

Abstract
The centromere regulates proper chromosome segregation and its dysfunction is implicated in chromosomal instability (CIN). However, relatively little is known about how centromere dysfunction occurs in cancer. Here we define the consequences of phosphorylation by Cyclin E1/CDK2 on a conserved Ser18 residue of centromere-associated protein CENP-A, an essential histone H3 variant that specifies centromere identity. Ser18 hyperphosphorylation in cells occurred upon loss of FBW7, a tumor suppressor whose inactivation leads to chromosomal instability (CIN). This event on CENP-A reduced its centromeric localization, increased CIN and promoted anchorage-independent growth and xenograft tumor formation. Overall, our results revealed a pathway that Cyclin E1/CDK2 activation coupled with FBW7 loss promotes CIN and tumor progression via CENP-A-mediated centromere dysfunction.
Keywords: FBW7, chromosomal instability, centromere, phosphorylation
Introduction
FBW7 is a well-established tumor suppressor (1). It belongs to the F-box family of proteins that are an essential component of the SCF (SKP1-CUL1-F-box protein) type of E3 ubiquitin ligase complex, which targets substrates for polyubiquitination followed by proteasome-mediated degradation (2). F-box proteins determine substrate specificity for the SCF ligase complex. There are many important characterized FBW7 E3 ligase substrates, including Cyclin E1 (3). Loss of FBW7 in cancer leads to aberrant accumulation of substrates, accounting for many tumor phenotypes observed in cell lines, xenograft or genetic mouse models, and human patients (3).
Chromosome instability (CIN) is a cancer hallmark that contributes to cancer progression, tumor heterogeneity and drug resistance (4,5). Notably, FBW7 depletion induces CIN in colon cancer cells, including mitotic defects, which can be rescued by co-depletion of Cyclin E1 (6). Cyclin E1/CDK2 kinase activity peaks at the G1/S cell cycle phase and is required for proper cell cycle progression into S phase (7). However, the underlying molecular mechanism by which Cyclin E1 contributes to CIN remains elusive. Chromosome stability requires the centromere, which is the specialized chromatin locus where the kinetochore is built. The centromere is enriched for CENtromere Protein A (CENP-A), an essential histone H3 variant that serves as a key epigenetic mark for centromere identity and propagation (8). CENP-A depletion displaces the downstream components from centromeres and kinetochores, resulting in chromosome missegregation (9); and CENP-A mislocalization to non-centromeric chromatin can lead to ectopic kinetochore and fragmented chromsoomes (10). Therefore, CENP-A must be tightly regulated to ensure proper centromere functions. Clinical evidence strongly correlates centromere gene misregulation with CIN and poor patient prognosis for several human cancer types (11,12). However, the roles and mechanisms of centromere misregulation are poorly understood in the context of cancer progression.
CENP-A must be replenished in each cell division and chromatin assembly at centromeres requires a dedicated pathway (13). Newly synthesized CENP-A protein binds to its chaperone and assembly factor Holiday Junction Recognition Protein (HJURP), which transiently localizes to centromeres at the time of new CENP-A incorporation, from late telophase to early G1 phase in part via MIS18 (14–17). Cell cycle kinases tightly regulate CENP-A deposition in human cells, mostly based on data from cancer cell lines (18,19). For example, phosphorylation of M18BP1 and HJURP by CDK1/2 prevents nucleosome assembly in S and G2 phases, and inhibition of CDK1/2 activity is required for CENP-A loading (18,19). Moreover, phosphorylation of CENP-A at Ser68 mediated by Cyclin B/CDK1 might also be important for proper CENP-A localization despite some debates (20–22). Finally, phosphorylation of CENP-A N-terminus at Ser16 and Ser18 residues has been implicated in chromosome segregation (23). However, the exact roles and pathways of CENP-A misregulation in cancer progression are poorly understood.
In this study, we show that FBW7 loss significantly compromises CENP-A deposition and reduces CENP-A levels at centromeres in human colon and breast cancer cell lines. FBW7 loss promotes excessive Cyclin E1/CDK2-mediated CENP-A phosphorylation at the Serine 18 (Ser18) residue in the N terminal tail. We show that human Cyclin E1/CDK2 is a bona fide CENP-A Ser18 kinase using in vitro and in vivo assays. Persistent CENP-A Ser18 phosphorylation caused by FBW7 loss results in increased frequencies of lagging chromosomes, chromosomal bridges and micronuclei formation, which could be rescued by co-depletion of Cyclin E1. In addition, the phosphor-mimetic CENP-A S18D mutant phenocopies FBW7 loss and promotes xenograft tumor growth. We suggest a novel mechanism by which FBW7 loss contributes to CIN and tumorigenesis.
Materials and Methods
Cell Culture
Wild-type and DLD1 FBW7−/− cell lines were kindly provided by Dr. Bert Volgestein (Johns Hopkins University) in 2004 and were cultured in RPMI supplemented with 10% fetal bovine serum (FBS). MCF10A cells were cultured in MEBM (Lonza) containing 52 μg/ml Bovine Pituitary Extract, 500 ng/ml hydrocortisone, 10 ng/ml hEGF, 5 μg/ml insulin (MEGM Bullet Kit, Lonza Corporation) and 100ng/ml cholera toxin (Sigma-Aldrich). MDA-MB-453 cells were maintained in Dulbecco’s modified Eagle medium (DMEM) with 10% FBS. These were obtained from ATCC from 2013-2016. All cells were maintained at 37°C in 5% CO2 incubator. For synchronization, cells were arrested 18 hr (DLD1) or 20 hr (DLD1 FBW7−/−) in 330 nM nocodazole, collected by mitotic shake-off and released by thorough washout. FACS analysis was performed as previously described (24).
Antibodies and Plasmids
Antibodies used in this study are listed in Table S1. The phospho-CENPA (Ser18) antibody was generated and provided as a kind gift by Cell Signaling Technology. HA-tagged WT or mutant FBW7 and Cyclin E1 were described previously (24). HA-CCNE1 R130A mutant was generated by site-directed mutagenesis and confirmed by sequencing. Full length HA tagged CENP-A was amplified by PCR with a 5′ primer that introduced an BamHI site and an HA tag and a 3′ primer that introduced an SalI site. The PCR product was digested with BamHI and SalI and cloned into pLenti CMV GFP vector (Addgene). Mutant CENP-A constructs were generated by site-directed mutagenesis and confirmed by sequencing.
siRNAs, Lentiviral shRNA Vectors and Generation of Stable Cell Lines
Non-Targeting siRNA No. 2 (D0012100220) and Cyclin E1 smart-pool siRNA (LQ-003213-00-0002) were obtained from Dharmacon. Lentiviral FBW7, CENP-A, Cyclin E1, Cyclin A, CDK1, CDK2 and Cyclin B shRNAs were obtained from the Broad Institute TRC shRNA library. Lentiviral shRNA virus packaging, retrovirus packaging and subsequent infections were performed as previously (25). Target sequences are listed as below:
Ctrl shRNA: AACAGTCGCGTTTGCGACTGG
FBW7 (#1): CCAGTCGTTAACAAGTGGAAT
FBW7 (#2): CCAGAGACTGAAACCTGTCTA
CENP-A (694): CCGAGTTACTCTCTTCCCAAA
CENP-A (695): GCCTATCTCCTCACCTTACAT
CENP-A (697): GCAGCAGAAGCATTTCTAGTT
Cyclin E1 (#1): CGACATAGAGAACTGTGTCAA
Cyclin E1 (#2): CCTCCAAAGTTGCACCAGTTT
Cyclin A (#1): AAGGCAGCGCCCGTCCAACAA
Cyclin A (#2): AACTACATTG ATAGGTTCCTG
CDK2 (#1): GCCTTCCTACACGTTAGATTT
CDK2 (#2): CCGAGAGATCTCTCTGCTTAA
CDK1 (#1): GGATGTGCTTATGCAGGAT
CDK1 (#2): GGGGTTCCTAGTACTGCAA
Cyclin B: GTCAGTGAACAACTGCAGG
CENP-A-SNAP Loading Assays
DLD1 cells stably expressing CENP-A-SNAP proteins were synchronized to G1 using double thymidine block. Cells were released into normal media and BTP was added to quench the pre-existing CENP-A-SNAP pool. After removal of BTP, cells were allowed to grow for 11 hours before addition of TMRstar to label newly synthesized CENP-A-SNAP proteins as previously described (14). BTP and TMRstar were purchased from New England Biolabs.
Chromatin Extraction and Western Blot Analysis
Whole cell lysate extraction and immunoprecipitation experiments were performed as described (25). Chromatin bound fractions were extracted similarly as described (26). Specifically, cells were collected at 95% confluency for chromatin bound fraction extraction. Cell pellets containing nuclei were resuspended with 0.4 N H2SO4 followed by thorough resuspension and centrifugation. TCA solution (final concentration 33%) was added to the cleared supernatant to precipitate chromatin bound fraction followed by resuspension in EBC lysis buffer as described (25).
To determine the specificity of the pS18 CENP-A antibody raised in this study, HeLa cell nuclei were harvested and digested with MNase before chromatin extraction with salt as described(15). CENP-A chromatin was immunoprecipitated using either anti-HA monoclonal antibody after transfection of HA-tagged CENP-A constructs, or rabbit anti- total CENP-A polyclonal antibody for endogenous CENP-A proteins before Western blotting.
Immunofluorescence and Image Quantification
Immunofluorescence was conducted using methods described previously (27). Human mitotic chromosomes were prepared by cytospin as previously described (28). Integrated fluorescence intensity measurements were performed by using the methods described previously (29). Primary antibodes were listed in Table S1. Images were deconvolved and projected using SoftWoRx (GE Healthcare) for presentation. Raw images were analyzed using CRaQ v1.12 in Fiji for centromeric foci analysis as previously described (30).
GST Protein Purification and GST Pull Down
The GST-Skp2 was generated by fusing the first 90 residues of mouse Skp2 in frame with the GST tag as described (31). GST plasmids were transformed with BL21 competent cells followed by purification and pull down experiments as described previously (25).
In Vitro Kinase Assays and Mass Spectrometry Analysis
Active Cdk2/Cyclin kinases were purchased from Millipore and the protocol was followed as previously described (32). Following in vitro kinase assay, mass spectrometry analysis was performed similarly as previously described (25).
CRISPR-Cas9 for CENP-A Knockins
GeneArt seamless (Life Technologies) or G-blocks (IDT) were used to generate either the wild-type or phospho-mimic mutant of CENP-A (CENP-A S18D). See Supplementary method for amino-acid sequences that were designed for CRISPR Knockin. The human codon-optimized Cas9 (Addgene #52961) plasmid was obtained from Addgene. sgRNAs and right arm (CENP-A wild type and CENP-A S18D), left arm and insertion tag sequence were designed and constructed by using the method described previously (33). Potential off-target effects of sgRNA candidates were analyzed by the online tool CRISPR Design developed by Zhang’s laboratory (http://crispr.mit.edu/), and the sgRNA sequences with fewer off-target sites in human genome were selected for further analysis. For donor vectors, the requisite 15-bp end terminal homology between adjacent fragments or vector were designed and generated by IDT.
Synthesized double stranded left arm, right, SNAP tag as well as linearized donor vector was ligated using the GeneArt seamless enzyme (Life Technologies). Following ligation and transformation, DNA was prepared and sequenced to confirm the appropriate ligation. DLD1 cells were then transfected with FBW7 sgRNAs and ligated knockin vector with the SNAP tag followed by puromycin selection. Puromycin selected cells were divided into single cell culture followed by cell line expansion, genomic DNA extraction, PCR and sequence validation to confirm successful knockins.
FBW7 sgRNA sequence:
5′-CACCGCACCCTCTGCGGCGTGTCATGG-3′
5′-AAACTGACACGCCGCAGAGGGTGCC-3′
Soft Agar Assays and Xenograft Tumor Growth Analysis
Soft agar assays were peformed as previously described (34). For xenograft experiments, 5×105 cells with 10% matrigel were injected into the hind flank of 6 week old female NOD SCID gamma mice. Tumor sizes were measured every 3-4 days after initial tumor growth using a caliper. The tumor volume was then determined using the formula: L × W2 × 0.52, where L is the longest diameter and W is the shortest diameter of the measured growth. After 42 days, mice were euthanized, and tumors were harvested and weighed. All animal experiments were complied with National Institutes of Health guidelines and were approved by the University of North Carolina at Chapel Hill Animal Care and Use Committee.
Statistics
Results were reported as means ± SEM from three independent experiments where applicable. The imaging data were analyzed by GraphPad Prism7 (GraphPad software) unless otherwise indicated. Wilcoxon rank-sum test, two-tailed unpaired student’s t-test, Mann-Whitney U test and Fisher exact’s test were used to analyze statistical significance as indicated. P-value <0.05 was considered statistically significant.
Results
FBW7 Loss Leads to Reduced CENP-A Localization at the Centromere
To determine if FBW7 loss in cancer cells is accompanied by any centromere defects, we first examined CENP-A protein levels in chromatin fractions from isogenic DLD1 colon cancer cell lines with and without FBW7 knockout (24). Using a validated CENP-A antibody (Fig. S1A), we found that FBW7−/− cells displayed decreased CENP-A levels in the chromatin fraction compared to FBW7+/+, whereas total CENP-A in whole cell lysates was unchanged (Fig. 1A). Interestingly, CENP-A decreases in the chromatin fraction was accompanied by a simultaneous increase in CENP-A levels in the soluble fraction (Fig. 1B), accounting for the unchanged CENP-A levels in whole cell lysates. Importantly, ectopic reintroduction of FBW7 to DLD1 FBW7−/− cells decreased the level of the canonical FBW7 E3 ligase substrate Cyclin E1 and rescued CENP-A protein levels in the chromatin fraction, supporting a specific role for FBW7 in regulating CENP-A levels in chromatin (Figs. 1A and S1B). However, an FBW7 R465H hotspot mutation identified in various cancer types from patients, which is known to cause complete loss of FBW7 E3 ligase activity (35), failed to restore Cyclin E1 and chromatin-bound CENP-A protein levels (Fig. S1B).
Figure 1. FBW7 Loss Leads to Defects in CENP-A Centromeric Localization.
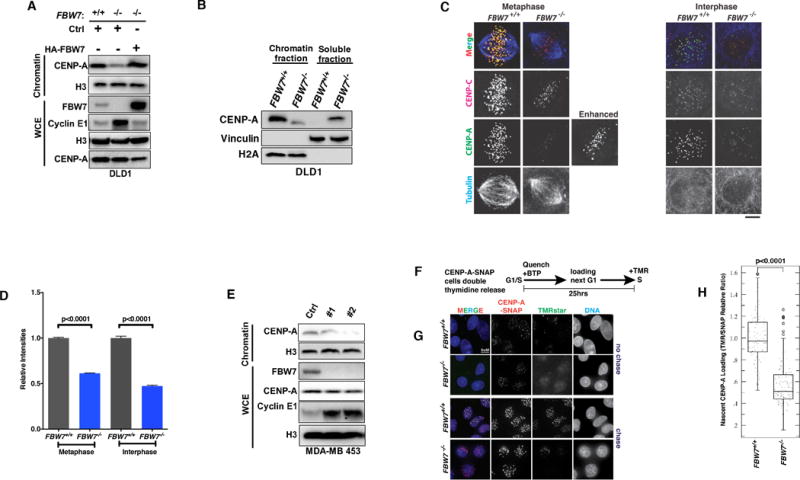
(A, B) Immunoblot of CENP-A using whole cell extracts, chromatin-bound and soluble fractions prepared from DLD1 cells (FBW7+/+), FBW7 knock-out (FBW7−/−) or FBW7−/− cells with reintroduction of exogenous FBW7.
(C, D) Representative images (C) and quantification (D) from FBW7+/+ or FBW7−/− DLD1 cells stained for CENP-A (green) or CENP-C (red). Cells were co-stained with anti-tubulin antibody (blue) to identify mitotic cells. Images are maximum-intensity projections of z stacks collected at 0.5-μM steps. Bars=5 μM. Results were statistically significant using unpaired student’s t-test. Data were shown as mean± SEM.
(E) Immunoblot of lysates from MDA-MB-453 infected with lentiviruses encoding FBW7 shRNA (#1 and #2) or control shRNA (Ctrl).
(F) Experimental scheme for CENP-A loading assay in DLD1 CENP-A-SNAP cells.
(G) DLD1 FBW7+/+ or FBW7−/− cells expressing CENP-A-SNAP were quenched with BTP followed by immediate TMRstar (green) incubation with no chase to assess blocking efficiency (top two panels), or quenched with BTP then allowed 11 hours chase time (bottom two panels) for new protein synthesis before labeling new CENP-A-SNAP with TMRstar. Anti-SNAP antibody was used to stain all CENP-A-SNAP (red). DNA was stained by DAPI (blue). Bars=5 μm.
(H) Box plots of CENP-A-SNAP loading at centromeres using the ratio of nascent (TMRstar) over total CENP-A-SNAP signals (anti-SNAP antibody). Mann-Whitney U test was used to determine statistical significance.
We then compared centromeric CENP-A levels between DLD1 FBW7 null and WT cells by immunofluorescence (IF) analysis, using CENP-C staining to identify kinetochores. Loss of FBW7 results in a marked reduction in CENP-A signal at centromeres compared to WT cells, in both metaphase and interphase (p<0.0001, student’s t-test) (Figs. 1C and 1D). FBW7 depletion using two independent shRNAs (#1 and #2) in the MDA-MB-453 breast cancer cell line and an immortalized breast epithelial cell line MCF-10A also showed similar results (Figs. 1E and S1C). There is no significant effect on total CENP-A levels, suggesting that CENP-A stability is not impacted (Figs. 1E and S1C). Notably, high levels of ectopic overexpression of FBW7 also caused a moderate drop in total CENP-A via an unknown mechanism (Fig. 1A).
Decreased CENP-A signals at centromeres could result from loss of pre-existing CENP-A or defective new CENP-A nucleosome assembly, which we addressed using a quench-chase-pulse SNAP-tagged CENP-A assay (Fig. 1F) (14). We first generated DLD1 FBW7+/+ and FBW7−/− clones that stably express CENP-A-SNAP with mild overexpression confirmed by western blotting and IF, or TMRstar labeling without BTP quenching (Figs. S1D and S1E). To determine if new CENP-A loading is impacted by FBW7 loss, the preexisting CENP-A-SNAP pool was quenched with BTP after a double thymidine block, and nascent protein was labeled with TMRstar for localization in the next G1 phase (14), in FBW7 WT and knockout cells, respectively (Fig. 1F). Notably, FBW7−/− cells displayed a significant decrease of 1.91 folds in TMRStar staining compared to FBW7+/+ cells (p<0.0001, Wilcoxon rank-sum test) (Figs. 1G and 1H).
The Cyclin E1/CDK2 Kinase Complex Mediates the CENP-A Reduction at Centromeres After FBW7 Loss
Several studies suggested that mitotic cyclins and CDK1/2 kinase activity are inhibitory for ectopic CENP-A loading by mediating phosphorylation of M18BP1, HJURP and the Ser68 residue of CENP-A itself (18–20). Cyclin E1 forms a functional kinase complex with CDK2 at the G1/S boundary to regulate cell cycle progression into S phase (36). FBW7 loss induces accumulation of Cyclin E1 (Fig. 1A), raising the possibility that FBW7 impacts CENP-A localization through Cyclin E1. To test this hypothesis, we determined the impact of Cyclin E1 depletion by siRNAs on CENP-A in FBW7−/− cells after fractionation (Fig. 2A). First, we observed that depletion of Cyclin E1 in FBW7−/− cells increased CENP-A levels in the chromatin fraction with a concomitant decrease in soluble fraction (Fig. 2A). Consistently, Cyclin E1 depletion increased centromeric CENP-A signals in both metaphase and interphase cells by IF and image quantification (p<0.0001, student-t test) (Figs. 2B and 2C). In a complementary set of experiments, depletion of Cyclin E1 by two independent shRNAs (#1 and #2) in FBW7−/− cells, to levels comparable to FBW7+/+ cells, consistently restored CENP-A protein levels in the chromatin fraction (Fig. 2D). It is worth noting that chromatin bound CENP-A levels in FBW7−/− cells with Cyclin E1 loss were higher than FBW7+/+ cells, which could be due to a combinatorial effect of FBW7 loss and Cyclin E1 depletion. Similarly, depletion of CDK2 by two different shRNAs also partially restored CENP-A levels at centromeres in FBW7 knockout cells in both Western blotting and IF experiments (Figs. S2A–C). Previous studies showed that both Cyclin E1 and Cyclin A bind CDK2 and regulate cell cycle progression (36,37). However, unlike Cyclin E1, Cyclin A depletion failed to restore CENP-A in chromatin and at centromeres in FBW7−/− cells (Figs. S2D–F). In addition to CENP-A, we also examined localization of the centromere and kinetochore components CENP-B and HEC1 by IF and image quantification. Interestingly, FBW7−/− cells also displayed decreased levels for CENP-B at centromeres, and to a lesser extent for the outer kinetochore protein HEC1, and these phenotypes are also ameliorated by Cyclin E1 depletion (p<0.0001 for all comparisons, student’s t-test) (Figs. 2E–2H).
Figure 2. Cyclin E1/CDK2 Kinase Complex Mediates the Reduction of Centromeric CENP-A Localization After FBW7 Loss.
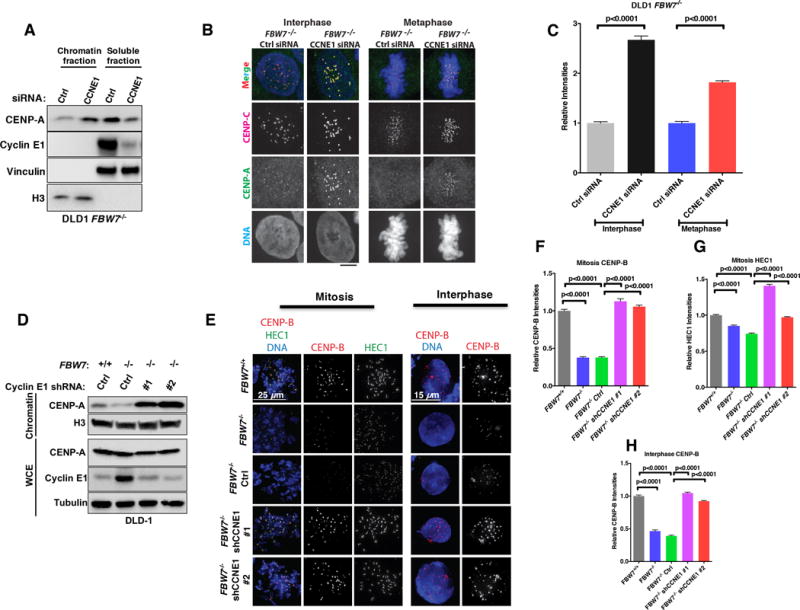
(A) Immunoblot analysis of lysates prepared from DLD1 FBW7−/− cells transfected with Cyclin E1 smartpool siRNA or control siRNA (Ctrl).
(B, C) Representative images (B) and Quantifications (C) from DLD1 FBW7−/− cells transfected with Cyclin E1 siRNA or control siRNA stained with CENP-A (green) or CENP-C (red). DNA was stained by DAPI (blue). Bars=5 μM.
(D) Immunoblot analysis of lysates prepared from DLD1 FBW7+/+ or FBW7−/− cells infected with lentivirus encoding either Cyclin E1 shRNAs (#1 and #2) or control shRNA (Ctrl).
(E, F, G, H) Representative images of mitotic cells after cytospin (left panel, E) and interphase cells by whole mount (right panel, E), and Quantifications (F-H) of DLD1 FBW7−/− cells infected with the lentivirus encoding either control shRNA (Ctrl) or Cyclin E1 shRNA (#1 and #2) stained for CENP-B (red) and HEC1 (green). DNA was stained by DAPI (blue). Bars=5 μM. Data were shown as mean± SEM.
Cyclin E1/CDK2 Kinase Complex Phosphorylates CENP-A at Ser18 In Vitro
In order to determine how Cyclin E1/CDK2 affects centromeric CENP-A localization, we tested the hypothesis that Cyclin E1/CDK2 directly phosphorylates CENP-A. We identified a canonical CDK substrate motif (S/TP) at the N-terminus of CENP-A at Ser18, which is well conserved across mammalian species (Fig. 3A). Previous research showed that CENP-A Ser18 is phosphorylated in HeLa and U2OS cells (23,38), which we confirmed by mass spectrometry (data not shown). We then determined if the Cyclin E1/CDK2 kinase can phosphorylate GST-tagged full-length CENP-A in vitro. Incubation with recombinant Cyclin E1/CDK2 kinase complex led to efficient CENP-A phosphorylation by radiography after separating proteins by SDS-PAGE, at a comparable level to a known Cyclin E1/CDK2 substrate Skp2 (Fig. 3B) (24). To determine if Cyclin E1/CDK2 phosphorylates CENP-A specifically at Ser18 residue or other adjacent sites, we mutated CENP-A at the Ser16, Ser18, Thr20, and Thr22 residues to alanines, either individually or together at all four residues (termed “4A”). We observed that only the Ser18 single mutation and 4A mutation abolished most CENP-A phosphorylation mediated by Cyclin E1/CDK2 (Fig. 3C). Mass spectrometry analysis of CENP-A proteins showed Ser18 as the major phosphorylation site after in vitro kinase assays (Fig. 3D).
Figure 3. The Cyclin E1/CDK2 Kinase Complex Phosphorylates CENP-A at Ser18 In Vitro.
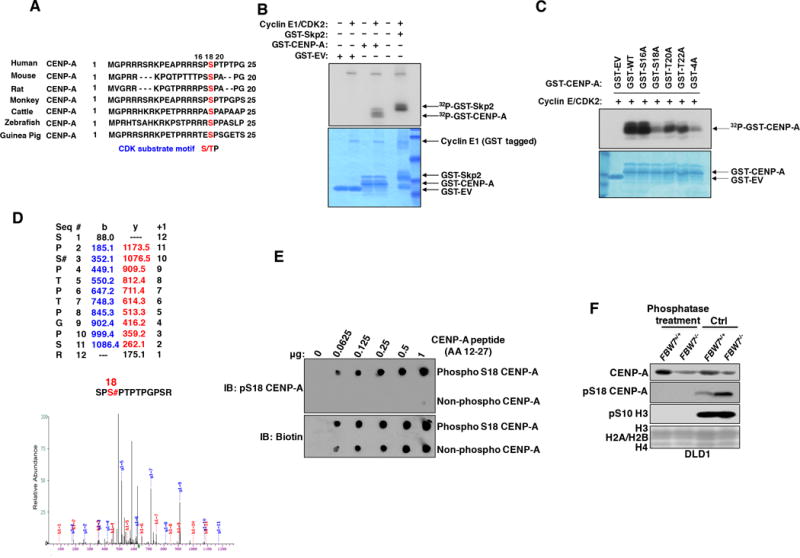
(A) N-terminal CENP-A amino acid sequence alignments in selected species.
(B, C) (upper panels) Autoradiography and (lower panels) SDS-PAGE Commassie Blue staining after in vitro Cyclin E1/CDK2 kinase assay using recombinant GST-CENP-A constructs and γ-32P-ATP. GST-tagged Cyclin E1 is indicated by an arrow. His tagged CDK2 is expected to be masked by GST due to similar molecular weight.
(D) Liquid chromatography-tandem mass spectrometry (LC-MS/MS) data for the excised CENP-A band showing CENP-A peptides phosphorylated at Ser18.
(E) Dot blot analysis of pS18 CENP-A and biotin antibodies for biotinylated CENP-A peptides (AA12-27) that are either WT (Non-phospho) or phosphorylated on Ser18 (phosphor S18) residue.
(F) Immunoblots of pS18-CENP-A in chromatin bound extracts from DLD1 FBW7+/+ or FBW7−/− treated with either CIP phosphatase (330 nM) or buffer only (Ctrl) at 37 degrees for 1 hour.
Next, we raised an antibody that specifically recognizes CENP-A phosphorylated at Ser18 (pS18-CENP-A). This antibody only recognizes CENP-A peptides with pS18 single or pS16/pS18 double phosphorylation, but not the unmodified or pS16 single phosphorylation peptide on dot plots (Figs. 3E and S3A). Moreover, CENP-A depletion by siRNA effectively reduced antibody signals by Western blotting using immunopreciptiated endogenous CENP-A (Fig. S3B). In addition, this antibody only recognized imunoprecipiated HA-tagged CENP-A WT proteins by western blotting but not after the Ser18 residue was mutated to an alanine (S18A) (Fig. S3C). Finally, FBW7−/− cells display significantly higher levels of pS18-CENP-A compared to FBW7+/+ cells and phosphatase treatment completely abrogated pS18-CENP-A signals in both cell lines (Fig. 3F), demonstrating the specificity of the antibody against phosphorylated CENP-A at Ser18.
Cyclin E1/CDK2 Kinase Regulates CENP-A Phosphorylation at Ser18 In Vivo
We then tested if increased pS18-CENP-A levels in FBW7−/− cells requires Cyclin E1, by comparing CENP-A Ser18 phosphorylation levels from telophase to early G1 in FBW7−/− cells. Cells were arrested at metaphase in nocodazole and released into normal media for 3 hours (Fig. S4A). Cyclin E1 depletion in FBW7 null cells resulted in significantly reduced pS18-CENP-A and increased total CENP-A levels in the chromatin fraction at each time point from telophase to early G1, while total CENP-A levels in whole cell extracts remained constant (Fig. 4A). Moreover, quantification of IF experiments showed that pS18-CENP-A signals at centromeres are higher in both interphase and mitotic cells in FBW7−/− cells compared to FBW7+/+ cells, but decreased significantly after Cyclin E1 depletion (Figs. S4B and S4C).
Figure 4. Cyclin E1/CDK2 Kinase Regulates CENP-A Phosphorylation at Ser18 In Vivo.
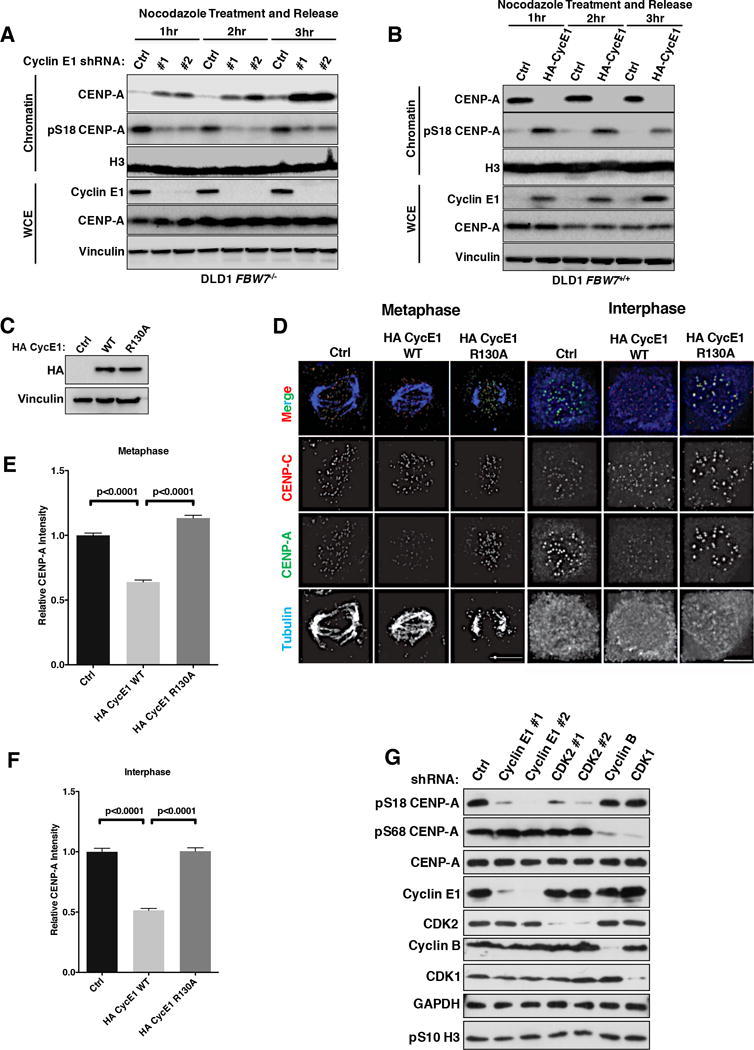
(A, B) Immunoblot analysis of lysates prepared from DLD1 FBW7−/− cells infected with (A) Cyclin E1 shRNAs (#1 and #2), or (B) FBW7+/+ cells infected with an HA-Cyclin E1 (HA-CycE1) lentivirus.
(C) Immunoblot analysis of whole cell extracts from DLD1 cells infected with the lentivirus encoding wild type HA Cyclin E1 (WT), HA Cyclin E1 R130A mutant (R130A) or empty vector control (Ctrl).
(D) Representative CENP-A (green) immunofluorescence images of both mitotic and interphase cells using cell lines as in (C). Centromeres were identified by CENP-C (red). Mitotic cells were identified by tubulin (blue).
(E, F) Quantitation of centromeric CENP-A signal intensities using the indicated DLD1 cells in (E) mitosis and (F) interphase. P values were calculated by unpaired student’s t-test. Data were shown as mean± SEM.
(G) Immunoblot analysis of pS18-, pS68- and total CENP-A in whole cell extracts from Hela cells transfected with the indicated siRNAs.
We then investigated if overexpression of Cyclin E1 enhances pS18-CENP-A levels in FBW7+/+ cells. We first arrested DLD1 FBW7+/+ cells in early S phase or metaphase with thymidine and nocodazole, respectively. CENP-A Ser18 phosphorylation in chromatin was highly enriched in thymidine-treated cells, coinciding with the peak of Cyclin E1 levels, but significantly diminished in cells treated with nocodazole (Fig. S4D). Similarly, in DLD1 FBW7+/+ cells, pS18-CENP-A signals at centromeres were low in G2 and most of mitosis, but started to increase in telophase cells by IF experiments (Figs. S4E and S4F).
Moreover, DLD1 FBW7+/+ cells with or without overexpression of HA-Cyclin E1 (HA-CycE1) were synchronized by nocodazole and released into normal media, respectively (24). After release from metaphase arrest, DLD1 cells entered into telophase/G1 within 3 hours (Fig. S4G). Cyclin E1 overexpression in DLD1 FBW7+/+ cells elevated levels of CENP-A Ser18 phosphorylation and simultaneously decreased chromatin-bound CENP-A (Fig. 4B). IF analysis confirmed that centromeric CENP-A levels were reduced to about 50% in Cyclin E1 overexpressing cells compared to control cells (p<0.0001, student’s t-test) (Figs. 4C–4F). To determine if the reduction in total centromeric CENP-A upon Cyclin E1 overexpression in DLD1 FBW7+/+ cells requires a functional Cyclin E1/CDK2 kinase, we infected DLD1 cells with either wild type (WT) Cyclin E1 or a Cyclin E1 R130A mutant that cannot bind CDK2 (39) (Fig. 4C). IF analysis showed that cells expressing WT Cyclin E1 displayed decreased total CENP-A at centromeres compared to the control cells, whereas the Cyclin E1 R130A mutant had no effect on centromeric CENP-A levels (Figs. 4D–4F).
To extend the observations in DLD1 cells, we also depleted Cyclin E1 or CDK2 by two independent shRNAs in HeLa cells, respectively, and confirmed the decrease of pS18-CENP-A (Fig. 4G). A recent study suggested that Cyclin B1/CDK1 phosphorylates CENP-A at Ser68 (20). Depletion of Cyclin E1 or CDK2 by shRNAs had no impact on CENP-A Ser68 phosphorylation levels by western blotting (Fig. 4G). Conversely, depletion of Cyclin B1 or CDK1 only specifically reduced levels of pS68-CENP-A, with no impact on pS18-CENP-A (Fig. 4G). Thus, CENP-A Ser18 and Ser68 phosphorylation in cells are regulated by Cyclin E1/CDK2 and Cyclin B/CDK1 kinases, respectively.
CENP-A Ser18 Phosphorylation Inhibits CENP-A Localization to Centromeres
Our data so far strongly suggested a connection between CENP-A Ser18 phosphorylation and reduced centromeric CENP-A localization. To test this hypothesis directly, we first overexpressed shRNA-resistant exogenous CENP-A WT or a phospho-mimetic S18D mutant in FBW7+/+ cells after depleting endogenous CENP-A (data not shown). Indeed, ectopically expressed WT CENP-A localized to centromeres labeled by the Anti-centromere antibody (ACA) in FBW7 proficient cells, whereas the S18D CENP-A mutant strongly decreased at centromeres by IF (Fig. 5A). Consistently, the CENP-A S18D mutant protein displayed decreased levels in chromatin compared to WT CENP-A by western blotting (Fig. 5B). Our cell fractionation experiments showed that a significant fraction of S18D CENP-A localized to the soluble fraction, while the WT CENP-A predominantly localized to chromatin (Fig. 5C). Conversely, we depleted endogenous CENP-A by shRNA followed by expression of WT or phosphorylation-deficient S18A CENP-A mutant protein from shRNA-resistant constructs in FBW7−/− cells. Notably, S18A CENP-A expressing cells displayed increased levels of chromatin-bound CENP-A protein and centromere localization compared to cells expressing WT CENP-A (Figs. 5D and 5E).
Figure 5. CENP-A Ser18 Phosphorylation Inhibits Centromeric Localization of CENP-A.
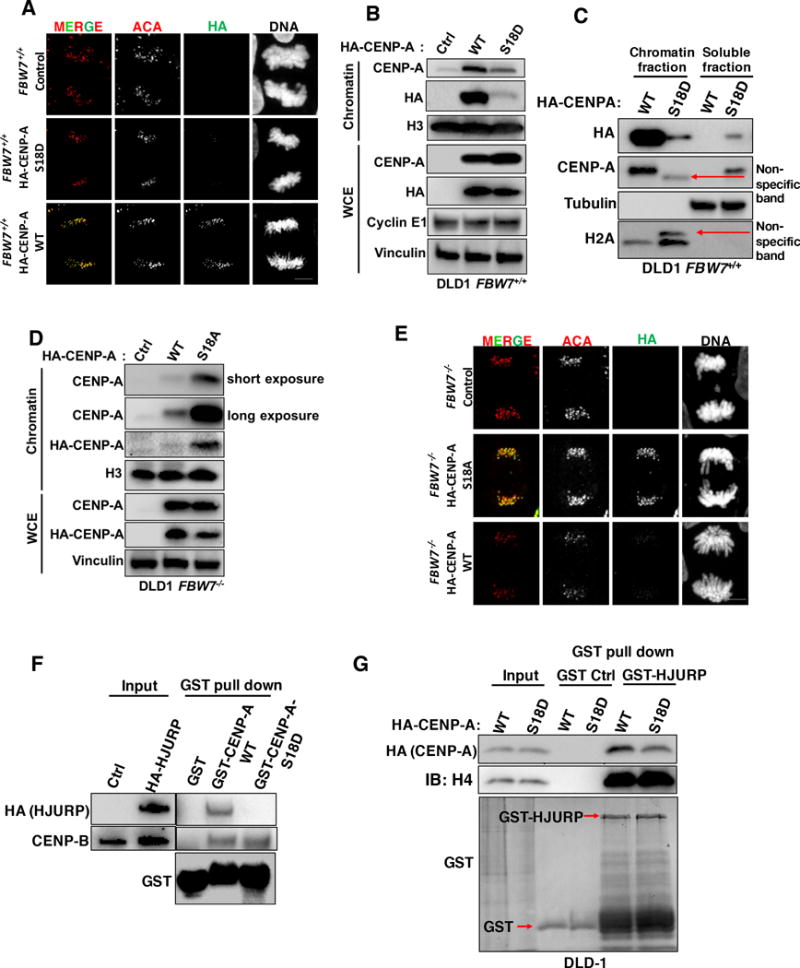
(A, E)Representative images of cells from (A) DLD1 FBW7+/+ cells infected with a CENP-A shRNA lentivirus followed by infection with either WT HA-CENP-A (WT), S18D, or empty vector (Ctrl). And (E) DLD1 FBW7−/− cells infected with a CENP-A shRNA lentivirus followed by infection with either WT HA-CENP-A (WT), S18A or empty vector (Ctrl) stained with HA (green), anti-centromere antibody (ACA) (red), and DAPI for DNA (grey). Bars=5 μm.
(B, D) Immunoblot analysis of whole cell extracts (WCE) and chromatin extracts using the same stable cell lines in (A) and (E).
(C) Immunoblot analysis of lysates from soluble and chromatin fractions using cells lines from (A). Arrows indicate the non-specific bands that may be due to secondary effects caused by ectopic expression of CENP-A S18D.
(F) Immunoblot analysis of lysates from GST pull-down (GST Control, GST-CENP-A WT or GST-CENP-A S18D) of 293T cells transfected with HA-tagged HJURP. CENP-B was also included for comparison.
(G) Immunoblot analysis of lysates from GST pull-down (GST Control or GST-HJURP) using DLD1 FBW7+/+ cells infected with a CENP-A shRNA lentivirus followed by infection with either WT HA-CENP-A (WT) or S18D.
Deposition of CENP-A at centromeres is mediated by the HJURP chaperone (15,16). We aimed to determine whether S18 phosphorylation affects CENP-A binding with HJURP. Our results showed that recombinant GST-CENP-A S18D mutant pulled down much less HJURP than GST-CENP-A WT (Fig. 5F), while both constructs pulled down similar amount of a different CENP-A binding protein CENP-B (Fig. 5F). In a complementary set of experiments, recombinant GST-HJURP pulled down less CENP-A S18D mutant than CENP-A WT (Fig. 5G). Furthermore, we analyzed recruitment of CENP-A to a LacO (Lac operon) array by an HJURP-LacI fusion protein in vivo (40). Chinese hamster ovary DG44 cells (Named as A03_1) containing LacO arrays at a single non-centromeric locus (Fig. S5A) (20) were co-transfected with mCherry-LacI-fused HJURP and EGFP-tagged WT, S18D or S18A mutant CENP-A, respectively. For EGFP-tagged WT or S18A CENP-A, we observed EGFP signals predominately at the LacO arrays as indicated by colocalization with mCherry-LacI-HJURP. However, a significantly higher proportion of cells expressing CENP-A S18D showed more diffuse EGFP signals at regions outside of the LacO arrays marked by mCherry-LacI-HJURP (Figs. S5B and S5C), implying that HJURP cannot efficiently recruit CENP-A S18D mutant proteins.
CENP-A Ser18 Phosphorylation Causes Mitotic Defects and CIN
Next we examined the effect of the S18D mutant on mitotic defects, including lagging chromosomes and chromosome bridges. Stable expression of S18D CENP-A in DLD1 FBW7+/+ cells led to increased bridging or lagging chromosomes compared to DLD1 FBW7+/+ control (p<0.01, Fisher’s exact test) (Figs. 6A and 6B). Conversely, stable expression of a CENP-A S18A phospho-deficient mutant, or stable Cyclin E1 depletion by shRNA in DLD1 FBW7−/− cells, significantly suppressed the frequencies of chromosome bridging and lagging phenotypes in FBW7−/− cells (p<0.01, Fisher’s exact test) (Figs. 6A and 6B). To further investigate the role of Ser18 phosphorylation in centromere function in other cell lines, we also stably expressed either CENP-A WT or S18D phospho-mimetic mutant proteins tagged with EGFP at low levels in HeLa cells (Fig. S6A). IF analysis showed that S18D EGFP-CENP-A had significantly weaker centromeric CENP-A localization than WT EGFP-CENP-A (Figs. S6B and S6C). HeLa cells expressing the S18D mutant also displayed significantly increased mitotic defects compared to the WT control (Figs. S6B and S6C).
Figure 6. Ser18 CENP-A Phosphorylation Leads to Mitotic Defects and Chromosomal Instability.
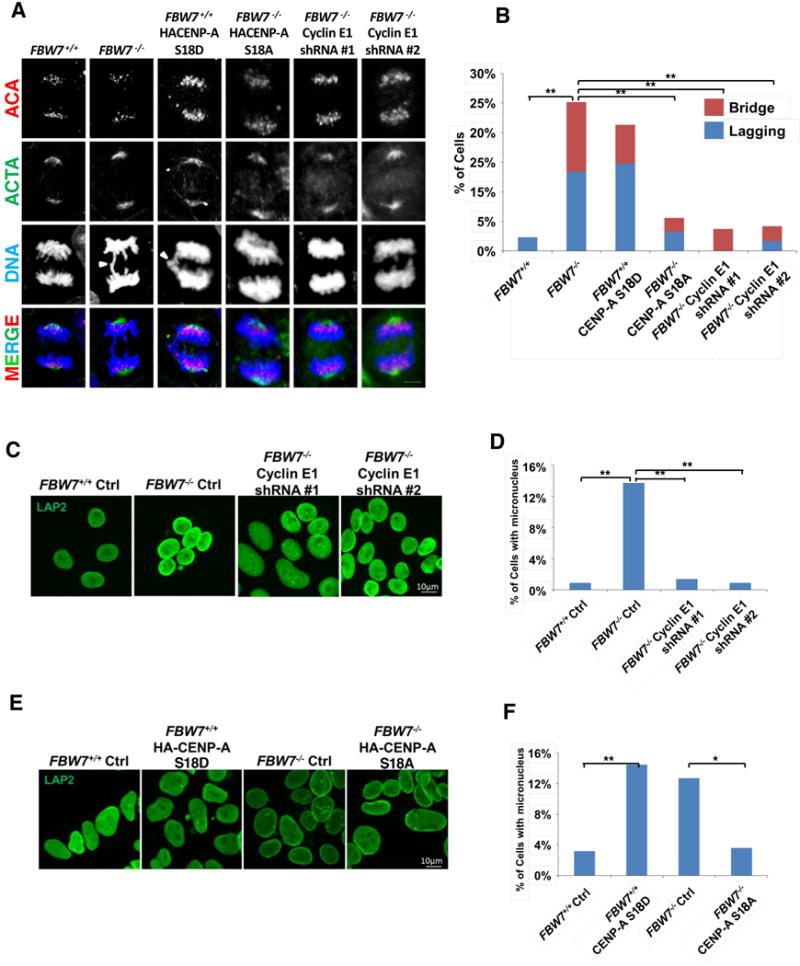
(A, B) Representative images (A) and quantification (B) of mitotic defects (chromatin bridges or lagging chromosomes) in DLD1 FBW7+/+ cells infected with the lentivirus encoding either CENP-A S18D or control, as well as DLD1 FBW7−/− cells infected with the lentivirus encoding CENP-A S18A, Cyclin E1 shRNAs (#1 and #2, respectively) or control. Cells were stained for ACA (red), actin (ACTA, green) and DNA by DAPI (blue). Bars=5 μm.
(C, D, E and F) Representative LAP2 antibody staining (green) and quantitation of micronuclei (MN) in various cell lines described and characterized in Figs. 2D, 5B and 5D. Bars=5 μm. Over 200 cells were quantified for each cell type. Statistical significance was determined by Fisher’s exact test. (*P < 0.05, **P < 0.01).
Micronucleus (MN) formation is a marker for increased CIN and cancer risk (6,41), and can be generated by lagging or bridging chromosomes during cell division (42,43). Staining for LAP2, a lamin- and chromatin-binding nuclear protein (44,45), revealed that DLD1 FBW7−/− cells display increased MN frequencies compared to FBW7+/+ cells, and that this phenotype is eliminated by Cyclin E1 depletion (p<0.01, Fisher’s exact test) (Figs. 6C and 6D). Strikingly, expression of phospho-mimetic S18D CENP-A in DLD1 FBW7+/+ cells significantly increased MN frequencies compared to FBW7+/+ controls (p<0.01, Fisher’s exact test), whereas expression of phospho-deficient S18A CENP-A in DLD1 FBW7−/− cells (DLD1 FBW7−/− S18A CENP-A) significantly reduced MN formation in DLD1 FBW7−/− (p<0.05, Fisher’s exact test) (Figs. 6E and 6F). Consistent with published results where loss of CENP-A by shRNA (DLD1 FBW7+/+ shCENP-A) also resulted in elevated MN frequencies (46) compared to controls (DLD1 FBW7+/+), the MN phenotype induced by CENP-A shRNA was only suppressed by expression of shRNA-resistant WT CENP-A (p<0.01, Fisher’s exact test) but not by S18D CENP-A (p>0.05, Fisher’s exact test) (Figs. S6D and S6E). These results suggest that reduced centromeric CENP-A localization due to Ser18 phosphorylation contributes to elevated levels of chromosome missegregation and MN formation.
CENP-A Ser18 Phosphorylation Promotes Tumor Progression
High levels of CIN predicts poor prognosis in cancer patients, and FBW7 loss or Cyclin E1 overexpression correlates with more aggressive tumor formation (47). To determine the clinical relevance of the findings, we first examined breast cancer samples for any potential correlation between Cyclin E1, CENPA pS18 and chromatin retention of CENP-A. We detected a correlation between high Cyclin E1 protein levels, increased chromatin-bound CENP-A Ser18 phosphorylation and decreased CENP-A levels in chromatin (Fig. 7A).
Figure 7. Ser18 CENP-A Phosphorylation Enhances Tumor Progression.
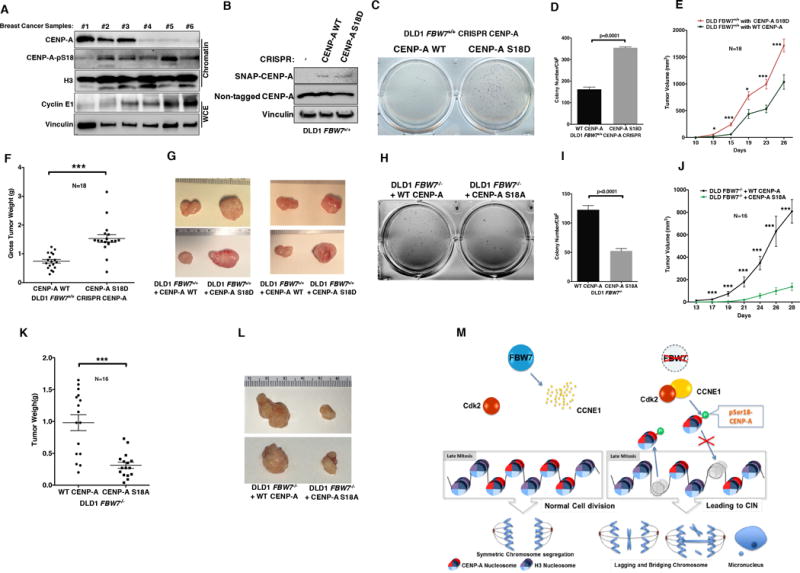
(A) Immunoblot analysis of CENP-A, pS18-CENP-A and Cyclin E1 using whole-cell and chromatin bound extracts prepared from breast cancer patient samples. Histone H3 and vinculin served as loading controls.
(B, C and D) (B) Immunoblot analysis of CENP-A, (C) anchorage-independent growth assays and (D) quantitation of DLD1 FBW7+/+ cells with endogenously tagged SNAP-CENPA WT or S18D mutant. Colony numbers in (D) are Mean ± SEM.
(E, F and G) The tumor growth curves of (E) tumor xenografts, (F) gross tumor weight at the time of necropsy and (G) representative images of tumor size using cell lines described in panel B. Tumor cells were transplanted subcutaneously into the bilateral dorsal region of female NOD/SCID mice. Results were statistically significant using unpaired student’s t-test. *** denotes p<0.005.
(H, I) (H) Anchorage independent growth assays and (I) quantitation of DLD1 FBW7−/− cells with exogenously tagged CENP-A WT or S18A mutant. Colony numbers in (I) are Mean ± SEM.
(J, K and L) (J) The tumor growth curves of tumor xenografts, (K) gross tumor weight at the time of necropsy and (L) representative image of tumor size for cell lines described in Figure 5D. Tumor cells were transplanted subcutaneously into the bilateral dorsal region of female NOD/SCID mice. Tumor volumes were measured with a caliper. Results were statistically significant using unpaired student’s t-test. *** denotes p<0.005.
(M) A model for the misregulation of CENP-A hyper-phosphorylation, reduced deposition and chromosome instability in FBW7 mutant cells.
To directly determine if CENP-A misregulation plays any role in tumor progression, we first compared anchorage-independent growth between cells expressing either WT or S18D phospho-mimetic CENP-A in DLD1 FBW7+/+ cells. We used CRISPR-Cas9 to replace one endogenous CENP-A locus with either a WT or S18D mutant CENP-A containing a SNAP tag (Figs. S7 and 7B). Notably, the CENP-A S18D mutant enhanced anchorage-independent growth and xenograft tumor growth compared to WT CENP-A (p<0.001, student’s t-test) (Figs. 7C–7G). Conversely, ectopic expression of the phosphorylation-deficient CENP-A S18A mutant significantly decreased anchorage-independent growth and xenograft tumor growth in DLD1 FBW7−/− cells compared to the cells expressing WT CENP-A as the control at a similar level (p<0.001, student’s t-test) (Fig. 5D, Figs. 7H–7L).
In summary, we demonstrated that Cyclin E1/CDK2 is necessary and sufficient for CENP-A Ser18 phosphorylation both in vitro and in cultured cells. Defective CENP-A localization at the centromere due to excessive phosphorylation of CENP-A at Ser18 enhances CIN, including chromosome missegregation and MN formation, and promotes anchorage-independent growth and tumor progression (Fig. 7M).
Discussion
CIN is characteristic of most human cancers, but the mechanisms leading to CIN and its role in cancer progression are poorly understood. In the present study, we identify a novel mechanism by which FBW7 loss promotes CIN and tumorigenesis through centromere misregulation. Specifically, we provide insight into mechanisms responsible for centromere misregulation in the cancer context and its roles during tumorigenesis, by connecting loss of FBW7 and excess Cyclin E1 with aberrant CENP-A Ser18 phosphorylation and reduced localization at the centromere. In summary, we identify an important new function for aberrant Cyclin E1/CDK2 activation in cancer, distinguishable from its well-established role in the G1/S transition (36).
Given the critical role of CENP-A in maintaining genome stability, assembly of new CENP-A at centromeres is tightly regulated to ensure faithful chromosomal segregation (48). Previous research, mostly done using other cancer cell lines, showed that the timing of CENP-A assembly is controlled by both CDK1 and CDK2 activities (19), which inhibit premature assembly of new CENP-A during S, G2 and most of mitosis. CDK1/2 and PLK1 impact the timing of new CENP-A deposition by phosphorylating M18BP1 and HJURP in HeLa and other cell lines (18,19,49). Another study in HeLa suggested that phosphorylation of CENP-A at Ser68 by Cyclin B1/CDK1 prevents premature CENP-A loading (20), despite some debates (21,22). While these studies highlighted important “normal” regulation that ensures proper centromere assembly, their roles in any clearly defined cancer model or in normal primary cells are not clear. We envision that under normal condition, Cyclin E1 levels are reduced by FBW7-mediated proteolysis and remain low from G2 to early G1 phases, to ensure that CENP-A is not hyperphosphorylated at Ser18 (among other functions) and to permit normal CENP-A assembly during late mitosis/G1. Here we identify CENP-A Ser18 hyper-phosphorylation after FBW7 loss or Cyclin E1 overexpression in cancers as another mechanism for regulating CENP-A centromeric localization, which contributes to mitotic errors and CIN.
FBW7 loss also leads to decreased centromeric CENP-B and HEC1 levels, which could be ameliorated by Cyclin E1 depletion. CENP-A Ser18 phosphorylation may also inhibit recruitment or function of other kinetochore components. For example, it is possible that pSer18 reduces CENP-A interactions with CENP-B (46,50), despite that we did not observe significant difference in CENP-B binding between CENP-A WT versus S18D proteins or peptides (Fig. 5 and not shown). However, there is also a possibility that Cyclin E1 directly regulates CENP-B and HEC1 independently from CENP-A phosphorylation, as CENP-C localization appears largely normal in FBW7−/− cells. While these important questions merit further investigations for fully understanding how FBW7 mutation or Cyclin E1 overexpression impacts CENP-A assembly, we emphasize that we identified Cyclin E1/CDK2 as a bona fide CENP-A Ser18 kinase, and its novel function in regulating CIN in cancer progression through centromere dysfunction relevant to many cancers with FBW7 mutation and Cyclin E1 overexpression.
Supplementary Material
Acknowledgments
The authors thank members of the Zhang and Wei laboratories for helpful discussions, and Dr. David Pellman for advices and critical readings. We would like to thank Dr. Bing Zhou for help on FACS analysis, Drs. Elizabeth M. Haynes, C. Robert Bagnell, Jr. and Josh Lawrimore for help on confocal imaging analysis, Dr. Seiji Sato and Dr. Nana Rokutanda for reagents, suggestions and comments on the CRISPR Knock in experiments. The authors thank Dr. Lars Jansen for providing SNAP-CENP-A plasmids.
Financial Support: This work has been supported by a NIH K99/R00 CA160351, V Scholar Award, Kimmel Scholar Award and University Cancer Research Fund (Q. Zhang), NIH R01 GM094777 (W. Wei) and NIH R01 GM119011 (G. H. Karpen).
Footnotes
Conflict of interest statement: The authors declare that they have no conflict of interest.
References
- 1.Davis RJ, Welcker M, Clurman BE. Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer cell. 2014;26:455–64. doi: 10.1016/j.ccell.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Z, Inuzuka H, Zhong J, Wan L, Fukushima H, Sarkar FH, et al. Tumor suppressor functions of FBW7 in cancer development and progression. FEBS letters. 2012;586:1409–18. doi: 10.1016/j.febslet.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Z, Liu P, Inuzuka H, Wei W. Roles of F-box proteins in cancer. Nature reviews Cancer. 2014;14:233–47. doi: 10.1038/nrc3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McGranahan N, Burrell RA, Endesfelder D, Novelli MR, Swanton C. Cancer chromosomal instability: therapeutic and diagnostic challenges. EMBO reports. 2012;13:528–38. doi: 10.1038/embor.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holland AJ, Cleveland DW. Losing balance: the origin and impact of aneuploidy in cancer. EMBO reports. 2012;13:501–14. doi: 10.1038/embor.2012.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rajagopalan H, Jallepalli PV, Rago C, Velculescu VE, Kinzler KW, Vogelstein B, et al. Inactivation of hCDC4 can cause chromosomal instability. Nature. 2004;428:77–81. doi: 10.1038/nature02313. [DOI] [PubMed] [Google Scholar]
- 7.Bertoli C, Skotheim JM, de Bruin RA. Control of cell cycle transcription during G1 and S phases. Nature reviews Molecular cell biology. 2013;14:518–28. doi: 10.1038/nrm3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allshire RC, Karpen GH. Epigenetic regulation of centromeric chromatin: old dogs, new tricks? Nat Rev Genet. 2008;9:923–37. doi: 10.1038/nrg2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goshima G, Kiyomitsu T, Yoda K, Yanagida M. Human centromere chromatin protein hMis12, essential for equal segregation, is independent of CENP-A loading pathway. J Cell Biol. 2003;160:25–39. doi: 10.1083/jcb.200210005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heun P, Erhardt S, Blower MD, Weiss S, Skora AD, Karpen GH. Mislocalization of the Drosophila centromere-specific histone CID promotes formation of functional ectopic kinetochores. Developmental cell. 2006;10:303–15. doi: 10.1016/j.devcel.2006.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang W, Mao JH, Zhu W, Jain AK, Liu K, Brown JB, et al. Centromere and kinetochore gene misexpression predicts cancer patient survival and response to radiotherapy and chemotherapy. Nat Commun. 2016;7:12619. doi: 10.1038/ncomms12619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu Z, Huang G, Sadanandam A, Gu S, Lenburg ME, Pai M, et al. The expression level of HJURP has an independent prognostic impact and predicts the sensitivity to radiotherapy in breast cancer. Breast cancer research : BCR. 2010;12:R18. doi: 10.1186/bcr2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stellfox ME, Bailey AO, Foltz DR. Putting CENP-A in its place. Cell Mol Life Sci. 2013;70:387–406. doi: 10.1007/s00018-012-1048-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jansen LE, Black BE, Foltz DR, Cleveland DW. Propagation of centromeric chromatin requires exit from mitosis. The Journal of cell biology. 2007;176:795–805. doi: 10.1083/jcb.200701066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dunleavy EM, Roche D, Tagami H, Lacoste N, Ray-Gallet D, Nakamura Y, et al. HJURP is a cell-cycle-dependent maintenance and deposition factor of CENP-A at centromeres. Cell. 2009;137:485–97. doi: 10.1016/j.cell.2009.02.040. [DOI] [PubMed] [Google Scholar]
- 16.Foltz DR, Jansen LE, Bailey AO, Yates JR, 3rd, Bassett EA, Wood S, et al. Centromere-specific assembly of CENP-a nucleosomes is mediated by HJURP. Cell. 2009;137:472–84. doi: 10.1016/j.cell.2009.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayashi T, Fujita Y, Iwasaki O, Adachi Y, Takahashi K, Yanagida M. Mis16 and Mis18 are required for CENP-A loading and histone deacetylation at centromeres. Cell. 2004;118:715–29. doi: 10.1016/j.cell.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 18.Muller S, Montes de Oca R, Lacoste N, Dingli F, Loew D, Almouzni G. Phosphorylation and DNA binding of HJURP determine its centromeric recruitment and function in CenH3(CENP-A) loading. Cell reports. 2014;8:190–203. doi: 10.1016/j.celrep.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 19.Silva MC, Bodor DL, Stellfox ME, Martins NM, Hochegger H, Foltz DR, et al. Cdk activity couples epigenetic centromere inheritance to cell cycle progression. Developmental cell. 2012;22:52–63. doi: 10.1016/j.devcel.2011.10.014. [DOI] [PubMed] [Google Scholar]
- 20.Yu Z, Zhou X, Wang W, Deng W, Fang J, Hu H, et al. Dynamic phosphorylation of CENP-A at Ser68 orchestrates its cell-cycle-dependent deposition at centromeres. Developmental cell. 2015;32:68–81. doi: 10.1016/j.devcel.2014.11.030. [DOI] [PubMed] [Google Scholar]
- 21.Fachinetti D, Logsdon GA, Abdullah A, Selzer EB, Cleveland DW, Black BE. CENP-A Modifications on Ser68 and Lys124 Are Dispensable for Establishment, Maintenance, and Long-Term Function of Human Centromeres. Developmental cell. 2017;40:104–13. doi: 10.1016/j.devcel.2016.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang K, Yu Z, Liu Y, Li G. Ser68 Phosphorylation Ensures Accurate Cell-Cycle-Dependent CENP-A Deposition at Centromeres. Developmental cell. 2017;40:5–6. doi: 10.1016/j.devcel.2016.12.015. [DOI] [PubMed] [Google Scholar]
- 23.Bailey AO, Panchenko T, Sathyan KM, Petkowski JJ, Pai PJ, Bai DL, et al. Posttranslational modification of CENP-A influences the conformation of centromeric chromatin. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:11827–32. doi: 10.1073/pnas.1300325110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lau AW, Inuzuka H, Fukushima H, Wan L, Liu P, Gao D, et al. Regulation of APC(Cdh1) E3 ligase activity by the Fbw7/cyclin E signaling axis contributes to the tumor suppressor function of Fbw7. Cell Res. 2013;23:947–61. doi: 10.1038/cr.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng X, Zhai B, Koivunen P, Shin SJ, Lu G, Liu J, et al. Prolyl hydroxylation by EglN2 destabilizes FOXO3a by blocking its interaction with the USP9x deubiquitinase. Genes & development. 2014;28:1429–44. doi: 10.1101/gad.242131.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shechter D, Dormann HL, Allis CD, Hake SB. Extraction, purification and analysis of histones. Nat Protoc. 2007;2:1445–57. doi: 10.1038/nprot.2007.202. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki A, Badger BL, Wan X, DeLuca JG, Salmon ED. The architecture of CCAN proteins creates a structural integrity to resist spindle forces and achieve proper Intrakinetochore stretch. Developmental cell. 2014;30:717–30. doi: 10.1016/j.devcel.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang W, Colmenares SU, Karpen GH. Assembly of Drosophila centromeric nucleosomes requires CID dimerization. Molecular cell. 2012;45:263–9. doi: 10.1016/j.molcel.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suzuki A, Badger BL, Salmon ED. A quantitative description of Ndc80 complex linkage to human kinetochores. Nat Commun. 2015;6:8161. doi: 10.1038/ncomms9161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bodor DL, Rodriguez MG, Moreno N, Jansen LE. Analysis of protein turnover by quantitative SNAP-based pulse-chase imaging. Curr Protoc Cell Biol. 2012 doi: 10.1002/0471143030.cb0808s55. Chapter 8:Unit8. [DOI] [PubMed] [Google Scholar]
- 31.Gao D, Inuzuka H, Tseng A, Chin RY, Toker A, Wei W. Phosphorylation by Akt1 promotes cytoplasmic localization of Skp2 and impairs APCCdh1-mediated Skp2 destruction. Nature cell biology. 2009;11:397–408. doi: 10.1038/ncb1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Inuzuka H, Tseng A, Gao D, Zhai B, Zhang Q, Shaik S, et al. Phosphorylation by casein kinase I promotes the turnover of the Mdm2 oncoprotein via the SCF(beta-TRCP) ubiquitin ligase. Cancer Cell. 2010;18:147–59. doi: 10.1016/j.ccr.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013;154:1370–9. doi: 10.1016/j.cell.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang J, Wang C, Chen X, Takada M, Fan C, Zheng X, et al. EglN2 associates with the NRF1-PGC1alpha complex and controls mitochondrial function in breast cancer. The EMBO journal. 2015;34:2953–70. doi: 10.15252/embj.201591437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature. 2011;471:104–9. doi: 10.1038/nature09732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koff A, Giordano A, Desai D, Yamashita K, Harper JW, Elledge S, et al. Formation and activation of a cyclin E-cdk2 complex during the G1 phase of the human cell cycle. Science. 1992;257:1689–94. doi: 10.1126/science.1388288. [DOI] [PubMed] [Google Scholar]
- 37.Xu M, Sheppard KA, Peng CY, Yee AS, Piwnica-Worms H. Cyclin A/CDK2 binds directly to E2F-1 and inhibits the DNA-binding activity of E2F-1/DP-1 by phosphorylation. Molecular and cellular biology. 1994;14:8420–31. doi: 10.1128/mcb.14.12.8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Logsdon GA, Barrey EJ, Bassett EA, DeNizio JE, Guo LY, Panchenko T, et al. Both tails and the centromere targeting domain of CENP-A are required for centromere establishment. The Journal of cell biology. 2015;208:521–31. doi: 10.1083/jcb.201412011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singer JD, Gurian-West M, Clurman B, Roberts JM. Cullin-3 targets cyclin E for ubiquitination and controls S phase in mammalian cells. Genes Dev. 1999;13:2375–87. doi: 10.1101/gad.13.18.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barnhart MC, Kuich PH, Stellfox ME, Ward JA, Bassett EA, Black BE, et al. HJURP is a CENP-A chromatin assembly factor sufficient to form a functional de novo kinetochore. The Journal of cell biology. 2011;194:229–43. doi: 10.1083/jcb.201012017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nersesyan A, Kundi M, Atefie K, Schulte-Hermann R, Knasmuller S. Effect of staining procedures on the results of micronucleus assays with exfoliated oral mucosa cells. Cancer Epidemiol Biomarkers Prev. 2006;15:1835–40. doi: 10.1158/1055-9965.EPI-06-0248. [DOI] [PubMed] [Google Scholar]
- 42.Fenech M, Kirsch-Volders M, Natarajan AT, Surralles J, Crott JW, Parry J, et al. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis. 2011;26:125–32. doi: 10.1093/mutage/geq052. [DOI] [PubMed] [Google Scholar]
- 43.Zhang CZ, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, et al. Chromothripsis from DNA damage in micronuclei. Nature. 2015;522:179–84. doi: 10.1038/nature14493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buendia B, Santa-Maria A, Courvalin JC. Caspase-dependent proteolysis of integral and peripheral proteins of nuclear membranes and nuclear pore complex proteins during apoptosis. J Cell Sci. 1999;112(Pt 11):1743–53. doi: 10.1242/jcs.112.11.1743. [DOI] [PubMed] [Google Scholar]
- 45.Gant TM, Harris CA, Wilson KL. Roles of LAP2 proteins in nuclear assembly and DNA replication: truncated LAP2beta proteins alter lamina assembly, envelope formation, nuclear size, and DNA replication efficiency in Xenopus laevis extracts. J Cell Biol. 1999;144:1083–96. doi: 10.1083/jcb.144.6.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fachinetti D, Folco HD, Nechemia-Arbely Y, Valente LP, Nguyen K, Wong AJ, et al. A two-step mechanism for epigenetic specification of centromere identity and function. Nature cell biology. 2013;15:1056–66. doi: 10.1038/ncb2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walther A, Johnstone E, Swanton C, Midgley R, Tomlinson I, Kerr D. Genetic prognostic and predictive markers in colorectal cancer. Nature reviews Cancer. 2009;9:489–99. doi: 10.1038/nrc2645. [DOI] [PubMed] [Google Scholar]
- 48.Black BE, Cleveland DW. Epigenetic centromere propagation and the nature of CENP-a nucleosomes. Cell. 2011;144:471–9. doi: 10.1016/j.cell.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McKinley KL, Cheeseman IM. Polo-like kinase 1 licenses CENP-A deposition at centromeres. Cell. 2014;158:397–411. doi: 10.1016/j.cell.2014.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fachinetti D, Han JS, McMahon MA, Ly P, Abdullah A, Wong AJ, et al. DNA Sequence-Specific Binding of CENP-B Enhances the Fidelity of Human Centromere Function. Developmental cell. 2015;33:314–27. doi: 10.1016/j.devcel.2015.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.