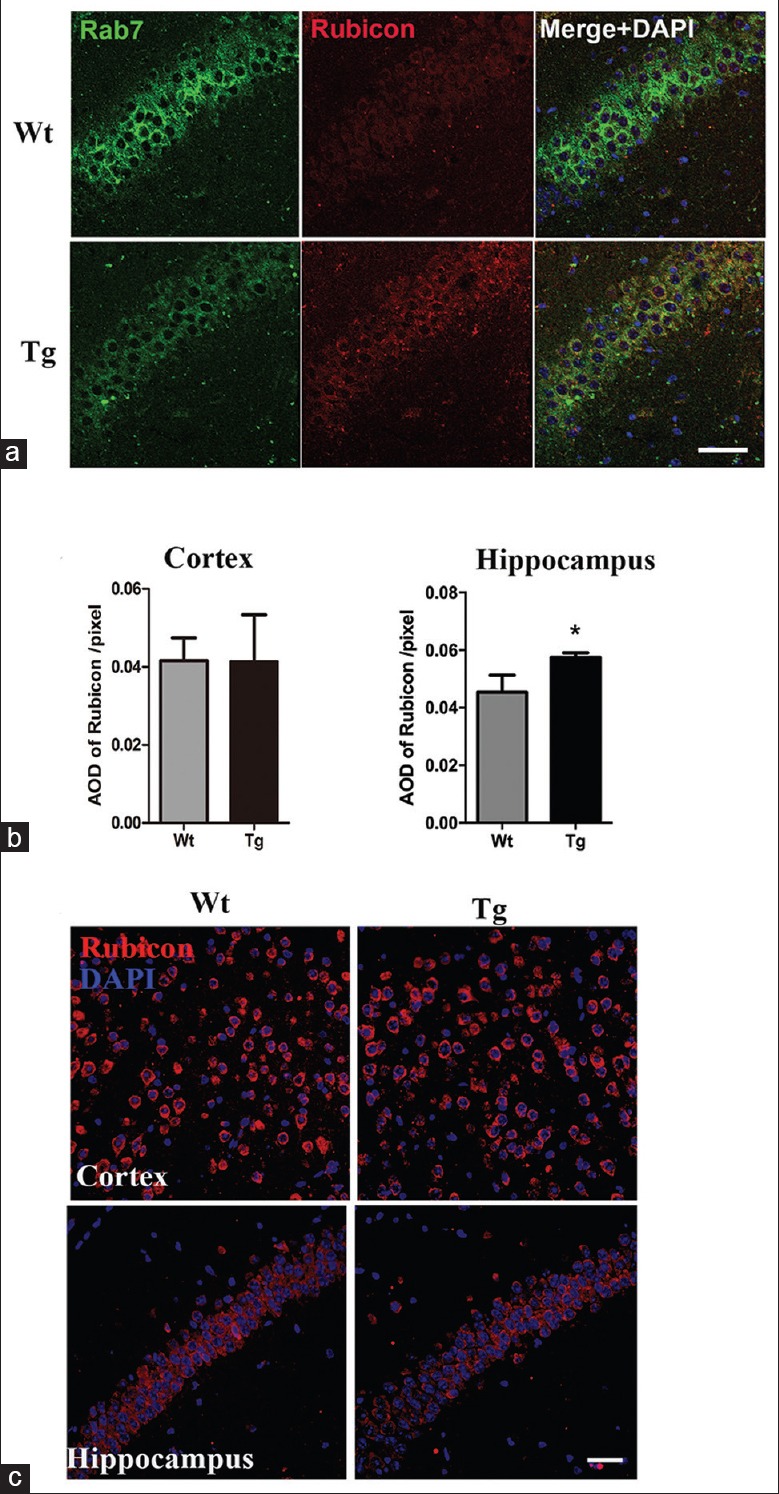
Rubicon in 7- and 12-month-old APP/PS1 mice. (a) Double-labeled immunofluorescence detection of Rubicon (red) and Rab7 (green) in 12-month-old Wt and Tg hippocampus (n = 4). Scale bar = 50 μm. (b) Quantitative analysis of AOD of Rubicon in 12-month-old APP/PS1 cortex and hippocampus. *P < 0.01. (c) Immunofluorescence of Rubicon (red) in 7-month-old cortex and hippocampus of Wt and Tg mice (n = 4). Scale bar = 40 μm. AOD: Average optical density; Wt: Wild type; Tg: Transgenic.
Alteration of Rab7-related class III phosphatidylinositol 3-kinase complex components in 12-month-old APP/PS1 mice
As depicted above, the expression pattern of Rab7 in the cortex and hippocampus of 12-month-old APP/PS1 mice was opposite. Quantitative analysis of Western blotting demonstrated that Rab7, Beclin1, and UVRAG were significantly increased in the APP/PS1 cortex (t = 5.367, P = 0.0330 for Rab7, t = 6.149, P = 0.0254 for Beclin1, and t = 8.362, P = 0.0140 for UVRAG) [Figure 4a and 4b], while Rab7 and Beclin1 were obviously decreased in the APP/PS1 hippocampus (t = 4.570, P = 0.0447 for Rab7 and t = 6.708, P = 0.0215 for Beclin1) [Figure 4c and 4d].
Figure 4.

Alternations of Rab7-related PI3KC3 complex components in 12-month-old APP/PS1 mice. (a and b) Western blotting and quantitative analysis of Rab7, UVRAG, and Beclin1 in Wt and APP/PS1 (Tg) cortex. (c and d) Western blotting and quantitative analysis of Rab7, UVRAG, and Beclin1 in Wt and Tg hippocampus. β-actin is used as a loading control (n = 4) *P < 0.05. (e and f) Double immunofluorescence-labeled Beclin1 (green) and UVRAG (red) in the cortex (e) and hippocampus (f) of Wt and Tg mice (n = 4). Merged images show co-localized Beclin1 and UVRAG in yellow. Scale bar = 30 μm. PI3KC3: Class III phosphatidylinositol 3-kinase; Wt: Wild type; Tg: Transgenic.
It has been reported that UVRAG can bind to Beclin1 and stabilize the Beclin1–PI3KC3 complex, which can activate Rab7 and induce autophagy.[16,17] As shown in Figure 4e, Beclin1 and UVRAG co-localized with each other, and were both increased in the cortex of APP/PS1 mice relative to controls [Figure 4e]. It is worth noting that the number of Beclin1-positive cells decreased markedly in the hippocampus of AD mice [Figure 4f]. Moreover, distribution of UVRAG in hippocampus of APP/PS1 mice is obviously concentrated in somata [Figure 4f], which may be a result of neurite dystrophy in AD hippocampal neurons.
In vitro study indicated that Rubicon could negatively regulate Rab7 activation.[18] Our staining revealed co-localization of Rubicon and Rab7 in the cortex (data not shown) and hippocampus of APP/PS1 mice [Figure 3a]. Compared with that of Wt mice, quantitative analysis of average optical density (AOD) of Rubicon revealed no obvious change in cortex (t = 0.045, P = 0.9647), while a prominent increase was noticed in hippocampus of APP/PS1 mice (t = 3.950, P = 0.0055) [Figure 3b].
Endosomal–autophagic–lysosomal dysregulation and alteration of Rab7-related class III phosphatidylinositol 3-kinase complex components in 7-month-old APP/PS1 mice
To reveal whether changes of the EAL pathway are dynamic in Aβ pathogenesis, we detected markers of EAL pathway and Rab7-related PI3KC3 components in 3-month-old APP/PS1 mice without neuronal Aβ accumulation and 7-month-old transgenic mice with early intracellular Aβ deposits [Supplementary Figure 1 (366.5KB, tif) ]. No obvious EAL pathway marker alterations were observed in 3-month-old transgenic brain (data not shown). Different from 12-month-old mice, all markers of EAL pathway, including Rab7, were significantly upregulated in the cortex (t = 3.863, P = 0.0083 for Rab7, t = 4.062, P = 0.0066 for Rab5, and t = 7.324, P = 0.0030 for LC3BII/I in quantitative analysis of Western blotting) and hippocampus (t = 5.119, P = 0.0022 for Rab7, t = 8.131, P = 0.0002 for Rab5, and t = 8.273, P = 0.0002 for LC3B II/I in quantitative analysis of Western blotting) of 7-month-old transgenic brain [Figure 5a, 5c and 5d]. LC3B was mainly distributed in neuronal somata, similar to that of 12-month-old APP/PS1 group [Figure 5a]. Both UVRAG and Beclin1 were elevated in APP/PS1 cortex (t = 5.097, P = 0.0022 for UVRAG and t = 3.310, P = 0.0162 for Beclin1 in the quantitation of Western blotting) and hippocampus (t = 5.619, P = 0.0014 for UVRAG and t = 8.038, P = 0.0002 for Beclin1 in the quantitation of Western blotting), accompanied by an increase of UVRAG-Beclin1 co-localization [Figure 5b and 5d]. Meanwhile, the expression of Rubicon remained stable in 7-month-old mice [Figure 3c].
Figure 5.

Alternations of EAL pathway and Rab7-related proteins in 7-month-old APP/PS1 mice. (a) Immunostaining of Rab5, Rab7, LC3B, and Lamp1 in cortex and hippocampus of Wt and Tg mice (n = 4). Scale bar = 50 μm. (b) Double immunofluorescence-labeled Beclin1 (green) and UVRAG (red) in cortex and hippocampus of Wt and Tg mice (n = 4). Scale bar = 40 μm. (c) Representative Western blotting images of EAL markers, UVRAG and Beclin1, in 7-month-old Tg cortex and hippocampus. β-actin as a loading control (n = 4). (d) Quantitative analysis of proteins in Wt and Tg cortex and hippocampus. *P < 0.05, †P < 0.01, ‡P < 0.001. EAL: Endosomal–autophagic–lysosomal; Wt: Wild type; Tg: Transgenic.
DISCUSSION
In the present study, we found different EAL dysregulation patterns in presymptomatic and symptomatic stages of AD in APP/PS1 mice. Recent studies have revealed that APP, the precursor of Aβ, is processed via EAL pathway.[2] At 7 months old, when Aβ began to accumulate, all EAL pathway markers (Rab5, Rab7, and LC3B, Lamp1) are increased in the cortex and hippocampus of APP/PS1 mice, which indicates the activation of EAL pathway for clearance of upcoming Aβ aggregates. Similar results were found in the cortex of 12-month-old mice, except that Rab7, a key regulator of late endosome maturation and fusion in the EAL pathway, was downregulated in the hippocampus, accompanied by a reduction of its activating PI3KC complex components (UVRAG, Beclin1) and enhancement of its inhibiting component (Rubicon). Based on our in vitro study, we speculated that Rab7 and PI3KC complex components may play pivotal roles in EAL dysregulation and Aβ pathogenesis in AD mice.
Rab7, a small Rab-family GTPase, is mainly located in late endosomes, autophagosomes, and lysosomes. Catalysis of guanine-nucleotide exchange factor is to transform Rab7 from a GDP-bound to a GTP-bound form for activation.[10] GTP-bound Rab7 regulates the early-to-late endosome transition and the maturation of late endosomes and late AVs.[22] GTP-Rab7 also regulates trafficking of vesicles by interacting with dynein/dynactin/p150glued and FYCO1.[23] Thus, any abnormality in expression levels, activation, or transportation of Rab7 would disturb the equilibrium of autophagic flux, leading to dysregulation of the EAL pathway. Reversely, Rab7 expression and function could be regulated by Aβ pathogenesis. APP and its β-C-terminal-fragment (β-CTF) can activate Rab5, a Rab-family GTPase located in early endosomes.[11] Overexpression of Rab5 or its active mutant results in late endocytic dysregulation, but its null mutant reverses the early and late endosome abnormality.[24] Therefore, increased upstream endocytic uptake could alter Rab7 expression and function. In addition, intercellular accumulation of metabolites such as Aβ and β-CTF, ceramide, and cholesterol has been proven to induce late endosome dysfunction.[25] In fibroblasts from patients with Niemann–Pick disease type C, cholesterol accumulates in the late endosomes and lysosomes, sequesters Rab7 in cholesterol-rich structures, and impairs the normal function of Rab7 to mediate vesicle fusion with lysosomes.[26] Meanwhile, the abnormal autophagosomes and lysosomes in downstream of the autophagic flux could also induce the dysfunction of Rab7.
Increased expression of Rab5 in the APP/PS1 mouse cortex could represent accelerated endocytic uptake, or it could merely be a consequence of Rab7 dysfunction. In soluble Aβ-treated PC12 cells, increased and enlarged Rab5-positive endosomes appeared after blocking the late endosomes,[27] indicating that late endocytic dysregulation could, in turn, lead to abnormalities in early endosomes. Thus, we hypothesized that increased delivery of APP-rich vesicles to the autophagy–lysosomal pathway leads to the upregulation of Rab5 and Rab7, which then accelerates amyloidogenesis in the cortex. Continuous deposition of Aβ eventually causes a functional deficit of Rab7 and impaired fusion of the late endosome/AVs with lysosomes in the hippocampus.
In an attempt to clarify the molecular mechanism of Rab7 dysfunction in APP/PS1 mice, we investigated the expression of two Rab7-related proteins, UVRAG and Beclin1, in the cortex and hippocampus. Beclin1 is a core component of the PI3KC3 complex, coordinating with VPS34 and other autophagy-related proteins to induce autophagosome nucleation. In fact, decreased expression of Beclin1 was identified in affected brain regions of patients with AD.[28] Also notably, recent studies have revealed new functions of Beclin1 in regulating membrane traffic pathways, including those involved in endocytosis and autophagy.[29,30] Beclin1, possibly through its association with endosomes,[30] promotes surface internalization and sorting of APP to Rab7-positive late endosomes and endolysosomes.[31] A deficiency of Beclin1 was found to impair endosome maturation and eventually led to neurodegeneration.[30]
UVRAG is another protein that can bind to the Beclin1–PI3KC3 complex and induce autophagy.[16,29,30,31] Independent of Beclin1, UVRAG can interact with C-VPS/HOPS and activate Rab7 by promoting the GDP-to-GTP transition and accelerating autophagosome/endosome maturation and endosome fusion.[17] In our study, increased Beclin1–UVRAG co-localization was observed in the cortex of AD mouse model, implying aberrant activation of Rab7, with subsequently increased endocytosis and greater induction of autophagy.
Rubicon has been proved to serve as a negative regulator of endosomal maturation and autophagy. Rubicon binding sequesters UVRAG from C-VPS/HOPS and inhibits the activation of Rab7. In contrast, activated Rab7 could compete with UVRAG for binding to Rubicon, thus releasing UVRAG from Rubicon and promoting autophagy.[15,18] In endosomal maturation, these components of the PI3KC3 complex function as a feed-forward loop. In hippocampus of 12-month-old AD mice, decreased Rab7 and increased Rubicon facilitated UVRAG and Rubicon binding, then disrupted the normal function of Rab7 in endocytosis and autophagy, and finally impairs maturation of the late endosome and fusion of endosomes with lysosomes.
A recent study analyzed autophagic flux in CA1 neurons of the hippocampus of patients with early- and late-stage AD. The results suggested that during the early disease response, autophagosome formation and lysosome biogenesis are increased and their fusion is not altered; however, autophagic flux is progressively impaired due to a deficiency of cargo clearance as the disease progresses.[32] That study implied that autophagic flux is a dynamic condition and can change during the course of disease. Lesions of neurons in the hippocampus and cortex may not happen simultaneously in APP/PS1 mice, or EAL pathway involved in different regions does not share the same mechanism. Early in the neurodegenerative process of neurons of APP/PS1 hippocampus, accelerated endocytosis leads to upregulation of Rab7 and the EAL pathway, and increased recruitment of endosomes and autophagosomes in neurons. As the disease progresses to the later stage, there is a marked accumulation of AVs due to a deficiency in lysosomal clearance, or other factors, and to exhaustion of Rab7 and its related proteins, resulting in late endocytic dysfunction and impaired endosomal–lysosomal fusion [Figure 6]. All of the above could potentially help us understand the difference between cortex and hippocampus in expression and Rab7 and other markers in 12-month-old AD mice.
Figure 6.

Proposed models of EAL pathway and PI3KC3 component involvement in AD. PI3KC3: Class III phosphatidylinositol 3-kinase; EAL: Endosomal–autophagic–lysosomal; AD: Alzheimer's disease.
The mechanisms of EAL pathway involved in AD pathogenesis seem way more complicated than we thought. In further clinical applications, autophagy-regulating therapy should not simply induce or block autophagy globally, as this might not restore the proper autophagic flux.
In summary, our data showed that dysregulation of EAL pathway could change along with the disease's progression and varies in different brain regions. Rab7 and its related PI3KC3 complex components may be involved in EAL dysregulation in AD. Understanding the underlying mechanism of this dysregulation has important implications for developing potential therapeutic methods by targeting autophagy in neurodegenerative diseases.
Supplementary information is linked to the online version of the paper on the Chinese Medical Journal website.
Financial support and sponsorship
This work was supported by a grant of National Natural Science Foundation of China (No. 81271406).
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Yuan-Yuan Ji
REFERENCES
- 1.Bird TD. Genetic factors in Alzheimer's disease. N Engl J Med. 2005;352:862–4. doi: 10.1056/NEJMp058027. doi: 10.1056/NEJMp058027. [DOI] [PubMed] [Google Scholar]
- 2.Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120:4081–91. doi: 10.1242/jcs.019265. doi: 10.1242/jcs.019265. [DOI] [PubMed] [Google Scholar]
- 3.Wei Z, Chen XC, Song Y, Pan XD, Dai XM, Zhang J, et al. Amyloid β protein aggravates neuronal senescence and cognitive deficits in 5XFAD mouse model of Alzheimer's disease. Chin Med J. 2016;129:1835–44. doi: 10.4103/0366-6999.186646. doi: 10.4103/0366-6999.186646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH, et al. Macroautophagy – A novel beta-amyloid peptide-generating pathway activated in Alzheimer's disease. J Cell Biol. 2005;171:87–98. doi: 10.1083/jcb.200505082. doi: 10.1083/jcb.200505082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kandalepas PC, Sadleir KR, Eimer WA, Zhao J, Nicholson DA, Vassar R, et al. The Alzheimer's β-secretase BACE1 localizes to normal presynaptic terminals and to dystrophic presynaptic terminals surrounding amyloid plaques. Acta Neuropathol. 2013;126:329–52. doi: 10.1007/s00401-013-1152-3. doi: 10.1007/s00401-013-1152-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–97. doi: 10.1038/nm.3232. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- 7.Jaeger PA, Wyss-Coray T. All-you-can-eat: Autophagy in neurodegeneration and neuroprotection. Mol Neurodegener. 2009;4:16. doi: 10.1186/1750-1326-4-16. doi: 10.1186/1750-1326-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nixon RA, Yang DS. Autophagy failure in Alzheimer's disease – Locating the primary defect. Neurobiol Dis. 2011;43:38–45. doi: 10.1016/j.nbd.2011.01.021. doi: 10.1016/j.nbd.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chua CE, Gan BQ, Tang BL. Involvement of members of the Rab family and related small GTPases in autophagosome formation and maturation. Cell Mol Life Sci. 2011;68:3349–58. doi: 10.1007/s00018-011-0748-9. doi: 10.1007/s00018-011-0748-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang T, Ming Z, Xiaochun W, Hong W. Rab7: Role of its protein interaction cascades in endo-lysosomal traffic. Cell Signal. 2011;23:516–21. doi: 10.1016/j.cellsig.2010.09.012. doi: 10.1016/j.cellsig.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 11.Xu W, Weissmiller AM, White JA, 2nd, Fang F, Wang X, Wu Y, et al. Amyloid precursor protein-mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. J Clin Invest. 2016;126:1815–33. doi: 10.1172/JCI82409. doi: 10.1172/JCI82409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eskelinen EL. Maturation of autophagic vacuoles in mammalian cells. Autophagy. 2005;1:1–10. doi: 10.4161/auto.1.1.1270. doi: 10.4161/auto.1.1.1270. [DOI] [PubMed] [Google Scholar]
- 13.Ginsberg SD, Mufson EJ, Alldred MJ, Counts SE, Wuu J, Nixon RA, et al. Upregulation of select Rab GTPases in cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer's disease. J Chem Neuroanat. 2011;42:102–10. doi: 10.1016/j.jchemneu.2011.05.012. doi: 10.1016/j.jchemneu.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jain N, Ganesh S. Emerging nexus between RAB GTPases, autophagy and neurodegeneration. Autophagy. 2016;12:900–4. doi: 10.1080/15548627.2016.1147673. doi: 10.1080/15548627.2016.1147673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin MG, Zhong Q. Interaction between small GTPase Rab7 and PI3KC3 links autophagy and endocytosis: A new Rab7 effector protein sheds light on membrane trafficking pathways. Small GTPases. 2011;2:85–8. doi: 10.4161/sgtp.2.2.15256. doi: 10.4161/sgtp.2.2.15256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, et al. Autophagic and tumour suppressor activity of a novel beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8:688–99. doi: 10.1038/ncb1426. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- 17.Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol. 2008;10:776–87. doi: 10.1038/ncb1740. doi: 10.1038/ncb1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun Q, Westphal W, Wong KN, Tan I, Zhong Q. Rubicon controls endosome maturation as a rab7 effector. Proc Natl Acad Sci U S A. 2010;107:19338–43. doi: 10.1073/pnas.1010554107. doi: 10.1073/pnas.1010554107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang M, Wang X, Jiang F, Wang W, Vincent I, Bu B, et al. Mitotic epitopes are incorporated into age-dependent neurofibrillary tangles in Niemann-Pick disease type C. Brain Pathol. 2010;20:367–77. doi: 10.1111/j.1750-3639.2009.00286.x. doi: 10.1111/j.1750-3639.2009.00286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barrachina M, Maes T, Buesa C, Ferrer I. Lysosome-associated membrane protein 1 (LAMP-1) in Alzheimer's disease. Neuropathol Appl Neurobiol. 2006;32:505–16. doi: 10.1111/j.1365-2990.2006.00756.x. doi: 10.1111/j.1365-2990.2006.00756.x. [DOI] [PubMed] [Google Scholar]
- 21.Torres M, Jimenez S, Sanchez-Varo R, Navarro V, Trujillo-Estrada L, Sanchez-Mejias E, et al. Defective lysosomal proteolysis and axonal transport are early pathogenic events that worsen with age leading to increased APP metabolism and synaptic abeta in transgenic APP/PS1 hippocampus. Mol Neurodegener. 2012;7:59. doi: 10.1186/1750-1326-7-59. doi: 10.1186/1750-1326-7-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poteryaev D, Datta S, Ackema K, Zerial M, Spang A. Identification of the switch in early-to-late endosome transition. Cell. 2010;141:497–508. doi: 10.1016/j.cell.2010.03.011. doi: 10.1016/j.cell.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 23.Pankiv S, Alemu EA, Brech A, Bruun JA, Lamark T, Overvatn A, et al. FYCO1 is a rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol. 2010;188:253–69. doi: 10.1083/jcb.200907015. doi: 10.1083/jcb.200907015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cataldo AM, Mathews PM, Boiteau AB, Hassinger LC, Peterhoff CM, Jiang Y, et al. Down syndrome fibroblast model of Alzheimer-related endosome pathology: Accelerated endocytosis promotes late endocytic defects. Am J Pathol. 2008;173:370–84. doi: 10.2353/ajpath.2008.071053. doi: 10.2353/ajpath.2008.071053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lauritzen I, Pardossi-Piquard R, Bourgeois A, Pagnotta S, Biferi MG, Barkats M, et al. Intraneuronal aggregation of the β-CTF fragment of APP (C99) induces aβ-independent lysosomal-autophagic pathology. Acta Neuropathol. 2016;132:257–76. doi: 10.1007/s00401-016-1577-6. doi: 10.1007/s00401-016-1577-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huynh KK, Gershenzon E, Grinstein S. Cholesterol accumulation by macrophages impairs phagosome maturation. J Biol Chem. 2008;283:35745–55. doi: 10.1074/jbc.M806232200. doi: 10.1074/jbc.M806232200. [DOI] [PubMed] [Google Scholar]
- 27.Yuyama K, Yanagisawa K. Late endocytic dysfunction as a putative cause of amyloid fibril formation in Alzheimer's disease. J Neurochem. 2009;109:1250–60. doi: 10.1111/j.1471-4159.2009.06046.x. doi: 10.1111/j.1471-4159.2009.06046.x. [DOI] [PubMed] [Google Scholar]
- 28.Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118:2190–9. doi: 10.1172/JCI33585. doi: 10.1172/jci33585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex – At the crossroads of autophagy and beyond. Trends Cell Biol. 2010;20:355–62. doi: 10.1016/j.tcb.2010.03.002. doi: 10.1016/j.tcb.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McKnight NC, Zhong Y, Wold MS, Gong S, Phillips GR, Dou Z, et al. Beclin 1 is required for neuron viability and regulates endosome pathways via the UVRAG-VPS34 complex. PLoS Genet. 2014;10:e1004626. doi: 10.1371/journal.pgen.1004626. doi: 10.1371/journal.pgen.1004626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Swaminathan G, Zhu W, Plowey ED. BECN1/Beclin 1 sorts cell-surface APP/amyloid β precursor protein for lysosomal degradation. Autophagy. 2016;12:2404–19. doi: 10.1080/15548627.2016.1234561. doi: 10.1080/15548627.2016.1234561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bordi M, Berg MJ, Mohan PS, Peterhoff CM, Alldred MJ, Che S, et al. Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy. 2016;12:2467–83. doi: 10.1080/15548627.2016.1239003. doi: 10.1080/15548627.2016.1239003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Amyloid plaque deposition in 12- and 7-month-old APP/PS1 mice. (a) 12-month-old APP/PS1 mice exhibited a considerable amount of Aβ deposition, mainly in extracellular plaques in the hippocampus and cortex. Scale bar = 500 μm. (b) A significant elevation of Aβ 40/42 was confirmed by sandwich ELISA of the hemisphere homogenate from the APP/PS1 mice. (c) 7-month-old APP/PS1 mice began to develop intracellular Aβ accumulation, while 12-month-old APP/PS1 mice showed not only extracellular plaques but also intracellular Aβ deposits. Scale bar = 40 μm. Aβ: Amyloid-β.