

Abstract
Spindlin1 (SPIN1) is a transcriptional coactivator with critical functions in embryonic development and emerging roles in cancer. SPIN1 harbors three Tudor domains, two of which engage the tail of histone H3 by reading the H3–Lys-4 trimethylation and H3–Arg-8 asymmetric dimethylation marks. To gain mechanistic insight into how SPIN1 functions as a transcriptional coactivator, here we purified its interacting proteins. We identified an uncharacterized protein (C11orf84), which we renamed SPIN1 docking protein (SPIN·DOC), that directly binds SPIN1 and strongly disrupts its histone methylation reading ability, causing it to disassociate from chromatin. The Spindlin family of coactivators has five related members (SPIN1, 2A, 2B, 3, and 4), and we found that all of them bind SPIN·DOC. It has been reported previously that SPIN1 regulates gene expression in the Wnt signaling pathway by directly interacting with transcription factor 4 (TCF4). We observed here that SPIN·DOC associates with TCF4 in a SPIN1-dependent manner and dampens SPIN1 coactivator activity in TOPflash reporter assays. Furthermore, knockdown and overexpression experiments indicated that SPIN·DOC represses the expression of a number of SPIN1-regulated genes, including those encoding ribosomal RNA and the cytokine IL1B. In conclusion, we have identified SPIN·DOC as a transcriptional repressor that binds SPIN1 and masks its ability to engage the H3–Lys-4 trimethylation activation mark.
Keywords: epigenetics, histone methylation, histone modification, transcription repressor, transcriptional coactivator
Introduction
Posttranslational modifications (PTMs)5 on histone tails directly affect chromatin structure and function. Combinations of histone PTMs constitute a “histone code” that functions as a signal platform to recruit specific effector molecules that contain specialized “reader” domains (1, 2). Lysine residues serve as the primary modification sites on histone tails, and they can be acetylated, methylated, and ubiquitylated. Acetylated lysine residues are read by Bromo and YEATS (YNK7, ENL, AF-9, and TFIIF small subunit) domains, and methylated lysine residues are read by Chromo, PHD (plant homeodomain), Tudor, MBT (malignant brain tumor) and PWWP (Pro-Trp-Trp-Pro) domains, to name just a few (3, 4). There are hundreds of epigenetic effector molecules that harbor reader domains and play critical roles in both the activation and repression of gene expression. These effector molecules usually do not function alone but often recruit protein complexes to histone tails to orchestrate the desired effect on chromatin. Here we are interested in understanding how the Tudor domain-containing protein SPIN1 functions as a transcriptional coactivator by identifying and characterizing proteins that bind it.
SPIN1 was originally described as an abundant maternal transcript deposited in mouse oocytes for early embryo development (5) and subsequently identified in a screen for genes involved in ovarian cancer (6). It is a member of a family of highly related proteins (SPIN1, 2A, 2B, 3, and 4) that all harbor three Tudor domains and are broadly expressed in different tissues (7). SPIN1-null mice exhibit early postnatal lethality and display a defect in meiosis (8). Overexpression of SPIN1 induces transformation of NIH3T3 cells that acquire the ability to grow in soft agar and in nude mice and also leads to perturbations of the cell cycle and chromosomal instability (9–11). SPIN1 protein levels are elevated in a number of different cancers (12, 13). Because of the emerging role of SPIN1 in transformation and cancer, there is a growing interest in developing small-molecule inhibitors that target this effector molecule. Indeed, we and others have identified compounds that block the SPIN1–H3K4me3 interaction and repress the transcriptional coactivator function of SPIN1 (14, 15).
SPIN1 is almost solely composed of three tandem Tudor domains (16). SPIN1 was first identified as a potential H3K4me3-binding protein in a screen for proteins that bound specifically to recombinant nucleosomes that were methylated at either the H3K9 or H3K4 positions (17). Subsequent studies validated this finding and mapped a direct interaction between the second Tudor domain of SPIN1 and the H3K4me3 mark (18), which was then characterized structurally at the atomic level (19). Recently, it was found that the first Tudor domain recognizes the H3R8me2a mark (20), making SPIN1 a bivalent reader of the histone code. As a reader of the H3K4me3 histone mark, SPIN1 functions as a transcription coactivator of rRNA genes (18), genes regulated by the MAZ (Myc-associated zinc finger protein) transcription factor (13), and Wnt/TCF4 target genes (12). Furthermore, recent RNA sequencing analysis of transcripts that are dysregulated by SPIN1 knockdown or small-molecule inhibitor treatment has expanded the number of genes that are regulated by SPIN1 (14).
In this study, we endeavored to gain a better understanding of how SPIN1 is regulated by its interacting protein(s). We identified a previously uncharacterized protein (C11orf84) as a SPIN1 docking protein (SPIN·DOC). SPIN·DOC directly interacts with SPIN1 and regulates its recruitment to chromatin. Overexpression of SPIN·DOC blocks SPIN1 chromatin association and impairs its coactivator function. Our functional studies revealed that the disruption of SPIN1 histone reading activity by SPIN·DOC is important for the attenuation of Wnt/TCF4 signaling. We thus provide new insights regarding the regulation of gene expression by SPIN1 and the functional significance of SPIN1–SPIN·DOC complex in the context of histone code reading.
Results
SPIN·DOC interacts with SPIN1 and its family members
To expand our understanding of how SPIN1 functions as a transcriptional regulator, we purified the protein complex that associates with it. To do this, we carried out tandem affinity purification from 293T cells stably expressing the SFB (S protein, 2× FLAG, and streptavidin-binding peptide)–tagged SPIN1 and then mock-transfected 293T cells. To investigate the possible existence of different types of SPIN1 protein complexes, SPIN1-binding proteins were isolated from both soluble and chromatin fractions and identified by MS analysis. On the basis of sequence coverage and the number of unique peptides, MS analysis revealed that C11orf84, a functionally uncharacterized protein, is the most abundant SPIN1-associated protein (Fig. 1A). Importantly, using a high-throughput affinity purification/MS approach, Gygi and co-workers (21) also identified SPIN1 as a C11orf84-interacting protein. They therefore validated this interaction using the reciprocal approach. We thus renamed this protein SPIN·DOC, for SPIN1 docking protein, to reflects its likely cellular function.
Figure 1.

SPIN1 interacts with SPIN·DOC in cells. A, lists of SPIN1-associated proteins identified in both the chromatin and soluble fraction by mass spectrometry analysis. B, GFP-SPIN1 and FLAG-SPIN·DOC was transiently transfected alone or together into HEK293T cells. Cell lysates were immunoprecipitated with α-FLAG or α-GFP and Western-blotted (WB) with the indicated antibodies. MW, molecular weight. C, HEK293T cells were transiently transfected with FLAG-SPIN·DOC, and whole-cell lysates were immunoprecipitated with α-FLAG or IgG control antibodies. Western blot analysis was performed with α-FLAG and α-SPIN1 antibodies. D, HEK293T cells were transiently transfected with GFP-SPIN, and whole-cell lysates were immunoprecipitated with α-GFP or IgG control antibodies. Western blot analysis was performed with α-FLAG and α-SPIN·DOC antibodies. E, HEK293T cells were subjected to knockdown with SPIN1 siRNA (or control (Con) siRNA) for 24 h, and then the whole-cell lysates were subjected to immunoprecipitation with α-SPIN·DOC or IgG control antibodies. Western blot analysis was performed using α-SPIN1. F, HEK293T cells were co-transfected with FLAG-SPIN·DOC and GFP-SPIN family members (SPIN1, 2A, 2B, 3, and 4), respectively. Whole-cell lysates were immunoprecipitated with α-GFP or IgG control antibodies and blotted with the indicated antibodies. In all depicted experiments, the input represent 2% of the sample used in the co-IP.
To further verify that SPIN·DOC associates with SPIN1, we performed co-immunoprecipitation (co-IP) experiments using ectopically expressing GFP-SPIN1 and FLAG-SPIN·DOC in HEK293T cells (Fig. 1B). Next, we demonstrated that ectopically expressed FLAG-SPIN·DOC (Fig. 1C) and GFP-SPIN1 (Fig. 1D) bind endogenous SPIN1 and SPIN·DOC, respectively. Last, we established that endogenous SPIN·DOC protein co-IPs endogenous SPIN1 (Fig. 1E). Because endogenous SPIN1 (30 kDa) runs close to the IgG light chain on SDS-PAGE, we knocked down SPIN1 and showed the absence of the SPIN1 protein interaction, proving that the Western blot signal seen at 30 kDa in the co-IP experiment is endogenous SPIN1 and not the IgG light chain.
SPIN1 is highly related to four additional family members - SPIN2A, 2B, 3, and 4. Interestingly, the affinity purification/MS study performed by Gygi et al. (21) identified not only SPIN1 in the C11orf84 protein complex but also SPIN2B and SPIN3. In the SPIN1 complex that we purified, we found SPIN·DOC and SPIN2A (Fig. 1A). These findings suggest that SPIN·DOC may bind all the members of the SPIN family of proteins. To test this, we performed co-IP experiments using ectopically expressing GFP-SPIN family members and FLAG-SPIN·DOC and indeed found that SPIN·DOC interacts strongly with all SPIN proteins (Fig. 1F). Taken together, the two independent MS studies and the co-IPs experiments demonstrate that SPIN·DOC is a bona fide SPIN1 binding partner.
SPIN·DOC inhibits SPIN1 methyl reading function
SPIN1 binds methyl marks (H3K4me2a, H3R8me2a, and H4K20me3) through its Tudor domains (18, 20, 22). We hypothesized that SPIN1 could either recruit SPIN·DOC to chromatin as part of its protein complex or that SPIN·DOC could block the interaction of SPIN1 with methylated histone tails. To begin to address this issue, we performed in vitro pulldown assays from transfected cell lysates using biotinylated histone tail peptides immobilized on beads. Rather unexpectedly, we found that, when SPIN·DOC is overexpressed, we observed a major inhibition of SPIN1 binding to both the H3K4me3 and H4K20me3 peptides (Figs. 2, A and B, bottom panels). Recently, we identified a small molecule (EML631) that interacts with the second Tudor domain of SPIN1 and blocks its ability to read the H3K4me3 histone mark (14). The biotinylated form of EML631 is able to pull down SPIN1 from a cell lysate. We observed that overexpression of SPIN·DOC also impedes the interaction of SPIN1 with EML631 (Fig. 2C), similar to the methyl–peptide interactions. Furthermore, the interaction between SPIN1 and SPIN·DOC is relatively stable, as even at a 1 m salt concentration there is no decrease in the efficiency of the co-IP (Fig. 2D). These in vitro results are consistent with the idea that the second Tudor domain of SPIN1 is structurally deformed or masked by the SPIN·DOC interaction, and we next explored this possibility in cells.
Figure 2.

SPIN·DOC inhibits the SPIN1 methyl reading function. GFP-SPIN1 and FLAG-SPIN·DOC were transiently transfected alone or co-transfected into HEK293T cells. A–C, the whole-cell lysates were then subjected to pulldown analysis with biotinylated H3K4me3 peptides (A), biotinylated H4K20me3 peptides (B), or biotinylated EML631 (C). MW, molecular weight; WB, Western blot. D, whole-cell lysates were immunoprecipitated with α-GFP or IgG control antibodies in the presence of 20 mm Tris-HCl (pH 8.0), 1 mm EDTA, and 0.5% Nonidet P-40 with increasing concentrations of NaCl as indicated. The respective pulldowns and co-IPs were subjected to SDS-PAGE and Western blot analysis using the indicated antibodies. The input represents 2% of the lysate that was used.
SPIN·DOC disrupts SPIN1 binding to chromatin
SPIN1 is a nuclear protein. Past studies that investigated the changes of protein subnuclear localization during the cell cycle have used a fractionation approach to separate soluble nuclear proteins from chromatin bound proteins (23). This soluble/chromatin fractionation approach has also been used to investigate the impact of mutating histone code reader modules on chromatin association (24). Here we used this fractionation approach to determine whether the overexpression of SPIN·DOC will force SPIN1 out of the chromatin fraction. Indeed, GFP-SPIN1 is present in both the soluble nuclear fraction and the chromatin isolate (Fig. 3A). When FLAG-SPIN·DOC was co-expressed with GFP-SPIN1, we observed a redistribution of GFP-SPIN1 from the chromatin fraction to the soluble fraction (Fig. 3A). To further establish whether ectopic expression of SPIN·DOC influences SPIN1 binding to chromatin, we performed a recently developed cell-based assay. In situ imaging experiments using specific fixation and detergent extraction conditions have been used to demonstrate that small-molecule inhibitors of the BDR4 and TRIM24 Bromo domains are able to promote the disassociation of these effector molecules from chromatin (25). Similarly, when SPIN·DOC is ectopically expressed in cells, it forces GFP-SPIN1 off chromatin, where it is more susceptible to extraction with Triton X-100, causing a reduced GFP signal (Fig. 3B).
Figure 3.

SPIN·DOC disrupts SPIN1 binding to chromatin in cells. A, GFP-SPIN1 and FLAG-SPIN·DOC were transfected or co-transfected into HeLa cells, and cell lysates were biochemically separated into soluble and chromatin-enriched fractions that were then analyzed by Western blotting (WB) with the indicated antibodies. Whole-cell extract (WCE) is shown to control for total GFP-SPIN1 and FLAG-SPIN·DOC. Histone H3 and β-actin levels are shown as controls for the integrity of the fractionation. MW, molecular weight. B, GFP-SPIN1 and FLAG-SPIN·DOC were transfected or co-transfected into HeLa cells. Shown is a representative image of GFP localization determined in cells with or without Triton X-100 treatment prior to fixation and visualization by fluorescence microscopy. DAPI staining is shown to indicate the nuclei of cells. C, HEK293T cells were transfected or co-transfected with SPIN·DOC and the GFP-SPIN family (SPIN1, 2A, 2B, 3, and 4). The whole-cell lysates were then subjected to pulldown analysis with biotinylated H3K4me3 peptides. The pulldowns were subjected to SDS-PAGE and Western blot analysis using α-GFP antibodies.
As we showed in Fig. 1F, SPIN·DOC binds all five SPIN family members. Using the H3K4me3 peptide pulldown approach, we find that SPIN·DOC blocks the reader ability of all SPIN family members, not just SPIN1 (Fig. 3C). SPIN·DOC is thus a general regulator of the SPIN protein family of effector molecules. These results (the peptide block and the chromatin block) suggest that SPIN·DOC likely functions as negative regulator of SPIN1-mediated transcriptional activation by blocking the SPIN1 histone methyl-binding ability. To investigate this possibility, we first focused on the well-characterized role of SPIN1 in WNT/TCF4-mediated transcriptional regulation.
SPIN·DOC represses SPIN1-mediated Wnt/TCF4 signaling in cells
It has been reported that SPIN1 directly interacts with TCF4 and activates Wnt signaling through its histone methylation reader activity (12, 20). We first confirmed the interaction between TCF4 and SPIN1 using a co-IP approach (Fig. 4A). In addition, we showed that tagged SPIN·DOC can co-immunoprecipitate TCF4 (Fig. 4B). When we knock down endogenous SPIN1 using siRNA, we lose the ability of tagged SPIN·DOC to co-immunoprecipitate TCF4 (Fig. 4C, bottom panel), which demonstrates that the TCF4–SPIN·DOC interaction is bridged by SPIN1. Because TCF4 is bound by the SPIN1–SPIN·DOC complex, we next measured Wnt-responsive TOPflash luciferase reporter activity in T778 cells upon SPIN1 or SPIN·DOC overexpression. This in vitro reporter system has been demonstrated to be responsive to the coactivator activity of SPIN1 (20). As expected, SPIN1 overexpression activated the Wnt signaling pathway, but when SPIN·DOC is ectopically overexpressed, the coactivator activity of SPIN1 is dampened in a dose-dependent manner (Fig. 4D). Furthermore, we confirmed that chemical inhibition (EML631) of SPIN1, which, like SPIN·DOC, blocks the H3K4me3 reader ability of SPIN1, has the same effect on the activity of Wnt signaling (Fig. 4E). Taken together, these results indicate that SPIN·DOC does not compete for the SPIN1/TCF4 binding site but, rather, functions to destabilize the SPIN1–TCF complex on a partially chromatinized reporter plasmid by preventing the association of SPIN1 with the H3K4me3 activation mark.
Figure 4.

SPIN·DOC blocks SPIN1-mediated Wnt/TCF-4 signaling. A and B, GFP-SPIN1 and Myc-TCF4 (A) and Myc-TCF4 and FLAG-SPIN·DOC (B) were transiently co-transfected into HEK293T cells. The whole-cell lysates were immunoprecipitated and blotted with the indicated antibodies. MW, molecular weight; WB, Western blot. C, HEK293T cells were transfected with SPIN1 siRNA or control siRNA. The whole-cell lysates were immunoprecipitated with a-FLAG or IgG control antibodies, and then the indicated proteins were identified by Western blot analysis. D, Wnt-responsive luciferase reporter assays were performed in transfected T778 cells in the presence of GFP-SPIN1 and increasing amounts of FLAG-SPIN·DOC plasmid DNA. E, Wnt-responsive luciferase reporter assays were performed with GFP-SPIN1–transfected T778 cells that were incubated with or without EML631 (15, 30 mm). Error bars show S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001; two-tailed Student's t test. The input represents 2% of the lysate that was used in every experiment.
SPIN·DOC down-regulates SPIN1 target gene expression
We next investigated whether SPIN·DOC functions as a transcriptional repressor of known endogenous SPIN1 targets. SPIN1 has been reported to regulate rRNA gene expression as a transcriptional coactivator (18) and to activate WNT/TCF4 signaling (12, 20). In addition, we recently identified a number of additional target genes that are regulated by SPIN1 (14). To determine whether SPIN·DOC regulates SPIN1 coactivator activity in cells at endogenous genes, we knocked down SPIN·DOC in T778 liposarcoma cells and performed RT-qPCR analysis on a panel of reported SPIN1 target genes (rRNA, IL1B, BST2, C1QTNF1, ALDH1A3, and IFI44L1) (Fig. 5A). The reduction in endogenous SPIN·DOC levels resulted in increased expression of all known SPIN1 targets tested, supporting the role of SPIN·DOC as a transcriptional repressor. Furthermore, as we have reported previously (14), all members of this gene set can be activated with the overexpression of ectopic SPIN1. However, with the co-expression of SPIN·DOC, SPIN1 is no longer able to robustly increase the expression of this gene set (Fig. 5B). Finally, using a ChIP-qPCR approach, we observed that GFP-SPIN1 was recruited to the promoter region of rRNA and IL1B, and the recruitment was dramatically blocked by co-expression of FLAG-SPIN·DOC (Fig. 5C). Thus, SPIN·DOC disturbs the binding of SPIN1 to chromatin, thereby largely neutralizing the activator effects of SPIN1 on its target genes.
Figure 5.
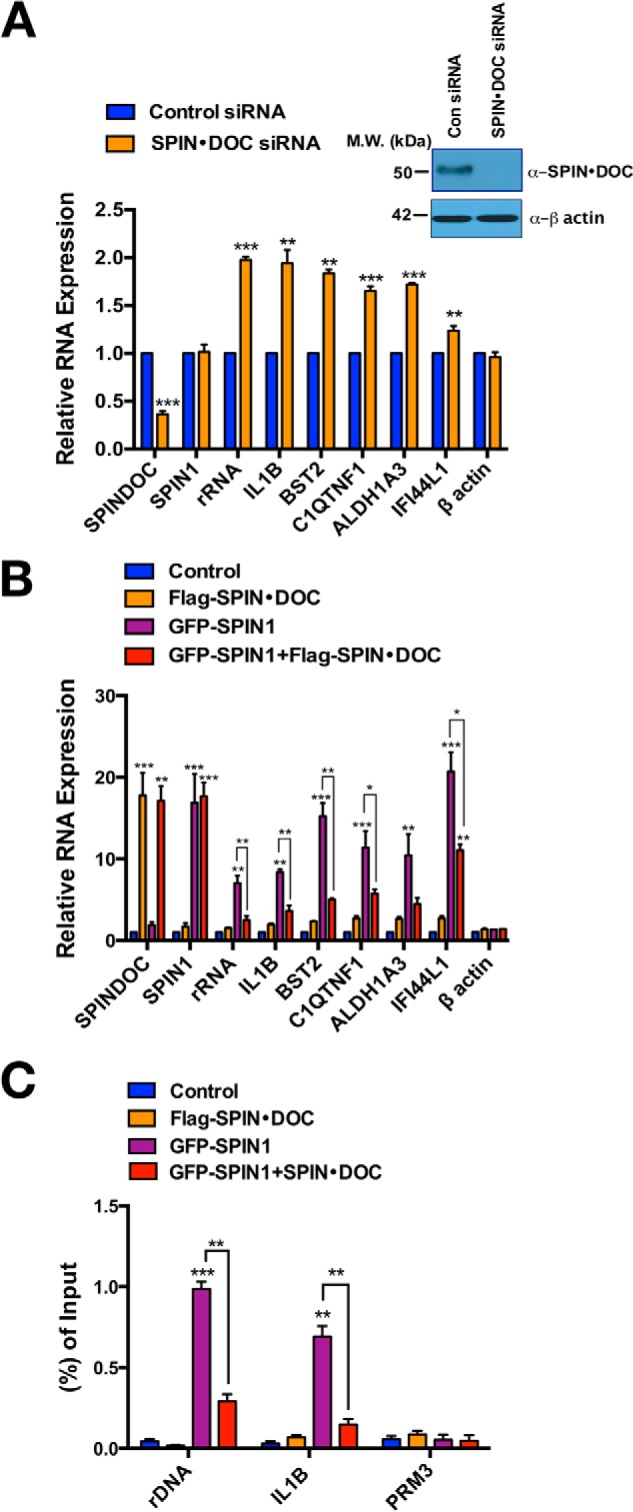
SPIN·DOC represses SPIN1-regulated target gene expression. A, the mRNA levels of SPIN1 target genes, in control and SPIN·DOC knockdown T778 cells, analyzed by RT-qPCR. The inset shows efficient knockdown of SPIN·DOC protein levels as detected by Western blot analysis. MW, molecular weight; Con, control. B, GFP-SPIN1 and FLAG-SPIN·DOC were transfected or co-transfected into T778 cells. SPIN1-regulated genes were analyzed by RT-qPCR. Gene expression was normalized to β-actin levels. C, the effect of SPIN·DOC on blocking SPIN1 chromatin association was assessed by α-GFP ChIP-qPCR analysis at two active loci (rDNA and IL1B) and a control locus (PRM3). ChIP-qPCR data are shown as a ratio to input. The mean value for the control groups was arbitrarily set as 1. All data represents the average of three independent experiments. Error bars indicate S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001; two-tailed Student's t test.
Discussion
The deregulation of SPIN1 levels was originally reported in a screen for genes involved in ovarian cancer (6). Subsequently, it was found that the overexpression of SPIN1 induces transformation of NIH3T3 cells and leads to perturbations of the cell cycle and chromosomal instability (9–11). SPIN1 protein levels are elevated in a number of different cancers (12, 13). Thus, there are ongoing efforts to identify small-molecule inhibitors of this effector molecule. SPIN1 harbors three Tudor domains that read methylation marks on histone tails, and it is this reader ability that has been the target of small-molecule inhibitor development (14, 15). A deep mechanistic understanding of how SPIN1 functions as a transcriptional coactivator is still lacking.
ChIP sequencing analysis revealed that SPIN1 occupies the promoters of a large number of genes with a high degree of overlap with the H3K4me3 transcriptional activation mark (13). RNA sequencing analysis of SPIN1 knockdown cells or cells treated with a SPIN1 inhibitor finds that only the minority of SPIN1-occupied genes are differentially regulated by SPIN1 loss (14), which indicates that SPIN1-mediated transcriptional control may be determined by additional regulatory factors. We therefore identified the stable protein complex that associates with SPIN1. In this study, we discovered the functionally uncharacterized protein SPIN·DOC as a major SPIN1 binding protein. This is very likely a direct interaction and not mediated by a linker protein because no other SPIN1-interacting protein is represented by as many peptides in the MS analysis. We found that the SPIN·DOC interaction blocks the reader ability of SPIN1 and attenuates its coactivator activity at a number of known target genes.
This is not the first finding of a Tudor domain interacting protein that obstructs the histone code reading ability of a chromatin effector molecule. Indeed, it was recently reported that the Tudor-interacting repair regulator (TIRR) directly binds the tandem Tudor domains of 53BP1, and it impaired their ability to bind the H4K20me2 mark (26, 27). Overexpression of TIRR disrupts 53BP1 function by masking the histone methyl-lysine binding function of 53BP1, as revealed by NMR-based structural studies (26). Importantly, the 53BP1–TIRR interaction is regulated by DNA damage–induced phosphorylation. The SPIN1–SPIN·DOC interaction seems to be mechanistically similar, although we have yet to determine how it is regulated.
When we set about purifying the SPIN1 protein complex, we were expecting to identify proteins that could help explain why SPIN1 functioned as a coactivator. Instead, we discovered a stably associated repressor protein, SPIN·DOC. This finding raised a number of interesting questions. First, it is likely that SPIN1 needs to dissociate from SPIN·DOC to be a fully functional transcriptional coactivator and may be regulated by a specific signal transduction pathway and the subsequent posttranslational modification of either SPIN1 or SPIN·DOC. Second, once SPIN·DOC disassociates from SPIN1, a secondary activator complex may be assembled, and it would be of value to isolate the SPIN1 protein complex from SPIN·DOC knockout or knockdown cells. Indeed, without the presence of SPIN·DOC, SPIN1 may be at liberty to form its activator complex. Third, structural studies of the SPIN1/SPIN·DOC complex would be of great value so that we can understand how the reader function of SPIN1 is blocked; are the Tudor domains of SPIN1 just shielded by SPIN·DOC, or are the aromatic cages within the Tudor domains deformed in some way to prevent the reading of the histone code?
Experimental procedures
SPIN1 complex purification and MS analysis
The tandem affinity purification of the SPIN1 protein complex is based on a detailed protocol described previously (28). Briefly, 1 × 108 HEK293T cells stably expressing tagged SPIN1 were lysed with NETN buffer (20 mm Tris-HCl (pH 8.0), 100 mm NaCl, 1 mm EDTA, and 0.5% Nonidet P-40, containing 1 μg/ml each of pepstatin A and aprotinin) for 30 min. Crude lysates were subjected to centrifugation at 16,000 × g at 4 °C for 30 min, and the pellet was digested with TurboNuclease (Accelagen) for 10 min in digesting buffer (50 mm Tris (pH 8), 1 mm MgCl2, and protease inhibitor) to extract chromatin-bound proteins. The supernatants were cleared at 16,000 × g to remove debris from chromatin-bound protein fractions. Both fractions were then incubated with streptavidin-conjugated beads (Amersham Biosciences) for 2 h at 4 °C. The beads were washed three times with NETN buffer, and the bead-bound proteins were eluted with NETN buffer containing 2 mg/ml biotin (Sigma). The elutes were incubated with S protein beads (Novagen). The beads were again washed three times with NETN buffer and subjected to SDS-PAGE. The protein band containing the entire sample was excised, and MS analyses were performed by the TAPLIN Biological Mass Spectrometry Facility of Harvard University. Mass spectrometry raw data were processed and further analyzed using the MUSE (Minkowski distance-based unified scoring environment) algorithm as described previously (29) to assign quality scores to the identified protein–protein interactions.
Cell culture and RNA interference
T778, HeLa, and HEK293T cells were purchased from the ATCC. All cell lines used in this study were tested for mycoplasma by using MycoAlertTM (Lonza) and were found to be uncontaminated. T778 cells were cultured in RPMI 1640 medium supplemented with 10% FBS, 1% penicillin/streptomycin, and l-glutamine. HeLa and HEK293T cells were cultured in DMEM supplemented with 10% FCS, 1% penicillin/streptomycin, nonessential amino acids, and l-glutamine. All cells were maintained in a humidified 37 °C incubator with 5% CO2. T778 cells were transfected with C11orf84 siRNA (Silencer® Select pre-designed siRNA, Ambion/Life Technologies, catalog no. 4392420; sense, AGGCAGACCUAUGACCAAAtt; antisense, UUUGGUCAUAGGUCUGCCUct) or control siRNA (Silencer® Select negative control 1 siRNA, Ambion/Life Technologies, catalog no. 4390843) using polyethyleneimine (PEI) for 24 h according to the instructions of the manufacturer.
Quantitative real-time PCR (RT-qPCR)
Total RNA was extracted from cells using TRIzol® reagent (Invitrogen) and reverse-transcribed using a Superscript III First Strand Synthesis Kit (Invitrogen). Quantitative RT-PCR was performed with the Applied Biosystems 7900HT RT-PCR instrument using iTaq Universal SYBR Green Supermix (Bio-Rad) with primers for the indicated genes. All primers used in this study were recently reported elsewhere (14). Gene expression was calculated following normalization to GAPDH levels using the comparative cycle threshold method and is shown as -fold relative to the expression of each gene in the control cells.
In vitro pulldown assay with GFP fusion proteins
HEK 293T cells were transiently transfected and cotransfected with GFP-SPIN1, 2A, 2B, 3, 4, and FLAG-C11orf84 using PEI according to the instructions of the manufacturer. Cells were lysed in 1× mild buffer (50 mm Tris HCl (pH 7.5), 150 mm NaCl, 0.1% Nonidet P-40, 5 mm EDTA, 5 mm EGTA, and 15 mm MgCl2) containing protease inhibitor mixture (Roche). 30 μl of streptavidin-agarose beads (Millipore) were prewashed with 1× mild buffer and incubated with 10 mg of biotinylated histone peptides for 2 h with rocking at 4 °C. After three washes with 500 μl of buffer to remove unbound histone peptides, the histone peptide–streptavidin-agarose mixture was incubated with cell lysates overnight with rocking at 4 °C. After three washes with 500 μl of binding buffer, 30 μl of 2× protein loading buffer was added to the beads and boiled. The samples were loaded on an SDS-PAGE and detected by Western blotting.
Chip-qPCR
T778 cells were transiently transfected and cotransfected with GFP-SPIN1 and FLAG-C11orf84 using PEI. As controls, cells were transfected with the empty GFP vector. The ChIP assay was performed following the EZ-ChipTM (Millipore, catalog no. 17-371) assay kit protocol. Briefly, cells were cross-linked with 1% formaldehyde for 10 min at room temperature, and the reaction was stopped with 125 mm glycine. Chromatin was sheared by using a Bioruptor sonication device (Diagenode) and subjected to immunoprecipitation overnight at 4 °C by using 2 μg of GFP antibody (Life Technologies, catalog no. A6455). Immune complexes were incubated overnight with 30 ml of a mixture of protein A/G-agarose at 4 °C. After reverse cross-linking was performed, the DNA was eluted and purified using a PCR purification kit (Qiagen). The primer sequences used for qPCR analyses have been reported previously and were as follows: PRM3, forward, 5′-GAAGTTATCCTGACTCACAC-3′; PRM3 reverse, 5′-CCAGAGCCCAGGCCACAGCC-3′; IL1B promoter (−199 to −109) forward, 5′-AACGATTGTCAGGAAAACAATG-3′; IL1B promoter (−199 to −109) reverse, 5′-CTGGTTCATGGAAGGGC-3′; rDNA loci (12,855-12,970) forward, 5′-ACCTGGCGCTAAACCATTCGT-3′; and rDNA loci (12,855–12,970) reverse, 5′-GGACAAACCCTTGTGTCGAGG-3′.
In situ cell extraction
In situ fractionation was modified from a protocol described previously (25). GFP-SPIN1 and FLAG-C11orf84 transiently transfected HeLa cells were washed with ice-cold PBS and then with freshly prepared cytoskeleton buffer (10 mm PIPES (pH 6.8), 300 mm sucrose, 100 mm NaCl, and 3 mm MgCl2). Immediately thereafter, non-chromatin-bound proteins were extracted by addition of cold cytoskeleton buffer supplemented with 0.25% Triton X-100 for 5 min at 4 °C. The cells were then fixed with 4% paraformaldehyde for 10 min at room temperature and imaged.
Small-scale biochemical fractionation
Small-scale biochemical fractionation was modified from a protocol described previously (23). In brief, GFP-SPIN1 and FLAG-C11orf84 co-transfected HEK293T cells were collected, washed with PBS, and resuspended in buffer A (10 mm HEPES (pH 7.9), 10 mm KCl, 1.5 mm MgCl2, 0.34 m sucrose, 10% glycerol, 1 mm DTT, and a complete protease inhibitor mixture tablet (Roche)). Triton X-100 (0.1% final concentration) was added, and the cells were incubated for 5 min on ice. Nuclei were collected by centrifugation at 1500 × g (4 °C, 5 min). The supernatant (S1) was clarified by centrifugation at 20,000 × g (4 °C, 5 min). The nuclei were washed once with buffer A and lysed in buffer B (3 mm EDTA, 0.2 mm EGTA, 1 mm DTT, and a complete protease inhibitor mixture tablet). The soluble fraction was combined with S1 and boiled at 100 °C with SDS sample buffer. Chromatin was washed with buffer B, resuspended in SDS sample buffer, and boiled at 100 °C for 5 min.
Co-immunoprecipitation and Western blotting
Cells were lysed with 1× mild buffer (20 mm Tris-HCl (pH 8.0), 150 mm NaCl, 1 mm EDTA, and 0.5% Nonidet P-40) containing protease inhibitor mixture on ice for 30 min. Clear cell lysates were incubated with either protein A-agarose beads coupled with the indicated primary antibody or streptavidin-Sepharose beads (Amersham Biosciences) for 3 h at 4 °C. Beads were then washed and boiled in 2× Laemmli buffer and separated on SDS-PAGE. PVDF membranes were blocked in 5% milk in TBST (20 mm Tris-Cl at pH 7.4, 150 mm NaCl, 0.1% Tween 20) buffer and then probed with antibodies as indicated.
Luciferase reporter assay
Wnt-responsive luciferase reporter assays in T778 cells were carried out in 96-well plates in triplicate as described previously (30). In brief, the plasmid DNA samples transfected per well were 20 ng of TOPflash, 1 ng of Renilla, 50 ng of GFP-SPIN1, and 50∼150 ng FLAG-C11orf84. After 24 h, firefly and Renilla luciferase activities were measured using a Dual-Luciferase assay kit (Promega). TOPflash luciferase activity was normalized to that of Renilla. The TOPflash and TCF4 vectors were a gift from Haitao Li.
Author contributions
M. T. B. and N. B. conceived the project. The MS studies were overseen by J. C., and he edited the manuscript. M. G. established the stable cell line that expressed SFB-tagged SPIN1 and purified the protein complexes. X. L. performed the mass spectrometry data analysis to remove nonspecific interacting proteins. N. B. performed all other experiments. G. S. provided EML631 and discussed and edited the manuscript. M. T. B. and N. B. wrote the manuscript.
M. T. B. is a cofounder of EpiCypher. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PTM
- posttranslational modification
- H3K4me3
- H3–Lys-4 trimethylation
- H3R8me2a
- H3–Arg-8 asymmetric dimethylation
- SPIN·DOC
- SPIN1 docking protein
- IP
- immunoprecipitation
- H4K20me3
- H3–Lys-20 trimethylation
- RT-qPCR
- quantitative real-time PCR
- TIRR
- Tudor-interacting repair regulator
- PEI
- polyethyleneimine.
References
- 1. Jenuwein T., and Allis C. D. (2001) Translating the histone code. Science 293, 1074–1080 [DOI] [PubMed] [Google Scholar]
- 2. Strahl B. D., and Allis C. D. (2000) The language of covalent histone modifications. Nature 403, 41–45 [DOI] [PubMed] [Google Scholar]
- 3. Yun M., Wu J., Workman J. L., and Li B. (2011) Readers of histone modifications. Cell Res. 21, 564–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Su Z., and Denu J. M. (2016) Reading the combinatorial histone language. ACS Chem. Biol. 11, 564–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Oh B., Hwang S. Y., Solter D., and Knowles B. B. (1997) Spindlin, a major maternal transcript expressed in the mouse during the transition from oocyte to embryo. Development 124, 493–503 [DOI] [PubMed] [Google Scholar]
- 6. Yue W., Sun L. Y., Li C. H., Zhang L. X., and Pei X. T. (2004) Screening and identification of ovarian carcinomas related genes. Chin. J. Cancer 23, 141–145 [PubMed] [Google Scholar]
- 7. Staub E., Mennerich D., and Rosenthal A. (2002) The Spin/Ssty repeat: a new motif identified in proteins involved in vertebrate development from gamete to embryo. Genome Biol. 3, RESEARCH0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chew T. G., Peaston A., Lim A. K., Lorthongpanich C., Knowles B. B., and Solter D. (2013) A Tudor domain protein SPINDLIN1 interacts with the mRNA-binding protein SERBP1 and is involved in mouse oocyte meiotic resumption. PLoS ONE 8, e69764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gao Y., Yue W., Zhang P., Li L., Xie X., Yuan H., Chen L., Liu D., Yan F., and Pei X. (2005) Spindlin1, a novel nuclear protein with a role in the transformation of NIH3T3 cells. Biochem. Biophys. Res. Commun. 335, 343–350 [DOI] [PubMed] [Google Scholar]
- 10. Zhang P., Cong B., Yuan H., Chen L., Lv Y., Bai C., Nan X., Shi S., Yue W., and Pei X. (2008) Overexpression of spindlin1 induces metaphase arrest and chromosomal instability. J. Cell Physiol. 217, 400–408 [DOI] [PubMed] [Google Scholar]
- 11. Yuan H., Zhang P., Qin L., Chen L., Shi S., Lu Y., Yan F., Bai C., Nan X., Liu D., Li Y., Yue W., and Pei X. (2008) Overexpression of SPINDLIN1 induces cellular senescence, multinucleation and apoptosis. Gene 410, 67–74 [DOI] [PubMed] [Google Scholar]
- 12. Wang J. X., Zeng Q., Chen L., Du J. C., Yan X. L., Yuan H. F., Zhai C., Zhou J. N., Jia Y. L., Yue W., and Pei X. T. (2012) SPINDLIN1 promotes cancer cell proliferation through activation of WNT/TCF-4 signaling. Mol. Cancer Res. 10, 326–335 [DOI] [PubMed] [Google Scholar]
- 13. Franz H., Greschik H., Willmann D., Ozretić L., Jilg C. A., Wardelmann E., Jung M., Buettner R., and Schüle R. (2015) The histone code reader SPIN1 controls RET signaling in liposarcoma. Oncotarget 6, 4773–4789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bae N., Viviano M., Su X., Lv J., Cheng D., Sagum C., Castellano S., Bai X., Johnson C., Khalil M. I., Shen J., Chen K., Li H., Sbardella G., and Bedford M. T. (2017) Developing Spindlin1 small-molecule inhibitors by using protein microarrays. Nat. Chem. Biol. 13, 750–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wagner T., Greschik H., Burgahn T., Schmidtkunz K., Schott A. K., McMillan J., Baranauskienė L., Xiong Y., Fedorov O., Jin J., Oppermann U., Matulis D., Schüle R., and Jung M. (2016) Identification of a small-molecule ligand of the epigenetic reader protein Spindlin1 via a versatile screening platform. Nucleic Acids Res. 44, e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhao Q., Qin L., Jiang F., Wu B., Yue W., Xu F., Rong Z., Yuan H., Xie X., Gao Y., Bai C., Bartlam M., Pei X., and Rao Z. (2007) Structure of human spindlin1: tandem Tudor-like domains for cell cycle regulation. J. Biol. Chem. 282, 647–656 [DOI] [PubMed] [Google Scholar]
- 17. Bartke T., Vermeulen M., Xhemalce B., Robson S. C., Mann M., and Kouzarides T. (2010) Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell 143, 470–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang W., Chen Z., Mao Z., Zhang H., Ding X., Chen S., Zhang X., Xu R., and Zhu B. (2011) Nucleolar protein Spindlin1 recognizes H3K4 methylation and stimulates the expression of rRNA genes. EMBO Rep. 12, 1160–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang N., Wang W., Wang Y., Wang M., Zhao Q., Rao Z., Zhu B., and Xu R. M. (2012) Distinct mode of methylated lysine-4 of histone H3 recognition by tandem Tudor-like domains of Spindlin1. Proc. Natl. Acad. Sci. U.S.A. 109, 17954–17959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Su X., Zhu G., Ding X., Lee S. Y., Dou Y., Zhu B., Wu W., and Li H. (2014) Molecular basis underlying histone H3 lysine-arginine methylation pattern readout by Spin/Ssty repeats of Spindlin1. Genes Dev. 28, 622–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huttlin E. L., Ting L., Bruckner R. J., Gebreab F., Gygi M. P., Szpyt J., Tam S., Zarraga G., Colby G., Baltier K., Dong R., Guarani V., Vaites L. P., Ordureau A., Rad R., et al. (2015) The BioPlex Network: a systematic exploration of the human interactome. Cell 162, 425–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shanle E. K., Shinsky S. A., Bridgers J. B., Bae N., Sagum C., Krajewski K., Rothbart S. B., Bedford M. T., and Strahl B. D. (2017) Histone peptide microarray screen of chromo and Tudor domains defines new histone lysine methylation interactions. Epigenetics Chromatin 10, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Méndez J., and Stillman B. (2000) Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol. Cell Biol. 20, 8602–8612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen S., Yang Z., Wilkinson A. W., Deshpande A. J., Sidoli S., Krajewski K., Strahl B. D., Garcia B. A., Armstrong S. A., Patel D. J., and Gozani O. (2015) The PZP domain of AF10 senses unmodified H3K27 to regulate DOT1L-mediated methylation of H3K79. Mol. Cell 60, 319–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhan Y., Kost-Alimova M., Shi X., Leo E., Bardenhagen J. P., Shepard H. E., Appikonda S., Vangamudi B., Zhao S., Tieu T. N., Jiang S., Heffernan T. P., Marszalek J. R., Toniatti C., Draetta G., et al. (2015) Development of novel cellular histone-binding and chromatin-displacement assays for bromodomain drug discovery. Epigenetics Chromatin 8, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Drané P., Brault M. E., Cui G., Meghani K., Chaubey S., Detappe A., Parnandi N., He Y., Zheng X. F., Botuyan M. V., Kalousi A., Yewdell W. T., Münch C., Harper J. W., Chaudhuri J., et al. (2017) TIRR regulates 53BP1 by masking its histone methyl-lysine binding function. Nature 543, 211–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang A., Peng B., Huang P., Chen J., and Gong Z. (2017) The p53-binding protein 1-Tudor-interacting repair regulator complex participates in the DNA damage response. J. Biol. Chem. 292, 6461–6467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li X., Wang W., Wang J., Malovannaya A., Xi Y., Li W., Guerra R., Hawke D. H., Qin J., and Chen J. (2015) Proteomic analyses reveal distinct chromatin-associated and soluble transcription factor complexes. Mol. Syst. Biol. 11, 775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li X., Han H., Zhou M. T., Yang B., Ta A. P., Li N., Chen J., and Wang W. (2017) Proteomic analysis of the human tankyrase protein interaction network reveals its role in pexophagy. Cell Rep. 20, 737–749 [DOI] [PubMed] [Google Scholar]
- 30. Wang Y., Fu Y., Gao L., Zhu G., Liang J., Gao C., Huang B., Fenger U., Niehrs C., Chen Y. G., and Wu W. (2010) Xenopus skip modulates Wnt/β-catenin signaling and functions in neural crest induction. J. Biol. Chem. 285, 10890–10901 [DOI] [PMC free article] [PubMed] [Google Scholar]