

Abstract
Objective:
To review recent research advances on tau, a major player in Alzheimer's disease (AD) pathogenesis, a biomarker for AD onset, and potential target for AD therapy.
Data Sources:
This review was based on a comprehensive search using online literature databases, including PubMed, Web of Science, and Google Scholar.
Study Selection:
Literature search was based on the following keywords: Alzheimer's disease, tau protein, biomarker, cerebrospinal fluid (CSF), therapeutics, plasma, imaging, propagation, spreading, seeding, prion, conformational templating, and posttranslational modification. Relevant articles were carefully reviewed, with no exclusions applied to study design and publication type.
Results:
Amyloid plaques enriched with extracellular amyloid beta (Aβ) and intracellular neurofibrillary tangles comprised of hyperphosphorylated tau proteins are the two main pathological hallmarks of AD. Although the Aβ hypothesis has dominated AD research for many years, clinical Aβ-targeting strategies have consistently failed to effectively treat AD or prevent AD onset. The research focus in AD has recently shifted to the role of tau in AD. In addition to phosphorylation, tau is acetylated and proteolytically cleaved, which also contribute to its physiological and pathological functions. Emerging evidence characterizing pathological tau propagation and spreading provides new avenues for research into the molecular and cellular mechanisms underlying AD pathogenesis. Techniques to detect tau at minute levels in CSF and blood have been developed, and improved tracers have facilitated tau imaging in the brain. These advances have potential to accurately determine tau levels at early diagnostic stages in AD. Given that tau is a potential therapeutic target, anti-tau immunotherapy may potentially be a viable treatment strategy in AD intervention.
Conclusion:
Detecting changes in tau and targeting tau pathology represent a promising lead in the diagnosis and treatment of AD.
Keywords: Alzheimer's Disease, Biomarker, Immunotherapy, Tau Imaging, Tau Protein, Transcellular Propagation
INTRODUCTION
Alzheimer's disease (AD) is the leading cause of age-related dementia, and approximately 47 million people currently suffer from dementia worldwide. With the current aging population, the number of dementia cases is projected to increase to more than 131 million by 2050.[1] Amyloid plaques composed of extracellular amyloid beta (Aβ) and intracellular neurofibrillary tangles (NFTs) containing hyper-phosphorylated tau (p-tau) represent the two main pathological hallmarks of AD.[2] The biggest obstacle in AD treatment is the lack of a basic understanding of AD etiology. Given the pathological role of Aβ plaques in AD, the β-amyloid (or Aβ cascade) hypothesis has dominated the field of AD research for many years.[3] However, targeting Aβ has not resulted in successful reversion or attenuation of AD symptoms. Therefore, research in AD therapeutics has shifted toward the role of tau in AD pathogenesis. Although tau pathology is considered secondary to Aβ accumulation, pathological changes in tau correlate more accurately with disease progression and cognitive changes.[4] Indeed, tau may contribute to the pathogenic AD onset through Aβ-dependent and Aβ-independent mechanisms. New findings describing the ability of tau to propagate transcellularly have recently attracted much attention. Biomarkers aimed at early detection of pathological changes in tau and potential immunotherapies targeting tau are reviewed.
BIOCHEMICAL AND PHYSIOLOGICAL PROPERTIES OF TAU
Human tau is encoded by the MAPT gene on chromosome 17q21.31,[5] comprising 16 exons that are alternatively spliced on exons 2, 3, and 10 to generate 6 tau protein isoforms ranging from 352 to 441 amino acids in the adult human brain[6] [Figure 1]. Tau consists of three domains [Figure 2]: an assembly domain, a projection domain, and a proline-rich domain.[7] Tau is predominantly expressed in neurons and with lower expression in glia cells[8] and can also be found in the extracellular environment.[9] Given its prominent localization on microtubule-enriched axonal processes, tau is often used as an axonal marker,[10] while recent studies also report a physiological role for tau in dendrites.[11]
Figure 1.

Alternative splicing of the MAPT gene and tau isoforms. Tau is encoded by the MAPT gene comprising 16 exons. Exons 0 and 1 encode the 5’ untranslated sequence of MAPT mRNA while exon 14 encodes part of the 3’ untranslated region. Exons 4a, 6, and 8 are only transcribed in peripheral tissues. Exons 2 and 3 encode 29-amino acid residue inserts near the amino-terminus, while exons 9–12 encode microtubule binding domain repeats near the carboxyl-terminus. Alternative splicing of MAPT results in 6 isoforms ranging from 352 to 441 amino acids. Tau isoforms are named based on the number of amino-terminal inserts (0N, 1N, and 2N) or carboxyl-terminal microtubule binding domain repeats (3R and 4R).
Figure 2.

Domains of the protein structure of tau. Tau consists of 3 domains: The projection domain resides in the amino-terminus, the proline-rich domain resides in the middle part, and the assembly domain is situated in the carboxyl-terminus. The assembly domain is composed of repeat domains and flanking regions and supports microtubule assembly. The projection domain extends away from microtubules and does not bind to microtubules. The proline-rich domain contains up to seven PXXP motifs, which are interaction motifs that bind SH3-domain protein components.
Tau is also extensively modified posttranslationally and has been reported to be modified by phosphorylation, acetylation, glycosylation, glycation, deamidation, isomerization, nitration, methylation, ubiquitylation, sumoylation, and truncation.[12] The largest tau isoform (tau 441) can be phosphorylated at numerous serine/threonine and tyrosine residues.[13] Ser/thr motifs preceding a proline residue are phosphorylated by proline-directed protein kinases (PDPKs) such as glycogen synthase kinase-3β (GSK-3β), cyclin-dependent-like kinase-5 (CDK5), and dual specificity tyrosine-phosphorylation-regulated kinase 1A. Proline-independent ser/thr residues are phosphorylated by non-PDPKs such as calcium/calmodulin-activated PKII, microtubule affinity-regulated kinase 110, PKA, and casein kinase 1.[13] Protein phosphatase 2A (PP2A) accounts for approximately 70% of all tau dephosphorylation activity in the human brain.[14]
Under physiological conditions, tau is highly soluble and natively unfolded in the cytoplasm. At the cellular level, tau regulates microtubule dynamics and facilitates reorganization of the microtubule cytoskeleton and mediates different functions depending on its subcellular location. In mature neurons, tau is concentrated in the axonal process where it interacts with microtubules through its assembly domain, thereby stabilizing microtubules and promoting microtubule assembly.[15] In addition, tau regulates axon elongation and maturation[16] and axonal transport via different mechanisms.[17] Although only a small proportion of tau is localized to dendrites, dendritic tau is thought to be involved in regulating synaptic plasticity.[11] Moreover, nuclear tau may play an important role in maintaining the integrity of genomic DNA, cytoplasmic RNA, and nuclear RNA.[18] Tau-deficient mouse models have also implicated roles for tau in regulating neuronal activity, neurogenesis, and iron transport.[19]
TAU-MEDIATED NEURODEGENERATION
As a pathological hallmark of AD, NFTs are intracellular inclusions composed of hyper-p-tau arranged in paired helical filaments (PHFs).[20,21] Although the role of p-tau in AD pathology is thought to be secondary to Aβ accumulation in the brain, a growing body of evidence suggests that tau has unique roles in AD that are Aβ independent. Tau aggregates and tau missorting impair axonal transport, mitochondrial function, and cytoskeletal dynamics and elevate oxidative stress in AD in an Aβ-independent manner.
POSTTRANSLATIONAL MODIFICATIONS OF TAU IN ALZHEIMER'S DISEASE PATHOGENESIS
Tau phosphorylation
In healthy human brain, 1.9 moles of phosphate is on average associated with 1.0 mole of tau, whereas tau derived from pathological filaments in AD patients comprises 6–8 moles of phosphate per mole of tau.[22] Tau hyperphosphorylation in AD is derived from increased phosphorylation and/or decreased dephosphorylation. Tau phosphoregulation by PDPKs such as GSK-3β and CDK5 have been long implicated in AD pathogenesis;[23] however, evidence regarding alterations in their activity and levels in AD brain remain controversial.[23,24] Because PP2A-dependent tau dephosphorylation is compromised in AD, elevations in p-tau in AD brain may be due to reduced tau dephosphorylation.[25]
Hyper-p-tau in AD could exert pathological effects via several mechanisms: (1) Alterations in microtubule stability: Phosphorylation reduces tau affinity for tubulin causing p-tau to detach from microtubules.[26,27] (2) Tau phosphorylation promotes tau aggregation.[28] In addition to tangle formation associated with aggregation, this may have two additional effects in synaptic function and turnover. (3) Synaptic dysfunction: p-tau is aberrantly trafficked from axons to form aggregates in the somatodendritic compartment, thereby interfering with normal synaptic transmission.[29] (4) P-tau impairs proteosomal degradation and autophagy: As a small, unfolded, and short-lived cytosolic protein, tau is normally an ideal substrate for proteasomal degradation. P-tau aggregation reduces autophagy and subsequent protease cleavage, thereby inhibiting tau turnover.[30]
Tau acetylation
Tau can be acetylated by the P300/CBP acetyltransferase, a 300 kDa CREB-binding protein at several Lys residues within its flanking region[31] or repeat domain.[32] Tau can also be deacetylated by sirtuin 1 and histone deacetylase 6.[31] Tau acetylation at certain sites may have distinct effects on tau aggregation and degradation. For instance, acetylation at Lys259, Lys290, Lys321, or Lys353 facilitates tau degradation by suppressing tau phosphorylation and aggregation; tau acetylation is reduced at these sites in human AD brain and tau-transgenic mice.[32] However, tau acetylation at Lys280 is elevated in patients at early and moderate AD Braak stages, thus preventing tau degradation to promote p-tau accumulation.[31,33] More recently, tau acetylation at Lys174 has been identified as an early pathological event in AD brain. Tau-acetylation at this site reduces tau turnover, which is critical for tau-mediated toxicity.[34,35]
Tau O-GlcNAcylation
Tau N-glycosylation in human AD brain facilitates tau hyperphosphorylation, thus promoting its accumulation in PHF tangles.[36] In contrast, O-GlcNAcylation (a form of O-glycosylation) negatively regulates tau phosphorylation.[37] Interestingly, p-tau contains less O-GlcNAc than non-p-tau in human brain (up to 4-fold less), and downregulating O-GlcNAcylation can increase p-tau both in vitro and in vivo.[38]
Tau processing through proteolysis
Tau protein can undergo enzymatic and nonenzymatic cleavage in vitro and in vivo. Enzymes such as caspases, calpains, thrombin, and cathepsins cleave tau.[39] Experiments in vitro indicate that caspases 1, 3, 6, 7, and 8 can cleave tau at different sites, resulting in the generation of truncated tau species which are primed for subsequent phosphorylation.[40,41] Calpain activity is regulated by a calcium-dependent heat-stable inhibitor, calpastatin. In AD brain, calpastatin levels are found to be significantly decreased and calpains activity is aberrantly enhanced. Calpain-dependent tau cleavage generates a 17 kDa tau fragment found in pathological NFTs,[39] indicating that tau cleavage may facilitate aggregation of full-length tau.
Other enzymes involved in tau cleavage include asparagine endopeptidase (AEP)[42] and caspase 2.[43] AEP can cleave endogenous tau at Asn255 and Asn368 independently of caspases and calpains, generating neurotoxic fragments that form insoluble fibrils in vitro.[42] Caspase 2 cleaves tau at Asp314 generating a truncated 35 kDa fragment, which mislocalizes to dendritic spines, and also aberrantly promotes mislocalization of endogenous tau to spines.[43]
Tau aggregation
Tau aggregates range from soluble tau oligomers to insoluble tau deposits found in dystrophic neurites, neuropil threads, and NFTs in cell bodies. Tau aggregates are ubiquitous to AD and are key mediators of tau-mediated neurodegeneration.[44] Although the exact mechanism underlying tau aggregation is unknown, aberrant posttranslational modification may promote tau aggregation. Given that tau hyperphosphorylation precedes aggregation, and tau aggregates are obligately phosphorylated in AD brain, phosphorylation has been suggested as a primary trigger for tau aggregation. However, phosphorylation at certain sites renders tau refractory to aggregation, and tau aggregation can occur in vitro in a phosphorylation-independent manner.[45] Thus, phosphorylation itself is not sufficient to drive aggregation, but may help enhance ongoing multimerization events.
Although mechanisms underlying tau aggregation in AD brain remain unclear, aggregation appears to be necessary for tau-mediated neurodegeneration. Tau transgenic mice overexpressing an aggregate-prone tau variant developed AD-like tau pathology and cognitive deficits while tau pathology was absent in mice with expressing an aggregate-refractory tau variant.[46] Because NFT burden correlates tightly with AD severity, tau aggregation was initially presumed to drive NFT toxicity. Evidence from human AD brain and transgenic mice suggest that most neurons likely die independently of NFT formation. Moreover, neurons comprising NFT are able to survive for an extended period.[47,48] Interestingly, abrogating tau expression improved memory while NFTs remained present in a repressible tau transgenic mouse model, indicating that NFTs may not be necessary or sufficient for neurodegeneration.[49] An alternative explanation has been proposed where NFTs may act as a protective cellular response whereby toxic tau species are sequestered to limit intracellular damage. Since synapse loss, dysfunction, and cognitive deficits occur long before NFT formation, it has been suggested that soluble tau species (especially tau oligomers) may be the main cause of neurodegeneration. However, the exact oligomeric tau species that drive neurodegenerative processes have yet to be defined.
Mechanisms of tau-mediated neurodegeneration
Although the Aβ cascade hypothesis places tau downstream of Aβ aggregation and toxicity, growing evidence suggests that tau dysfunction may have a more significant role in mediating neurodegeneration independently of Aβ, which is discussed below.
Missorting of tau and synaptic loss
In AD, tau hyperphosphorylation can aberrantly drive p-tau into postsynaptic spines, thereby increasing dendritic tau levels and causing synaptic dysfunction.[11] Although Aβ also triggers synaptic dysfunction, reduction of tau levels can mitigate Aβ neurotoxicity, indicating that tau mediates Aβ toxicity in dendrites. Potential mechanisms underlying tau synaptotoxicity include Fyn-dependent phosphorylation of glutamate receptor subunits; tau is required for targeting the Src-family kinase Fyn to dendritic spines to phosphorylate the NMDA receptor subunit NR2B, thereby stabilizing NR2B interactions with postsynaptic density protein 95 that potentiate Aß-induced glutamate excitotoxicity.[11]
Tau dysfunction and impaired axonal transport
Tau hyperphosphorylation and aggregation lead to abrogation of its normal cellular function. Both hyperphosphorylation and aggregation lead to impaired axonal tau transport by reducing tau binding to microtubules, causing microtubule disassembly. Importantly, kinesin motor-mediated axonal transport is impaired in AD, where p-tau is reported to impair kinesin-dependent axonal cargo trafficking by blocking transport of the adaptor-molecule c-Jun N-terminal kinase interacting protein 1 from the cell body to axons.[50]
Mitochondrial dysfunction
Mitochondria dysfunction has been previously implicated in AD pathogenesis. Studies both in vitro and in vivo indicate a pathogenic role for p-tau in dysregulating mitochondria fission and fusion, thus inducing deficiencies in mitochondria respiratory function and consequent ATP generation.[51] Interactions between filamentous actin (F-actin) and the mitochondria fission protein dynamin-related protein 1 (DRP1) block translocation of DRP1 to mitochondria, thus promoting mitochondrial elongation. This suggests a role for actin in mediating tau-dependent mitochondria dysfunction.[51] In addition, overexpression of full-length tau promotes mitochondria elongation, while overexpression of truncated tau fragments induces mitochondrial fragmentation.[52]
Cytoskeletal dysfunction and oxidative stress
Cytoskeletal dysfunction in AD is pathologically characterized by F-actin-enriched intracellular aggregates known as Hirano bodies.[53] Previous studies indicate that tau is capable of inducing actin bundles which stabilize F-actin, thus reducing actin turnover and dynamics.[54] This has significant consequences for oxidative stress: AD brain is characterized by excessive oxidative stress, and transgenic tau mouse models show elevations in oxidative stress indicators such as free-radical damage and increased reactive oxygen species.[55] Tau dysregulation in neurons impairs the ability to cope with excessive oxidative stress. For instance, the stress response protein repressor element 1-silencing transcription factor is absent in neurons affected by abnormal tau.[55] Further, pathogenic tau species have also been shown to block the transport of peroxisomes loaded with antioxidants, thereby limiting cellular distribution of antioxidants.[56] These events leave neurons vulnerable to oxidative stress leading to DNA damage, an early event seen in AD pathogenesis.[57]
TRANSCELLULAR TAU PROPAGATION HYPOTHESIS
Progressive pathological tau severity (Braak staging) has been characterized based on postmortem autopsies by Braak and Braak.[58] Neurofibrillary pathology initiates within the anteromedial temporal lobe, progressing to the hippocampus proper and multimodal association areas, eventually spreading to secondary and primary cortices. An updated scheme recently proposed that the locus coeruleus (LC) is the first brain region to feature pathological changes in tau.[59] Pathological tau spreading in AD was previously thought to be cell-autonomous, where tau dispersion would take place between cells comprising similar gene expression profiles. According to this “selective vulnerability theory,” these cell types are intrinsically more vulnerable to tau-mediated pathogenesis and show functional and morphological abnormalities earlier in AD pathogenesis.[60] Alternatively, the “transcellular tau propagation hypothesis” describes transcellular tau spreading as a “prion-like” process. In this model, tau aggregates or aggregate-prone seeds escape the cell of origin and gain access to neighboring-naïve cells to nucleate intracellular aggregation through conformational templating. Newly transduced cells can then proceed to propagate misfolded tau to other naive cells[61] [Figure 3]. Templated propagation was first conceived from experiments in human and animal models of transmissible spongiform encephalopathies, including Creutzfeldt–Jakob disease, Kuru disease in human, and scrapie in sheep. Pathogenic protein species from affected individuals in these disorders have been shown to be transmissible to naïve animals, with the capacity to be repeatedly passaged to other animals.[62] The term “prion” was coined to describe a transmissible agent which demonstrated properties of a protein and was infectious.[63] Application of the prion concept to tau propagation is a groundbreaking concept which may apply to a variety of proteotoxic neurodisorders including AD, Parkinson's disease, and amyotrophic lateral sclerosis. However, since the prion hypothesis has not yet been definitively shown in human tauopathies, the concept in association with AD and related tauopathies remains controversial.[60]
Figure 3.
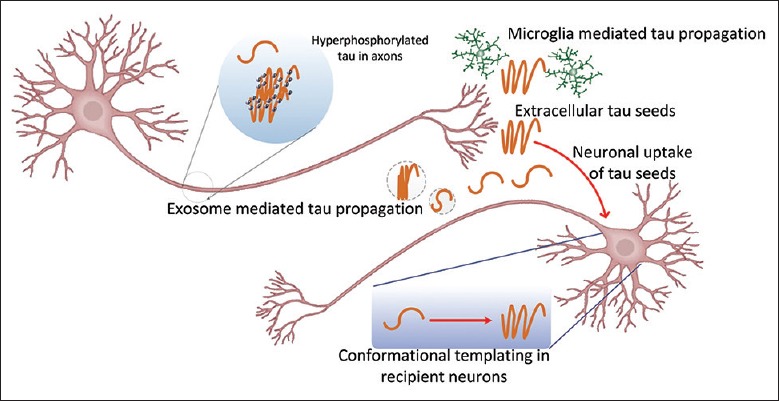
Proposed model for tau transcellular propagation. Hyperphosphorylated tau becomes misfolded, whereby tau aggregates in the neuronal axon, thus forming nucleating tau seeds that initiate transcellular propagation. Tau seeds can be secreted into the extracellular matrix in the form of tau-containing exosomes in a manner dependent on neuronal activity. Extracellular tau seeds can enter neurons via bulk endocytosis and promote intracellular tau aggregation by altering its conformational structure (conformational templating). Newly affected cells can proceed to propagate misfolded tau to other naive cells, thus propagating tau pathology. Microglia are also implicated in the tau transcellular propagation process.
Tau propagation experiments in vitro show exogenous truncated tau aggregates (fibrils) can enter cells and promote intracellular fibrillization of full-length tau, which normally does not aggregate spontaneously.[64] Newly formed fibrils could be subsequently transduced into cells in co-culture and further aggregate truncated tau monomers in vitro. Studies using preformed tau fibrils (PFFs) indicate that both full-length and truncated tau aggregates can facilitate tau propagation. Use of 35S radiolabeled tau in pulse-chase experiments indicates that new tau aggregates are formed largely by recruiting endogenous tau. However, PFFs induced robust tau propagation in cells expressing P301L mutant tau, thus demonstrating that P301L tau mutations enhance endogenous tau seeding.[65]
Similar observations of tau propagation in vivo have recently been described[66,67,68,69,70,71,72,73] [Table 1]. In these studies, mutant human tau aggregate seeds induce tau propagation in transgenic mice expressing human tau; brain extracts lacking tau aggregates fail to reproduce this phenomenon, indicating that tau seeds are required to induce pathological spreading in vivo.
Table 1.
Evidence of tau transcellular propagation in mouse models
| Tau in mouse models | Tau seeds | Mouse model | Effects | Reference |
|---|---|---|---|---|
| WT 2N4R human tau | NFT-containing extracts from aged P301S mouse brain | ALZ17 mice | Filamentous tau pathology in young mice spread time dependently | Clavaguera et al.[66] |
| Murine tau | P301S mice brain extracts | Nontransgenic mice | Tau threads and coiled bodies confined to injection sites | Clavaguera et al.[66] |
| P301L human tau | – | FVB-Tg4510 mice | Tau pathology spread from EC to synaptically connected regions | de Calignon et al.[67] |
| 1N4R P301S human tau | Tau aggregates from brain extracts | PS19 mice | Tau inclusions showed contralateral spreading to the hippocampus | Ahmed et al.[68] |
| 1N4R P301S human tau | PFFs | PS19 mice | Tau propagation is dependent on synaptic connectivity | Iba et al.[69] |
| 1N4R P301S human tau | PFFs | P301S mice | Altered neuronal network activity and impaired basal synaptic transmission and plasticity | Stancu et al.[70] |
| WT 2N4R human tau | AGD, PSP, CBD brain extracts | ALZ17 mice | Reconstitution of argyrophilic tau inclusions and hallmark lesions of each disease | Clavaguera et al.[71] |
| 1N4R P301S human tau | CBD/AD brain extracts | PS19 mice | Distinct pattern for tau propagation of each disease | Boluda et al.[72] |
| Human tau and murine tau | – | AAV-h-tau mice | Propagation was attenuated in AAV-h-tau mice pharmacologically depleted of microglia | Asai et al.[73] |
| 1N4R P301S human tau | – | PS19 mice | Presence of tau-containing exosome and inhibition of exosome synthesis reduce propagation | Asai et al.[73] |
–: Not applicable; WT: Wild type; NFT: Neurofibrillary tangle; PFFs: Preformed tau fibrils; PSP: Progressive supranuclear palsy; CBD: Corticobasal degeneration; AGD: Argyrophilic grain disease; AD: Alzheimer’s disease; EC: Entorhinal cortex.
Although most studies used brain extracts of unknown composition, PFFs comprising full-length tau or truncated tau from PS19 mouse models (overexpressing 1N4R mutant human tau P301S driven by a murine prion protein promoter) also demonstrate successful tau propagation in vivo, indicating that tau fibrils are sufficient to mediate propagation.[69] One concern regarding injection models is the possibility that tau seeds may freely diffuse away from the injection site. Interestingly, tau injection into the hippocampus or striatum/overlaying cortex consistently induced tau pathology in the LC, a noradrenergic nucleus in the brainstem distal from the injection site. Despite widespread transmission to distal sites, direct injection into the striatum failed to develop tau pathology at the injection site even 3 months postinjection.[69] This suggests that tau spreading in these models may depend on synaptic connectivity rather than spatial proximity. Moreover, tau inclusions in vivo show anterograde and retrograde transport[68] based on tau propagation in both efferent and afferent nuclei of the hippocampal formation.
Inducible tau transgenic mouse models such as FVB-Tg (tetO-TauP301L) 4510 have been established, where tau expression is confined to the entorhinal cortex (EC) to mimic early tau pathology in AD.[67] This model enables the characterization of pathological tau propagation from the medial entorhinal cortex to synaptically connected regions.[67] Using this model, tau pathology was observed to originate in the EC and spread to downstream synaptic pathways such as the dentate gyrus (DG), CA1-3 in the hippocampus, and cingulate cortex. However, recent evidence questions the notion that tau lesions initiate from the LC to other regions in PS19 mouse models.[74] Tau injection within the LC in PS19 mice induced tau propagation through the major afferent and efferent pathways within the LC; however, regions affected in early stages of AD such as the hippocampus and EC were devoid of tau lesions.
Although tau propagation has been reproduced in vivo in several models, the functional consequence is not well understood. Most studies observed no signs of neurodegeneration, despite evidence of tau propagation observed. This suggests that propagated tau species may not be neurotoxic.[67] However, recent evidence from primary neuronal cultures, organotypic hippocampal slices, and P301S mice describe altered neuronal network activity and impaired basal synaptic transmission and plasticity, suggesting that tau transduction may have some deleterious effects on neuronal function.[70]
Similar to prions, tau comprises unique conformational species or “strains.”[75] Stable expression of the tau repeat domain in cultured cells can indefinitely propagate distinctive amyloid conformations in a clonal fashion; reintroduction of tau from these lines into naive cells can reestablish identical clonal cell lines. Successive introduction of two different tau strains into P301S transgenic mice for three generations in vivo and subsequent immunopurification and reexposure of these tau strains to cultured cells can regenerate the original tau strains in culture. Moreover, tau isolates from patients with different tauopathies are shown to be associated with different tau strains. These findings provide a potential explanation for the distinctive pathological patterns observed in various tauopathies.[71] Distinct patterns for tau propagation have been observed following intracerebral injection of brain extracts from differing tauopathies into young PS19 mice.[72] These features might reflect the presence of distinctive pathogenic tau strains associated with differing tauopathies.
Despite evidence supporting tau spreading through a transsynaptic or transneuronal propagation pathway, recent work has challenged this view.[73] AAV-mediated human tau (h-tau) expression in neurons by injection into the medial EC in 4-month-old mice produces hyperphosphorylated human tau, where tau oligomers can propagate to the DG granule cell layer at 28 days postinjection. Intriguingly, propagation was attenuated in AAV-h-tau mice pharmacologically depleted of microglia, indicating a role for microglia in the propagation process. Mechanistic studies demonstrate that microglia can phagocytose and secrete tau in exosomes more efficiently than neurons and astrocytes. Furthermore, microglia-derived exosomes can transduce tau into neurons more efficiently than exosome-free tau in vivo. The presence of tau-containing exosomes has been confirmed in PS19 mouse models while systemic inhibition of exosome synthesis can decrease tau propagation, suggesting that exosomes may be involved in tau propagation. Taken together, these findings implicate microglia as a key contributor to tau propagation through phagocytic tau uptake and release of tau-containing exosomes.[76]
The potential mechanism underlying tau propagation is slowly gaining clarity. Extracellular tau seeds were originally thought to enter neurons via bulk endocytosis.[77] However, recent evidence reveals that tau aggregates can facilitate cell entry through clathrin-mediated endocytosis. BIN1, which normally inhibits endocytic flux, is genetically associated with late-onset AD and it is downregulated in AD. Loss of BIN1 is linked to increased endocytic trafficking, which promotes tau propagation.[78] Internalized tau can damage endosomal membranes, and subsequent tau leakage into the cytoplasm can mediate tau propagation. Furthermore, transduced neuronal tau aggregates can be secreted into the extracellular matrix in manner dependent on neuronal activation.[79]
Unsolved questions
Despite recent progress supporting the tau propagation hypothesis, many basic questions remain. For instance, the nature of tau seed remains largely unknown. Although most studies utilize tau fibrils as nucleating seeds,[64,65] evidence suggests that tau trimers represent the minimal oligomer size required to seed tau polymerization.[80] This hypothesis has been substantiated in human neurons derived from induced pluripotent stem cells.[81] However, some studies demonstrate that tau monomers are sufficient to seed polymerization.[82] Studies also suggest that low-molecular-weight aggregates, and short/long fibrils, but not monomers, are taken up by neurons.[77,83] More recently, a rare phosphorylated high-molecular-weight tau species has been shown to be absorbed and propagated in neurons based on studies using brain extracts from transgenic mice and human brain, as well as recombinant tau in vitro and in vivo.[84] Interestingly, tau propagation can also be induced using preaggregated Aβ, suggesting a possible seeding mechanism induced by alternate aggregate species.[85]
Altogether, the question remains whether tau seeding can be altered by tau mutations derived from familial tauopathies. A majority of the studies described so far performed in vivo and in vitro employ mutant tau. Some studies suggest that tau seeding frequency is affected by the mutation status of the nucleating seed and the recipient cells.[65] However, tau mutations are only found in a small group of tauopathic patients; no mutations have yet been described in AD patients. Since mutant tau only appears in a small number of cases of sporadic tauopathy, studies focused on wild-type tau in human AD may be more relevant to AD pathogenesis.[86]
Whether seeding is tau-isoform dependent is also unclear. While some studies suggest that species-specific seeding barriers prevent tau seeding of other isoforms,[87,88] other studies report that 4R oligomers from individuals with PSP can seed both 3R and 4R tau in vitro.[89] Although the issue remains highly speculative, seeding barriers may vary according to seeding species and the context-specific nature of exogenous tau.
TAU IN ALZHEIMER'S DISEASE DIAGNOSTICS
Detection of tau in cerebrospinal fluid and plasma as a biomarker for Alzheimer's disease diagnostics
Although Aβ and tau are both considered key biomarkers in AD, conflicting views still remain regarding their temporal relevance during the progression of AD. Current models suggest that Aβ dysregulation precedes alterations in tau;[90] this is supported by clinical studies characterizing sporadic and familial AD.[91] Interestingly, tau pathology histologically characterized using the AT8 antibody can be detected in individuals as young as 6 years old in autopsies.[59] This implies that subcortical tau deposition may initiate a pathological AD-associated cascade, in which extracellular and aggregated Aβ may only be produced under pathological conditions by neurons expressing aberrant tau.[59] Recent studies have proposed that changes in pathogenic Aβ and tau can occur independently but may synergistically aggravate neurodegenerative progression.[92,93] Indeed, a recently established model of biomarker dynamics in sporadic AD shows that total tau and p-tau appear earlier than Aβ1–42 in cerebrospinal fluid (CSF). However, in amyloid or APOE4-positive individuals, elevations in Aβ1–42 preceded elevations in p-tau and total tau.[94] This raises the possibility that Aβ and APOE4 status may accelerate the progressive appearance of preexisting elevations in pathological tau in AD. Taken together, tau may be a good biomarker in early detection of AD.
Recent evidence characterizing the progression of mild cognitive impairment (MCI) in AD shows that the alterations in Aβ42 are apparent at the time of MCI diagnosis and maintain during disease progression, whereas total tau and p-tau progressively increase from the time of diagnosis and further elevate with dementia. This suggests that tau may be a more accurate indicator of AD progression compared to Aβ during MCI to late AD onset.[95] Consistent with this, longitudinal study comparing CSF biomarkers and amyloid PET revealed that CSF Aβ42/t-tau (total tau) and Aβ42/p-tau had better diagnostic capacity compared to CSF Aβ biomarkers only, where Aβ42/t-tau showed the highest accuracy of all CSF/PET biomarkers.[96] This supports the use of CSF tau as a biomarker for early detection and diagnosis in AD and for use in clinical evaluation of AD drug trials.
Accurate tau measurements in CSF are facilitated by recent technological developments in protein detection. Although enzyme-linked immunosorbent assay (ELISA) systems for measuring CSF t-tau and p-tau have been available for decades,[97] new methods such as xMAP technology, have recently been used in the AD neuroimaging initiative (ADNI) program which characterizes concurrent changes in Aβ1–42, t-tau and p-tau.[98] The xMAP technology flow cytometry platform involves covalent coupling of a capture antibody to spectrally defined microspheres, resulting in improved throughput rates, smaller sample size, reduced assay time, and comparable accuracy, thus making xMAP a potential alternative to conventional ELISA.[99]
Although detection of tau in CSF is feasible, a minimally invasive biopsy method such as plasma detection would be preferable. Detection of biomarkers in plasma, however, requires methods with high sensitivity and accuracy in detecting neurological components.[100] Single-molecule ELISA (digital ELISA) involves the enrichment of proteins of low abundance by immunocapture using microscopic beads in 50 femtoliter reaction chambers designed to hold single beads, coupled with fluorescence imaging to detect single-protein molecules.[101] Using this method, lower detection thresholds of 0.02 pg/ml of tau have been achieved, which represents a 1000-fold increase in sensitivity compared to conventional ELISA.[102] However, the current use of digital ELISA systems is costly and time-consuming.
Electrochemical biosensors have also been developed for diagnostic applications. Recently, a multielectrode electrochemical biosensor to detect full-length 2N4R tau features a detection limit of 0.03 pmol/L, with comparable detection fidelity in the presence of albumin or human serum and faster processing (<1 h) compared to digital ELISA.[103] Given the rapid development of biosensor technology, it is quite likely that similar methods will be adapted to detect p-tau and other tau isoforms/species in CSF and serum. Moreover, other blood-based biomarkers such as brain-derived tau exosomes are also being developed, which may be useful in detecting tau oligomers or other tau species that are prone to aggregation and propagation.[76]
Tau as an Alzheimer's disease biomarker in clinical practice
Reductions in Aβ42 and elevations in total and p-tau in CSF have been implicated as potential biomarkers in AD patients according to NIH-NIA criteria. Although CSF biomarkers are currently being developed and have been characterized in AD, use of these biomarkers in diagnosis and prognostic prediction is still pending, since no clear consensus has been reached on their application in the clinic. Although use of biomarkers in combination is superior to use of single biomarkers, no optimized standards have been established as an “AD-positive profile.” More specifically, no consensus has been established with respect to biomarker staging during the progression of AD in live patients. Given the variation seen in AD neuropathology and cognitive decline, combined with the lack of a defined etiology for AD-associated dementia in the clinic, setting standards for biomarkers in AD continues to encounter difficulties.[104]
Another concern limiting the widespread use of CSF tau biomarkers in clinical practice is the lack of CSF-based calibration standards. Without calibration standards, wide variations are seen between research groups using different immunoassays, with varied interpretation of clinical results obtained. The involvement of the ADNI study committee in standardizing methodologies, in conjunction with other organizations and laboratories, has helped establish globally accepted standardized protocols. Using defined detailed test procedures, independent laboratories may limit variability and generate results with greater consistency.[105]
DEVELOPMENT OF TAU IMAGING TECHNOLOGY
Requirements in the clinical application of tau tracers
In clinical practice, an ideal tau tracer should demonstrate high sensitivity, good selectivity, and appropriate half-life. Several obstacles need to be overcome to achieve successful tau imaging in vivo.[106] Given that NFTs are located intracellularly, tracers are required to show competent penetration across the blood–brain barrier and neuronal membrane. Because Aβ and tau comprise beta sheets, combined with the fact that Aβ concentrations inside the central nervous system are much higher than that of tau, a tau tracer should have a higher affinity for tau to achieve tau-specific signals using minimal tracer concentrations. With six different tau isoforms expressed, all bearing different conformations at various stages of disease progression, these factors add an additional layer of complexity to the design of ideal tau tracers.
Currently available tau tracers
Currently available tau tracers fall into 4 groups: the nonselective tracer FDDNP,[107] arylquinoline derivatives (THK-523, THK-5105, THK-5117, and THK-5331), pyrido-indole derivatives (T807 and T808), and phenyl/pyridinylbutadienyl-benzothiazoles/benzothiazoliums derivative PBB3. Table 2 shows a comparison of their performance in human PET studies.[107,108,109,110,111,112,113,114]
Table 2.
Comparison of current tau tracers
| Tracer | Radio-tracer | Spatial distribution pattern in human | Selectivity Tau/Aβ | Affinity Tau isoforms | Critical weakness | Reference |
|---|---|---|---|---|---|---|
| FDDNP | 18F | Temporal, parietal, posterior cingulate, and frontal regions | Nonselective | 3R + 4R | Nonselective | Small et al.[107] |
| THK-523 | 18F | NA | 10 folds | 3R + 4R No detection of non-AD tau | Defluorination | Fodero-Tavoletti et al.[108] |
| THK-5105 | 18F | Temporal, parietal, posterior cingulate, frontal, and mesial temporal cortices | 25 folds | 3R + 4R | Off-target binding in brainstem, thalamus, and subcortical white matter Relatively slower kinetics | Okamura et al.[109] |
| THK-5117 | 18F | Temporal lobe, parietal cortices, orbitofrontal, posterior cingulate cortices | No Aβ binding | 3R + 4R | Off-target binding in subcortical white matter, cerebellum | Harada et al.[110] |
| THK-5351 | 18F | Temporal lobe | No Aβ binding | 3R + 4R | Off-target binding in basal ganglia | Harada et al.[111] |
| PBB3 | 11C | Temporal lobe | 50 folds | 3R + 4R, able to detect non-AD tau | Short half-life retention in dural venous sinuses | Maruyama et al.[112] |
| T807 | 18F | Inferior temporal, lateral parietal, and posterior cingulate cortices | 27 folds | 3R + 4R, low affinity for non-AD tau | Retention in basal ganglia, anterior midbrain, venous sinuses, and choroid plexus | Chien et al.[113] |
| T808 | 18F | Temporal lobes, frontal lobe | 27 folds | 3R + 4R | Defluorination | Chien et al.[114] |
NA: Not applicable; AD: Alzheimer’s disease.
Despite claims these tracers are selective, no autopsy studies have yet confirmed their selectivity. In addition, sample sizes used in these trials were relatively small. Larger clinical studies will be required to test their sensitivity, selectivity, and long-term safety. Moreover, tau tracers that can recognize other non-AD tau isoforms (3R-tau or 4R tau) and neurotoxic species such as tau oligomers would expand their applicability in the clinic.
Use of current tau tracers in preliminary stages of testing may hold great potential for facilitating early diagnosis in AD. Several studies have used T807 to investigate pathological tau spreading in normal aging populations and AD. While little tau pathology was found in young cohorts, more tau emerged in medial temporal lobe in aged individuals. Elevations in Aβ corresponded to the spreading of tau pathology to the lateral temporal lobe and neocortex, accurately correlating with changes in cognition during normal aging, and transitional phases from MCI to AD.[115] This highlights the value of tau tracers during in vivo Braak staging and suggests that tau pathology in regions beyond the medial temporal lobe may be strong indicators for advanced AD onset. Interestingly, in vivo imaging technology based on single-chain antibody fragments (scFvs) that selectively bind to oligomeric tau or specific p-tau species is currently being developed. This would be ideal for its concurrent use in treatment and diagnostics.[116,117] If successful, tau imaging may be an effective prognostic method to diagnose early-stage neurodegenerative disorders.
TARGETING TAU IN ALZHEIMER'S DISEASE TREATMENT
Although most drug development efforts so far have focused on Aβ, interest in tau as a drug target is currently growing. Several approaches are being employed which target various aspects of tau pathogenesis, including modulation of tau gene expression, modulation of posttranslational modifications, tau immunotherapy, and microtubule stabilization.[28] Table 3 summarizes current drug development strategies employed.[118,119,120,121,122,123,124,125]
Table 3.
Current drug development effort targeting tau
| Drug name | Mechanism of action | Status | Outcome | Reference |
|---|---|---|---|---|
| MAPT gene expression modulator | ||||
| Antisense oligonucleotides | NA | |||
| Splicing modulators | NA | |||
| Tau protein posttranslational modification modulators | ||||
| TRx 0237 (LMTX™) | Tau aggregation inhibitor | Phase 3 | Failed to improve cognition | Gauthier et al.[118] |
| Tideglusib (NP-12) | GSK-3 inhibitor | Phase 2b | Missed primary endpoint | Lovestone et al.[119] |
| Lithium chloride | GSK-3 inhibitor | Phase 2 | Forlenza et al.[120] | |
| Valproic acid | GSK-3 inhibitor | Phase 2 | Tariot et al.[121] | |
| Metformin | Reduce tau phosphorylation | NA | Promote tau aggregation | Barini et al.[122] |
| Microtubule-stabilizing drugs | ||||
| Epothilone D | Microtubule-stabilizing | Discontinued | Alzforum[123] | |
| Davunetide | Microtubule-stabilizing | Discontinued | ||
| Modulators of PP2A/O-GlcNAcase (Asn120290) | NA | |||
| Promote tau clearance by enhancing autophagy | NA | |||
| Immunotherapy | ||||
| AADvac1 | Synthetic peptide targeting pathological tau protein | Phase 1 | Favorably safe characteristics and good immunogenicity | Novak et al.[124] |
| ACI-35 | Synthetic peptide targeting pathological tau protein | Phase 1 | NA | Agadjanyan et al.[125] |
| BMS-986168 | Monoclonal antibody | Phase 2 | NA | Alzforum[123] |
| C2N 8E12 | Monoclonal antibody | Phase 2 | NA | Alzforum[123] |
| RO 7105705 | Monoclonal antibody | Phase 1 | NA | Alzforum[123] |
NA: Not applicable; PP2A: Protein phosphatase 2A; GSK: Glycogen synthase kinase.
CONCLUSION AND FUTURE DIRECTIONS
Transcellular tau propagation offers new paradigms in our current understanding of AD pathogenesis. Evidence from preclinical in vitro and in vivo models of tau-mediated neurodegeneration have explained, at least in part, pathogenic events observed in AD brain. However, detailed mechanisms and the relevance of these changes to clinical AD cases will be required to fully understand AD and to design and develop future disease-modifying therapies and diagnostic tools. Although detection of tau in CSF and plasma is currently being developed as a diagnostic biomarker, efforts should be made to fully facilitate its implementation in the clinic. Likewise, advances in tau imaging have been made, and with the current evaluation of several tau tracers in clinical trials, use of tau as a neurodegenerative biomarker seems encouraging. Results from tau imaging studies in humans are anticipated to further consolidate or extend our understanding of the tau transcellular propagation hypothesis. Efforts to develop tau immunotherapies are of equal importance; anti-tau vaccines (AADvac) and other active/passive immunotherapies currently undergoing clinical trials show good potential as future therapies.
Financial support and sponsorship
This work was supported by grants from the National Natural Science Foundation of China (No. 81671352, 91232709), the National Key Project of Research and Development Plan (No. 2016YFC1306404), the National Institute of Health (No. R21 AG048519, R01 AG021173, R01 AG038710, R01 AG044420, R01 NS046673, RF1 AG056130, and RF1 AG056114), the Tanz Family Fund as well as scholarship from China Scholarship Council (No. 201608350068).
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
We would like to thank Timothy Y. Huang for providing advice for revision.
Footnotes
Edited by: Qiang Shi
REFERENCES
- 1.International AsD. World Alzheimer Report 2016: Improving Healthcare for People Living with Dementia. 2016. [Last accessed on 2017 Jun 06]. Available from: https://www.alz.co.uk/research/world-report-2016 .
- 2.Futch HS, Croft CL, Truong VQ, Krause EG, Golde TE. Targeting psychologic stress signaling pathways in Alzheimer's disease. Mol Neurodegener. 2017;12:49. doi: 10.1186/s13024-017-0190-z. doi: 10.1186/s13024-017-0190-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Efthymiou AG, Goate AM. Late onset Alzheimer's disease genetics implicates microglial pathways in disease risk. Mol Neurodegener. 2017;12:43. doi: 10.1186/s13024-017-0184-x. doi: 10.1186/s13024-017-0184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agadjanyan MG, Zagorski K, Petrushina I, Davtyan H, Kazarian K, Antonenko M, et al. Humanized monoclonal antibody armanezumab specific to N-terminus of pathological tau: Characterization and therapeutic potency. Mol Neurodegener. 2017;12:33. doi: 10.1186/s13024-017-0172-1. doi: 10.1186/s13024-017-0172-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miyamoto T, Stein L, Thomas R, Djukic B, Taneja P, Knox J, et al. Phosphorylation of tau at Y18, but not tau-fyn binding, is required for tau to modulate NMDA receptor-dependent excitotoxicity in primary neuronal culture. Mol Neurodegener. 2017;12:41. doi: 10.1186/s13024-017-0176-x. doi: 10.1186/s13024-017-0176-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beevers JE, Lai MC, Collins E, Booth HDE, Zambon F, Parkkinen L, et al. MAPT genetic variation and neuronal maturity alter isoform expression affecting axonal transport in iPSC-derived dopamine neurons. Stem Cell Reports. 2017;9:587–99. doi: 10.1016/j.stemcr.2017.06.005. doi: 10.1016/j.stemcr.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mukrasch MD, Bibow S, Korukottu J, Jeganathan S, Biernat J, Griesinger C, et al. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 2009;7:e34. doi: 10.1371/journal.pbio.1000034. doi: 10.1371/journal.pbio.1000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leyns CEG, Holtzman DM. Glial contributions to neurodegeneration in tauopathies. Mol Neurodegener. 2017;12:50. doi: 10.1186/s13024-017-0192-x. doi: 10.1186/s13024-017-0192-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamada K, Cirrito JR, Stewart FR, Jiang H, Finn MB, Holmes BB, et al. In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci. 2011;31:13110–7. doi: 10.1523/JNEUROSCI.2569-11.2011. doi: 10.1523/jneurosci.2569-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zempel H, Dennissen FJA, Kumar Y, Luedtke J, Biernat J, Mandelkow EM, et al. Axodendritic sorting and pathological missorting of tau are isoform-specific and determined by axon initial segment architecture. J Biol Chem. 2017;292:12192–207. doi: 10.1074/jbc.M117.784702. doi: 10.1074/jbc.M117.784702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–97. doi: 10.1016/j.cell.2010.06.036. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 12.Martin L, Latypova X, Terro F. Post-translational modifications of tau protein: Implications for Alzheimer's disease. Neurochem Int. 2011;58:458–71. doi: 10.1016/j.neuint.2010.12.023. doi: 10.1016/j.neuint.2010.12.023. [DOI] [PubMed] [Google Scholar]
- 13.Hanger DP, Anderton BH, Noble W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol Med. 2009;15:112–9. doi: 10.1016/j.molmed.2009.01.003. doi: 10.1016/j.molmed.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 14.Gong CX, Lidsky T, Wegiel J, Zuck L, Grundke-Iqbal I, Iqbal K, et al. Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain. Implications for neurofibrillary degeneration in Alzheimer's disease. J Biol Chem. 2000;275:5535–44. doi: 10.1074/jbc.275.8.5535. doi: 10.1074/jbc.275.8.5535. [DOI] [PubMed] [Google Scholar]
- 15.Mandelkow EM, Mandelkow E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med. 2012;2:a006247. doi: 10.1101/cshperspect.a006247. doi: 10.1101/cshperspect. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lewis TL, Jr, Courchet J, Polleux F. Cell biology in neuroscience: Cellular and molecular mechanisms underlying axon formation, growth, and branching. J Cell Biol. 2013;202:837–48. doi: 10.1083/jcb.201305098. doi: 10.1083/jcb.201305098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rodríguez-Martín T, Cuchillo-Ibáñez I, Noble W, Nyenya F, Anderton BH, Hanger DP, et al. Tau phosphorylation affects its axonal transport and degradation. Neurobiol Aging. 2013;34:2146–57. doi: 10.1016/j.neurobiolaging.2013.03.015. doi: 10.1016/j.neurobiolaging.2013.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Violet M, Delattre L, Tardivel M, Sultan A, Chauderlier A, Caillierez R, et al. A major role for tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front Cell Neurosci. 2014;8:84. doi: 10.3389/fncel.2014.00084. doi: 10.3389/fncel.2014.000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2016;17:22–35. doi: 10.1038/nrn.2015.1. doi: 10.1038/nrn.2015.1. [DOI] [PubMed] [Google Scholar]
- 20.Choi SH, Kim YH, Quinti L, Tanzi RE. Kim DY 3D culture models of Alzheimer's disease: A road map to a “cure-in-a-dish”. Mol Neurodegener. 2016;11:75. doi: 10.1186/s13024-016-0139-7. doi: 10.1186/s13024-016-0139-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caillet-Boudin ML, Buée L, Sergeant N, Lefebvre B. Regulation of human MAPT gene expression. Mol Neurodegener. 2015;10:28. doi: 10.1186/s13024-015-0025-8. doi: 10.1186/s13024-015-0025-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang JZ, Xia YY, Grundke-Iqbal I, Iqbal K. Abnormal hyperphosphorylation of tau: Sites, regulation, and molecular mechanism of neurofibrillary degeneration. J Alzheimers Dis. 2013;33(Suppl 1):S123–39. doi: 10.3233/JAD-2012-129031. doi: 10.3233/jad-2012-129031. [DOI] [PubMed] [Google Scholar]
- 23.Wilkaniec A, Czapski GA, Adamczyk A. Cdk5 at crossroads of protein oligomerization in neurodegenerative diseases: Facts and hypotheses. J Neurochem. 2016;136:222–33. doi: 10.1111/jnc.13365. doi: 10.1111/jnc.13365. [DOI] [PubMed] [Google Scholar]
- 24.Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer's disease. J Neurochem. 2008;104:1433–9. doi: 10.1111/j.1471-4159.2007.05194.x. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iqbal K, Alonso Adel C, Chen S, Chohan MO, El-Akkad E, Gong CX, et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta. 2005;1739:198–210. doi: 10.1016/j.bbadis.2004.09.008. doi: 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 26.Alonso A, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci U S A. 2001;98:6923–8. doi: 10.1073/pnas.121119298. doi: 10.1073/pnas.121119298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alonso Adel C, Mederlyova A, Novak M, Grundke-Iqbal I, Iqbal K. Promotion of hyperphosphorylation by frontotemporal dementia tau mutations. J Biol Chem. 2004;279:34873–81. doi: 10.1074/jbc.M405131200. doi: 10.1074/jbc.M405131200. [DOI] [PubMed] [Google Scholar]
- 28.Iqbal K, Liu F, Gong CX. Tau and neurodegenerative disease: The story so far. Nat Rev Neurol. 2016;12:15–27. doi: 10.1038/nrneurol.2015.225. doi: 10.1038/nrneurol.2015.225. [DOI] [PubMed] [Google Scholar]
- 29.Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–81. doi: 10.1016/j.neuron.2010.11.030. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, Dunmore J, et al. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest. 2007;117:648–58. doi: 10.1172/JCI29715. doi: 10.1172/jci29715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 2010;67:953–66. doi: 10.1016/j.neuron.2010.08.044. doi: 10.1016/j.neuron.2010.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cook C, Carlomagno Y, Gendron TF, Dunmore J, Scheffel K, Stetler C, et al. Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Hum Mol Genet. 2014;23:104–16. doi: 10.1093/hmg/ddt402. doi: 10.1093/hmg/ddt402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cohen TJ, Guo JL, Hurtado DE, Kwong LK, Mills IP, Trojanowski JQ, et al. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun. 2011;2:252. doi: 10.1038/ncomms1255. doi: 10.1038/ncomms1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Min SW, Chen X, Tracy TE, Li Y, Zhou Y, Wang C, et al. Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat Med. 2015;21:1154–62. doi: 10.1038/nm.3951. doi: 10.1038/nm.3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sohn PD, Tracy TE, Son HI, Zhou Y, Leite RE, Miller BL, et al. Acetylated tau destabilizes the cytoskeleton in the axon initial segment and is mislocalized to the somatodendritic compartment. Mol Neurodegener. 2016;11:47. doi: 10.1186/s13024-016-0109-0. doi: 10.1186/s13024-016-0109-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu F, Zaidi T, Iqbal K, Grundke-Iqbal I, Merkle RK, Gong CX, et al. Role of glycosylation in hyperphosphorylation of tau in Alzheimer's disease. FEBS Lett. 2002;512:101–6. doi: 10.1016/s0014-5793(02)02228-7. doi: 10.1016/S0014-5793(02)02228-7. [DOI] [PubMed] [Google Scholar]
- 37.Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX. O-GlcNAcylation regulates phosphorylation of tau: A mechanism involved in Alzheimer's disease. Proc Natl Acad Sci U S A. 2004;101:10804–9. doi: 10.1073/pnas.0400348101. doi: 10.1073/pnas.0400348101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu F, Shi J, Tanimukai H, Gu J, Gu J, Grundke-Iqbal I, et al. Reduced O-glcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer's disease. Brain. 2009;132:1820–32. doi: 10.1093/brain/awp099. doi: 10.1093/brain/awp099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Garg S, Mandelkow EM, Mandelkow E. Proteolytic processing of tau. Biochem Soc Trans. 2010;38:955–61. doi: 10.1042/BST0380955. doi: 10.1042/bst0380955. [DOI] [PubMed] [Google Scholar]
- 40.Rissman RA, Poon WW, Blurton-Jones M, Oddo S, Torp R, Vitek MP, et al. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest. 2004;114:121–30. doi: 10.1172/JCI20640. doi: 10.1172/jci20640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Horowitz PM, Patterson KR, Guillozet-Bongaarts AL, Reynolds MR, Carroll CA, Weintraub ST, et al. Early N-terminal changes and caspase-6 cleavage of tau in Alzheimer's disease. J Neurosci. 2004;24:7895–902. doi: 10.1523/JNEUROSCI.1988-04.2004. doi: 10.1523/jneurosci.1988-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Z, Song M, Liu X, Kang SS, Kwon IS, Duong DM, et al. Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer's disease. Nat Med. 2014;20:1254–62. doi: 10.1038/nm.3700. doi: 10.1038/nm.3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao X, Kotilinek LA, Smith B, Hlynialuk C, Zahs K, Ramsden M, et al. Caspase-2 cleavage of tau reversibly impairs memory. Nat Med. 2016;22:1268–76. doi: 10.1038/nm.4199. doi: 10.1038/nm.4199. [DOI] [PubMed] [Google Scholar]
- 44.Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8:663–72. doi: 10.1038/nrn2194. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 45.Schneider A, Biernat J, von Bergen M, Mandelkow E, Mandelkow EM. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry. 1999;38:3549–58. doi: 10.1021/bi981874p. doi: 10.1021/bi981874p. [DOI] [PubMed] [Google Scholar]
- 46.Mocanu MM, Nissen A, Eckermann K, Khlistunova I, Biernat J, Drexler D, et al. The potential for beta-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous tau in inducible mouse models of tauopathy. J Neurosci. 2008;28:737–48. doi: 10.1523/JNEUROSCI.2824-07.2008. doi: 10.1523/jneurosci.2824-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1:a006189. doi: 10.1101/cshperspect.a006189. doi: 10.1101/cshperspect. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, et al. Caspase activation precedes and leads to tangles. Nature. 2010;464:1201–4. doi: 10.1038/nature08890. doi: 10.1038/nature08890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van der Jeugd A, Hochgräfe K, Ahmed T, Decker JM, Sydow A, Hofmann A, et al. Cognitive defects are reversible in inducible mice expressing pro-aggregant full-length human tau. Acta Neuropathol. 2012;123:787–805. doi: 10.1007/s00401-012-0987-3. doi: 10.1007/s00401-012-0987-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Falzone TL, Gunawardena S, McCleary D, Reis GF, Goldstein LS. Kinesin-1 transport reductions enhance human tau hyperphosphorylation, aggregation and neurodegeneration in animal models of tauopathies. Hum Mol Genet. 2010;19:4399–408. doi: 10.1093/hmg/ddq363. doi: 10.1093/hmg/ddq363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.DuBoff B, Götz J, Feany MB. Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron. 2012;75:618–32. doi: 10.1016/j.neuron.2012.06.026. doi: 10.1016/j.neuron.2012.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Quintanilla RA, Dolan PJ, Jin YN, Johnson GV. Truncated tau and aβ cooperatively impair mitochondria in primary neurons. Neurobiol Aging. 2012;33:619.e25–35. doi: 10.1016/j.neurobiolaging.2011.02.007. doi: 10.1016/j.neurobiolaging.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Furgerson M, Fechheimer M, Furukawa R. Model hirano bodies protect against tau-independent and tau-dependent cell death initiated by the amyloid precursor protein intracellular domain. PLoS One. 2012;7:e44996. doi: 10.1371/journal.pone.0044996. doi: 10.1371/journal.pone.0044996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.He HJ, Wang XS, Pan R, Wang DL, Liu MN, He RQ, et al. The proline-rich domain of tau plays a role in interactions with actin. BMC Cell Biol. 2009;10:81. doi: 10.1186/1471-2121-10-81. doi: 10.1186/1471-2121-10-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu T, Aron L, Zullo J, Pan Y, Kim H, Chen Y, et al. REST and stress resistance in ageing and Alzheimer's disease. Nature. 2014;507:448–54. doi: 10.1038/nature13163. doi: 10.1038/nature13163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kou J, Kovacs GG, Höftberger R, Kulik W, Brodde A, Forss-Petter S, et al. Peroxisomal alterations in Alzheimer's disease. Acta Neuropathol. 2011;122:271–83. doi: 10.1007/s00401-011-0836-9. doi: 10.1007/s00401-011-0836-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Coppedè F, Migliore L. DNA damage and repair in Alzheimer's disease. Curr Alzheimer Res. 2009;6:36–47. doi: 10.2174/156720509787313970. doi: 10.2174/156720509787313970. [DOI] [PubMed] [Google Scholar]
- 58.Braak H, Braak E. Staging of Alzheimer's disease-related neurofibrillary changes. Neurobiol Aging. 1995;16:271–8. doi: 10.1016/0197-4580(95)00021-6. doi: 10.1016/0197-4580(95)00021-6. [DOI] [PubMed] [Google Scholar]
- 59.Braak H, Del Tredici K. The preclinical phase of the pathological process underlying sporadic Alzheimer's disease. Brain. 2015;138:2814–33. doi: 10.1093/brain/awv236. doi: 10.1093/brain/awv236. [DOI] [PubMed] [Google Scholar]
- 60.Walsh DM, Selkoe DJ. A critical appraisal of the pathogenic protein spread hypothesis of neurodegeneration. Nat Rev Neurosci. 2016;17:251–60. doi: 10.1038/nrn.2016.13. doi: 10.1038/nrn.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lewis J, Dickson DW. Propagation of tau pathology: Hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol. 2016;131:27–48. doi: 10.1007/s00401-015-1507-z. doi: 10.1007/s00401-015-1507-z. [DOI] [PubMed] [Google Scholar]
- 62.Prusiner SB. Cell biology. A unifying role for prions in neurodegenerative diseases. Science. 2012;336:1511–3. doi: 10.1126/science.1222951. doi: 10.1126/science.1222951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–44. doi: 10.1126/science.6801762. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 64.Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284:12845–52. doi: 10.1074/jbc.M808759200. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guo JL, Lee VM. Seeding of normal tau by pathological tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem. 2011;286:15317–31. doi: 10.1074/jbc.M110.209296. doi: 10.1074/jbc.M110.209296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–13. doi: 10.1038/ncb1901. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.de Calignon A, Polydoro M, Suárez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, et al. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron. 2012;73:685–97. doi: 10.1016/j.neuron.2011.11.033. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ahmed Z, Cooper J, Murray TK, Garn K, McNaughton E, Clarke H, et al. A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: The pattern of spread is determined by connectivity, not proximity. Acta Neuropathol. 2014;127:667–83. doi: 10.1007/s00401-014-1254-6. doi: 10.1007/s00401-014-1254-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VM, et al. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci. 2013;33:1024–37. doi: 10.1523/JNEUROSCI.2642-12.2013. doi: 10.1523/jneurosci.2642-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stancu IC, Vasconcelos B, Ris L, Wang P, Villers A, Peeraer E, et al. Templated misfolding of tau by prion-like seeding along neuronal connections impairs neuronal network function and associated behavioral outcomes in tau transgenic mice. Acta Neuropathol. 2015;129:875–94. doi: 10.1007/s00401-015-1413-4. doi: 10.1007/s00401-015-1413-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A. 2013;110:9535–40. doi: 10.1073/pnas.1301175110. doi: 10.1073/pnas.1301175110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Boluda S, Iba M, Zhang B, Raible KM, Lee VM, Trojanowski JQ, et al. Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer's disease or corticobasal degeneration brains. Acta Neuropathol. 2015;129:221–37. doi: 10.1007/s00401-014-1373-0. doi: 10.1007/s00401-014-1373-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. 2015;18:1584–93. doi: 10.1038/nn.4132. doi: 10.1038/nn.4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Iba M, McBride JD, Guo JL, Zhang B, Trojanowski JQ, Lee VM, et al. Tau pathology spread in PS19 tau transgenic mice following locus coeruleus (LC) injections of synthetic tau fibrils is determined by the LC's afferent and efferent connections. Acta Neuropathol. 2015;130:349–62. doi: 10.1007/s00401-015-1458-4. doi: 10.1007/s00401-015-1458-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 2014;82:1271–88. doi: 10.1016/j.neuron.2014.04.047. doi: 10.1016/j.neuron.2014.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang Y, Balaji V, Kaniyappan S, Krüger L, Irsen S, Tepper K, et al. The release and trans-synaptic transmission of Tau via exosomes. Mol Neurodegener. 2017;12:5. doi: 10.1186/s13024-016-0143-y. doi: 10.1186/s13024-016-0143-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wu JW, Herman M, Liu L, Simoes S, Acker CM, Figueroa H, et al. Small misfolded tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J Biol Chem. 2013;288:1856–70. doi: 10.1074/jbc.M112.394528. doi: 10.1074/jbc.M112.394528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Calafate S, Flavin W, Verstreken P, Moechars D. Loss of bin1 promotes the propagation of tau pathology. Cell Rep. 2016;17:931–40. doi: 10.1016/j.celrep.2016.09.063. doi: 10.1016/j.celrep.2016.09.063. [DOI] [PubMed] [Google Scholar]
- 79.Wu JW, Hussaini SA, Bastille IM, Rodriguez GA, Mrejeru A, Rilett K, et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci. 2016;19:1085–92. doi: 10.1038/nn.4328. doi: 10.1038/nn.4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mirbaha H, Holmes BB, Sanders DW, Bieschke J, Diamond MI. Tau trimers are the minimal propagation unit spontaneously internalized to seed intracellular aggregation. J Biol Chem. 2015;290:14893–903. doi: 10.1074/jbc.M115.652693. doi: 10.1074/jbc.M115.652693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Usenovic M, Niroomand S, Drolet RE, Yao L, Gaspar RC, Hatcher NG, et al. Internalized tau oligomers cause neurodegeneration by inducing accumulation of pathogenic tau in human neurons derived from induced pluripotent stem cells. J Neurosci. 2015;35:14234–50. doi: 10.1523/JNEUROSCI.1523-15.2015. doi: 10.1523/jneurosci.1523-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Michel CH, Kumar S, Pinotsi D, Tunnacliffe A, St George-Hyslop P, Mandelkow E, et al. Extracellular monomeric tau protein is sufficient to initiate the spread of tau protein pathology. J Biol Chem. 2014;289:956–67. doi: 10.1074/jbc.M113.515445. doi: 10.1074/jbc.M113.515445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jackson SJ, Kerridge C, Cooper J, Cavallini A, Falcon B, Cella CV, et al. Short fibrils constitute the major species of seed-competent tau in the brains of mice transgenic for human P301S tau. J Neurosci. 2016;36:762–72. doi: 10.1523/JNEUROSCI.3542-15.2016. doi: 10.1523/jneurosci.3542-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Takeda S, Wegmann S, Cho H, DeVos SL, Commins C, Roe AD, et al. Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight tau derived from Alzheimer's disease brain. Nat Commun. 2015;6:8490. doi: 10.1038/ncomms9490. doi: 10.1038/ncomms9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vasconcelos B, Stancu IC, Buist A, Bird M, Wang P, Vanoosthuyse A, et al. Heterotypic seeding of tau fibrillization by pre-aggregated abeta provides potent seeds for prion-like seeding and propagation of Tau-pathology in vivo. Acta Neuropathol. 2016;131:549–69. doi: 10.1007/s00401-015-1525-x. doi: 10.1007/s00401-015-1525-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Caillierez R, Bégard S, Lécolle K, Deramecourt V, Zommer N, Dujardin S, et al. Lentiviral delivery of the human wild-type tau protein mediates a slow and progressive neurodegenerative tau pathology in the rat brain. Mol Ther. 2013;21:1358–68. doi: 10.1038/mt.2013.66. doi: 10.1038/mt.2013.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nonaka T, Watanabe ST, Iwatsubo T, Hasegawa M. Seeded aggregation and toxicity of α-synuclein and tau: Cellular models of neurodegenerative diseases. J Biol Chem. 2010;285:34885–98. doi: 10.1074/jbc.M110.148460. doi: 10.1074/jbc.M110.148460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dinkel PD, Siddiqua A, Huynh H, Shah M, Margittai M. Variations in filament conformation dictate seeding barrier between three- and four-repeat tau. Biochemistry. 2011;50:4330–6. doi: 10.1021/bi2004685. doi: 10.1021/bi2004685. [DOI] [PubMed] [Google Scholar]
- 89.Gerson JE, Sengupta U, Lasagna-Reeves CA, Guerrero-Muñoz MJ, Troncoso J, Kayed R, et al. Characterization of tau oligomeric seeds in progressive supranuclear palsy. Acta Neuropathol Commun. 2014;2:73. doi: 10.1186/2051-5960-2-73. doi: 10.1186/2051-5960-2-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jack CR, Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010;9:119–28. doi: 10.1016/S1474-4422(09)70299-6. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tracking the insidious course of Alzheimer’s disease. Nat Med. 2012;18:1342–3. doi: 10.1038/nm.2922. doi: 10.1038/nm.2922. [DOI] [PubMed] [Google Scholar]
- 92.Duyckaerts C. Tau pathology in children and young adults: Can you still be unconditionally baptist? Acta Neuropathol. 2011;121:145–7. doi: 10.1007/s00401-010-0794-7. doi: 10.1007/s00401-010-0794-7. [DOI] [PubMed] [Google Scholar]
- 93.Musiek ES, Holtzman DM. Origins of Alzheimer's disease: Reconciling cerebrospinal fluid biomarker and neuropathology data regarding the temporal sequence of amyloid-beta and tau involvement. Curr Opin Neurol. 2012;25:715–20. doi: 10.1097/WCO.0b013e32835a30f4. doi: 10.1097/WCO.0b013e32835a30f4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Young AL, Oxtoby NP, Daga P, Cash DM, Fox NC, Ourselin S, et al. A data-driven model of biomarker changes in sporadic Alzheimer's disease. Brain. 2014;137:2564–77. doi: 10.1093/brain/awu176. doi: 10.1093/brain/awu176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jack CR, Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. Tracking pathophysiological processes in Alzheimer's disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207–16. doi: 10.1016/S1474-4422(12)70291-0. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Palmqvist S, Zetterberg H, Mattsson N, Johansson P, Minthon L, et al. Alzheimer's Disease Neuroimaging Initiative. Detailed comparison of amyloid PET and CSF biomarkers for identifying early Alzheimer disease. Neurology. 2015;85:1240–9. doi: 10.1212/WNL.0000000000001991. doi: 10.1212/WNL.0000000000001991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vanmechelen E, Vanderstichele H, Davidsson P, Van Kerschaver E, Van Der Perre B, Sjögren M, et al. Quantification of tau phosphorylated at threonine 181 in human cerebrospinal fluid: A sandwich ELISA with a synthetic phosphopeptide for standardization. Neurosci Lett. 2000;285:49–52. doi: 10.1016/s0304-3940(00)01036-3. doi:10.1016/S0304-3940(00)01036-3. [DOI] [PubMed] [Google Scholar]
- 98.Weiner MW, Veitch DP, Aisen PS, Beckett LA, Cairns NJ, Green RC, et al. The Alzheimer's disease neuroimaging initiative: A review of papers published since its inception. Alzheimers Dement. 2013;9:e111–94. doi: 10.1016/j.jalz.2013.05.1769. doi: 10.1016/j.jalz.2013.05.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Reijn TS, Rikkert MO, van Geel WJ, de Jong D, Verbeek MM. Diagnostic accuracy of ELISA and xMAP technology for analysis of amyloid beta(42) and tau proteins. Clin Chem. 2007;53:859–65. doi: 10.1373/clinchem.2006.081679. doi: 10.1373/clinchem.2006.081679. [DOI] [PubMed] [Google Scholar]
- 100.Blennow K, Zetterberg H. Understanding biomarkers of neurodegeneration: Ultrasensitive detection techniques pave the way for mechanistic understanding. Nat Med. 2015;21:217–9. doi: 10.1038/nm.3810. doi: 10.1038/nm.3810. [DOI] [PubMed] [Google Scholar]
- 101.Rissin DM, Kan CW, Campbell TG, Howes SC, Fournier DR, Song L, et al. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat Biotechnol. 2010;28:595–9. doi: 10.1038/nbt.1641. doi: 10.1038/nbt.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Randall J, Mörtberg E, Provuncher GK, Fournier DR, Duffy DC, Rubertsson S, et al. Tau proteins in serum predict neurological outcome after hypoxic brain injury from cardiac arrest: Results of a pilot study. Resuscitation. 2013;84:351–6. doi: 10.1016/j.resuscitation.2012.07.027. doi: 10.1016/j.resuscitation.2012.07.027. [DOI] [PubMed] [Google Scholar]
- 103.Wang SX, Acha D, Shah AJ, Hills F, Roitt I, Demosthenous A, et al. Detection of the tau protein in human serum by a sensitive four-electrode electrochemical biosensor. Biosens Bioelectron. 2017;92:482–8. doi: 10.1016/j.bios.2016.10.077. doi: 10.1016/j.bios.2016.10.077. [DOI] [PubMed] [Google Scholar]
- 104.Lehmann S, Gabelle A, Paquet C. Can we rely only on ratios of cerebrospinal fluid biomarkers for AD biological diagnosis? Alzheimers Dement. 2015;11:1125–6. doi: 10.1016/j.jalz.2014.09.003. doi: 10.1016/j.jalz.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 105.Kang JH, Korecka M, Figurski MJ, Toledo JB, Blennow K, Zetterberg H, et al. The Alzheimer's disease neuroimaging initiative 2 biomarker core: A review of progress and plans. Alzheimers Dement. 2015;11:772–91. doi: 10.1016/j.jalz.2015.05.003. doi: 10.1016/j.jalz.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Saint-Aubert L, Lemoine L, Chiotis K, Leuzy A, Rodriguez-Vieitez E, Nordberg A, et al. Tau PET imaging: Present and future directions. Mol Neurodegener. 2017;12:19. doi: 10.1186/s13024-017-0162-3. doi: 10.1186/s13024-017-0162-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Small GW, Kepe V, Ercoli LM, Siddarth P, Bookheimer SY, Miller KJ, et al. PET of brain amyloid and tau in mild cognitive impairment. N Engl J Med. 2006;355:2652–63. doi: 10.1056/NEJMoa054625. doi: 10.1056/NEJMoa054625. [DOI] [PubMed] [Google Scholar]
- 108.Fodero-Tavoletti MT, Okamura N, Furumoto S, Mulligan RS, Connor AR, McLean CA, et al. 18F-THK523: A novel in vivo tau imaging ligand for Alzheimer's disease. Brain. 2011;134:1089–100. doi: 10.1093/brain/awr038. doi: 10.1093/brain/awr038. [DOI] [PubMed] [Google Scholar]
- 109.Okamura N, Furumoto S, Fodero-Tavoletti MT, Mulligan RS, Harada R, Yates P, et al. Non-invasive assessment of Alzheimer's disease neurofibrillary pathology using 18F-THK5105 PET. Brain. 2014;137(Pt 6):1762–71. doi: 10.1093/brain/awu064. doi: 10.1093/brain/awu064. [DOI] [PubMed] [Google Scholar]
- 110.Harada R, Okamura N, Furumoto S, Furukawa K, Ishiki A, Tomita N, et al. [(18)F]THK-5117 PET for assessing neurofibrillary pathology in Alzheimer's disease. Eur J Nucl Med Mol Imaging. 2015;42:1052–61. doi: 10.1007/s00259-015-3035-4. doi: 10.1007/s00259-015-3035-4. [DOI] [PubMed] [Google Scholar]
- 111.Harada R, Okamura N, Furumoto S, Furukawa K, Ishiki A, Tomita N, et al. 18F-THK5351: A Novel PET radiotracer for imaging neurofibrillary pathology in Alzheimer disease. J Nucl Med. 2016;57:208–14. doi: 10.2967/jnumed.115.164848. doi: 10.2967/jnumed.115.164848. [DOI] [PubMed] [Google Scholar]
- 112.Maruyama M, Shimada H, Suhara T, Shinotoh H, Ji B, Maeda J, et al. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron. 2013;79:1094–108. doi: 10.1016/j.neuron.2013.07.037. doi: 10.1016/j.neuron.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chien DT, Bahri S, Szardenings AK, Walsh JC, Mu F, Su MY, et al. Early clinical PET imaging results with the novel PHF-tau radioligand [F-18]-T807. J Alzheimers Dis. 2013;34:457–68. doi: 10.3233/JAD-122059. doi: 10.3233/jad-122059. [DOI] [PubMed] [Google Scholar]
- 114.Chien DT, Szardenings AK, Bahri S, Walsh JC, Mu F, Xia C, et al. Early clinical PET imaging results with the novel PHF-tau radioligand [F18]-T808. J Alzheimers Dis. 2014;38:171–84. doi: 10.3233/JAD-130098. doi: 10.3233/jad-130098. [DOI] [PubMed] [Google Scholar]
- 115.Schwarz AJ, Yu P, Miller BB, Shcherbinin S, Dickson J, Navitsky M, et al. Regional profiles of the candidate tau PET ligand 18F-AV-1451 recapitulate key features of Braak histopathological stages. Brain. 2016;139:1539–50. doi: 10.1093/brain/aww023. doi: 10.1093/brain/aww023. [DOI] [PubMed] [Google Scholar]
- 116.Tian H, Davidowitz E, Lopez P, He P, Schulz P, Moe J, et al. Isolation and characterization of antibody fragments selective for toxic oligomeric tau. Neurobiol Aging. 2015;36:1342–55. doi: 10.1016/j.neurobiolaging.2014.12.002. doi: 10.1016/j.neurobiolaging.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Krishnaswamy S, Lin Y, Rajamohamedsait WJ, Rajamohamedsait HB, Krishnamurthy P, Sigurdsson EM, et al. Antibody-derived in vivo imaging of tau pathology. J Neurosci. 2014;34:16835–50. doi: 10.1523/JNEUROSCI.2755-14.2014. doi: 10.1523/jneurosci.2755-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gauthier S, Feldman HH, Schneider LS, Wilcock GK, Frisoni GB, Hardlund JH, et al. Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer's disease: A randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet. 2016;388:2873–84. doi: 10.1016/S0140-6736(16)31275-2. doi: 10.1016/s0140-6736(16)31275-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lovestone S, Boada M, Dubois B, Hüll M, Rinne JO, Huppertz HJ, et al. A phase II trial of tideglusib in Alzheimer's disease. J Alzheimers Dis. 2015;45:75–88. doi: 10.3233/JAD-141959. doi: 10.3233/jad-141959. [DOI] [PubMed] [Google Scholar]
- 120.Forlenza OV, De-Paula VJ, Diniz BS. Neuroprotective effects of lithium: Implications for the treatment of Alzheimer's disease and related neurodegenerative disorders. ACS Chem Neurosci. 2014;5:443–50. doi: 10.1021/cn5000309. doi: 10.1021/cn5000309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tariot PN, Schneider LS, Cummings J, Thomas RG, Raman R, Jakimovich LJ, et al. Chronic divalproex sodium to attenuate agitation and clinical progression of Alzheimer disease. Arch Gen Psychiatry. 2011;68:853–61. doi: 10.1001/archgenpsychiatry.2011.72. doi: 10.1001/archgenpsychiatry.2011.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Barini E, Antico O, Zhao Y, Asta F, Tucci V, Catelani T, et al. Metformin promotes tau aggregation and exacerbates abnormal behavior in a mouse model of tauopathy. Mol Neurodegener. 2016;11:16. doi: 10.1186/s13024-016-0082-7. doi: 10.1186/s13024-016-0082-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Alzforum. Treating Tau: Finally, Clinical Candidates are Stepping into the Ring. 2017. [Last accessed on 2017 Oct 15]. Available from: http://www.alzforum.org/news/conference-coverage/treating-tau-finally-clinical-candidates-are-stepping-ring .
- 124.Novak P, Schmidt R, Kontsekova E, Zilka N, Kovacech B, Skrabana R, et al. Safety and immunogenicity of the tau vaccine AADvac1 in patients with Alzheimer's disease: A randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Neurol. 2017;16:123–34. doi: 10.1016/S1474-4422(16)30331-3. doi: 10.1016/S1474-4422(16)30331-3. [DOI] [PubMed] [Google Scholar]
- 125.Agadjanyan MG, Petrovsky N, Ghochikyan A. A fresh perspective from immunologists and vaccine researchers: Active vaccination strategies to prevent and reverse Alzheimer's disease. Alzheimers Dement. 2015;11:1246–59. doi: 10.1016/j.jalz.2015.06.1884. doi: 10.1016/j.jalz.2015.06.1884. [DOI] [PMC free article] [PubMed] [Google Scholar]