

Abstract
Recent structural studies suggest that GLUT1 (glucose transporter 1)-mediated sugar transport is mediated by an alternating access transporter that successively presents exofacial (e2) and endofacial (e1) substrate-binding sites. Transport studies, however, indicate multiple, interacting (allosteric), and co-existent, exo- and endofacial GLUT1 ligand-binding sites. The present study asks whether these contradictory conclusions result from systematic analytical error or reveal a more fundamental relationship between transporter structure and function. Here, homology modeling supported the alternating access transporter model for sugar transport by confirming at least four GLUT1 conformations, the so-called outward, outward-occluded, inward-occluded, and inward GLUT1 conformations. Results from docking analysis suggested that outward and outward-occluded conformations present multiple β-d-glucose and maltose interaction sites, whereas inward-occluded and inward conformations present only a single β-d-glucose interaction site. Gln-282 contributed to sugar binding in all GLUT1 conformations via hydrogen bonding. Mutating Gln-282 to alanine (Q282A) doubled the Km(app) for 2-deoxy-d-glucose uptake and eliminated cis-allostery (stimulation of sugar uptake by subsaturating extracellular maltose) but not trans-allostery (uptake stimulation by subsaturating cytochalasin B). cis-Allostery persisted, but trans-allostery was lost in an oligomerization-deficient GLUT1 variant in which we substituted membrane helix 9 with the equivalent GLUT3 sequence. Moreover, Q282A eliminated cis-allostery in the oligomerization variant. These findings reconcile contradictory conclusions from structural and transport studies by suggesting that GLUT1 is an oligomer of allosteric, alternating access transporters in which 1) cis-allostery is mediated by intrasubunit interactions and 2) trans-allostery requires intersubunit interactions.
Keywords: allosteric regulation, glucose transport, membrane transport, oligomerization, structure-function
Introduction
Glucose plays a crucial role in mammalian energy metabolism serving as a preferred metabolic substrate in brain and exercising skeletal muscle (1). However, the molecular mechanism by which glucose enters and exits cells is the subject of considerable controversy (2). The blood–brain barrier and glial and erythrocyte sugar transport are mediated by the transport protein GLUT1 (3). Recent structural studies suggest that GLUT1-mediated sugar transport is mediated by a carrier that alternately presents exofacial (e2) and endofacial (e1) substrate-binding sites (4–8). Transport studies, on the other hand, demonstrate multiple, interacting, co-existent exo- and endofacial ligand-binding sites (9–16) and e1 ligand-induced sugar occlusion within GLUT1 (17). The present study asks whether these apparently contradictory conclusions are mutually exclusive and thus indicative of some form of systematic error in analysis or, rather, are revealing of a more fundamental relationship between transporter structure and function.
GLUT1 comprises 492 amino acids, is a member of the major facilitator superfamily (MFS)2 of proteins and shares the common MFS fold (18, 19) of 12 transmembrane domains, cytoplasmic N and C termini and a long, partially structured intracellular loop connecting membrane spanning helices 6 and 7 (20). GLUT1 has been crystalized in an inward open (e1 or endofacial) conformation (6). Additional members of the MFS family have been crystalized in outward open (e2 or exofacial) and outward (e2o) and inward (e1o) partially occluded conformations. These members include human GLUT3 (e2, e2o (5)), GLUT5 (e1, e2 (4)), and the bacterial xylose transporter XylE (e1, e1o, e2o (7, 21)). Each structure supports the hypothesis that the sugar translocation pathway consists of eight amphipathic, membrane spanning α-helices (H1, H2, H4, H5, H7, H8, H10, and H11) coordinated by a scaffold of four hydrophobic α-helices (H3, H6, H9, and H12). The N- and C-terminal membrane-spanning α-helices share a similar topology and are related by a 2-fold symmetry (22).
Two competing hypotheses have been presented to explain glucose transporter behavior: the simple, alternating access carrier that sequentially presents mutually exclusive exofacial and endofacial substrate-binding sites (18, 19) or the fixed-site transporter with co-existent exofacial and endofacial sugar-binding sites (9, 23). Initial studies using purified GLUT1 suggested that exofacial and endofacial inhibitors bind at mutually exclusive binding sites, thereby supporting the simple carrier model (24, 25). Erythrocyte sugar transport and ligand-binding studies (13–16) and studies using nonreduced, purified transporter (15, 16) suggest GLUT1 behaves like a fixed-site transporter with interacting, cooperative binding sites.
Cell membrane resident GLUT1 forms noncovalent homodimers and tetramers (26–29). Purified, nonreduced GLUT1 forms a mixture of dimeric and tetrameric species, whereas reduced purified GLUT1 is largely dimeric. GLUT1 and GLUT3 co-expressed in the same cells do not form heterocomplexes (29, 30), but substitution of GLUT3 membrane spanning α-helix 9 (H9) into GLUT1 shifts the GLUT1 population from a tetrameric/dimeric mixture with high transport capacity to a dimeric population with reduced transport capacity, suggesting that H9 is involved in GLUT1 oligomerization. Conversely, substitution of GLUT1 H9 into GLUT3 converts GLUT3 into a tetramer and increases its transport capacity to GLUT1 levels, confirming the pivotal role of H9 in determining GLUT quaternary structure and catalytic function (29).
Ligand-binding studies with nonreduced and reduced GLUT1 provide further insights into the subunit organization of the transport complex (27). The sugar transport inhibitor cytochalasin B (CB) and intracellular sugar compete for binding at the GLUT1 endofacial sugar-binding site (31). Purified tetrameric GLUT1 binds 0.5 mol CB/mol GLUT1, whereas purified dimeric GLUT1 binds one mol CB/mol GLUT1 (27). This contradiction is explained by the suggestion (28) that each subunit of tetrameric GLUT1 undergoes the e2 ⇌ e1 catalytic cycle, but at any instant two subunits must present the e1 conformation and two must adopt the e2 conformation. In dimeric GLUT1, each subunit is functionally unconstrained by its neighbor and free to adopt either the e1 or e2 conformation. Thus, both subunits of dimeric GLUT1 bind CB when [CB] is saturating.
This suggestion is reinforced by demonstrations of functional coupling between GLUT1 ligand- and substrate-binding sites. trans-Allostery is observed when low concentrations of GLUT1 endofacial site inhibitors (e.g. forskolin or CB) increase the affinity of the external site for transported sugar (12). As inhibitor concentration is further increased, transport is inhibited. Exofacial cis-allostery is observed when extracellular maltose (a nontransportable disaccharide that binds at the exofacial sugar-binding site), stimulates sugar uptake at low maltose concentrations but inhibits uptake as its concentration is raised (13). Endofacial cis-allostery is seen when endofacial ligand binding (e.g. forskolin) increases the affinity of GLUT1 for a second e1 ligand (e.g. CB) (11, 12). These findings suggest that GLUT1 presents multiple, co-existent endo- and exofacial ligand-binding sites and/or that oligomerization promotes subunit cooperativity.
This study interrogates crystal and homology-modeled GLUT1 structures to ask whether GLUT1 can present multiple substrate and ligand interaction sites. Then using insights gained from this analysis, we mutagenize GLUT1 to examine its impact on cis- and trans-allostery. We conclude that exofacial cis-allostery is an intramolecular phenomenon resulting from cross-talk between multiple, co-existent ligand interaction sites present in the exofacial cavity of each GLUT1 protein, whereas trans-allostery and endofacial cis-allostery require ligand-induced subunit–subunit interactions.
Results
Homology-modeled GLUT1 structures
GLUT1 and GLUT3 structures have been described previously (5, 6). The current study presents and interrogates three homology-modeled GLUT1 structures—the so-called outward, outward-occluded, and inward-occluded conformations of GLUT1, plus the experimentally derived inward conformation of GLUT1 (Protein Data Bank code 4PYP (6); Fig. 1). These conformations present a striking physical correspondence to the proposed kinetic intermediates in the alternating access carrier catalytic cycle named e2, e2o, e1o, and e1 (1, 32, 33), argue strongly for sugar movements through a central translocation pore, and are henceforth termed GLUT1-e2, GLUT1-e2o, GLUT1-e1o, and GLUT1-e1.
Figure 1.

Homology-modeled GLUT1 conformations. A, GLUT1 is shown in cartoon representation normal to the bilayer plane (horizontal orange lines). Membrane spanning α-helices (2, 3, 4, 7, 11, and 12) are indicated, and the locations of the interstitium and cytoplasm are highlighted. Four conformations are depicted: exofacial (GLUT1-e2), exofacial-occluded (GLUT1-e2o), endofacial-occluded (GLUT1-e1o), and endofacial (GLUT1-e1). B, a second depiction of GLUT1-e1 is shown along the bilayer normal from the cytoplasmic side. Membrane spanning α-helices (noted by numbers 1–12) are indicated. C, representation of ligand-interaction cavities present in all four GLUT1 conformations shown normal to the bilayer plane. N- (N-Term) and C-terminal (C-Term) halves (helices 1–6 and 7–12, respectively, shown in gray in cartoon representation) of each conformation are indicated. Solvent-exposed residues in the ligand interaction cavities of each conformation are shown as surface maps colored cyan. Residues common to all four cavities are shown as surface maps colored red and include: N-terminal residues Gly-26 and Thr-30 (of helix 1) and Gln-161, Ile-164, Val-165, and Ile-168 (of helix 5) and C-terminal residues Gln-282, Gln-283, Ile-287, Asn-288, Phe-291, and Tyr-292 (of helix 7); Asn-317 (of helix 8); Phe-379 and Trp-388 (of helix 10); and Asn-411, Trp-412, and Asn415 (of helix 11).
Docking analysis of GLUT1–substrate interactions
Docking analysis first requires the location of GLUT1 pockets of sufficient size to permit ligand entry and coordination. Cavity analysis of all four homology-modeled GLUT1 conformations suggests the existence of a translocation pore that transitions from one contiguous with the interstitium but excluded from cytosol in GLUT1-e2 through intermediate, occluded cavity forms in GLUT1-e2o and GLUT1-e1o to a cavity contiguous with the cytosol but excluded from the interstitium in GLUT1-e1 (Fig. 1). Translocation pore volume increases in the occluded state. Computed cavity volumes (Fig. 1) are GLUT1-e2 = 2,850.8 Å3, GLUT1-e2o = 4,397.5 Å3, GLUT1-e1o = 3,029.4 Å3, and GLUT1-e1 = 2,845.9 Å3. For comparison, the molecular volume of β-d-glucose (β-d-Glc) based on its self-diffusion coefficient is 433 Å3 (34). Loop 6-7 was not included in cavity calculations for the GLUT1-e1 conformation. The side chains of several residues line the cavities in all four conformations (Fig. 1C).
Glide software was developed to optimize ligand docking to rigid protein structures using co-crystallized ligand–protein complexes as comparative standards (35, 36). Glide scores for computed ligand/protein pairs are useful for selecting the best docked poses but can under- or overestimate ΔG for binding by 2 kcal/mol (30-fold) (37).
Transport studies show that β-d-Glc binding at the GLUT1 exofacial sugar-binding site involves H-bonding to pyranose ring C1, C3, and C4 oxygens (38, 39). Similarly, ligand binding at the endofacial sugar-binding site involves H-bonding with OH groups at C3 and C4 in the pyranose ring and is inhibited by bulky substitutions at C6 (39). Docking analysis of β-d-Glc interactions with GLUT1-e2, GLUT1-e2o, GLUT1-e1o, and GLUT1-e1 conformations is shown in Fig. 2 (A and B). The illustrated interactions conform to the aforementioned stereospecificity of GLUT1-ligand binding. Gln-282 is the one residue whose side chain interacts with β-d-Glc in all four GLUT1 conformations. Fig. 2C shows the relative positions of β-d-Glc in the XylE-e2o-β-d-Glc co-crystal structure (7) and of β-d-Glc docked to the homology-modeled GLUT1-e2o structure following alignment of the two protein structures. The agreement is excellent and exceeds the resolution (2.9 Å) of the XyleE-e2o structure.
Figure 2.

β-d-Glc docking to homology-modeled GLUT1 conformations. A, each GLUT1 conformation is shown complexed with β-d-Glc (shown in red as a space-filling representation). The location of GLUT1 Gln-282 is shown in cyan in space-filling format. Conformation nomenclature is indicated beneath each structure where β-d-Glc is represented by the letter S, and occluded β-d-Glc (by convention) is represented by the letter S in parentheses. B, β-d-Glc docking to GLUT1 conformations in which Glc is shown as a 2D structure, coordinating GLUT1 residues are shown as circles and are colored according to their properties: green, hydrophobic, cyan, polar; red, negative. The GLUT1 backbone is shown as ribbons, solvent-exposed regions of β-d-Glc are indicated by gray-shaded circles, and H-bonds shared between amino acid side chain amines, carbonyls, or hydroxyls with β-d-Glc and their directionality are represented as red arrows. C, alignment of XylE-e2o containing a co-crystallized β-d-Glc (Protein Data Bank code 4GBZ (7); XyleE with homology-modeled GLUT1-e2o containing its docked β-d-Glc (GLUT1-e2o(S)). Both proteins are hidden to show the proximity of co-crystallized and docked sugars. XylE-bound β-d-Glc lacks hydrogens, and its carbons are colored yellow. GLUT1-bound β-d-Glc carbons are colored cyan. The black scale bar indicates the length of a single C–C bond (0.154 Å).
Additional glucose interaction sites
β-d-Glc docking to GLUT1-e2 reveals additional potential interaction sites, which we call intermediate and peripheral sites (Fig. 3A). The intermediate site persists in GLUT1-e2o but not in e1o and e1, suggesting that an extra sugar (in addition to the core or transported sugar) is excluded in e1o and e1 states. If occupancy of the intermediate site modifies GLUT1 catalytic behavior, this could explain how extracellular sugar allosterically modulates sugar uptake. How it affects exit is more difficult to explain (12, 13). β-d-Glc docking to GLUT1-e1o and GLUT1-e1 (Fig. 3, C and D) suggests that each conformation presents a single interaction envelope. This precludes simultaneous occupancy of endofacial conformations by two intracellular sugars.
Figure 3.

GLUT1 presents additional β-d-Glc binding sites. GLUT1 is oriented as in Fig. 1A. A, β-d-Glc (in red) docking to GLUT1-e2 reveals three potential sites termed peripheral, intermediate, and core. Computed glide scores (GSs) for ligand binding are as follows: peripheral GS = −5.1 kcal/mol, intermediate GS = −5.1 kcal/mol, core GS = −4.9 kcal/mol. B, β-d-Glc (in red) docking to GLUT1-e2o reveals two potential sites termed peripheral and core. Computed GSs for ligand binding are as follows: peripheral GS = −6.0 kcal/mol, core GS = −5.8 kcal/mol. C, β-d-Glc (in red) docking to GLUT1-e1o reveals one potential site with computed GS for ligand binding = −5.1 kcal/mol. D, β-d-Glc (in red) docking to GLUT1-e1. Computed GSs for ligand binding are as follows: core 1 GS = −5.4 kcal/mol.
Interpretation of Km(app) for transport is model-dependent and includes both binding and translocation rate constants (33). Glide scores for β-d-Glc interaction at these sites range from −4.9 to −6 kcal/mol, suggesting Kd(app) for β-d-Glc binding of 18–135 μm. Kd(app) for β-d-Glc binding to GLUT1 is 0.5 mm (14). Computed Kd(app) for xylose docking to each of the eight known XylE structures ranges from 4 to 90 μm (2), yet Kd(app) for xylose binding to XylE is 0.4 mm (7).
Docking analysis of GLUT1–inhibitor interactions
Maltose and CB are nontransported inhibitors of GLUT1-mediated sugar transport acting at exofacial and endofacial sites, respectively (15). Molecular docking of β-maltose to GLUT1-e2 suggests two maltose interaction domains: 1) the core β-d-Glc site and 2) an outer location comprising peripheral and intermediate β-d-Glc interaction sites (Fig. 4A). These sites do not sterically clash, suggesting that GLUT1-e2 can form a complex with two maltose molecules. GLUT1-e2o can also accommodate a core β-d-Glc or core β-maltose plus an outer β-maltose (Fig. 4B). Glide scores for maltose interaction with core and outer sites range from −6.1 to −3.4 kcal/mol corresponding to Kd(app) ≈ 15 μm to 2 mm. Maltose stimulates then inhibits GLUT1-mediated sugar uptake with K0.5 values of 32 μm and 3.2 mm, respectively (13), indicating close agreement between Glide scores and Kd(app) when the interfering exofacial ligand is nontransportable.
Figure 4.
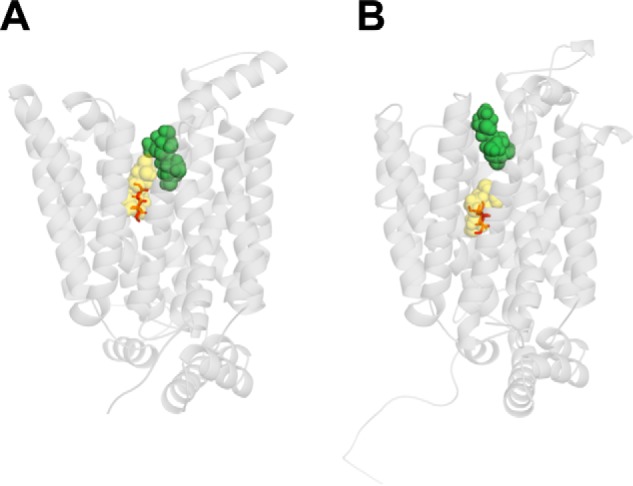
Maltose binding to the exofacial conformation of GLUT1. GLUT1 is oriented as in Fig. 1A. A, β-maltose binding. Maltose (a disaccharide comprising two glucose units joined with an α(1→4) bond) can occupy two sites in GLUT1-e2: a site (shown in yellow) comprising the core β-d-Glc site and extending into additional space or a site (shown in green) comprising intermediate and peripheral β-d-Glc sites. β-d-Glc is indicated as a stick figure occupying its core site. Glide scores for maltose-binding core and intermediate sites are −6.1 and −5.6 kcal/mol, respectively. B, maltose occupies two sites in GLUT1-e2o comprising core (yellow) and peripheral (green) sites. Glide scores for maltose binding at core and peripheral sites are −3.4 and −5.0 kcal/mol, respectively. β-d-Glc is indicated as a stick figure occupying the core site.
Docking analysis of CB-GLUT1-e1 interactions suggests two possible orientations for CB coordination (Fig. 5). Each sterically clashes with the core GLUT1-e1 β-d-Glc interaction envelope (Fig. 5), thus explaining competition between CB and β-d-Glc for binding. Glide scores for CB interaction with GLUT1-e1 are consistent with Kd(app) for CB binding of 0.1 to 5 μm. Kd(app) for CB binding to GLUT1 ranges from 150 to 180 nm (24, 40).
Figure 5.
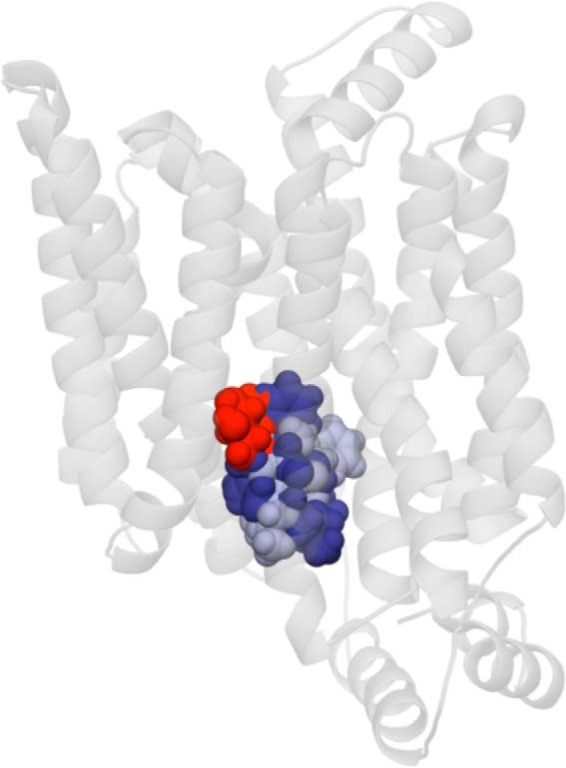
CB interaction sites in GLUT1-e1. GLUT1 is oriented as in Fig. 1A. CB adopts two overlapping coordinations in GLUT1-e1. These are shown as space-filling molecules in dark blue (CB site 1) and light blue (CB site 2). GS for CB binding to sites 1 and 2 are −7.2 and −6.6 kcal/mol, respectively. Both CB sites suggest steric hindrance with the core β-d-Glc binding site (shown as a space-filling molecule in red).
Effects of inhibitors on sugar transport
The predicted involvement of Gln-282 in β-d-Glc coordination by all GLUT1 conformations suggests that this residue plays a central role in sugar transport. We therefore mutagenized Gln-282 to alanine. The concentration dependence of the initial rate of 2-deoxy-d-glucose (2DG) uptake by HEK293 cells expressing wtGLUT1 or GLUT1 containing the Q282A mutation (GLUT1Q282A) is well-approximated by Michaelis–Menten kinetics (Fig. 6A). Vmax for net 2DG uptake (wtGLUT1 = 3.18 × 10−12 ± 0.25 × 10−12 mol/μg protein/min) is unaffected by the Q282A mutation (Vmax = 3.38 × 10−12 ± 0.28 × 10−12 mol/μg protein/min). However, Km(app) for net sugar uptake by wtGLUT1 (0.89 ± 0.18 mm) is doubled in GLUT1Q282A (Km(app) = 1.59 ± 0.28 mm). Cell-surface GLUT1 biotinylation studies confirm that HEK293 cells express similar levels of wtGLUT1 and GLUT1Q282A (Fig. 6B). Replicate analysis (n = 5; Fig. 6C) reveals that Km(app) is significantly increased in GLUT1Q282A (paired t test, p = 0.0046), but Vmax is unchanged (p = 0.2036).
Figure 6.

Sugar transport in HEK293 cells heterologously expressing wtGLUT1 or GLUT1Q282A. A, Michaelis–Menten kinetics of zero-trans 2DG uptake in cells expressing wtGLUT1 (○) or GLUT1Q282A (●). 2DG uptake in μmol/μg cell protein/min is plotted versus [2DG] in mm. Each data point is the mean ± S.E. of three or more duplicate measurements and is corrected for 2DG uptake in mock-transfected cells. The curves were computed by nonlinear regression assuming Michaelis–Menten uptake kinetics (Equation 1) and have the following constants: wtGLUT1 (●): Vmax = 3.2 ± 0.02 pmol/μg protein/min, Km(app) = 0.89 ± 0.18 mm, R2 = 0.884, standard error of regression = 0.31 pmol/μg protein/min; GLUT1Q282A (○): Vmax = 3.4 ± 0.3 pmol/μg protein/min, Km(app) = 1.59 ± 0.28 mm, R2 = 0.926, standard error of regression = 0.24 pmol/μg protein/min. B, cell surface expression of wtGLUT1 and GLUT1Q282A in HEK293 cells. The mobility of molecular weight markers is indicated at the left of the blot which shows GLUT1 levels present in biotinylated membrane proteins collected from untransfected (UTF), wtGLUT1-expressing (WT), and GLUT1Q282A-expressing (Q282A) HEK293 cells. C, Km(app) but not Vmax for 2DG transport is affected in GLUT1Q282A. The results of five separate experiments are shown as scatter-dot plots for both Km(app) and Vmax. The results are shown as means ± S.E. Paired t test analysis (dashed lines indicate paired measurements) indicates that Vmax is not significantly affected by the Q282A mutation (p = 0.2036) but that Km(app) increases 2-fold (p = 0.0046).
Two types of “allostery” have been described for GLUT1-mediated sugar import. cis-Allostery is obtained when extracellular inhibitors (e.g. maltose) stimulate sugar uptake at low concentrations but inhibit uptake at higher concentrations (13). trans-Allostery describes sugar uptake stimulation by subsaturating levels of endofacial inhibitors (e.g. CB, forskolin) (12).
cis-Allostery is eliminated in GLUT1Q282A
Subsaturating extracellular maltose (10–50 μm) stimulates wtGLUT1-mediated 2DG uptake (Fig. 7A), but higher concentrations inhibit transport. Low concentrations of maltose are without effect on sugar uptake by GLUT1Q282A, but higher concentrations inhibit transport (Fig. 7A).
Figure 7.
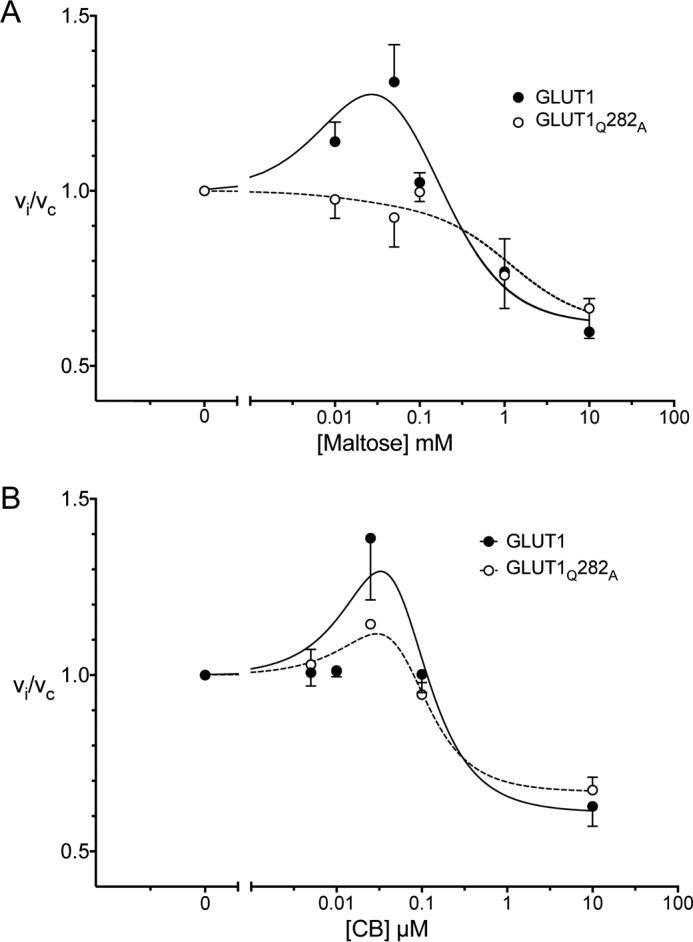
cis- and trans-Allostery in wtGLUT1 (●) and GLUT1Q282A (○). A, cis-allostery. Concentration dependence of maltose modulation of 2DG influx is shown. Normalized 2DG uptake (vi/vc) is plotted as a function of [maltose] (mm) on a log scale. The curves drawn through the points (solid lines for wtGLUT1 (●) and dashed lines for GLUT1Q282A (○)) were computed by nonlinear regression using Equation 2 and have the following constants: wtGLUT1 (●), K1 = 0.0028, K2 = 0.31 mm−1, K3 = 0.197 mm−1, K4 = 1.62 mm−2, R2 = 0.582, standard error of regression = 0.147; GLUT1Q282A (○), K1 = 0.028, K2 = 1.83 mm−1, K3 = 1.911 mm−1, K4 = 1.62 mm−2, R2 = 0.582, standard error of regression = 0.147. B, trans-allostery. Concentration dependence of CB modulation of 2DG influx is shown. Normalized 2DG uptake (vi/vc) is plotted as a function of [CB] (μm) on a log scale. The curves drawn through the points (solid lines for wtGLUT1 (●) and dashed lines for GLUT1Q282A (○)) were computed by nonlinear regression using Equation 2 and have the following constants: wtGLUT1 (●), K1 = 0.0041 μm2, K2 = 0.073 μm, K3 = 2 × 10−12 μm, K4 = 1.64, R2 = 0.637, standard error of regression = 0.179; GLUT1Q282A (○), K1 = 0.0050 μm2, K2 = 0.039 μm, K3 = 8.3 × 10−14 μm, K4 = 1.495, R2 = 0.849, standard error of regression = 0.067.
trans-Allostery persists in GLUT1Q282A
CB stimulates 2DG uptake at low concentrations (0.025 μm CB) in both wtGLUT1 and GLUT1Q282A but inhibits transport at higher concentrations (Fig. 7B).
cis- and trans-Allostery in a GLUT1-oligomerization mutant
GLUT1 forms a mixture of homotetramers and homodimers in red cell membranes and in CHO and HEK293 cells (26, 29). GLUT1 tetramerization (but not sugar transport) is eliminated in a GLUT1/GLUT3 chimera in which the GLUT1 membrane-spanning helix 9 is substituted with the GLUT3 membrane-spanning helix 9 sequence (29). We used this tetramerization-deficient mutant (GLUT1(GLUT3-H9)) to ask whether cis- or trans-allostery require GLUT1 tetramerization.
Subsaturating levels of maltose stimulate both GLUT1- and GLUT1(GLUT3-H9)-mediated 2DG uptake, whereas higher concentrations of maltose inhibit uptake (Fig. 8A). Introduction of the Q282A mutation to the GLUT1(GLUT3-H9) background eliminates sugar uptake stimulation by subsaturating [maltose] (Fig. 8A). Although cis-allostery persists in the oligomerization-deficient mutant, subsaturating [CB] no longer stimulates 2DG uptake in GLUT1(GLUT3-H9)- or GLUT1(GLUT3-H9)Q282A-expressing HEK293 cells (Fig. 8B).
Figure 8.
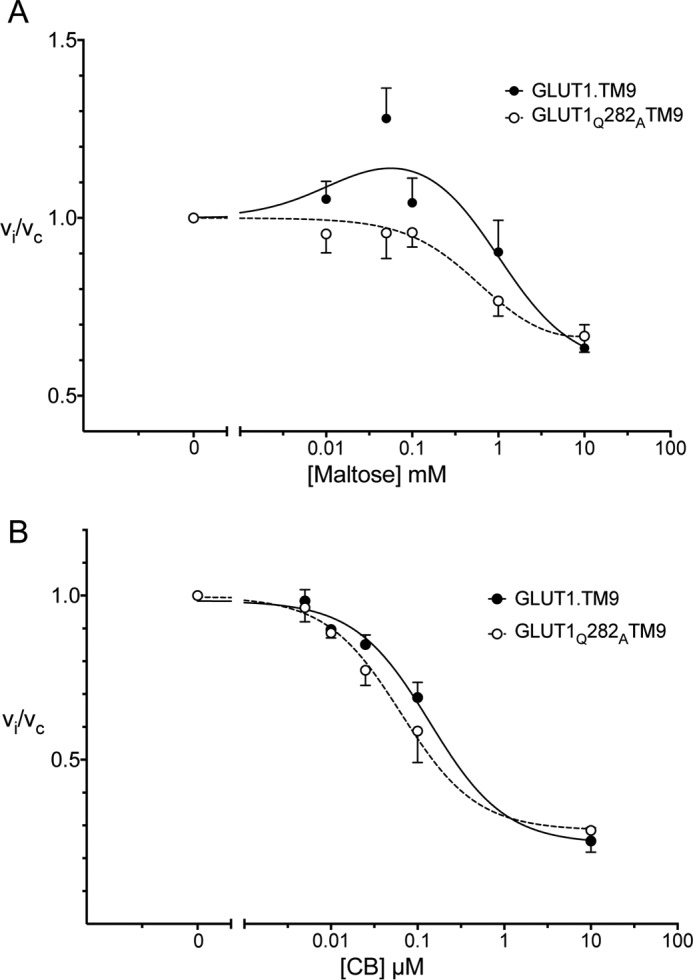
cis- and trans-Allostery in a GLUT1 oligomerization-deficient background. GLUT1(GLUT3-H9) and GLUT1(GLUT3-H9)Q282A expressed in HEK293 cells were tested for their ability to mediate cis- and trans-allostery. A, cis-allostery. Concentration dependence of maltose modulation of 2DG influx. Normalized 2DG uptake (vi/vc) is plotted versus [maltose] (mm) on a log scale. The curves drawn through the points (solid lines for GLUT1(GLUT3-H9) and dashed lines for GLUT1(GLUT3-H9)Q282A) were computed by nonlinear regression using Equation 2 and have the following constants: GLUT1(GLUT3-H9) (●), K1 = 0.0022, K2 = 2.052 mm−1, K3 = 1.707 mm−1, K4 = 1.72 mm−2, R2 = 0.656, standard error of regression = 0.137; GLUT1(GLUT3-H9)Q282A (○), K1 = 0.081, K2 = 0.63 mm−1, K3 = 0.626 mm−1, K4 = 1.528 mm−2, R2 = 0.747, standard error of regression = 0.080. B, trans-allostery. Concentration dependence of CB modulation of 2DG influx. Normalized 2DG uptake (vi/vc) is plotted versus [CB] (μm) on a log scale. The curves drawn through the points (solid lines for GLUT1(GLUT3-H9) and dashed lines for GLUT1(GLUT3-H9)Q282A) were computed by nonlinear regression using Equation 3 and have the following constants: GLUT1(GLUT3-H9) (●), K1 = 0.984 μm/s, K2 = 0.740 μm/s, K3 = 0.138 μm, R2 = 0.963, standard error of regression = 0.058; GLUT1(GLUT3-H9)Q282A (○), K1 = 0.996 μm/s, K2 = 0.711 μm/s, K3 = 0.065 μm, R2 = 0.904, standard error of regression = 0.093.
The effects of stimulating levels of maltose (10 and 50 μm) and CB (25 nm) on 2DG uptake in GLUT1, GLUT1Q282A, GLUT1(GLUT3-H9), and GLUT1(GLUT3-H9)Q282A are summarized in Fig. 9. Stimulation by maltose but not by CB is eliminated in GLUT1Q282A. Stimulation by maltose but not by CB persists in GLUT1(GLUT3-H9), and all stimulations are lost in GLUT1(GLUT3-H9)Q282A.
Figure 9.

Scatter plots of the effects of maltose (10 and 50 μm) and CB (25 nm) on 0. 1 mm 2DG uptake in cells expressing GLUT1 (A), GLUT1Q282A (B), GLUT1(GLUT3-H9) (C), and GLUT1(GLUT3-H9)Q282A (D). Normalized uptake (vi/vc) is plotted versus concentrations of maltose and CB applied during 2DG uptake measurements. The results are shown as the averages of paired replicates (n = 4) and means ± S.E. of multiple experiments (n ≥ 3). The data were examined by unpaired t test analysis comparing the effect of treatment to no treatment (vi/vc = 1 as indicated by the dashed horizontal line), and the computed significance levels are indicated above the points for treatments resulting in p < 0.05.
Discussion
As more sugar transporter structures become available, the weight of evidence supporting the alternating access transporter model for sugar transport grows. Each study (4, 6, 7, 21, 22, 41) has interpreted transporter structures in the context of this model (18, 19, 32) in which the transporter cycles between conformations presenting either an exofacial cavity to extracellular sugars or an edofacial cavity to cytoplasmic sugars. Sugar binding to exofacial (e2) or endofacial (e1) conformations promotes gating transitions that occlude the bound sugar from the interstitium (forming the e2o state) or from the cytoplasm (forming the e1o state). Occlusion triggers rigid body movements leading to e2o conversion to e1o and vice versa. The trans-gate then opens, releasing bound sugar at the opposite side of the membrane. The catalytic cycle concludes via the reverse sequence of conformational changes with or without bound sugar as cargo.
This interpretation of the structural data has excited criticism (2, 9, 11–16) for three reasons: 1) we do not yet have crystal structures of each GLUT conformation complexed with a transported sugar; 2) available sugar transport data demonstrate that GLUT1 simultaneously presents exofacial and endofacial ligand-binding sites; and 3) transport and ligand-binding studies demonstrate that the transporter interacts with more than one exofacial ligand (e.g. β-d-Glc plus maltose) and more than one endofacial ligand (e.g. CB plus forskolin) at any instant.
Ligand interaction sites
We therefore asked whether available crystal structures support the idea that e2 and e1 forms of GLUT1 can simultaneously bind multiple ligands in exo- or endofacial cavities. Although a crystal structure for GLUT1-e1 is available (6), it was necessary to model GLUT1-e2, GLUT1-e2o, and GLUT1-e1o structures using the human GLUT3 (Protein Data Bank code 4ZWC) structure (5) and the XylE e2-occluded and e1-occluded structures (7, 21), respectively.
Molecular docking reveals that the GLUT1-e2 exofacial cavity presents three potential, nonoverlapping β-d-Glc interaction sites and two nonoverlapping maltose interaction sites. β-d-Glc interaction envelopes are located at peripheral, intermediate, and core locations within the exofacial cavity. One maltose interaction envelope overlaps with the core β-d-Glc interaction envelope, and the second is more peripherally located in the exofacial cavity. Docking suggests that GLUT1-e2 can simultaneously accommodate core β-d-Glc and peripheral maltose. These three β-d-Glc sites may represent progressive steps in β-d-Glc binding or three co-existent interaction sites. The latter hypothesis is consistent with the observation that the transporter can bind extracellular maltose and transported sugar simultaneously (see Ref. 13 and this study).
We next asked whether GLUT1-e1 or GLUT1-e2 simultaneously interact with endofacial and exofacial ligands. Molecular docking analysis indicates that this is highly improbable. Finally, we asked whether GLUT1-e1 interacts with more than one endofacial ligand simultaneously. GLUT1-e1 presents a single potential, CB interaction envelope that accommodates CB in either of 2 possible but mutually exclusive orientations. Both orientations sterically clash with the GLUT1-e1 β-d-Glc interaction envelope, providing a rationale for competition between intracellular β-d-Glc and CB for binding to GLUT1 (25). CB interaction envelopes also sterically clash with the forskolin interaction envelope,3 explaining competition between CB and forskolin for binding to GLUT1 (11).
A model for allostery
cis- and trans-Allostery (sugar import stimulation at low [inhibitor] followed by inhibition at high [inhibitor]) require that GLUT1 must bind inhibitors at least two sites. This is readily explicable for cis-allostery (maltose-dependent) because GLUT1-e2 presents two co-existent, exofacial maltose interaction sites, and β-d-Glc competes with maltose for binding at both sites. trans-(CB-dependent) allostery is more difficult to explain because GLUT1-e1 presents only one CB interaction site. We therefore conclude that each GLUT1 molecule is an alternating access transporter capable of exofacial cis-allostery but incapable of trans-allostery when catalyzing sugar import. How then do we explain trans-allostery?
Previous work (26, 27, 29, 42–46) demonstrates that GLUT1 forms mixtures of dimeric and tetrameric GLUT1 complexes. When purifying RBC GLUT1, the ratio of dimeric:tetrameric GLUT1 is affected by cellular redox status with reducing conditions favoring the dimeric form (26, 27). GLUT1 cysteines 347 and 421 may form mixed disulfides under nonreducing conditions (26, 27), and GLUT1 transmembrane helix 9 contains GLUT1-specific sequence essential for tetramerization (29). Reduced, dimeric GLUT1 presents 1 CB binding site/GLUT1 molecule, whereas nonreduced, tetrameric GLUT1 presents only 0.5 CB binding sites/GLUT1 molecule (26, 27, 45). Extracellular reductant inhibits RBC sugar import by 80–90% (26, 27) and eliminates trans- but not cis-allostery (12).
The molecular mechanisms by which Gln-282 and membrane spanning helix 9 (TM9) promote cis- and trans-allostery, respectively, are unknown. However, our observations support the following model. Dimeric GLUT1 comprises two physically associated but functionally independent GLUT1 molecules. Each subunit displays cis- but not trans-allostery in net sugar uptake and binds 1 molecule of CB, and because transport is rate-limited by conformational changes between unliganded e1 and e2 states (relaxation (32)), transport is characterized by a low kcat. Tetrameric GLUT1 comprises a noncovalent dimer of two associated and functionally coupled GLUT1 molecules. Intradimer subunit interactions produce a functional, anti-parallel arrangement of subunits. If one GLUT1 molecule presents an e2 conformation, its cognate partner in the dimer must present an e1 conformation and vice versa. When an e2 subunit of any dimer interacts with extracellular sugar to undergo the eS2 → eS1 conformational change, its cognate partner undergoes the e1 → e2 conformational change thereby coupling transport via one subunit to the regeneration of an e2 conformation in the cognate subunit, bypassing slow relaxation, and accelerating net sugar transport. Because only two subunits in tetrameric GLUT1 can present the e1 conformation, the stoichiometry of CB binding is 0.5 mol CB/mol GLUT1. Each subunit functions as an allosteric alternating access transporter in import mode. trans-Allostery in sugar import obtains when one e1 subunit interacts with high affinity with an endofacial ligand (e.g. CB or forskolin). The dimer presenting this liganded e1 conformation is locked in an inhibited state, but its occupancy is communicated to the adjacent dimer, increasing the affinity of that dimer for extracellular β-d-Glc or kcat for transport. As the endofacial ligand concentration is raised, the remaining free e1 subunit in the adjacent dimer is occupied, and both dimers are inhibited. Endofacial cis-allostery obtains when the affinity of an unliganded e1 subunit in one dimer is increased by occupancy of the e1 subunit of the adjacent dimer.
In conclusion, GLUT1 functions as an oligomer of allosteric, alternating access transporters. cis- and trans-Allostery require intra- and intersubunit interactions, respectively. Each GLUT1 molecule appears to present a core, catalytic sugar-binding site. The exofacial conformer of GLUT1 presents at least one and possibly two additional sugar interaction sites whose occupancy allosterically affects transport via the catalytic site. trans-Allostery requires at least one subunit to bind an endofacial ligand and one to bind an extracellular, imported sugar. Preventing GLUT1–GLUT1 interactions in an oligomerization-deficient mutant eliminates trans- but not cis-allostery. Mutating Gln-282 to alanine eliminates cis-allostery but not trans-allostery.
Experimental procedures
Reagents
[3H]2DG was purchased from American Radiolabeled Chemicals. Unlabeled 2DG, maltose, CB, and phloretin were purchased from Sigma–Aldrich. All primers were purchased from Integrated DNA Technologies. Herculase polymerase, XL1-Blue Competent cells, and QuikChange multisite-directed mutagenesis kits were obtained from Agilent Technologies. SuperSignal Pico West, NeutrAvidin Gel, micro-BCA kits, spin columns, and EZ-Link Sulfo-NHS-ss-Biotin were from Pierce.
Solutions
PBS comprised 140 mm NaCl, 10 mm Na2HPO4, 3.4 mm KCl, 1.84 mm KH2PO4, pH 7.3. Solubilization buffer comprised PBS medium with 0.5% Triton X-100 and 5 mm MgCl2. Stop solution comprised PBS-Mg medium plus CB (CB; 10 μm) and phloretin (100 μm). Sample buffer contained 0.125 m Tris-HCl (pH 6.8), 4% SDS, 20% glycerol, and 50 mm DTT. Transfer buffer comprised 12 mm Tris Base, 96 mm glycine, 20% methanol.
Antibodies
A custom-made (New England Peptide) affinity-purified goat polyclonal antibody (C-Ab) raised against a peptide corresponding to GLUT4 C-terminal residues 498–509 was used at 1:10,000 dilution (47). Horseradish peroxidase-conjugated donkey anti-goat secondary antibody (Jackson ImmunoResearch) was used at 1:50,000 dilution.
Tissue culture
HEK293 cells were maintained in DMEM supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin in a 37 °C humidified 5% CO2 incubator as described previously (48). All of the experiments were performed with confluent cells. The plates were subcultured into 12-well plates at a ratio of 1:2–1:5 2–4 days prior to transfections. Passages 4–20 were used for all experiments.
Mutagenesis
GLUT1-encoding cDNA was inserted into the EcoRV–NotI restriction sites of PCDNA 3.1(+). As described previously (29), the C-terminal 13 amino acids of this GLUT1 construct are substituted using the C-terminal 13 amino acids of GLUT4 to facilitate detection of heterologously expressed GLUT1 against a low level background expression of endogenous GLUT1. Mutagenesis was as described previously (49) using QuikChange multisite-directed mutagenesis kits and was verified by sequencing. The GLUT1 construct in which H9 is substituted with GLUT3 H9 sequence was described previously (29).
Transient transfection
The cells (70–90% confluence) were transfected with 2 μg (12-well plates) or 5 μg of DNA/well (6-well plates). Transfections were performed 36–48 h prior to analysis of sugar uptake or protein expression. Sugar uptake and cell-surface biotinylation measurements were performed in tandem. Heterologous expression of GLUT1, GLUT3, GLUT1/GLUT3, and GLUT4 chimeras and their associated mutations in HEK293 cells was as described previously (29, 49).
Cell-surface expression measurements
Three days post-transfection, 6-well plates of HEK cells were washed twice with ice-cold PBS and incubated on ice with ice-cold PBS containing 5 mm EZ-Link Sulfo-NHS-ss-Biotin for 30 min with gentle rocking. The reactions were quenched by adjusting each well to 12.5 mm Trizma (Tris base). The cells were harvested and resuspended in biotin lysis buffer, and lysates were bound to NeutrAvidin gel in spin columns according to kit instructions. Protein was eluted from spin columns using reductant, and the eluate protein concentration was determined spectrophotometrically. Normalized loads were analyzed by Western blotting.
Western blotting
GLUT1 expression in whole cell lysates and cell surface expression by biotinylation were analyzed by Western blot as previously described (50). Total and isolated Biotinylated proteins were normalized for total protein concentration by BCA and resolved by SDS-PAGE on a 10% NuPage gel in NuPage running buffer. The gels were transferred onto PVDF membranes blocked with 3% bovine serum albumin in PBS-T, probed with primary antibody overnight at 4 °C, probed with secondary antibody for 1 h at room temperature, and developed using SuperSignal Pico West Chemiluminescent substrate. Blots were imaged on a FujiFilm LAS-3000, and relative band densities were quantitated using ImageJ32 software.
2-Deoxy-d-glucose uptake
2DG uptake was measured as described previously (29). Briefly, 36–48 h post-transfection, confluent 12-well plates of HEK-293 cells were serum- and glucose-starved for 2 h at 37 °C in FBS and penicillin/streptomycin-free DMEM lacking glucose. The cells were washed with 1.0 ml of DPBS-Mg at 37 °C for 15 min and then exposed to 0.4 ml of [3H]2DG uptake solution at various 2DG concentrations (0.1–20 mm) for 5 min at 37 °C. Uptake was stopped by the addition of 1 ml of ice-cold stop solution. The cells were washed twice with ice-cold stop solution and lysed with Triton lysis buffer. Total protein content was analyzed in duplicate by BCA. Each sample was counted in duplicate by liquid scintillation spectrometry. Each mutant was analyzed in triplicate on at least three separate occasions. cis- and trans-allostery experiments measured 0.1 mm 2DG uptake in cells exposed to [maltose] or [CB], respectively.
Homology modeling
We modeled GLUT1 e2, e2-occluded, and e1-occluded structures respectively using the human GLUT3 (Protein Data Bank code 4ZWC) structure (5) and the XylE e2-occluded (Protein Data Bank code 4GBZ) and e1-occluded (Protein Data Bank code 4JA3) structures (7, 21). We removed ligands and used chain A as the template for each modeled structure. Sequence alignments were generated using ClustalX. Homology models were built using Modeler 9.9 and analyzed using PROCHECK. The GLUT1 e1 structure (Protein Data Bank code 4PYP (6)) was used directly.
Cavity analysis
Cavities for ligand docking were calculated using the CastP server (http://sts.bioe.uic.edu/castp/) (51),4 and the grid was centered on the residues forming the cavity.
Stochastic docking
β-d-Glucose, maltose and CB structures were obtained from ZINC (http://zinc.docking.org; Ref. 52).4 The WZB117 structure was generated using the 3D structure generator Corina from Molecular Networks GmbH (http://www.molecular-networks.com).4 Docking was performed using the Schrodinger software suite. No restraints were used during the docking. The protein structure was preprocessed with the Protein Preparation Wizard, bond orders were assigned, hydrogens were added, and the H-bond network was optimized. The system was energy-minimized using the OPLS 2005 force field. Ligand structures were prepared with the LigPrep module, and the pKa of the ligands was calculated using the Epik module. Computational docking was performed by the GLIDE module in standard-precision mode and default values for grid generation. Grids were mapped using CastP cavity analysis and ligand positions from the original crystal structures.
Data analysis
Data analysis was undertaken using GraphPad Prism (version 7.0c; GraphPad Software, Inc.). Curve fitting was by nonlinear regression using the following equations. For concentration dependence of 2-deoxy-d-glucose uptake,
| (Eq. 1) |
where [S] is [2DG], and Vmax and Km(app) have the usual meaning. Exofacial cis-allostery and endofacial trans-allostery are expressed as normalized 2DG uptake, which is described by the following,
| (Eq. 2) |
where vi/vc is uptake in the presence of inhibitor divided by uptake in the absence of inhibitor, [I] is the concentration of cis- or trans-inhibitor, and interpretation of constants is model-dependent and described by Lloyd et al.5 When simple saturable inhibition of transport is observed, normalized 2DG uptake is described by Equation 3,
| (Eq. 3) |
where vi/vc is uptake in the presence of inhibitor divided by uptake in the absence of inhibitor, K1 is uptake in the absence of inhibitor I, K2 is the difference between K1 and uptake in the presence of saturating [I], and K3 is Ki(app) for uptake inhibition by I.
Author contributions
K. P. L. conducted most of the experiments, analyzed the results, modeled and analyzed GLUT1 structures and, wrote most of the paper. O. A. O. conducted cell-surface biotinylation experiments. J. K. D. Z. contributed to construction of the oligomerization deficient GLUT1 mutants. A. C. conceived the idea for the project and, with K. P. L., analyzed the results and wrote the paper.
Acknowledgment
We thank Andrew Simon for reviewing the manuscript.
This work was supported by National Institutes of Health Grants DK36081 and DK44888. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
O. A. Ojelabi and A. Carruthers, unpublished docking analysis.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
K. P. Lloyd, O. A. Ojelabi, A. H. Simon, J. K. De Zutter, and A. Carruthers, submitted for publication.
- MFS
- major facilitator superfamily
- CB
- cytochalasin B
- Glc
- glucose
- 2DG
- 2-deoxy-d-glucose
- GS
- glide score.
References
- 1. Cura A. J., and Carruthers A. (2012) Role of monosaccharide transport proteins in carbohydrate assimilation, distribution, metabolism, and homeostasis. Compr. Physiol. 2, 863–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cunningham P., and Naftalin R. J. (2014) Reptation-induced coalescence of tunnels and cavities in Escherichia coli XylE transporter conformers accounts for facilitated diffusion. J. Membr. Biol. 247, 1161–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mueckler M., Caruso C., Baldwin S. A., Panico M., Blench I., Morris H. R., Allard W. J., Lienhard G. E., and Lodish H. F. (1985) Sequence and structure of a human glucose transporter. Science 229, 941–945 [DOI] [PubMed] [Google Scholar]
- 4. Nomura N., Verdon G., Kang H. J., Shimamura T., Nomura Y., Sonoda Y., Hussien S. A., Qureshi A. A., Coincon M., Sato Y., Abe H., Nakada-Nakura Y., Hino T., Arakawa T., Kusano-Arai O., et al. (2015) Structure and mechanism of the mammalian fructose transporter GLUT5. Nature 526, 397–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Deng D., Sun P., Yan C., Ke M., Jiang X., Xiong L., Ren W., Hirata K., Yamamoto M., Fan S., and Yan N. (2015) Molecular basis of ligand recognition and transport by glucose transporters. Nature 526, 391–396 [DOI] [PubMed] [Google Scholar]
- 6. Deng D., Xu C., Sun P., Wu J., Yan C., Hu M., and Yan N. (2014) Crystal structure of the human glucose transporter GLUT1. Nature 510, 121–125 [DOI] [PubMed] [Google Scholar]
- 7. Sun L., Zeng X., Yan C., Sun X., Gong X., Rao Y., and Yan N. (2012) Crystal structure of a bacterial homologue of glucose transporters GLUT1–4. Nature 490, 361–366 [DOI] [PubMed] [Google Scholar]
- 8. Yan N. (2017) A glimpse of membrane transport through structures: advances in the structural biology of the GLUT glucose transporters. J. Mol. Biol. 429, 2710–2725 [DOI] [PubMed] [Google Scholar]
- 9. Baker G. F., and Naftalin R. J. (1979) Evidence of multiple operational affinities for d-glucose inside the human erythrocyte membrane. Biochim. Biophys. Acta 550, 474–484 [DOI] [PubMed] [Google Scholar]
- 10. Naftalin R. J. (1988) Pre-steady-state uptake of d-glucose is inconsistent with the mobile carrier model. Trends Biochem. Sci. 13, 425–428 [DOI] [PubMed] [Google Scholar]
- 11. Robichaud T., Appleyard A. N., Herbert R. B., Henderson P. J., and Carruthers A. (2011) Determinants of ligand binding affinity and cooperativity at the GLUT1 endofacial site. Biochemistry 50, 3137–3148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cloherty E. K., Levine K. B., and Carruthers A. (2001) The red blood cell glucose transporter presents multiple, nucleotide-sensitive sugar exit sites. Biochemistry 40, 15549–15561 [DOI] [PubMed] [Google Scholar]
- 13. Hamill S., Cloherty E. K., and Carruthers A. (1999) The human erythrocyte sugar transporter presents two sugar import sites. Biochemistry 38, 16974–16983 [DOI] [PubMed] [Google Scholar]
- 14. Cloherty E. K., Heard K. S., and Carruthers A. (1996) Human erythrocyte sugar transport is incompatible with available carrier models. Biochemistry 35, 10411–10421 [DOI] [PubMed] [Google Scholar]
- 15. Carruthers A., and Helgerson A. (1991) Inhibitions of sugar transport produced by ligands binding at opposite sides of the membrane: evidence for simultaneous occupation of the carrier by maltose and cytochalasin B. Biochemistry 30, 3907–3915 [DOI] [PubMed] [Google Scholar]
- 16. Helgerson A. L., and Carruthers A. (1987) Equilibrium ligand binding to the human erythrocyte sugar transporter: evidence for two sugar-binding sites per carrier. J. Biol. Chem. 262, 5464–5475 [PubMed] [Google Scholar]
- 17. Blodgett D. M., and Carruthers A. (2005) Quench-flow analysis reveals multiple phases of GluT1-mediated sugar transport. Biochemistry 44, 2650–2660 [DOI] [PubMed] [Google Scholar]
- 18. Widdas W. F. (1952) Inability of diffusion to account for placental glucose transfer in the sheep and consideration of the kinetics of a possible carrier transfer. J. Physiol. 118, 23–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jardetzky O. (1966) Simple allosteric model for membrane pumps. Nature 211, 969–970 [DOI] [PubMed] [Google Scholar]
- 20. Reddy V. S., Shlykov M. A., Castillo R., Sun E. I., and Saier M. H. Jr. (2012) The major facilitator superfamily (MFS) revisited. FEBS J. 280, 3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Quistgaard E. M., Löw C., Moberg P., Trésaugues L., and Nordlund P. (2013) Structural basis for substrate transport in the GLUT-homology family of monosaccharide transporters. Nat. Struct. Mol. Biol. 20, 766–768 [DOI] [PubMed] [Google Scholar]
- 22. Abramson J., Smirnova I., Kasho V., Verner G., Kaback H. R., and Iwata S. (2003) Structure and mechanism of the lactose permease of Escherichia coli. Science 301, 610–615 [DOI] [PubMed] [Google Scholar]
- 23. Baker G. F., and Widdas W. F. (1973) The asymmetry of the facilitated transfer system for hexoses in human red cells and the simple kinetics of a two component model. J. Physiol. 231, 143–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Baldwin S. A., Baldwin J. M., Gorga F. R., and Lienhard G. E. (1979) Purification of the cytochalasin B binding component of the human erythrocyte monosaccharide transport system. Biochim. Biophys. Acta 552, 183–188 [DOI] [PubMed] [Google Scholar]
- 25. Gorga F. R., and Lienhard G. E. (1981) Equilibria and kinetics of ligand binding to the human erythrocyte glucose transporter: evidence for an alternating conformation model for transport. Biochemistry 20, 5108–5113 [DOI] [PubMed] [Google Scholar]
- 26. Zottola R. J., Cloherty E. K., Coderre P. E., Hansen A., Hebert D. N., and Carruthers A. (1995) Glucose transporter function is controlled by transporter oligomeric structure: a single, intramolecular disulfide promotes GLUT1 tetramerization. Biochemistry 34, 9734–9747 [DOI] [PubMed] [Google Scholar]
- 27. Hebert D. N., and Carruthers A. (1992) Glucose transporter oligomeric structure determines transporter function: reversible redox-dependent interconversions of tetrameric and dimeric GLUT1. J. Biol. Chem. 267, 23829–23838 [PubMed] [Google Scholar]
- 28. Hebert D. N., and Carruthers A. (1991) Cholate-solubilized erythrocyte glucose transporters exist as a mixture of homodimers and homotetramers. Biochemistry 30, 4654–4658 [DOI] [PubMed] [Google Scholar]
- 29. De Zutter J. K., Levine K. B., Deng D., and Carruthers A. (2013) Sequence determinants of GLUT1 oligomerization: analysis by homology-scanning mutagenesis. J. Biol. Chem. 288, 20734–20744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Burant C. F., and Bell G. I. (1992) Mammalian facilitative glucose transporters: evidence for similar substrate recognition sites in functionally monomeric proteins. Biochemistry 31, 10414–10420 [DOI] [PubMed] [Google Scholar]
- 31. Basketter D. A., and Widdas W. F. (1978) Asymmetry of the hexose transfer system in human erythrocytes: comparison of the effects of cytochalasin B, phloretin and maltose as competitive inhibitors. J. Physiol. 278, 389–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lieb W. R., and Stein W. D. (1974) Testing and characterizing the simple carrier. Biochim. Biophys. Acta 373, 178–196 [DOI] [PubMed] [Google Scholar]
- 33. Carruthers A. (1991) Mechanisms for the facilitated diffusion of substrates across cell membranes. Biochemistry 30, 3898–3906 [DOI] [PubMed] [Google Scholar]
- 34. Baker P. F., and Carruthers A. (1981) Sugar transport in giant axons of Loligo. J. Physiol. 316, 481–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Halgren T. A., Murphy R. B., Friesner R. A., Beard H. S., Frye L. L., Pollard W. T., and Banks J. L. (2004) Glide: a new approach for rapid, accurate docking and scoring: 2. Enrichment factors in database screening. J. Med. Chem. 47, 1750–1759 [DOI] [PubMed] [Google Scholar]
- 36. Friesner R. A., Banks J. L., Murphy R. B., Halgren T. A., Klicic J. J., Mainz D. T., Repasky M. P., Knoll E. H., Shelley M., Perry J. K., Shaw D. E., Francis P., and Shenkin P. S. (2004) Glide: a new approach for rapid, accurate docking and scoring: 1. Method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749 [DOI] [PubMed] [Google Scholar]
- 37. Friesner R. A., Murphy R. B., Repasky M. P., Frye L. L., Greenwood J. R., Halgren T. A., Sanschagrin P. C., and Mainz D. T. (2006) Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 49, 6177–6196 [DOI] [PubMed] [Google Scholar]
- 38. Barnett J. E., Holman G. D., and Munday K. A. (1973) Structural requirements for binding to the sugar-transport system of the human erythrocyte. Biochem. J. 131, 211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Barnett J. E., Holman G. D., Chalkley R. A., and Munday K. A. (1975) Evidence for two asymmetric conformational states in the human erythrocyte sugar-transport system. Biochem. J. 145, 417–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sogin D. C., and Hinkle P. C. (1980) Binding of cytochalasin B to human erythrocyte glucose transporter. Biochemistry 19, 5417–5420 [DOI] [PubMed] [Google Scholar]
- 41. Lemieux M. J., Song J., Kim M. J., Huang Y., Villa A., Auer M., Li X. D., and Wang D. N. (2003) Three-dimensional crystallization of the Escherichia coli glycerol-3-phosphate transporter: A member of the major facilitator superfamily. Protein Sci. 12, 2748–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Graybill C., van Hoek A. N., Desai D., Carruthers A. M., and Carruthers A. (2006) Ultrastructure of human erythrocyte GLUT1. Biochemistry 45, 8096–8107 [DOI] [PubMed] [Google Scholar]
- 43. Coderre P. E., Cloherty E. K., Zottola R. J., and Carruthers A. (1995) Rapid substrate translocation by the multisubunit, erythroid glucose transporter requires subunit associations but not cooperative ligand binding. Biochemistry 34, 9762–9773 [DOI] [PubMed] [Google Scholar]
- 44. Pessino A., Hebert D. N., Woon C. W., Harrison S. A., Clancy B. M., Buxton J. M., Carruthers A., and Czech M. P. (1991) Evidence that functional erythrocyte-type glucose transporters are oligomers. J. Biol. Chem. 266, 20213–20217 [PubMed] [Google Scholar]
- 45. Hinkle P. C., Sogin D. C., Wheeler T. J., Teleford J. N., and Vivo D. C. (1979) Studies of the glucose transporter from human erythrocytes reconstituted in liposomes. In Function and Molecular Aspect of Biomembrane Transport (Quagliariello E. E. A., SK, ed.) pp. 487–494, Elsevier/North-Holland Biomedical Press, Amsterdam [Google Scholar]
- 46. Jarvis S. M., Ellory J. C., and Young J. D. (1986) Radiation inactivation of the human erythrocyte nucleoside and glucose transporters. Biochim. Biophys. Acta 855, 312–315 [DOI] [PubMed] [Google Scholar]
- 47. Blodgett D. M., De Zutter J. K., Levine K. B., Karim P., and Carruthers A. (2007) Structural basis of GLUT1 inhibition by cytoplasmic ATP. J. Gen. Physiol. 130, 157–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ojelabi O. A., Lloyd K. P., Simon A. H., De Zutter J. K., and Carruthers A. (2016) WZB117 inhibits GLUT1-mediated sugar transport by binding reversibly at the exofacial sugar binding site. J. Biol. Chem. 291, 26762–26772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vollers S. S., and Carruthers A. (2012) Sequence determinants of GLUT1-mediated accelerated-exchange transport: analysis by homology-scanning mutagenesis. J. Biol. Chem. 287, 42533–42544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cura A. J., and Carruthers A. (2010) Acute modulation of sugar transport in brain capillary endothelial cell cultures during activation of the metabolic stress pathway. J. Biol. Chem. 285, 15430–15439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dundas J., Ouyang Z., Tseng J., Binkowski A., Turpaz Y., and Liang J. (2006), CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 34, W116–W118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Irwin J. J., Sterling T., Mysinger M. M., Bolstad E. S., and Coleman R. G. (2012) ZINC: a free tool to discover chemistry for biology. J. Chem. Inf. Model. 52, 1757–1768 [DOI] [PMC free article] [PubMed] [Google Scholar]