

Abstract
β-Amyloid (Aβ) aggregation is thought to initiate a cascade of neurodegenerative events in Alzheimer's disease (AD). Much effort is underway to develop strategies to reduce Aβ concentration or inhibit aggregation. Cathepsin B (CatB) proteolytically degrades Aβ into non-aggregating fragments but is potently inhibited by cystatin C (CysC). It has been suggested that decreasing CysC would facilitate Aβ clearance by relieving CatB inhibition. However, CysC binds Aβ and inhibits Aβ aggregation, suggesting that an intervention that increases CysC would prevent Aβ aggregation. Both approaches have been tested in animal models, yielding contradictory results, possibly because of the opposing influences of CysC on Aβ degradation versus aggregation. Here, we sought to develop a model that quantitatively predicts the effects of CysC and CatB on Aβ aggregation. Aβ aggregation kinetics in the absence of CatB or CysC was measured. The rate constant for Aβ degradation by CatB and the equilibrium constant for binding of CysC to Aβ were determined. We derived a mathematical model that combines material balances and kinetic rate equations. The model accurately predicted Aβ aggregation kinetics at various CatB and CysC concentrations. We derived approximate expressions for the half-times of degradation and aggregation and show that their ratio can be used to estimate, at any given Aβ, CatB, or CysC concentration, whether Aβ aggregation or degradation will result. Our results may be useful for designing experiments and interpreting results from investigations of manipulation of CysC concentration as an AD therapy.
Keywords: aggregation, amyloid-β (Aβ), cathepsin B (CTSB), fibril, kinetics, cystatin C
Introduction
Alzheimer's disease (AD)2 is the most common cause of dementia, estimated to affect over 44 million people worldwide. According to the amyloid cascade hypothesis, the initiating event in AD occurs when the membrane-bound amyloid precursor protein (APP) is sequentially cleaved into a small peptide fragment, β-amyloid (Aβ), and secreted into extracellular fluid (1, 2). Processing of APP by a β-site-cleaving enzyme (β-secretase) initiates the amyloid secretory pathway, followed by liberation of the peptide from its transmembrane domain by a γ-site-cleaving enzyme (γ-secretase). Specificity of the γ-secretase is broad, and cleavage generates various lengths of Aβ, of which the most common are Aβ40 and Aβ42 (3). After liberation (and possibly before secretion), Aβ self-associates into small, soluble oligomers that mature into larger fibrillar structures. Eventually, aggregates precipitate and deposit on neuronal tissue as amyloid plaque. Aggregates are thought to be neurotoxic (4), with smaller Aβ oligomeric intermediates believed to be more toxic than mature fibrils (5). For this reason, the kinetics of amyloid protein aggregation have been studied extensively (6–10). Although Aβ40 concentration is higher than Aβ42 in human AD brain tissue, Aβ42 is believed to be more aggregation-prone and more toxic (2).
Cystatin C (CysC) is a small (13.4 kDa) extracellular protein that is ubiquitously synthesized and is at relatively high concentrations in cerebrospinal fluid (CSF) (11). The link between CysC, Aβ, and AD was identified years ago with the discovery that CysC and Aβ co-deposit in cerebrovascular and senile plaques of AD patients (12, 13) and in amyloid deposits in the cerebrovasculature of patients with cerebral amyloid angiopathy (14–16). More recently, soluble CysC–Aβ complexes were detected in CSF from AD patients (17). CysC has been reported to bind Aβ and prevent further aggregation in vitro (18–20) and to protect cultured neurons from Aβ toxicity (21). Binding epitopes have been proposed based on molecular docking simulations and limited proteolysis experiments (22). CSF CysC levels were reported to be lower in AD than in healthy controls (23, 24), whereas plasma CysC levels were reportedly higher in AD (25), possibly because high levels are associated with an anti-inflammatory response. CysC levels may be low in patients with mild cognitive impairment but later increase as a general response to cell damage (25).
CysC is an inhibitor of cysteine proteases, including those in the cathepsin family. Cathepsin B (CatB), a cysteine-type protease of the lysosomal/endosomal pathways, is specifically inhibited by CysC with sub-nanomolar affinity (20, 26, 27). Although CatB is mainly active intracellularly, it can be secreted. Should CatB escape the cell (in the case of apoptosis or secretory pathway activation), CysC has the important role of suppressing proteolysis in the extracellular space (28). CatB has been shown to degrade Aβ, primarily through C-terminal truncation (29). Acidic pH favors rapid sequential conversion of Aβ42 into Aβ40 and then finally to the less-amyloidogenic Aβ38 (30). Reaction rates are slower at neutral pH but not completely diminished (30). CatB secretion from neuronal cells following stimulation by Aβ aggregates has been documented (31, 32), and secreted CatB is known to retain a significant fraction of its activity (33, 34). There is conflicting data with regard to the relationship of CatB to AD; CatB was expressed at lower levels in AD patient monocytes compared with healthy controls in one study (35), but in another study, CatB levels were higher in plasma of AD patients compared with controls (36).
Because of their roles in preventing fibrillation and in degrading Aβ, manipulation of CysC or CatB has been identified as a potential AD therapeutic. However, there is disagreement as to whether CysC or CatB levels should be modulated and whether the levels should be suppressed or enhanced. Some researchers have reported neuroprotective effects of CysC enhancement in animal models. Mi et al. (37) found that transgenic mice expressing human CysC at ∼2-fold higher levels than endogenous cystatins showed a marked decrease in Aβ fibril deposition. Kaeser et al. (38) showed that CysC overexpression reduced plaque formation in human APP-transgenic mice. In another study using CatB-deficient mice, overexpression of CysC reduced the total amyloid plaque load (39).
Other researchers have found evidence that CatB proteolysis of Aβ is an important neuroprotective mechanism and that high amounts of CysC interfere with Aβ degradation. CatB enhancement in transgenic AD mouse models reduced both the total amount of soluble Aβ42 and the ratio of Aβ42/Aβx, indicating CatB may preferentially degrade Aβ42 relative to other forms (30). Sun et al. (39) found mice overexpressing CatB lacked symptoms of cognitive impairment and premature mortality compared with CatB-free controls. These researchers also showed that expression of CysC could neutralize the neuroprotective effects of CatB. Similarly, Wang et al. (40) overexpressed neuronal CatB in transgenic mice and observed significantly lowered Aβ42 levels, without affecting APP processing. Transduction of neurons to express CatB accelerated Aβ degradation and suppressed its production in vitro and led to a reduction of both Aβ40 and Aβ42 (41). Deletion of stefin B (a cysteine protease inhibitor) in an AD mouse model reduced Aβ accumulation and inhibited development of learning and memory deficits (42).
Others interpret the outcome of studies in light of putative β-secretase activity by CatB (3, 29). Inhibition of CatB reduced plaque load and improved cognitive defects in transgenic mouse models; this was postulated to be due to reduced cleavage of APP into Aβ (43, 44). Wang et al. (45) found that exogenously-applied CysC reduced soluble Aβ levels in animal studies, which was attributed to CysC-mediated reduction in β-secretase activity.
Differing results in transgenic animal models may be caused by differences in concentrations of Aβ, CysC, and/or CatB. In particular, CysC plays two conflicting roles in the regulation of Aβ as follows: by binding to Aβ it can inhibit Aβ aggregation, but by binding to CatB it inhibits Aβ degradation. CysC and CatB interactions have been postulated to form key control mechanisms to regulate Aβ aggregation potential (39, 43, 47). The aim of this study was to quantify the effects of CysC and CatB on the fate of Aβ. We modeled the protein interactions by considering binding, degradation, and aggregation reactions, and then we measured each relevant thermodynamic and kinetic parameter. We sought to understand the sensitivity of Aβ aggregation to perturbations in CysC/CatB balance and to be able to predict the outcome simply by knowledge of total CysC and CatB concentrations.
Results
Aβ aggregation kinetics
We measured Aβ aggregation kinetics at three different concentrations using ThT, a dye that fluoresces when bound to amyloid fibrils. Briefly, Aβ40 (8, 28, and 46 μm) or Aβ42 (2.5, 10, and 20 μm) was loaded into microwells and heated to 37 °C. Sample fluorescence was continuously monitored for up to 72 h. Data for each of three replicates are shown in Fig. 1. Post-aggregation, Aβ40 at 28 μm was centrifuged, and the peptide content of the supernatant was measured. No peptide was detected (assay sensitivity <500 nm), so we concluded that all Aβs had converted to aggregated species. Therefore, at long time the fibrillar Aβ concentration (in equivalent monomer molar units) equaled the total Aβ concentration [Aβ]0. To convert ThT fluorescence intensity to concentration of fibrillar Aβ, we plotted the maximum ThT signal (at 72 h) against [Aβ]0 and fit the data to the polynomial shown in Equation 1,
| (Eq. 1) |
where ThTb is background signal, and a and b were determined by least-squares fitting (Table 1).
Figure 1.
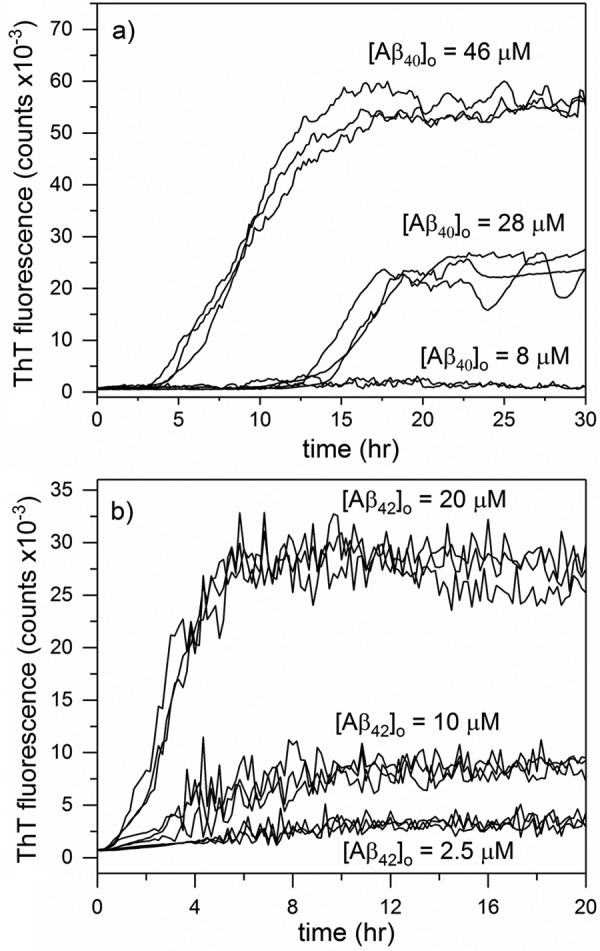
ThT fluorescence data of Aβ40 (a) or Aβ42 (b) aggregation. Three independent samples of each initial Aβ concentration were prepared, and fluorescence was measured every 10 min for up to 30 h at 37 °C.
Table 1.
Parameters for conversion of ThT fluorescence intensity to [Aβagg] (μm)
| aa | b | |
|---|---|---|
| Aβ40 | 33 ± 8 | 25 ± 10 |
| Aβ42 | 430 ± 35 | 44 ± 15 |
a Parameters a and b were determined by non-linear least-squares fitting of ThT fluorescence data at 72 h to ThT − ThTb = a[Aβ]0 + b[Aβ]02 (Equation 1), where [Aβ]0 is the total Aβ concentration in μm and [Aβagg] = [Aβ]0 at 72 h. ThTb was determined by averaging three sample wells containing ThT but no Aβ and was found to be 520 ± 40. Error estimates are 95% confidence intervals.
Equation 1 was used to convert ThT intensity at all time points to [Aβagg], where [Aβagg] is the concentration of aggregated Aβ expressed in equivalent monomer molar concentration. Aggregation data were fit to several different simple kinetic models, including the monomer partitioning, Gompertz, and autocatalytic models (supplemental Fig. S1) (6, 48). The best global fits were obtained by the autocatalytic model, a two-step model that includes a first-order irreversible conversion of monomer to aggregation-competent form and a second-order reaction between monomer and aggregate to produce more aggregate (6) as shown in Model 1.
This kinetic model has an analytical solution shown in Equation 2,
| (Eq. 2) |
where [Aβagg] is the monomer-equivalent concentration of Aβ aggregates at any time t. Although such a two-step model cannot account for oligomer or protofibril formation, protofibril association, fragmentation, or other events (9), it does capture the sigmoidal shape and concentration dependence of the data, and it can describe accumulation of ThT-positive aggregates. Aggregation data for six (Aβ40) or nine (Aβ42) independently prepared samples were globally fit to Equation 2 by least-squares regression to recover k1 and k2 for both Aβ40 and Aβ42 (Table 2). Best fit lines were generated and compared with each aggregation data set (Fig. 2). Although k2 was similar for the two Aβ forms, k1 was 5000-fold higher for Aβ42 than for Aβ40, consistent with its higher aggregation propensity.
Table 2.
Kinetic and thermodynamic parameters
Error estimates are 95% confidence intervals.
Figure 2.
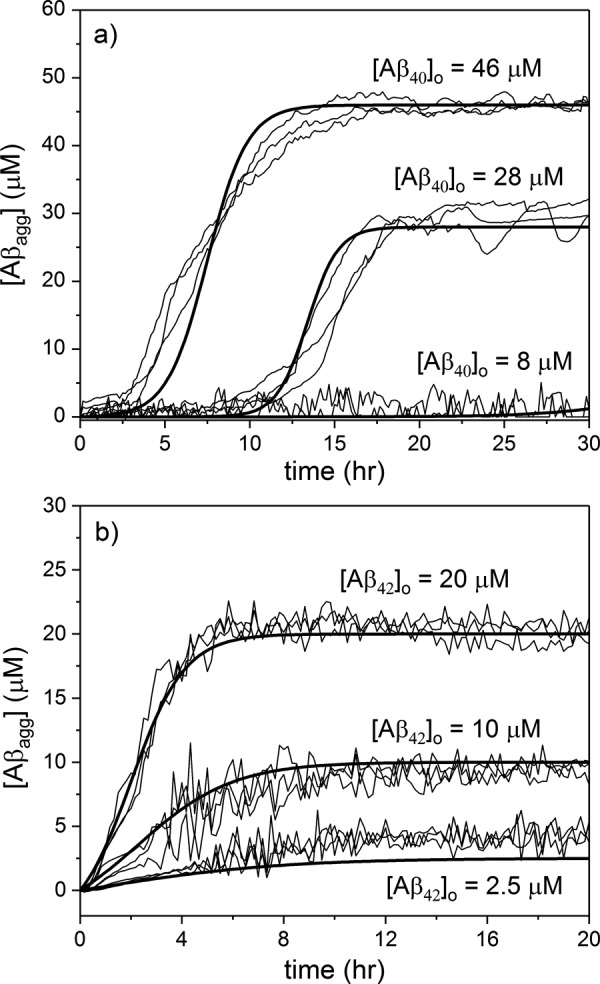
ThT signal was converted to [Aβagg] by an empirically-determined quadratic function. Autocatalytic aggregation model parameters k1 and k2 were derived from global fits to 28 and 46 μm (Aβ40) (a) and 2.5, 10, and 20 μm (Aβ42) data sets (b). Best fit lines (bold lines) were compared with data (thin lines). 8 μm (Aβ40)0 data are shown compared with model prediction. These data were not included in the data sets for parameter estimation. Because essentially no aggregation was detected, the information content is less, and the data put an upper but not lower bound on k1 and k2.
CysC–Aβ-binding interaction
CysC co-localizes with Aβ in AD plaques (12, 49). A few studies show that CysC inhibits Aβ fibril formation in vitro, possibly redirecting Aβ self-association toward amorphous non-fibrillar aggregates (12, 50). It is unknown whether CysC predominantly binds monomeric or aggregated Aβ. We designed a FRET assay to measure CysC–Aβ binding in solution (Fig. 3a). Data were normalized to the maximum observed FRET efficiency and expressed as a fraction of bound Aβ (Fig. 3b). Data from three independent series were fit globally to determine the binding constant, KD, and the cooperativity parameter n. n was found to be 0.94 ± 0.05, so the data were re-fit with n = 1, to obtain a final value for KD of 72 ± 6 nm (Table 2). Our estimate is slightly weaker than that derived from an ELISA method, where KD was estimated to be 10 nm (50).
Figure 3.
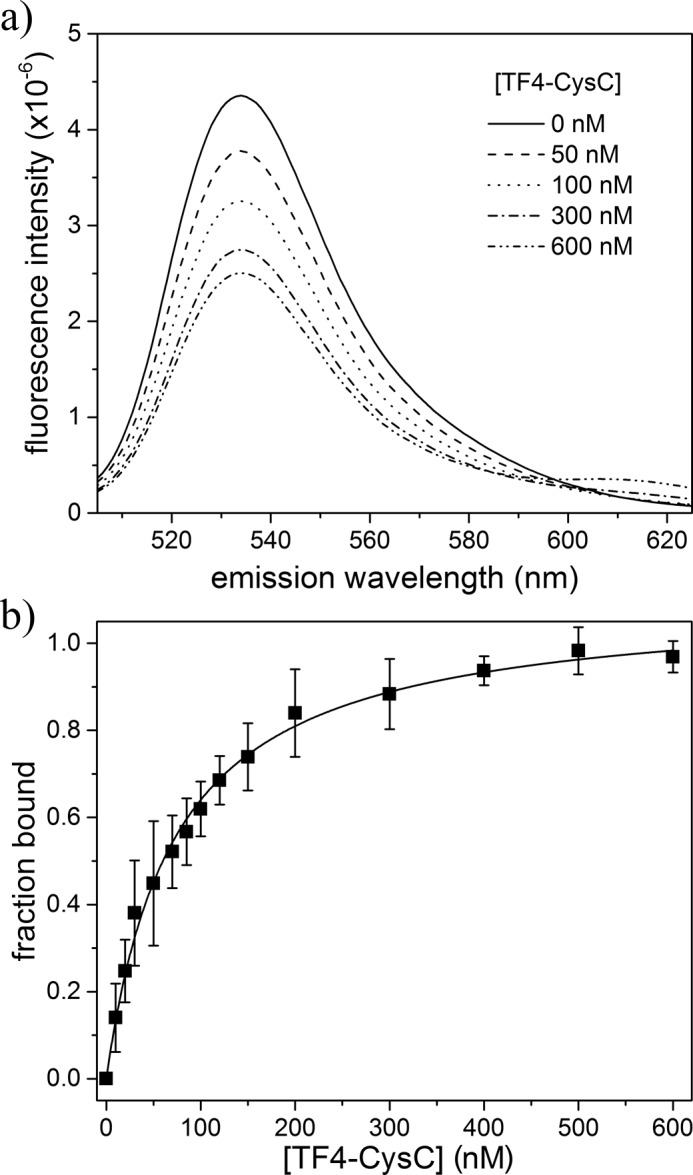
HF488-Aβ/TF4-CysC binding interaction by FRET analysis. a, mixture fluorescence was measured as a function of TF4-CysC, holding HF488-Aβ constant at 400 nm (excitation 488 nm). Average of three independent scans is shown for a representative set of mixtures. b, donor emission at 530 nm was used to calculate FRET efficiency and determine fraction of HF488-Aβ bound for each TF4-CysC concentration. Data points are the average of three independent trials. Error bars represent the sample standard deviation. Solid line is the Hill equation fit to the data, with n = 1.
CatB degradation of Aβ
At neutral pH, CatB proteolysis of Aβ proceeds via C-terminal truncation; products of Aβ42 degradation were reported to be Aβ40, Aβ38, and Aβ33 (30). Preliminary experiments in our laboratory were consistent with this finding (data not shown). To determine degradation rate constants, fixed concentrations of CatB were incubated with various amounts of Aβ40 or Aβ42 in physiological conditions (pH 7.4, 37 °C). Degradation over time was measured by dot-blot assay, where membranes were probed using antibodies specifically against the C terminus of Aβ40 or Aβ42 (Figs. 4a and 6, a and b). Measurement linearity was determined by the concentration standard curves (Fig. 4b), and only those samples in the linear concentration region were reported. Sample intensity was converted to Aβ concentration using the standard curves. Aβ40 concentration could reliably be determined as low as 50 nm, whereas Aβ42 could only be determined down to 200 nm. With Aβ40, concentration decreased over time as expected (Fig. 4a). Concentrations over short time periods (1 h) were plotted to determine the initial reaction rate (Fig. 5a), and initial rates were plotted against initial Aβ concentrations to determine the degradation rate constant kdeg (Fig. 5b and Table 2). Aβ42 degradation kinetics were measured in a similar manner (Fig. 6a), except Aβ42 blots were stripped and re-probed with anti-Aβ40 to show the generation of Aβ40 directly from Aβ42 as a result of CatB activity (Fig. 6b). Quantitative densitometry analyses at 600 nm Aβ42 are shown (Fig. 6, c and d), demonstrating the close temporal correspondence between loss of antibody binding to the C terminus of Aβ42 and gain of anti-Aβ40-specific antibody binding. Values of kdeg for Aβ40 and Aβ42 were not statistically different.
Figure 4.
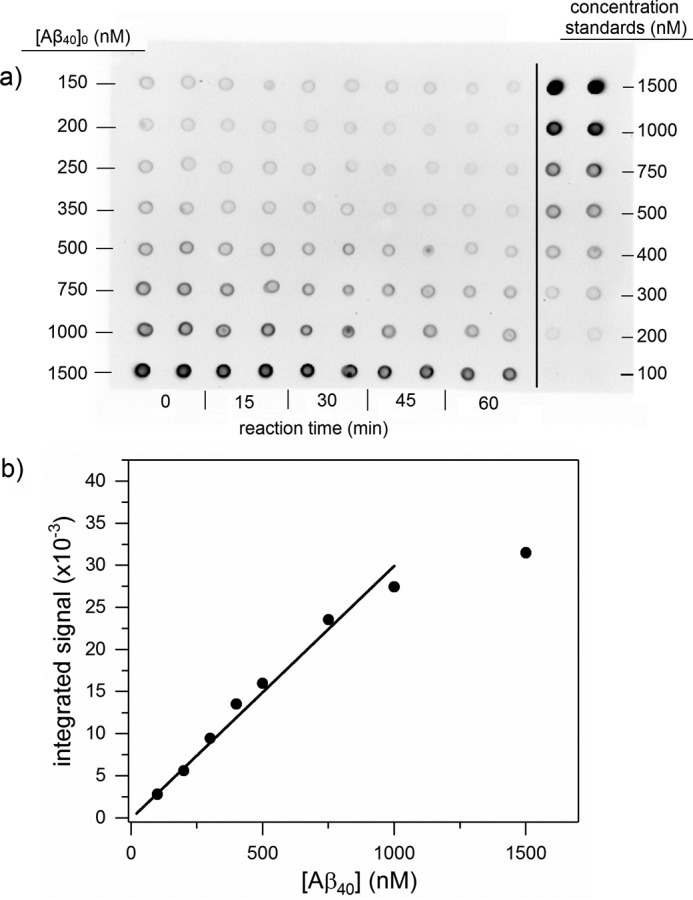
Aβ40 degradation by CatB. a, sample immunoblot showing Aβ40 degradation by CatB. Mixtures of 200 nm CatB and the listed [Aβ40]0 were incubated at 37 °C and pH 7.4. Samples were dotted onto the membrane in duplicate at each time point shown. Concentration standards were also dotted in duplicate. b, concentration standard curves were used to convert integrated signal to Aβ concentration. Signals near saturation were discarded from analysis due to non-linearity. Data points are the average of two dots in the concentration standards shown in a.
Figure 6.

CatB proteolysis of Aβ42. Mixtures of 200 nm CatB and Aβ42 at concentrations shown were incubated at 37 °C, pH 7.4. Samples were dotted onto the membrane at the given time points. Membranes were imaged using an antibody against Aβ42 (a) and then stripped and imaged again using an antibody against Aβ40 (b). Densitometric scans of the 600 nm sample before and after re-probing are shown in c and d, respectively. Data are the average of two values.
Figure 5.
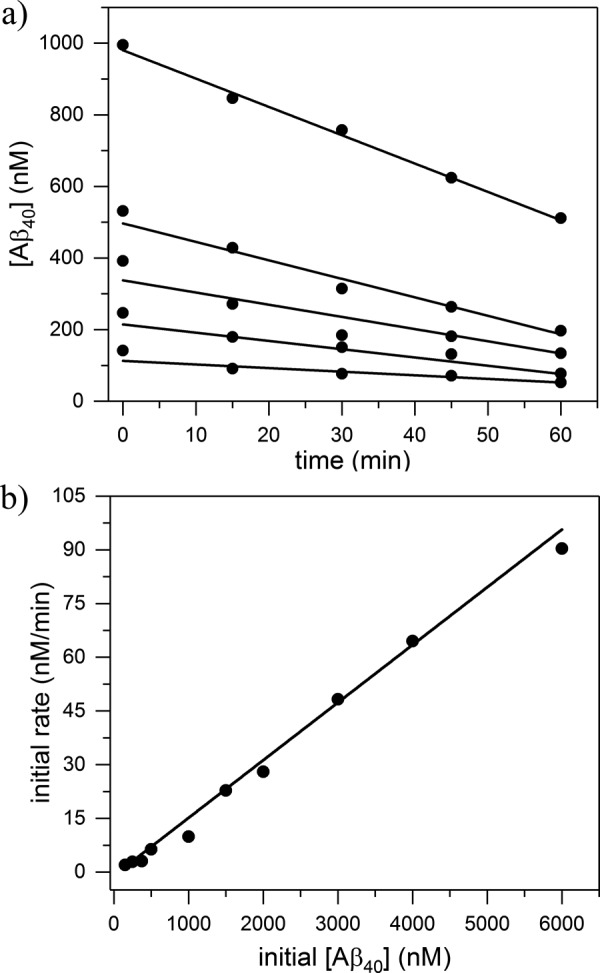
Aβ degradation kinetics. a, dot intensities of degradation samples were converted to Aβ concentrations using concentration standards and plotted as a function of time. b, initial rates were plotted against initial Aβ concentration. The slope of the linear fit was used to calculate kdeg for CatB degradation of Aβ as described under “Experimental procedures.” Each point is the mean value of at least two independent measurements.
CatB degradation of CysC
Optimal CatB proteolytic activity occurs in acidic conditions (pH <6), which is relevant for CatB contained within endocytic and lysosomal vesicles (51). Upon secretion to neutral CSF, CatB activity is expected to decrease, but not deactivate entirely (33, 34). Others have reported that secreted cathepsin D specifically degrades extracellular CysC in acidic to near-neutral tumor environments (52). We speculated that CatB could similarly degrade CysC, reducing CysC inhibitory capacity and promoting uncontrolled proteolysis. When considering the interaction between CatB and CysC, a complication arises due to coupling of the inhibitory and degradation reactions. To measure true degradation kinetics, it is necessary to first “turn off” inhibitory binding modes between CysC and CatB. Using site-directed mutagenesis, we produced and purified W106G CysC, a variant which has been shown to be a poor inhibitor of CatB (53). We confirmed by CatB activity assay that W106G inhibition constant is far weaker than WT (supplemental Fig. S3). The CD spectrum of W106G was identical to WT, confirming that loss of activity is due to specific loss of interaction, rather than loss of protein fold (supplemental Fig. S2).
We used an immunoblot assay to measure the kinetics of W106G CysC degradation by CatB at 37 °C for up to 2 h. There was no detectable CysC degradation at pH 7.4 (supplemental Fig. S4). We stripped and re-probed each membrane with a CysC N-terminal antibody (C-27) and a third antibody that does not overlap with the N or C terminus (Cyst24); neither showed significant degradation (data not shown). At pH 6.0, W106G degradation was noticeable (supplemental Fig. S4). WT was degraded much more slowly due to coupling of the inhibition reaction to degradation.
Model development
We developed a kinetic model to predict the effect of CysC and CatB on Aβ aggregation and degradation. CysC plays two distinct roles in this model: by binding to Aβ it inhibits Aβ aggregation, but by binding to CatB it inhibits Aβ degradation. We assumed all binding reactions are reversible and sufficiently fast to be assumed always at equilibrium, with equilibrium dissociation constants of KD (binding of CysC to Aβ40 or Aβ42) and KI (binding of CysC to CatB). We assumed that CysC binds only non-aggregated Aβ. Aβ is enzymatically degraded by CatB, by the rate constant kdeg. We assumed that CatB degrades Aβ monomers but does not degrade Aβ aggregates. Degradation of CysC by CatB was not included because it was negligible at pH 7.4.
The mathematical model includes coupled algebraic and differential equations. Material balance Equations 3 and 4 are derived assuming that the concentration of CatB bound to the substrate is negligibly small (Km ≫ [S], see the supplemental text for justification). Free CatB or CysC concentration [CatB] or [CysC], respectively, at any time is calculated as the initial concentration ([CysC]0 or [CatB]0, respectively) minus the protein that is bound in the CysC–CatB complex or (for CysC) to Aβ, where KD and KI are given in Table 2.
| (Eq. 3) |
| (Eq. 4) |
Aβ monomer material balances account for decreases due to binding to CysC, enzymatic degradation, or aggregation. Additionally, Aβ40 can be generated from Aβ42 degradation as shown in Equations 5 and 6.
| (Eq. 5) |
| (Eq. 6) |
The rate of production of degraded or aggregated Aβ is described by differential Equations 7–10,
| (Eq. 7) |
| (Eq. 8) |
| (Eq. 9) |
| (Eq. 10) |
with rate constants determined experimentally and summarized in Table 2. We assumed no cross-aggregation of Aβ40 and Aβ42. The set of equations is initialized by first simultaneous solution of the coupled material balance equations (Equations 3–6) given the initial concentrations of CysC, CatB, and Aβ. Then the differential equations (Equations 7–10) were solved numerically, with iterative solutions of the material balances as intact monomeric Aβ is lost.
We solved the model equations to calculate Aβ aggregation kinetic profiles at several different CysC and CatB concentrations. Specific conditions tested are shown in Table 3. We then measured Aβ aggregation kinetics at these conditions, using ThT fluorescence, and we compared experimental data to model simulations. There were no additional adjustments made to the model.
Table 3.
Model validation sample set
Aβ, CysC, and CatB were mixed to the final concentrations given and incubated at 37 °C with 10 μm ThT. Aβ aggregation over time is shown in Fig. 7 and is compared with model predictions.
| Sample | [Aβ40] | [Aβ42] | [CysC] | [CatB] |
|---|---|---|---|---|
| μm | μm | μm | μm | |
| i | 28 | |||
| ii | 28 | 12 | ||
| iii | 28 | 12 | 0.8 | |
| iv | 28 | 12 | 1.6 | |
| v | 28 | 1.6 | ||
| vi | 10 | |||
| vii | 10 | 4 | ||
| viii | 10 | 4 | 1 | |
| ix | 10 | 1 |
Model output and data are shown superimposed in Fig. 7. Model simulations accurately captured the experimental data. At 2.5-fold excess Aβ, CysC was able to significantly delay aggregation of Aβ40 but had only a minor impact on Aβ42 aggregation (cases ii and vii). In the absence of CysC, CatB degradation of Aβ was sufficiently fast to completely suppress Aβ aggregation (cases v and ix). When both CatB and CysC were added (at up to 15-fold excess CysC), the lag time for onset of aggregation was delayed, and the steady-state amount of aggregated Aβ was lowered due to degradation (cases iii, iv, and viii).
Figure 7.
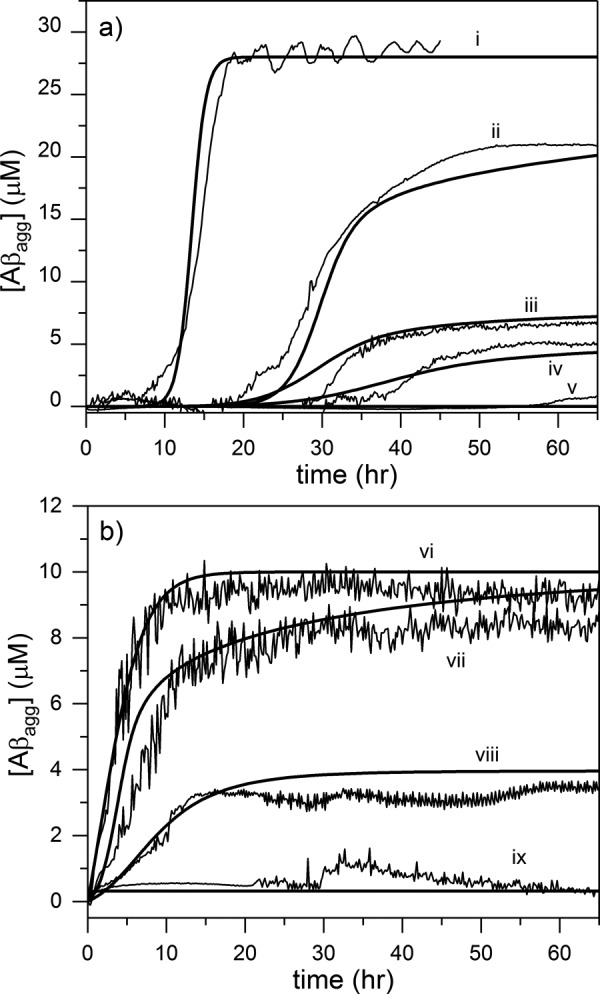
Comparison of model simulations with experimental measurements. a, Aβ40, and b, Aβ42 aggregation was monitored by ThT fluorescence at 37 °C, pH 7.4. Experimental data (thin lines) are plotted against model prediction (bold lines) for the following initial conditions: trace I, 28 μm Aβ40; trace ii, 28 μm Aβ40, 12 μm CysC; trace iii, 28 μm Aβ40, 12 μm CysC, 0.8 μm CatB; trace iv, 28 μm Aβ40, 12 μm CysC, 1.6 μm CatB; trace v, 28 μm Aβ40, 1.6 μm CatB; trace vi, 10 μm Aβ42; trace vii, 10 μm Aβ42, 4 μm CysC; trace viii, 10 μm Aβ42, 4 μm CysC, 1 μm CatB; trace ix, 10 μm Aβ42, 1 μm CatB. All data lines are the average of two independent samples except trace ii, in which a single representative sample is shown.
Comparison of time constants for degradation versus aggregation
We next sought to develop a simple parameter to predict whether Aβ aggregation or degradation would be the dominant outcome of Aβ–CysC–CatB interactions. We defined a parameter χ, as shown in Equation 11,
| (Eq. 11) |
where t50, deg is the time required for degradation of 50% of Aβ in the absence of any aggregation, and t50, agg is the time required for aggregation of 50% of Aβ in the absence of any degradation. χ → 0 if degradation is the primary outcome, and χ → ∞ if aggregation is the primary outcome. Aggregation time is calculated from Equation 12.
| (Eq. 12) |
To a first approximation, degradation time is shown in Equation 13,
| (Eq. 13) |
(see supplemental text for derivation).
We solved the full-scale numerical model for several different Aβ, CatB, and CysC concentrations and plotted the fraction of aggregated Aβ at long time from the simulations versus the calculated χ (Fig. 8a). The calculations collapse onto a set of overlapping curves. This plot falls into two clear regimes: aggregation-dominant (χ ≫ 1) and degradation-dominant (χ ≪ 1), with a sharp transition at χ = 1. This behavior was identical for Aβ40 and Aβ42, meaning that degradation and aggregation rates used to calculate χ are properly scaled. From this result, we can quantitatively predict whether Aβ will be completely degraded or will end up in toxic aggregated forms, simply from knowledge of initial concentrations of Aβ, CatB, and CysC.
Figure 8.
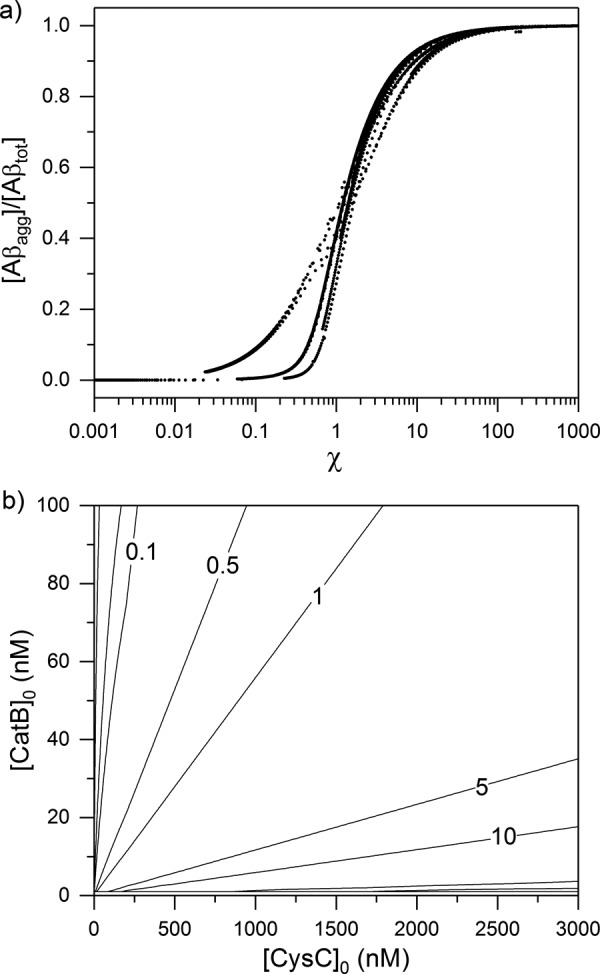
a, interaction model was iterated over various initial concentrations of CysC (0–3000 nm), CatB (0–100 nm), and Aβ (20 or 100 nm, for both Aβ40 and Aβ42). On each iteration, χ was calculated, the model was solved to steady state, and the fraction of aggregated Aβ compared with the total initial Aβ was determined. Each point represents one complete model solution given a unique set of initial conditions. b, example set of calculated χ values for initial Aβ40 concentration of 10 nm is shown on a contour diagram. Lines of constant χ are shown.
For illustration purposes, we calculated χ at 10 nm Aβ40 over a range of CatB and CysC concentrations (Fig. 8b). This figures shows, for example, that switching from aggregation (χ ∼5) to degradation (χ∼0.5) at constant CatB requires an ∼10-fold decrease in CysC.
Discussion
We developed a simple kinetic model to predict the outcome of interactions between CatB, CysC, and Aβ on Aβ aggregation and degradation. Several approximations and assumptions were made in developing the model that deserve brief comment.
ThT fluorescence is commonly used to measure aggregation of Aβ and other amyloidogenic proteins. We found that the maximum ThT intensity was not linear with total fibril mass, in agreement with other work (10, 54, 55). Non-linearity could arise due to differences in ThT-binding modes at high fibril concentration and accompanying changes in absorbance (56). Additionally, self-quenching effects have been demonstrated at high binding site occupancy (54), which is expected when total fibril mass is small compared with ThT concentration. To account for non-linearity, we fit a polynomial function to a plot of the maximum signal versus aggregate mass and then used this fit to correct all other aggregation samples.
Because we used a single experimental output, ThT fluorescence, as a measure of aggregation, we chose to describe the aggregation kinetics using a simple two-parameter model that captures the conversion of Aβ from ThT-negative to ThT-positive aggregates. The autocatalytic model was able to capture the shape and concentration dependence of the experimental data for both Aβ40 and Aβ42 and to return reasonable values for the two parameters. That said, the fact that the equations fit the data should not be interpreted as indicating that the mechanism is a correct description of the underlying self-association process. In particular, this simple model neglects Aβ oligomer formation, fibril elongation, fibril fragmentation, and other steps in the complex Aβ self-assembly process.
We assumed that CysC binds only the monomeric (unaggregated) form of Aβ. This reduces the complexity of the model, as it eliminates the need to account for different aggregate sizes and to determine the binding affinity as a function of aggregation state. There is some experimental support for this assumption. Mi et al. (17) found stable CysC–Aβ complexes in brain homogenates corresponding only to monomer–monomer interactions and only in non-demented control subjects. No such complexes were found in AD patients. The researchers speculated that Aβ–CysC binding was competing with Aβ self-association, with the latter dominating in AD patients. Other protein binders of Aβ are thought to modulate aggregation through more complex mechanisms. For example, transthyretin has been shown to preferentially bind small aggregates of Aβ rather than Aβ monomers (57), resulting in suppression of fibril elongation and lateral association but not initial nucleus formation (58). Although it is possible that CysC can bind Aβ aggregates, it was not necessary to invoke extra binding modes for the model to accurately simulate aggregation data. Our model is consistent with a mechanism whereby CysC sequesters monomeric Aβ, which is slowly released to maintain equilibrium as free Aβ is converted to aggregates or is degraded. It would be of interest to repeat FRET measurements of CysC binding to oligomeric Aβ, if technical difficulties, such as the heterogeneity and instability of the oligomeric Aβ preparation and the effect of aggregation state on donor emission, could be properly managed.
This model assumes that CatB does not degrade Aβ aggregates. Although Aβ fibrils are known to gain extensive resistance to proteolysis (59), CatB degradation of Aβ42 fibrils in vitro has been reported at pH 7.0 and 37 °C (30). However, we observed no degradation of ThT-positive Aβ aggregates. In Aβ/CysC/CatB co-incubation studies, sample fluorescence reached a plateau and did not decline (Fig. 7, traces iii, iv, and viii). Some samples maintained a plateau phase for nearly 48 h with no evidence of degradation. Lack of fibril degradation is likely due to a combination of experimental conditions favoring a very small effective concentration of CatB (due to CysC binding) and very low kdeg rate constant for fibril proteolysis (due to neutral pH).
Despite these approximations, we were able to accurately predict the aggregation profiles of Aβ40 or Aβ42 at several different concentrations of CatB and/or CysC (Fig. 7). Moreover, we developed a simplified parameter, χ, which can be quickly used to estimate whether specific concentration regimes will lead to predominantly Aβ aggregation or Aβ degradation. Knowing CysC and CatB levels in an AD animal model, for example, a researcher could find the corresponding value of χ and then determine whether conditions would support Aβ aggregation. CSF biomarker levels in human AD have been determined in multiple studies (23, 24, 36). Typical Aβ40 and Aβ42 CSF concentrations are 2 and 0.2 nm, respectively (36). CysC concentrations in CSF are reported to be from 200 to 5000 nm in CSF and 100–1000 nm in plasma (23, 24, 36). Total CatB (as determined by ELISA and not residual activity) is ∼0.3 nm in CSF and ∼6 nm in plasma (36). Using Aβ40 = 2 nm, Aβ42 =0.2 nm, CysC = 500 nm, and CatB = 0.5 nm, we obtain χ ∼10 for Aβ40 and χ ∼15,000 for Aβ42, showing a strong propensity for Aβ aggregation under typical CSF conditions. If CysC and CatB concentrations were at typical plasma levels (100 and 6 nm, respectively), χ ∼0.2 for Aβ40, the system moves to the degradation regime. It should be pointed out that the model was developed from experimental data obtained at concentrations higher than physiological for practical reasons. Analyses of χ at typical CSF and plasma conditions require extrapolation of the model to much lower concentrations of Aβ and CatB and should therefore be interpreted cautiously.
The model derived in this study could be expanded to accommodate more complex binding interactions, different cellular environments, and generation of Aβ from APP. For example, CysC is well-known to form domain-swapped dimers (18, 60–63), which lose the ability to inhibit CatB (64). CysC reversibly forms domain-swapped dimers during normal cellular trafficking, but only monomers are secreted (65). The disease-causing L68Q CysC mutation may domain-swap extracellularly, resulting in a pool of predominantly inactive CysC (66–68). If the domain-swapped dimer binds Aβ with the same affinity as monomeric CysC but does not inhibit CatB, CysC and CatB would no longer undermine the neuroprotective effects of one another. Additionally, we have observed oligomeric aggregates of CysC in vitro, which are not domain-swapped (18, 20). Non-swapped oligomers remain fully active against CatB, but they inhibit Aβ aggregation much more potently than CysC monomers. It is not clear whether these oligomers exist naturally; if so, oligomer formation could allow CysC to potently control both CatB activity and Aβ aggregation. Engineered CysC mutants also provide a method for testing the model. For example, W106G binds CatB but is a poor inhibitor of its enzymatic activity; we anticipate we would observe rapid degradation of Aβ by CatB if this mutant were used in place of wild-type CysC. We plan to carry out these experiments in the near future.
We considered only the protein interactions at neutral pH typical of extracellular fluids. Most CatB is normally restricted to intracellular compartments. The mildly acidic conditions of the endosomal/lysosomal system could alter considerably the outcome. In acidic cellular compartments, CatB activity is expected to increase, leading to fast kdeg rate constants for both Aβ and CysC substrates (33, 34). Additionally, the assumption of protease-resistant fibrils may be less valid at pH 6 than pH 7.4. However, Aβ aggregation is also accelerated at pH 6 (30). The kinetic model could easily be adapted to predict outcomes at acidic pH once experimental data are collected, allowing determination of k1, k2, kdeg, KI, and KD.
Finally, the model could be adapted to consider Aβ generation. Aβ generation from APP is a complex process resulting from many enzyme interactions (3, 69). It has been suggested that CatB may act as a β-secretase, truncating and priming APP for eventual liberation by a γ-secretase (43–45). This action would be a countervailing effect to CatB's degradation of Aβ.
Experimental procedures
Cystatin C preparation
Recombinant human CysC was produced and purified as described previously (20). Monomeric CysC was fractionated from any dimers or aggregates by membrane filtration and stored at 40 μm in PBSA-E buffer (10 mm phosphate, 150 mm NaCl, 0.02% w/v NaN3, 1 mm EDTA, pH 7.4) at 4 °C. W106G CysC was generated by site-directed mutagenesis (Agilent QuikChange II kit). W106G plasmids were transformed into BL21(DE3) cells (New England Biolabs). W106G was produced and purified as described for WT. Protein concentration was determined by BCA assay (Pierce), using known concentrations of WT as standards. After purification, W106G was stored at 28 μm in PBSA-E at 4 °C.
Aβ aggregation kinetics
Lyophilized Aβ40 (Bachem, HCl salt) and Aβ42 (Bachem, HCl salt) were reconstituted in denaturing buffer (8 m urea, 100 mm glycine, pH 10) to 2.8 mm, as described previously (9). Small volumes were aliquoted, snap-frozen, and stored at −80 °C. Immediately prior to use, aliquots were thawed and gently diluted to 200 μm in PBS (10 mm phosphate, 150 mm NaCl). ThT powder (Sigma) was dissolved to 100 μm in PBS and filtered through a 0.45-μm syringe filter. Freshly-diluted Aβ and ThT were combined in black 96-well plates to final concentrations of 8, 28, and 46 μm Aβ40 or 2.5, 10, and 20 μm Aβ42 and 10 μm ThT. The maximum fluorescence intensity is limited by ThT concentration, rather than Aβ (70). For this reason, we chose to hold ThT concentration constant to allow direct comparison of maximum ThT intensity. A clear plastic adhesive strip was used to seal each well and prevent evaporative loss. Fluorescence of ThT was continuously monitored for up to 72 h in a Tecan Infinite M200 plate reader (excitation 440 nm and emission 480 nm), with temperature controlled to 37 °C. Post-aggregation, three reaction mixtures (28 μm Aβ40) were removed and centrifuged at 14,000 rpm for 30 min at 4 °C. Residual soluble peptide concentration in the supernatant was determined by BCA assay, using freshly-dissolved Aβ monomers for concentration standards. Fibrillar Aβ concentration (in equivalent monomer molar concentration) was calculated from ThT fluorescence intensity, using maximum ThT fluorescence at 72 h for calibration.
Fluorescent labeling of CysC with Tidefluor 4
Tidefluor 4 succinimidyl ester (TF4 SE) was purchased from AAT Bioquest and dissolved in anhydrous N,N-dimethylformamide (DMF, Sigma) to 20 mm. CysC stock was concentrated to 140 μm by ultrafiltration (Amicon Ultra-0.5 ml, 3-kDa MWCO, Millipore) and dialyzed into PBS pH-adjusted to 6.5 with NaH2PO4. TF4 SE solution was added to CysC to final concentrations of 1.4 mm TF4 SE, 130 μm CysC, and 7% v/v DMF. Protein/dye mixture tube was wrapped in aluminum foil and gently shaken at 4 °C for 24 h to conjugate TF4 SE to CysC. Conjugation reaction conditions were chosen to selectively label the N terminus of CysC. Excess dye was removed by gel filtration (Bio-Spin P-6 gel column, Bio-Rad). CysC-TF4 conjugates were eluted in pH 7.4 PBS and stored wrapped in foil at 4 °C. Conjugation efficiency was determined from absorbance at 280 and 590 nm as shown in Equations 14 and 15.
| (Eq. 14) |
| (Eq. 15) |
Molar absorptivities of CysC and TF4 (ϵCysC, 280 and ϵTF4, 590) are 11,100 m−1 cm−1 (46) and 90,000 m−1 cm−1 (AAT Bioquest), respectively. CF, a correction factor for A280 of TF4, equals 0.436 (AAT Bioquest). Conjugation efficiency was 72%.
FRET measurements
Hilyte Fluor 488-labeled Aβ40 (HF488-Aβ40, Anaspec, AS-60491-01) was reconstituted in denaturing buffer (8 m urea, 100 mm glycine, pH 10) to 2.8 mm. Small volumes were aliquoted, snap-frozen, and stored at −80 °C. HF488-Aβ40 was thawed and diluted to 10 μm with PBS. Concentration of HF-Aβ40 was confirmed by absorbance (ϵ497 = 70,000 m−1 cm−1, Anaspec) and then adjusted to 800 nm by addition of PBS. TF4-CysC was diluted from stock to 2 μm. TF4-CysC and HF488-Aβ40 were mixed to final concentrations of 400 nm HF488-Aβ40 and 0–600 nm TF4-CysC. Samples were incubated at 37 °C for 0–60 min and then transferred to a 130-μl cuvette, loaded into the temperature-controlled sample pedestal of a QM-40 fluorometer (PTI, Inc.) and maintained at 37 °C. Fluorescence spectra of all samples were measured using excitation at 488 nm and emission scan of 510–630 nm. Spectra of the same sample taken at various times after mixing showed minor changes up to 10 min and then no difference thereafter (data not shown). Thus, all reported spectra were recorded after at least 10 min of co-incubation. The average of three spectra per sample was recorded. Emission of HF488-Aβ40 (FRET donor) at 530 nm was used to determine overall fraction of bound HF488-Aβ40 as shown in Equation 16.
| (Eq. 16) |
I530 and I530, 0 are the sample emission at 530 nm in the presence and absence of FRET acceptor (TF4-CysC), respectively. I530, sat is the sample emission under saturating conditions (large excess of TF4-CysC). The fraction of bound HF488-Aβ40 was fit to the Hill equation to derive the binding constant, KD and the cooperativity parameter n as shown in Equation 17.
| (Eq. 17) |
Cathepsin B-mediated degradation kinetics
CatB purified from human liver (16.5 μm, Enzo Life Science, BML-SE198-0025) was aliquoted and stored at −80 °C. CatB was activated by mixing equal volumes of CatB stock with activation buffer (10 mm phosphate, 150 mm NaCl, 0.005% v/v Tween 20, 10 mm DTT, pH 7.4) and incubating at room temperature for 15 min. CatB active-site concentration was determined by titration with the irreversible inhibitor, E-64. Stock E-64 (Sigma) was diluted to 1 μm in PBS-T (10 mm phosphate, 150 mm NaCl, 0.005% v/v Tween 20, pH 7.4). Activated CatB (200 nm) was combined with E-64 (0–1000 nm) and incubated at 37 °C for 10 min. Samples were diluted 10-fold at 37 °C PBS-T and then combined in equal volume with 200 μm Z-FR-AMC fluorogenic substrate (R&D Systems). Final concentrations were 10 nm CatB, 0–25 nm E-64, and 100 μm Z-FR-AMC. Fluorescence was monitored over time on a BioTek FLX800 plate reader (excitation 360 nm and emission 460 nm), maintaining samples at 37 °C. Initial rates were plotted against E-64 concentration, and CatB active-site concentration was determined from the x-intercept of the plot. The active-site concentration was found to be ∼95% of the total CatB concentration.
Kinetics of degradation of Aβ40, Aβ42, and CysC (WT and W106G) by CatB were determined by immunoblot assays. Stock Aβ40 and Aβ42 were thawed and diluted to 10 μm in PBS-T. CatB was activated as described and diluted to 400 nm in PBS-T. CatB and Aβ were combined to final concentrations of 200 nm CatB and 0–5 μm Aβ. In other experiments, activated CatB was mixed with WT or W106G CysC to final concentrations of 200 nm CatB and 0–1000 nm CysC, using either PBS-T (pH 7.4 experiments) or MES-T (25 mm MES, 1 mm EDTA, 1 mm DTT, 0.005% v/v Tween 20, pH 6.0 experiments) as diluent. Mixtures were incubated at 37 °C for up to 2 h. Every 15 min, 2 μl was spotted onto a nitrocellulose membrane and allowed to dry completely. Membranes were washed in TBS-T (20 mm Tris base, 150 mm NaCl, 0.05% v/v Tween 20, pH 8.0) and incubated overnight in blocking buffer (5% w/v nonfat dry milk in TBS-T). Membranes were incubated with antibody against the C terminus of either Aβ40 (Abcam, ab76317), Aβ42 (Abcam, ab10148), or CysC (Novus, Cyst13) at 2 μg/ml in TBS-T for 1 h at room temperature. The membranes were then washed in TBS-T and incubated with 2 μg/ml goat anti-rabbit (Novus, NBP1-75,325, Aβ blots) or goat anti-mouse (Novus, NB7539, CysC blots) HRP-conjugated secondary antibody in TBS-T for 1 h at room temperature. The membranes were then washed in TBS-T, and antigen was imaged by addition of chemiluminescent substrate (GE Healthcare). Images were taken on a ChemiDoc XRS+ (Bio-Rad). Blot images were background-subtracted and quantified using ImageJ. Initial degradation rates were plotted against the substrate concentration and data were fit to Equation 18,
| (Eq. 18) |
to determine the degradation rate constant kdeg.
Model building
All model equations were numerically integrated using ode15s solver package in MATLAB. Equations were input to the solver by a mass matrix to discriminate between material balance and ordinary differential equations. Initial conditions were determined by implicit solution to CysC, Aβ, and CatB material balance equations, given the total amount of each protein. Integration time steps were determined automatically by the solver package. Model equations were solved until steady state was reached.
Author contributions
T. J. P. and R. M. M. designed the study and wrote the paper. T. J. P. carried out all experiments. T. J. P. and R. M. M. analyzed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgment
We thank Jacob Mehlhoff for assistance in cloning and characterizing W106G CysC.
This work was supported by National Science Foundation Grants CBET-1262729 and CBET-1703237. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S4, text for derivation, and text for justification.
- AD
- Alzheimer's disease
- Aβ
- β-amyloid
- APP
- amyloid precursor protein
- CysC
- cystatin C
- CatB
- cathepsin B
- ThT
- thioflavin T
- DMF
- N,N-dimethylformamide
- Z-FR-AMC
- benzyloxycarbonyl-FR-amido-4-methylcoumarin.
References
- 1. Busciglio J., Gabuzda D. H., Matsudaira P., and Yankner B. A. (1993) Generation of β-amyloid in the secretory pathway in neuronal and nonneuronal cells. Proc. Natl. Acad. Sci. U.S.A. 90, 2092–2096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Selkoe D. J., and Hardy J. (2016) The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol. Med. 8, 595–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Andrew R. J., Kellett K. A., Thinakaran G., and Hooper N. M. (2016) A Greek tragedy: The growing complexity of Alzheimer amyloid precursor protein proteolysis. J. Biol. Chem. 291, 19235–19244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mattson M. P. (1997) Cellular actions of β-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol. Rev. 77, 1081–1132 [DOI] [PubMed] [Google Scholar]
- 5. Yang T., Li S., Xu H., Walsh D. M., and Selkoe D. J. (2017) Large soluble oligomers of amyloid β-protein from Alzheimer brain are far less neuroactive than the smaller oligomers to which they dissociate. J. Neurosci. 37, 152–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bernacki J. P., and Murphy R. M. (2009) Model discrimination and mechanistic interpretation of kinetic data in protein aggregation studies. Biophys. J. 96, 2871–2887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pellarin R., and Caflisch A. (2006) Interpreting the aggregation kinetics of amyloid peptides. J. Mol. Biol. 360, 882–892 [DOI] [PubMed] [Google Scholar]
- 8. Morris A. M., Watzky M. A., and Finke R. G. (2009) Protein aggregation kinetics, mechanism, and curve-fitting: a review of the literature. Biochim. Biophys. Acta 1794, 375–397 [DOI] [PubMed] [Google Scholar]
- 9. Murphy R. M., and Pallitto M. M. (2000) Probing the kinetics of β-amyloid self-association. J. Struct. Biol. 130, 109–122 [DOI] [PubMed] [Google Scholar]
- 10. Hellstrand E., Boland B., Walsh D. M., and Linse S. (2010) Amyloid β-protein aggregation produces highly reproducible kinetic data and occurs by a two-phase process. ACS Chem. Neurosci. 1, 13–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pate K. M., and Murphy R. M. (2017) Cerebrospinal fluid proteins as regulators of β-amyloid aggregation and toxicity. Isr. J. Chem. 57, 602–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Levy E., Sastre M., Kumar A., Gallo G., Piccardo P., Ghetti B., and Tagliavini F. (2001) Codeposition of cystatin C with amyloid-β protein in the brain of Alzheimer disease patients. J. Neuropathol. Exp. Neurol. 60, 94–104 [DOI] [PubMed] [Google Scholar]
- 13. Coria F., Castaño E. M., and Frangione B. (1987) Brain amyloid in normal aging and cerebral amyloid angiopathy is antigenically related to Alzheimer's disease β-protein. Am. J. Pathol. 129, 422–428 [PMC free article] [PubMed] [Google Scholar]
- 14. McCarron M. O., Nicoll J. A., Stewart J., Ironside J. W., Mann D. M., Love S., Graham D. I., and Grubb A. (2000) Absence of cystatin C mutation in sporadic cerebral amyloid angiopathy-related hemorrhage. Neurology 54, 242–244 [DOI] [PubMed] [Google Scholar]
- 15. Maruyama K., Kametani F., Ikeda S., Ishihara T., and Yanagisawa N. (1992) Characterization of amyloid fibril protein from a case of cerebral amyloid angiopathy showing immunohistochemical reactivity for both β protein and cystatin C. Neurosci. Lett. 144, 38–42 [DOI] [PubMed] [Google Scholar]
- 16. Vinters H. V., Secor D. L., Pardridge W. M., and Gray F. (1990) Immunohistochemical study of cerebral Alzheimer A4 peptide in cerebral microvessel walls colocalizes with γ trace in patients with leukoencephalopathy. Ann. Neurol. 28, 34–42 [DOI] [PubMed] [Google Scholar]
- 17. Mi W., Jung S. S., Yu H., Schmidt S. D., Nixon R. A., Mathews P. M., Tagliavini F., and Levy E. (2009) Complexes of amyloid-β and cystatin C in the human central nervous system. J. Alzheimers Dis. 18, 273–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Perlenfein T. J., Mehlhoff J. D., and Murphy R. M. (2017) Insights into the mechanism of cystatin C oligomer and amyloid formation and its interaction with β-amyloid. J. Biol. Chem. 292, 11485–11498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Selenica M. L., Wang X., Ostergaard-Pedersen L., Westlind-Danielsson A., and Grubb A. (2007) Cystatin C reduces the in vitro formation of soluble A-β1–42 oligomers and protofibrils. Scand. J. Clin. Lab. Invest. 67, 179–190 [DOI] [PubMed] [Google Scholar]
- 20. Perlenfein T. J., and Murphy R. M. (2016) Expression, purification, and characterization of human cystatin C monomers and oligomers. Protein Expr. Purif. 117, 35–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tizon B., Ribe E. M., Mi W., Troy C. M., and Levy E. (2010) Cystatin C protects neuronal cells from amyloid-β-induced toxicity. J. Alzheimers Dis. 19, 885–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Juszczyk P., Paraschiv G., Szymanska A., Kolodziejczyk A. S., Rodziewicz-Motowidlo S., Grzonka Z., and Przybylski M. (2009) Binding epitopes and interaction structure of the neuroprotective protease inhibitor cystatin C with β-amyloid revealed by proteolytic excision mass spectrometry and molecular docking simulation. J. Med. Chem. 52, 2420–2428 [DOI] [PubMed] [Google Scholar]
- 23. Zhong X. M., Hou L., Luo X. N., Shi H. S., Hu G. Y., He H. B., Chen X. R., Zheng D., Zhang Y. F., Tan Y., Liu X. J., Mu N., Chen J. P., and Ning Y. P. (2013) Alterations of CSF cystatin C levels and their correlations with CSF Aβ40 and Aβ42 levels in patients with Alzheimer's disease, dementia with Lewy bodies and the atrophic form of general paresis. PLoS ONE 8, e55328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Craig-Schapiro R., Kuhn M., Xiong C., Pickering E. H., Liu J., Misko T. P., Perrin R. J., Bales K. R., Soares H., Fagan A. M., and Holtzman D. M. (2011) Multiplexed immunoassay panel identifies novel CSF biomarkers for Alzheimer's disease diagnosis and prognosis. PLoS ONE 6, e18850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang R., Chen Z., Fu Y., Wei X., Liao J., and Liu X. (2017) Plasma cystatin C and high-density lipoprotein are important biomarkers of Alzheimer's disease and vascular dementia: a cross-sectional study. Front. Aging Neurosci. 9, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Björk I., Pol E., Raub-Segall E., Abrahamson M., Rowan A. D., and Mort J. S. (1994) Differential changes in the association and dissociation rate constants for binding of cystatins to target proteinases occurring on N-terminal truncation of the inhibitors indicate that the interaction mechanism varies with different enzymes. Biochem. J. 299, 219–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nycander M., Estrada S., Mort J. S., Abrahamson M., and Björk I. (1998) Two-step mechanism of inhibition of cathepsin B by cystatin C due to displacement of the proteinase occluding loop. FEBS Lett. 422, 61–64 [DOI] [PubMed] [Google Scholar]
- 28. Turk V., Stoka V., Vasiljeva O., Renko M., Sun T., Turk B., and Turk D. (2012) Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim. Biophys. Acta 1824, 68–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mackay E. A., Ehrhard A., Moniatte M., Guenet C., Tardif C., Tarnus C., Sorokine O., Heintzelmann B., Nay C., Remy J. M., Higaki J., Van Dorsselaer A., Wagner J., Danzin C., and Mamont P. (1997) A possible role for cathepsins D, E, and B in the processing of β-amyloid precursor protein in Alzheimer's disease. Eur. J. Biochem. 244, 414–425 [DOI] [PubMed] [Google Scholar]
- 30. Mueller-Steiner S., Zhou Y., Arai H., Roberson E. D., Sun B., Chen J., Wang X., Yu G., Esposito L., Mucke L., and Gan L. (2006) Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer's disease. Neuron 51, 703–714 [DOI] [PubMed] [Google Scholar]
- 31. Gan L., Ye S., Chu A., Anton K., Yi S., Vincent V. A., von Schack D., Chin D., Murray J., Lohr S., Patthy L., Gonzalez-Zulueta M., Nikolich K., and Urfer R. (2004) Identification of cathepsin B as a mediator of neuronal death induced by Aβ-activated microglial cells using a functional genomics approach. J. Biol. Chem. 279, 5565–5572 [DOI] [PubMed] [Google Scholar]
- 32. Yakovlev A. A., Kvichansky A. A., Lyzhin A. A., Khaspekov L. G., and Gulyaeva N. V. (2013) Glutamate treatment and preconditioning differently affect cathepsin B release and intracellular proteases in primary cultures of cerebellar granular cells. Neurochem. J. 7, 111–120 [Google Scholar]
- 33. Linebaugh B. E., Sameni M., Day N. A., Sloane B. F., and Keppler D. (1999) Exocytosis of active cathepsin B: Enzyme activity at pH 7.0, inhibition and molecular mass. Eur. J. Biochem. 264, 100–109 [DOI] [PubMed] [Google Scholar]
- 34. Werle B., Jülke B., Lah T., Spiess E., and Ebert W. (1997) Cathepsin B fraction active at physiological pH of 7.5 is of prognostic significance in squamous cell carcinoma of human lung. Br. J. Cancer 75, 1137–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tiribuzi R., Crispoltoni L., Porcellati S., Di Lullo M., Florenzano F., Pirro M., Bagaglia F., Kawarai T., Zampolini M., Orlacchio A., and Orlacchio A. (2014) Neurobiology of aging monocytes from patients with sporadic Alzheimer's disease. Neurobiol. Aging 35, 345–356 [DOI] [PubMed] [Google Scholar]
- 36. Sundelöf J., Sundström J., Hansson O., Eriksdotter-Jönhagen M., Giedraitis V., Larsson A., Degerman-Gunnarsson M., Ingelsson M., Minthon L., Blennow K., Kilander L., Basun H., and Lannfelt L. (2010) Higher cathepsin B levels in plasma in Alzheimer's disease compared to healthy controls. J. Alzheimers Dis. 22, 1223–1230 [DOI] [PubMed] [Google Scholar]
- 37. Mi W., Pawlik M., Sastre M., Jung S. S., Radvinsky D. S., Klein A. M., Sommer J., Schmidt S. D., Nixon R. A., Mathews P. M., and Levy E. (2007) Cystatin C inhibits amyloid-β deposition in Alzheimer's disease mouse models. Nat. Genet. 39, 1440–1442 [DOI] [PubMed] [Google Scholar]
- 38. Kaeser S. A., Herzig M. C., Coomaraswamy J., Kilger E., Selenica M.-L., Winkler D. T., Staufenbiel M., Levy E., Grubb A., and Jucker M. (2007) Cystatin C modulates cerebral β-amyloidosis. Nat. Genet. 39, 1437–1439 [DOI] [PubMed] [Google Scholar]
- 39. Sun B., Zhou Y., Halabisky B., Lo I., Cho S. H., Mueller-Steiner S., Devidze N., Wang X., Grubb A., and Gan L. (2008) Cystatin C-cathepsin B axis regulates amyloid β levels and associated neuronal deficits in an animal model of Alzheimer's disease. Neuron 60, 247–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang C., Sun B., Zhou Y., Grubb A., and Gan L. (2012) Cathepsin B degrades amyloid-β in mice expressing wild-type human amyloid precursor protein. J. Biol. Chem. 287, 39834–39841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Embury C. M., Dyavarshetty B., Lu Y., Wiederin J. L., Ciborowski P., Gendelman H. E., and Kiyota T. (2017) Cathepsin B improves β-amyloidosis and learning and memory in models of Alzheimer's disease. J. Neuroimmune Pharmacol. 12, 340–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang D. S., Stavrides P., Mohan P. S., Kaushik S., Kumar A., Ohno M., Schmidt S. D., Wesson D., Bandyopadhyay U., Jiang Y., Pawlik M., Peterhoff C. M., Yang A. J., Wilson D. A., St George-Hyslop P., et al. (2011) Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer's disease ameliorates amyloid pathologies and memory deficits. Brain 134, 258–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hook V. Y., Kindy M., and Hook G. (2008) Inhibitors of cathepsin B improve memory and reduce β-amyloid in transgenic Alzheimer disease mice expressing the wild-type, but not the Swedish mutant, β-secretase site of the amyloid precursor protein. J. Biol. Chem. 283, 7745–7753 [DOI] [PubMed] [Google Scholar]
- 44. Hook G., Hook V., and Kindy M. (2011) The cysteine protease inhibitor, E64d, reduces brain amyloid-β and improves memory deficits in Alzheimer's disease animal models by inhibiting cathepsin B, but not BACE1, β-secretase activity. J. Alzheimers Dis. 26, 387–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang X.-F., Liu D.-X., Liang Y., Xing L.-L., Zhao W.-H., Qin X.-X., Shang D.-S., Li B., Fang W.-G., Cao L., Zhao W.-D., and Chen Y.-H. (2016) Cystatin C shifts APP processing from amyloid-β production towards non-amyloidgenic pathway in brain endothelial cells. PLoS ONE 11, e0161093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wahlbom M., Wang X., Lindström V., Carlemalm E., Jaskolski M., and Grubb A. (2007) Fibrillogenic oligomers of human cystatin C are formed by propagated domain swapping. J. Biol. Chem. 282, 18318–18326 [DOI] [PubMed] [Google Scholar]
- 47. Nagai A., Ryu J. K., Terashima M., Tanigawa Y., Wakabayashi K., McLarnon J. G., Kobayashi S., Masuda J., and Kim S. U. (2005) Neuronal cell death induced by cystatin C in vivo and in cultured human CNS neurons is inhibited with cathepsin B. Brain Res. 1066, 120–128 [DOI] [PubMed] [Google Scholar]
- 48. Murphy R. M. (2013) in Tandem Repeats in Genes, Proteins, and Disease: Methods and Protocols (Hatters D. M. and Hannan A. J., eds) pp. 201–217, Humana Press Inc., Totowa, NJ [Google Scholar]
- 49. Mathews P. M., and Levy E. (2016) Cystatin C in aging and in Alzheimer's disease. Ageing Res. Rev. 32, 38–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sastre M., Calero M., Pawlik M., Mathews P. M., Kumar A., Danilov V., Schmidt S. D., Nixon R. A., Frangione B., and Levy E. (2004) Binding of cystatin C to Alzheimer's amyloid β inhibits in vitro amyloid fibril formation. Neurobiol. Aging 25, 1033–1043 [DOI] [PubMed] [Google Scholar]
- 51. Polgár L., and Csoma C. (1987) Dissociation of ionizing groups in the binding. J. Biol. Chem. 262, 14448–14453 [PubMed] [Google Scholar]
- 52. Laurent-Matha V., Huesgen P. F., Masson O., Derocq D., Prébois C., Gary-Bobo M., Lecaille F., Rebière B., Meurice G., Oréar C., Hollingsworth R. E., Abrahamson M., Lalmanach G., Overall C. M., and Liaudet-Coopman E. (2012) Proteolysis of cystatin C by cathepsin D in the breast cancer microenvironment. FASEB J. 26, 5172–5181 [DOI] [PubMed] [Google Scholar]
- 53. Hall A., Håkansson K., Mason R. W., Grubb A., and Abrahamson M. (1995) Structural basis for the biological specificity of cystatin C. J. Biol. Chem. 270, 5115–5121 [DOI] [PubMed] [Google Scholar]
- 54. Lindberg D. J., Wenger A., Sundin E., Wesén E., Westerlund F., and Esbjörner E. K. (2017) Binding of thioflavin-T to amyloid fibrils leads to fluorescence self-quenching and fibril compaction. Biochemistry 56, 2170–2174 [DOI] [PubMed] [Google Scholar]
- 55. Bourhim M., Kruzel M., Srikrishnan T., and Nicotera T. (2007) Linear quantitation of Aβ aggregation using thioflavin T: reduction in fibril formation by colostrinin. J. Neurosci. Methods 160, 264–268 [DOI] [PubMed] [Google Scholar]
- 56. Sulatskaya A. I., Kuznetsova I. M., and Turoverov K. K. (2011) Interaction of thioflavin T with amyloid fibrils: stoichiometry and affinity of dye binding, absorption spectra of bound dye. J. Phys. Chem. B. 115, 11519–11524 [DOI] [PubMed] [Google Scholar]
- 57. Yang D. T., Joshi G., Cho P. Y., Johnson J. A., and Murphy R. M. (2013) Transthyretin as both a sensor and a scavenger of β-amyloid oligomers. Biochemistry 52, 2849–2861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liu L., and Murphy R. M. (2006) Kinetics of inhibition of β-amyloid aggregation by transthyretin. Biochemistry 45, 15702–15709 [DOI] [PubMed] [Google Scholar]
- 59. Nordstedt C., Näslund J., Tjernberg L. O., Karlström A. R., Thyberg J., and Terenius L. (1994) The Alzheimer Aβ peptide develops protease resistance in association with its polymerization into fibrils. J. Biol. Chem. 269, 30773–30776 [PubMed] [Google Scholar]
- 60. Ekiel I., and Abrahamson M. (1996) Folding-related dimerization of human cystatin C. J. Biol. Chem. 271, 1314–1321 [DOI] [PubMed] [Google Scholar]
- 61. Ekiel I., Abrahamson M., Fulton D. B., Lindahl P., Storer A. C., Levadoux W., Lafrance M., Labelle S., Pomerleau Y., Groleau D., LeSauteur L., and Gehring K. (1997) NMR structural studies of human cystatin C dimers and monomers. J. Mol. Biol. 271, 266–277 [DOI] [PubMed] [Google Scholar]
- 62. Janowski R., Kozak M., Jankowska E., Grzonka Z., Grubb A., Abrahamson M., and Jaskolski M. (2001) Human cystatin C, an amyloidogenic protein, dimerizes through three-dimensional domain swapping. Nat. Struct. Biol. 8, 316–320 [DOI] [PubMed] [Google Scholar]
- 63. Janowski R., Abrahamson M., Grubb A., and Jaskolski M. (2004) Domain swapping in N-truncated human cystatin C. J. Mol. Biol. 341, 151–160 [DOI] [PubMed] [Google Scholar]
- 64. Hall A., Ekiel I., Mason R. W., Kasprzykowski F., Grubb A., and Abrahamson M. (1998) Structural basis for different inhibitory specificities of human cystatins C and D. Biochemistry 37, 4071–4079 [DOI] [PubMed] [Google Scholar]
- 65. Merz G. S., Benedikz E., Schwenk V., Johansen T. E., Vogel L. K., Rushbrook J. I., and Wisniewski H. M. (1997) Human cystatin C forms an inactive dimer during intracellular trafficking in transfected CHO cells. J. Cell Physiol. 173, 423–432 [DOI] [PubMed] [Google Scholar]
- 66. Asgeirsson B., Haebel S., Thorsteinsson L., Helgason E., Gudmundsson K. O., Gudmundsson G., and Roepstorff P. (1998) Hereditary cystatin C amyloid angiopathy: monitoring the presence of the Leu-68 Gln cystatin C variant in cerebrospinal fluids and monocyte cultures by MS. Biochem. J. 329, 497–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Calero M., Pawlik M., Soto C., Castaño E. M., Sigurdsson E. M., Kumar A., Gallo G., Frangione B., and Levy E. (2001) Distinct properties of wild-type and the amyloidogenic human cystatin C variant of hereditary cerebral hemorrhage with amyloidosis, Icelandic type. J. Neurochem. 77, 628–637 [DOI] [PubMed] [Google Scholar]
- 68. Hyytiä H., Ristiniemi N., Airas L., Pettersson K., and Hellman J. (2010) Development of an immunoassay for the detection of cystatin C dimers. J. Immunol. Methods 355, 14–20 [DOI] [PubMed] [Google Scholar]
- 69. Haass C., Kaether C., Thinakaran G., and Sisodia S. (2012) Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2, a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Xue C., Lin T. Y., Chang D., and Guo Z. (2017) Thioflavin T as an amyloid dye: fibril quantification, optimal concentration and effect on aggregation. R. Soc. Open Sci. 4, 160696. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.