

Abstract
Kidney podocytes represent a key constituent of the glomerular filtration barrier. Identifying the molecular mechanisms of podocyte injury and survival is important for better understanding and management of kidney diseases. KIBRA (kidney brain protein), an upstream regulator of the Hippo signaling pathway encoded by the Wwc1 gene, shares the pro-injury properties of its putative binding partner dendrin and antagonizes the pro-survival signaling of the downstream Hippo pathway effector YAP (Yes-associated protein) in Drosophila and MCF10A cells. We recently identified YAP as an essential component of the glomerular filtration barrier that promotes podocyte survival by inhibiting dendrin pro-apoptotic function. Despite these recent advances, the signaling pathways that mediate podocyte injury remain poorly understood. Here we tested the hypothesis that, similar to its role in other model systems, KIBRA promotes podocyte injury. We found increased expression of KIBRA and phosphorylated YAP protein in glomeruli of patients with biopsy-proven focal segmental glomerulosclerosis (FSGS). KIBRA/WWc1 overexpression in murine podocytes promoted LATS kinase phosphorylation, leading to subsequent YAP Ser-127 phosphorylation, YAP cytoplasmic sequestration, and reduction in YAP target gene expression. Functionally, KIBRA overexpression induced significant morphological changes in podocytes, including disruption of the actin cytoskeletal architecture and reduction of focal adhesion size and number, all of which were rescued by subsequent YAP overexpression. Conversely, constitutive KIBRA knockout mice displayed reduced phosphorylated YAP and increased YAP expression at baseline. These mice were protected from acute podocyte foot process effacement following protamine sulfate perfusion. KIBRA knockdown podocytes were also protected against protamine-induced injury. These findings suggest an important role for KIBRA in the pathogenesis of podocyte injury and the progression of proteinuric kidney disease.
Keywords: actin, focal adhesion, Hippo pathway, podocyte, yes-associated protein (YAP)
Introduction
Kidney podocytes are target cells for injury across a spectrum of proteinuric kidney conditions, from primary glomerular disorders like focal segmental glomerulosclerosis (FSGS)3 and membranous nephropathy to secondary processes, such as diabetic nephropathy and hypertensive nephrosclerosis (1). Despite recent advances in elucidating the molecular architecture of podocyte foot processes and their interdigitating slit diaphragm, the underlying mechanisms of podocyte injury and loss remain unclear. No cell-specific therapy is currently clinically available, and validated therapeutic targets are scarce (2, 3). Tools available to characterize putative targets have grown exponentially, including immortalized murine and human podocyte cell lines and several genetic experimental models. Variability in the endogenous expression level of target molecules and interacting partners across different cell lines makes cross-species validation of findings essential. This is most notably seen in the disparate expression levels of nephrin, synaptopodin, and Cd2ap between mouse and human podocyte lines with resulting differences in actin cytoskeleton dynamics and response to noxious stimuli (4–7). Similarly, rodent strain-dependent glomerular disease susceptibility mandates careful consideration in the choice of in vivo podocyte injury. Protamine sulfate perfusion is a useful modality for inducing acute podocyte injury in otherwise resistant C57/BL6 mice (8, 9). Disruption of the charge barrier with intracellular calcium influx results in quantifiable foot process effacement by electron microscopy (9). Depending on the target investigated, effacement can be rapidly reversed with heparin (10). These tools are being increasingly applied to the quest for mediators of podocyte injury and survival.
KIBRA (kidney brain protein), encoded by the Wwc1 gene, is an upstream regulator of the Hippo signaling pathway, a conserved kinase cascade from Drosophila to mammals that regulates organ size via growth inhibition and promotion of apoptosis (11, 12). KIBRA is a 125-kDa cytoplasmic protein containing two amino-terminal WW domains, an internal motif similar to the C2 domain of the Ca2+-sensing protein synaptotagmin, and a carboxyl-terminal glutamic acid stretch (13, 14). It was originally characterized by yeast two-hybrid screening as a putative binding partner for the dual compartment pro-injury molecule dendrin (11, 13, 15). Acting upstream of the core kinases LATS and MST, KIBRA promotes the phosphorylation, cytoplasmic sequestration, and inactivation of the Hippo target Yes-associated protein (YAP) (16). YAP functions as a transcriptional co-activator via preferential interactions with the TEAD (transcriptional enhancer–associated domain) family of transcription factors to drive expression of target genes essential for the processes of cell growth, differentiation, and survival (12, 16). Studies in MCF10A human mammary epithelial cells and Drosophila revealed that KIBRA overexpression drives phosphorylation of LATS and of YAP and Yorkie (the homologue of mammalian YAP in Drosophila), resulting in growth inhibition, whereas KIBRA silencing results in increased cell proliferation, migration, and survival (11, 17).
The Hippo pathway has been extensively studied in the oncology field, where the role of YAP as a potent oncogene has made it an attractive target for chemotherapeutic drug development (18–20). We previously demonstrated that YAP promotes podocyte survival by inhibiting dendrin pro-apoptotic signaling (21). We also showed that podocyte-specific deletion of Yap causes FSGS and progressive renal failure (22). YAP was more recently found to promote renal fibrosis in a murine unilateral ureteral obstruction model (23). Upstream of YAP, KIBRA expression was first documented in podocytes by Duning et al. (24), where KIBRA regulated cell polarity via interactions with the actin-bundling protein synaptopodin and the cell polarity protein PATJ. The relevance of KIBRA to human kidney disease and experimental models of podocyte injury remain unclear. Here, we determined expression levels of KIBRA and phosphorylated YAP (P-YAP) relative to total YAP protein in FSGS, a significant human podocytopathy, and explored the mechanistic role of KIBRA and YAP in regulating podocyte biology and morphology. We also evaluated P-YAP/YAP expression in constitutive KIBRA/WWc1 knockout mice and defined the functional consequences of reduced KIBRA expression in protamine sulfate-induced podocyte injury both in vivo and in vitro.
Results
KIBRA and P-YAP expression are increased in human FSGS
FSGS, a human podocytopathy, is increasing in prevalence worldwide for unclear reasons and is the most common primary glomerular disease leading to end-stage kidney disease in the United States (25–27). We previously showed that the expression of YAP was decreased in human FSGS and that podocyte-specific deletion of YAP induced development of FSGS in mice (22). To determine the clinical relevance of KIBRA/Wwc1 expression, we first determined its expression profile in human FSGS. Immunohistochemistry stainings of biopsy cases from the Mount Sinai Glomerular Disease Biorepository revealed that at baseline, KIBRA expression was low in the glomerular tuft in patients with normal glomeruli (Fig. 1, A and B). In contrast, significantly increased KIBRA protein expression was seen in the glomeruli of primary FSGS cases. Given that KIBRA was recently shown to increase P-YAP expression in human podocytes in vitro (28), we quantified P-YAP expression in FSGS glomeruli and found it to be significantly higher (p < 0.005) compared with normal glomeruli (Fig. 1, A and B). YAP expression was lower in FSGS, but the difference did not reach statistical significance (p = 0.183). Importantly, the ratio of P-YAP/YAP was significantly increased in FSGS tissue (p < 0.005) (Fig. 1B), suggesting enhanced YAP inactivation (29, 30). Immunofluorescence staining of normal and FSGS biopsies further demonstrated that in the unscarred segments of FSGS glomeruli, there was increased KIBRA expression, with KIBRA colocalization with the podocyte marker synaptopodin (Fig. 1C). Consistent with published data, synaptopodin expression in these unscarred glomerular segments was decreased compared with normal glomeruli (31). These data suggest an association between increased KIBRA glomerular expression and human podocytopathy that may be mediated by increases in YAP phosphorylation.
Figure 1.

Expression of KIBRA, P-YAP, and YAP in human glomerular disease. A and B, immunohistochemistry stainings of serial tissue sections from renal biopsies demonstrate a significantly higher number of KIBRA-positive and P-YAP–positive cells per glomerulus in FSGS cases than in normal kidney tissue, but no significant difference in YAP. The ratio of P-YAP to YAP was significantly increased in FSGS cases. C, representative immunofluorescence images of renal biopsy tissue show colocalization of KIBRA (green) with the podocyte-specific marker synaptopodin (Synpo, red) in FSGS. *, p < 0.05; **, p < 0.005. Scale bars, 50 μm.
KIBRA promotes phosphorylation of Hippo pathway members YAP and LATS in podocytes
Having found increased KIBRA and P-YAP expression in human glomerular disease, we sought to further explore the role of KIBRA in podocytes in the context of regulation of Hippo/YAP function. We established a stable line of murine KIBRA/Wwc1 overexpression (OE) podocytes using retrovirus vector (Fig. 2A). Clonal selection was not performed, allowing for use of a heterogeneous pool of KIBRA-overexpressing cells. Consistent with previously published data (11, 16, 17), we found that KIBRA/Wwc1 overexpression promoted phosphorylation of YAP at Ser-127 (P-YAP), leading to a significantly higher ratio of P-YAP to YAP (Fig. 2A). Similarly, the ratio of phosphorylated LATS (P-LATS) to total LATS was significantly greater in KIBRA/Wwc1 OE podocytes (Fig. 2B). Given that the phosphorylated form of LATS kinase is active whereas the phosphorylated form of YAP is inactive (29, 32), these results suggest that KIBRA/Wwc1 overexpression can induce LATS-mediated YAP inactivation in murine podocytes.
Figure 2.
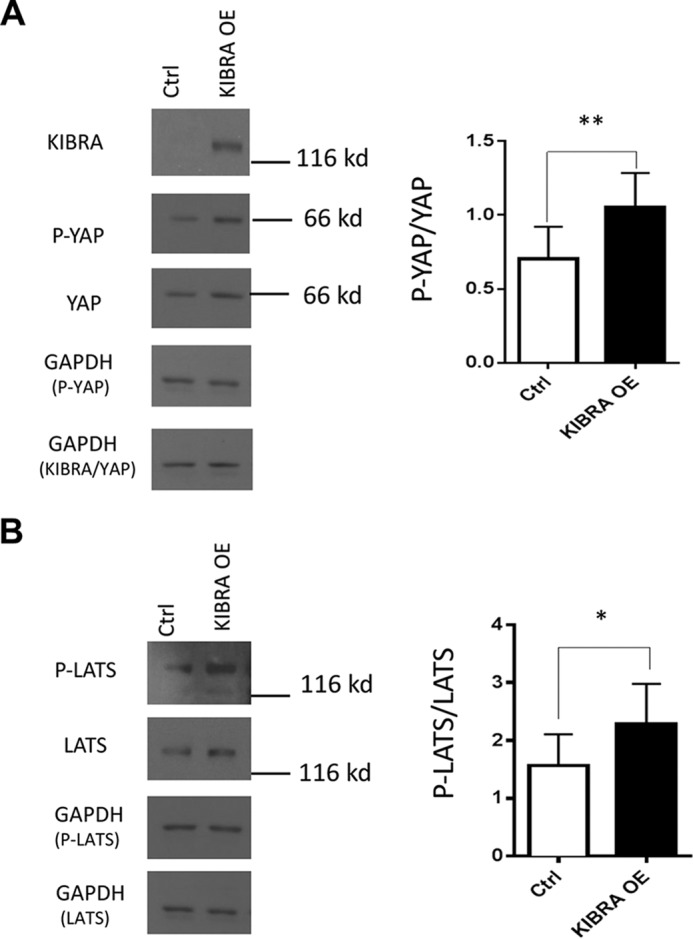
Effects of KIBRA overexpression on Hippo signaling pathway members YAP and LATS. A, representative Western blot showing ectopic overexpression of KIBRA in mouse podocytes. There was significantly greater expression of P-YAP (**) relative to total YAP expression in KIBRA OE podocytes. B, similarly, KIBRA overexpression promoted phosphorylation of LATS with increase in P-LATS/LATS versus control podocytes (*). Expression levels of P-YAP, total YAP, P-LATS, and LATS were normalized to GAPDH expression in all lysate samples. *, p < 0.05; **, p < 0.005. Error bars, S.E.
KIBRA reduces expression of YAP-associated genes
Because phosphorylation of YAP leads to its inactivation, we next determined whether KIBRA overexpression decreased the expression of YAP target genes. Quantitative PCR studies revealed significantly reduced transcription of YAP target, pro-growth gene Ki-67, but there were no significant differences in expression levels of Sox9, Birc5 (baculoviral inhibitor of apoptosis repeat-containing 5, also known as survivin), or Ctgf (connective tissue growth factor) (Fig. 3). Interestingly, KIBRA overexpression also led to significant decreases in the mRNA levels of all four TEAD transcription factors, which are YAP binding partners and co-activators, but not traditionally considered YAP targets. Yap gene expression itself was not significantly reduced in KIBRA OE podocytes, confirming that KIBRA-mediated antagonism of YAP does not primarily occur at the transcription level. Interestingly, gene expression of synaptopodin, an actin-bundling protein in podocytes (10) and a known binding partner of KIBRA (24), was significantly reduced with KIBRA overexpression (see Fig. 6). These results represent two potential pathways by which KIBRA promotes podocyte injury: 1) down-regulation of pro-growth gene expression and 2) disruption of the actin cytoskeleton that is essential to podocyte structure and function.
Figure 3.

Consequences of KIBRA overexpression on YAP-associated gene transcription. Quantitative PCR data reveal significantly decreased mRNA levels for the transcription factors TEAD 1–4 and YAP target Ki-67 in KIBRA OE podocytes versus controls. There were no significant differences between groups in the gene expression levels of YAP itself and other YAP targets. Transcription of synaptopodin was also significantly reduced in the setting of KIBRA overexpression (TEAD, transcriptional enhancer–associated domain; CTGF, connective tissue growth factor; Birc5, baculoviral inhibitor of apoptosis repeat-containing 5, also known as survivin; Synpo, synaptopodin) (*, p < 0.05; **, p < 0.005; N.S., not significant). Error bars, S.E.
Figure 6.
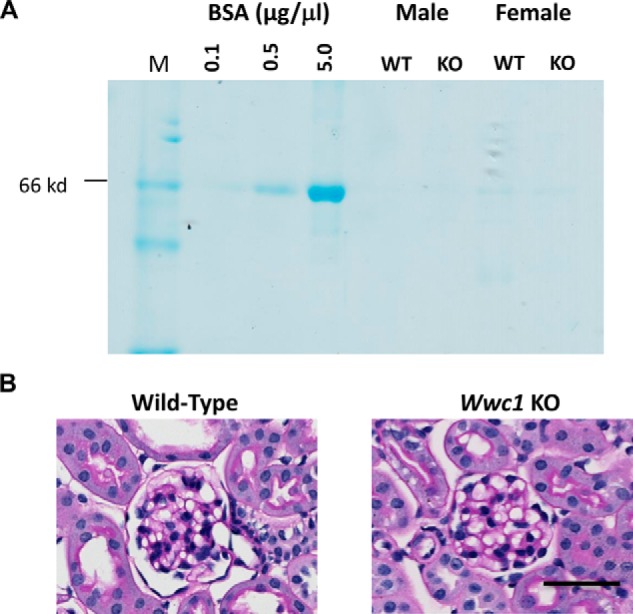
Assessment of baseline proteinuria and glomerular histology of constitutive KIBRA/Wwc1- KO mice. A, Coomassie staining of urine from two 9-week-old male and female pairs of KIBRA WT and KO mice reveals absence of proteinuria in either genotype at baseline. B, PAS staining reveals histologically normal glomeruli in both KIBRA WT and KO mice. Scale bar, 50 μm.
KIBRA-induced podocyte morphological and cytoskeletal alterations can be reversed by YAP overexpression
YAP is a known master regulator of biomechanical homeostasis, and its activity is highly interrelated to the actin cytoskeleton (33). Because KIBRA overexpression resulted in the decreased expression of YAP target genes but not YAP gene expression, we sought to explore whether YAP overexpression could antagonize the effects of KIBRA overexpression in podocytes. We overexpressed in podocytes KIBRA (KIBRA OE) alone or in combination with YAP (KIBRA-YAP OE). Overexpression was confirmed by Western blotting (Fig. 4A). These same cells were immunostained with phalloidin for F-actin and with anti-paxillin antibodies for focal adhesions to study the podocyte cytoskeleton (Fig. 4B). Actin stress fibers were more disorganized in the KIBRA OE podocytes than control, which was rescued by overexpressing YAP. The number and size of focal adhesions also decreased with KIBRA OE, which was similarly rescued by YAP OE. KIBRA OE promoted YAP cytoplasmic localization, whereas nuclear YAP was restored in KIBRA-YAP OE podocytes.
Figure 4.

Effects of KIBRA and YAP overexpression on podocyte cytoskeleton and YAP localization. A, representative Western blots showing ectopic overexpression of KIBRA alone and co-overexpression of KIBRA and YAP in murine podocytes. B, representative immunofluorescence images showing actin (phalloidin), focal adhesions (paxillin), YAP, and nuclei (Hoechst 33342) for control, KIBRA OE, and KIBRA/YAP OE podocytes. Insets within the paxillin images show zoomed-in morphological characteristics of representative focal adhesion sites. Scale bar, 100 μm.
We used high-content imaging to characterize the cell biological and morphological effects of KIBRA and concurrent KIBRA-YAP OE in podocytes. KIBRA OE cells displayed significantly smaller spreading area and a smaller number of projections as quantified by area/perimeter ratio. Co-expression of YAP with KIBRA rescued the cell size and cell area/perimeter ratio observed in control cells (Fig. 5A). KIBRA OE cells showed lower levels of contractile stress fibers, as measured by F-actin intensity per cell, which increased upon YAP co-overexpression in comparison with controls (Fig. 5B). KIBRA OE cells demonstrated significantly smaller nuclear size with a significantly elongated morphology compared with control cells (Fig. 5C), which mirrored the changes in cell size and actin cytoskeleton induced by KIBRA overexpression. YAP and KIBRA co-overexpression, however, returned the nuclear size and aspect ratio to levels similar to those of control cells. KIBRA OE cells also exhibited lower nuclear localization of YAP, whereas nuclear YAP localization was restored to the level of controls in KIBRA-YAP OE podocytes (Fig. 5D). Additionally, KIBRA OE podocytes had significantly fewer and smaller focal islands, which were reversed by YAP co-overexpression (Fig. 5E). These findings suggest that KIBRA may disrupt normal podocyte morphology, cell adhesion, and cytoskeletal integrity through antagonism of YAP, as YAP overexpression was able to reverse all KIBRA-mediated alterations.
Figure 5.

Quantitative analysis of KIBRA overexpression and YAP-KIBRA overexpression on podocyte morphology and cellular physiology. Shown are scatter plots of individual cell morphometrics of differentiated podocytes (Control), podocytes overexpressing KIBRA (KIBRA), and podocytes co-overexpressing KIBRA and YAP (KIBRA/YAP). A, KIBRA OE cells display a significantly smaller spreading area and a smaller number of projections as quantified by area/perimeter ratio. Co-expression of YAP with KIBRA rescues the cell size and cell area/perimeter ratio observed in control cells. B, KIBRA OE cells show lower levels of contractile stress fibers as measured by F-actin intensity per cell, which increases upon YAP co-overexpression. C, changes in nuclear morphology mirror the changes in cell size and actin cytoskeleton induced by KIBRA overexpression. KIBRA OE cells show significantly smaller nuclear size with a significantly elongated morphology compared with control cells. YAP and KIBRA co-overexpression returns nuclear size and aspect ratio to similar levels as control cells. KIBRA OE also exhibited lower nuclear localization of YAP (D) and significantly fewer and smaller focal islands (E). ****, p < 0.0001; ***, p < 0.001; **, p < 0.01; n.s., not significant; two-tailed Mann–Whitney test. Error bars, S.E.
Constitutive KIBRA KO mice have reduced P-YAP and increased YAP expression
Having demonstrated that P-YAP expression was increased relative to YAP in FSGS and that KIBRA overexpression directly induced greater P-YAP/YAP expression in vitro, we hypothesized that YAP phosphorylation would be reduced in the setting of KIBRA deletion. Constitutive Wwc1-KO mice (C57/Bl6) were obtained from Dr. Richard Huganir (Johns Hopkins University, Baltimore, MD). These mice have decreased fear response and memory but otherwise lack anatomical or functional abnormalities (34). We first confirmed that KIBRA/Wwc1-KO mice lacked a glomerular phenotype. Coomassie staining of urine samples from two 9-week-old male and female pairs of KIBRA/Wwc1-KO mice and WT littermates revealed no proteinuria in either genotype (Fig. 6A). PAS staining of kidney cortex samples showed histologically normal podocytes and glomeruli in both KIBRA/Wwc1-KO and WT mice (Fig. 6B).
We next performed immunohistochemistry staining of kidney sections from KIBRA/Wwc1-KO and WT mice following paraformaldehyde (PFA) perfusion. We found that consistent with our findings in human glomeruli (Fig. 1), KIBRA expression was relatively low in WT mice at baseline (Fig. 7A). P-YAP expression was significantly reduced (Fig. 7, A and B), and YAP expression was increased (Fig. 7, A and B) in KIBRA/Wwc1-KO versus WT littermates. This led to a significantly lower ratio of P-YAP to YAP (Fig. 7B).
Figure 7.

Expression of P-YAP and YAP in KIBRA/Wwc1-KO mice versus WT controls. A, despite low baseline expression of KIBRA WT mice, constitutive KIBRA/IWwc1-KO mice have significantly reduced P-YAP expression (B) and significantly increased YAP expression by comparison. The ratio of P-YAP to YAP is significantly reduced in KIBRA/IWwc1-KO versus WT mice. Scale bar, 20 μm (*, p < 0.05; **, p < 0.005). Error bars, S.E.
KIBRA deletion is protective in a model of acute podocyte foot process effacement
The findings of increased KIBRA glomerular expression in FSGS and disturbances of the actin cytoskeleton in KIBRA OE podocytes led us to determine whether KIBRA deletion would be protective against podocyte injury in vivo. We tested whether KIBRA/Wwc1-KO mice were protected against acute podocyte foot process effacement after protamine sulfate (PS) perfusion (10, 35). Scanning electron microscopy (SEM) after PS perfusion showed FP disruption in WT but not KIBRA/Wwc1-KO mice (Fig. 8A). At baseline, transmission electron microscopy (TEM) on kidney sections from WT and KO mice perfused with Hanks' balanced salt solution (HBSS) showed intact foot processes, without signs of foot process disruption in either group (Fig. 8B, left panels). However, following PS perfusion, WT mice had increased foot process effacement compared with KIBRA/Wwc1-KO littermates (Fig. 8B, right panels). Quantification of this observation confirmed that after PS perfusion, Wwc1-KO mice did not have a significant decrease in the number of FPs per glomerular basement membrane (GBM) length. Conversely, WT littermates had a marked reduction in FPs per μm GBM after PS perfusion (p < 0.005) (Fig. 8B). To visualize our in vivo findings at the cellular level, we next examined podocyte injury in human podocytes that have higher baseline expression levels of KIBRA than murine podocytes. Using an shRNA lentiviral approach to generate stably silenced cells (Fig. 9A), we found that KIBRA knockdown podocytes had more preservation of F-actin expression after protamine treatment when compared with control cells (Fig. 9B). Taken together, these data show that KIBRA silencing protects podocytes from actin cytoskeletal injury using both in vivo and in vitro models.
Figure 8.

KIBRA KO mice are protected from acute podocyte injury. A, SEM reveals that PS disrupts the three-dimensional structure and interdigitation of podocyte foot processes in WT glomeruli, whereas the foot processes of KO glomeruli are preserved. B, TEM shows normal appearing foot processes lining the glomerular basement membrane in both WT and KO mice following HBSS perfusion. However, representative images taken following PS perfusion demonstrate greater foot process effacement in WT glomeruli compared with KO glomeruli. Quantification reveals significant foot process effacement in WT mice after PS perfusion versus HBSS-perfused WT mice, whereas PS-perfused KO mice do not show significant reduction in foot process number versus HBSS-perfused KO mice (**, p < 0.005; N.S., not significant). Scale bars, 2 μm (A); 0.5 μm (B). Error bars, S.E.
Figure 9.
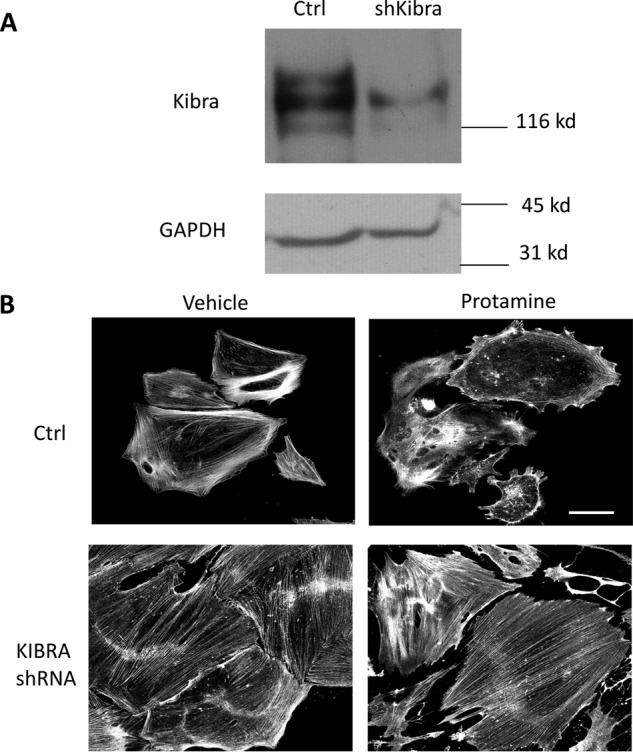
KIBRA knockdown podocytes are protected from protamine-induced injury. A, representative Western blot showing effective KIBRA knockdown in human podocytes via shRNA. B, after treatment with protamine sulfate, phalloidin staining reveals that KIBRA knockdown podocytes have preservation of actin stress fibers, whereas control podocytes have significant loss of actin fibers as compared with vehicle-treated cells. Scale bar, 100 μm.
Discussion
In this study, we have demonstrated that increased KIBRA expression in the podocyte is associated with human FSGS. In vitro, KIBRA promoted LATS phosphorylation resulting in YAP phosphorylation, cytoplasmic sequestration, and inactivation, characterized by decreased target gene expression. The finding that P-YAP expression was increased relative to total YAP expression in FSGS glomeruli supports these in vitro results and suggests that KIBRA may mediate podocyte injury and disease via inhibition of YAP signaling. KIBRA overexpression promoted deleterious morphological changes in podocytes, mainly through disorganization of the actin cytoskeleton and reduction in focal adhesion stability and size, all of which were reversed by subsequent YAP overexpression. These findings further underscore the closely linked yet opposing functions of KIBRA and YAP in podocyte biomechanical homeostasis. These findings are consistent with recent observations in Ewing sarcoma, where YAP/TEAD signaling also increases actin stress fiber and focal adhesion expression (36).
The role of KIBRA silencing was protective against acute protamine-induced podocyte injury both in vivo and in vitro. Our findings are consistent with reports of KIBRA pro-injury signaling in other model systems and are the first demonstration of an in vivo role for KIBRA in glomerular disease. In podocytes, the function of KIBRA appears similar to that of its binding partner dendrin, which we have extensively studied (37). Both KIBRA and dendrin null mice have no glomerular phenotype, and both promote podocyte injury. They both interact with YAP in podocytes, where KIBRA inhibits YAP signaling and YAP in turn inhibits dendrin. The functional interaction between KIBRA and dendrin under disease conditions remains unclear.
KIBRA expression in podocytes was first described in 2008 in the context of polarity signaling (24). In that study, KIBRA silencing in human podocytes impaired directed migration. KIBRA was also shown to be a binding partner for the actin-bundling protein synaptopodin. Downstream of KIBRA in the Hippo signaling pathway, YAP normally functions through binding to co-activators with subsequent activation of target genes upon its nuclear translocation in a dephosphorylated form. Despite increased YAP phosphorylation and cytoplasmic expression with KIBRA overexpression, we did not detect reduced expression of canonical YAP target genes that mediate cell survival, such as Ctgf, Sox9, and Birc5. Interestingly, we did however detect significant reduction in expression of the YAP target gene Ki-67. Outside its use as a proliferation marker, the relevance of this molecule to podocyte survival or actin cytoskeletal dynamics to our knowledge has not been explored. This is an area for further study, given the role of cell cycle dysregulation and the differential expression of cyclin-dependent kinases in podocyte apoptotic pathways (38–41). The Tead genes are not considered YAP target genes but rather YAP binding partners in enhancing pro-survival gene transcription (42–44). Indeed, interruption of YAP/TEAD interaction is the basis for potential therapeutic intervention in solid malignancies associated with increased YAP activation (18, 42). Interestingly, gene expression of Tead 1–4 and synaptopodin were also reduced with KIBRA overexpression, although it is unclear whether this occurred via YAP- dependent or -independent mechanisms. Intriguingly, Tead2 silencing in mammary epithelial cells has been shown to promote the cytoplasmic localization of YAP (45). It is possible that KIBRA antagonism of YAP is mediated via reduction in TEAD, in addition to being driven by phosphorylation of YAP via LATS kinase. Furthermore, TEAD2 also directly binds to zyxin, an actin-regulatory protein (45). Thus, reduced expression of synaptopodin and TEAD may contribute to KIBRA-induced alterations of normal podocyte morphology and increase the risk of foot process effacement and detachment from the glomerular basement membrane in vivo. Reduction of KIBRA was conversely demonstrated to have an actin-stabilizing effect against protamine treatment in vitro, which accounted for protection in KIBRA/Wwc1 mice against the deleterious effects of protamine perfusion. The importance of KIBRA to podocyte structural stability is underscored by our findings of increased KIBRA expression in FSGS, a disorder that is heterogeneous yet uniformly characterized by marked podocyte foot process effacement. The finding that YAP could rescue KIBRA OE podocytes from actin cytoskeletal disruption suggests that KIBRA-mediated injury is dependent on inhibition of YAP function through its cytoplasmic sequestration. Future studies will be needed to determine the potential therapeutic benefit of YAP agonists in KIBRA-mediated podocyte injury.
In summary, our findings highlight the role of KIBRA in promoting podocyte injury through inhibition of YAP signaling and disruption of normal actin cytoskeletal dynamics. Further investigation will be required to identify the factors that enhance KIBRA expression under disease conditions. The interaction of KIBRA with its pro-injury binding partner dendrin in progressive proteinuric kidney disease also remains to be determined. These essential studies would be necessary pretexts before determining whether inhibition of KIBRA signaling could in the future be part of a targeted therapeutic strategy to treat certain glomerular disorders. Although our focus in this study is on the glomerular podocyte, it is important to keep in mind that normal homeostasis of the glomerular capillary is maintained by the integrated functions of all of its layers, including the endothelium and basement membrane, in addition to the podocyte (46). Given that the main phenotype of KIBRA constitutive KO mice was memory impairment and that no other systemic effects were observed, it is possible that the design of a KIBRA inhibitor drug without the ability to cross the blood–brain barrier in the future could offer potential treatment for glomerular disease and spare patients adverse neurological side effects.
Experimental procedures
Antibodies
Antibodies used in this study were as follows: synaptopodin (G1; kind gift from Dr. Peter Mundel, Massachusetts General Hospital, Charlestown, MA), KIBRA (rabbit anti-KIBRA Novus Biologicals; IHC), KIBRA (rabbit anti-KIBRA, Cell Signaling; Western blotting), YAP (rabbit anti-YAP, Cell Signaling; Western blotting), P-YAP-Ser-127 (rabbit anti-P-YAP-Ser-127, Cell Signaling; Western blotting) P-YAP-Ser-179, D9W2I (rabbit anti-YAP, Cell Signaling; IHC), YAP (rabbit anti-YAP1, Novus Biologicals; IHC), LATS (rabbit anti-LATS, Cell Signaling), P-LATS-Thr-1079 (rabbit anti-P-LATS-Thr-1079, Cell Signaling), Alexa Fluor 488 goat anti-rabbit IgG (Life Technologies), and Alexa Fluor 594 goat anti-mouse IgG (Life Technologies).
KIBRA overexpression and silencing
Overexpression
pBabe-puro and pBabe-puro-Kibra were purchased from Addgene. The pBabe-puro plasmids along with the helper plasmids pUMVC and VSVG were transfected into HEK293 cells at 70% confluence using FuGENE 6 (Promega) to generate viral supernatant.
Silencing
pLKO.1 lentiviral shRNA plasmids were purchased from Addgene for scramble control and from Sigma-Aldrich for KIBRA. We used 5′-CCGGCCTTCACCAGAAGACCTTAAGCTCGAGCTTAAGGTCTTCTGGTGAAGGTTTTTG-3′ (hk59) sequence and 5′-CCGGTTGTCACAGTACAGCTAATTTCTCGAGAAATTAGCTGTACTGTGACAATTTTTG-3′ (hk60) to target KIBRA. The pLKO.1 plasmids (KIBRA shRNA or control shRNA) along with the helper plasmids psPAX2 and pMD2.G were transfected into HEK293 cells at 70% confluence using FuGENE 6 (Promega) to generate viral supernatant.
Podocyte infection
Wild-type undifferentiated podocytes were infected for 24 h and then selected. Noninfected podocytes cells were removed by selection in 2 μg/ml puromycin (Sigma). Podocytes were selected for 1 week, after which 1 μg/ml puromycin was used as the maintenance dose.
Three sets of cell lysate from KIBRA OE and control podocytes were analyzed via Western blot for P-YAP, YAP, P-LATS, and LATS expression and quantified with ImageJ.
RNA extraction and quantitative PCR
RNA was extracted from podocytes using the RNeasy purification kit (Qiagen) according to the manufacturer's protocol. 0.5–1 μg of total RNA was used to generate cDNA using the SuperScript III first-strand synthesis kit (Life Technologies) according to the manufacturer's protocol. 1 μl of cDNA was amplified in triplicate using Power SYBR Green quantitative PCR master mix on an ABI 7500 real-time PCR system (Applied Biosystems). Light cycler analysis software was used to determine crossing points using the second derivative method. Data were normalized to housekeeping genes (GAPDH) and presented as -fold increase compared with RNA isolated from the control group using the 2−ΔΔCT method.
Proteinuria
Albuminuria was screened using a 10% SDS-polyacrylamide gel followed by Coomassie Blue staining. 1 μl of bovine serum albumin standards of 0.1, 0.5, and 5 μg/μl was used, and 5 μl of urine was used from each sample to qualitatively assess the presence of albuminuria in KIBRA KO and WT mice.
Imaging and morphometric analyses
Morphometric analyses were carried out as described previously (47). Briefly, cells were fixed with 4% paraformaldehyde in PBS; permeabilized with 0.5% Triton X-100; blocked with 4% bovine serum albumin; and stained for paxillin (mouse anti-paxillin, Abcam), YAP (rabbit anti-YAP, Cell Signaling), and F-actin (phalloidin, Life Technologies) for characterization of focal adhesion metrics, YAP localization, and the actin cytoskeleton, respectively. Cells were imaged on a Zeiss LSM 880 superresolution confocal microscope with Airyscan at ×400 magnification. Actin cytoskeletal integrity in control and KIBRA OE podocytes was scored by a blinded expert using phalloidin images for two independent experiments. Multichannel images consisting of DAPI, phalloidin, paxillin, and YAP channels were aligned and stitched using the ZEN 2014 software. Image analysis and quantification was performed using Broad Institute's CellProfiler suite (48). Briefly, nuclei were identified as 10–60-pixel objects in the DAPI channel. The corresponding cytosol and whole-cell objects were outlined using the innate propagation algorithm, by identifying the nuclei and utilizing the contrast-enhanced phalloidin channel to define cell boundaries. Once the nuclei, cytosol, and cell boundary objects were identified, mean intensity, shape, and size metrics were quantified for each segmented object in the appropriate channel. Nuclear YAP/DAPI ratio for a given cell was defined as the integrated YAP intensity divided by the integrated DAPI intensity within the nuclear object. All measurements were exported directly to CSV files and were subsequently analyzed using MATLAB to generate histograms.
Human renal biopsies
Paraffin-embedded human renal biopsy samples of three patients with FSGS were obtained from the Department of Pathology at the Icahn School of Medicine at Mount Sinai. Additionally, three nephrectomy cases were obtained as normal controls. The study of human renal tissues was performed according to the Icahn School of Medicine's institutional review board protocols entitled “Pathogenesis of Glomerular Diseases” and “Glomerular Diseases Biorepository.” Informed consent was obtained from all patients enrolled in the latter prospective study.
Mice perfusion, kidney histology, and electron microscopic imaging of kidney tissues
To harvest the kidneys, mice were anesthetized first with isoflurane and then injected with ketamine/xylazine. For assessment of baseline glomerular histology, two 9-week-old male and female KO and WT pairs underwent pericardial perfusion with 50 ml of a filtered 3% PFA solution in PBS at room temperature. For quantification of P-YAP and YAP expression, two female WT, one male WT, two female KO, and one male KO (all age 8 weeks) underwent pericardial perfusion with 50 ml of a filtered 3% PFA solution in PBS at room temperature For PS experiments, 13-week-old WT (n = 5) and KO (n = 5) mice were either perfused with HBSS for 15 min followed by 50 ml of 3% PFA for kidney fixation or with 2 mg/ml PS in HBSS for 15 min followed by 50 ml of 3% PFA. Kidneys were harvested following perfusion, sectioned transversely into thirds, and stored in solutions of 2.5% glutaraldehyde (for EM) or 3% PFA (for PAS and IHC staining). Slides from paraffin-embedded kidney sections were used for IHC staining. SEM was performed using a JEOL 1011 electron microscope, and TEM was performed using a Hitachi H7000 transmission electron microscope.
Immunohistochemistry staining
Immunohistochemistry staining was performed with Discovery ULTRA autostainer from Roche Diagnostics. The procedure platform of RUO Discovery Multimer was used as a staining module. 2% BSA in 1× PBS was used as a blocking reagent and incubated in 37 °C for 32 min. The targeted signals were detected using anti-Kibra (NBP1–92053) rabbit polyclonal IgG from Novus Biologicals Biotechnology, anti-P-YAP (Ser-127, D9W2I-13008) rabbit monoclonal IgG in the dilution, and anti-YAP (NB110-58358) rabbit polyclonal IgG in the dilution. For human tissue samples, the following primary antibody dilutions were used: anti-KIBRA (1:50), anti-P-YAP (1:200), and anti-YAP (1:400). For mouse tissue samples, the following primary antibody dilutions were used: anti-KIBRA (1:400), anti-P-YAP (1:200), and anti-YAP (1:400). The primary antibodies were incubated in 37 °C for 60 min. Discovery OmniMap anti-rabbit HRP (RUO) was used as secondary antibody and incubated for 32 min. The Biotin-free Discovery ChromoMap DAB kit (RUO) from Roche Diagnostics was used as a detection system. Hematoxylin was used as the counterstain for cell nuclei. Bright-field microscopy was performed on stained whole slides using a Zeiss Axioimager wide-field microscope and Zeiss Zen Blue software was used to acquire and process images. KIBRA-positive, P-YAP–positive, and YAP-positive dots and total cell number were counted by a blinded observer. Approximately 10–15 glomeruli were quantified for each human and mouse kidney analyzed.
Foot process quantification
The degree of FP effacement was assessed in a series of images taken at ×3,000 magnification by a blinded expert. 5 images per glomerulus and 3 glomeruli per mouse × 4–5 mice/group yielded a total of 240–300 images for quantification. These images were analyzed in a blinded fashion by two independent observers using ImageJ software, where the length of the GBM on each image and the number of FPs per μm GBM were quantified.
Statistics
For imaging and morphometric analysis, normality was determined using the Shapiro–Wilk test using p ≥ 0.05. If the distribution was normal, a two-tailed t test was performed with a 95% confidence interval. For data sets with non-normal distribution, the Wilcoxon–Mann–Whitney rank sum test with a non-parametric 95% confidence interval was used. All statistical analyses were performed using MATLAB. All other results are presented as mean ± S.E. Two-tailed Student's t test using Prism software was used for comparisons between groups where p < 0.05 was considered statistically significant.
Study approval
All mouse studies were approved by the institutional animal care and use committee at the Icahn School of Medicine at Mount Sinai. Frozen sections from human biopsy material were obtained through the Icahn School of Medicine at Mount Sinai under two institutional review board–approved protocols entitled “Pathogenesis of Glomerular Disease” and “Glomerular Disease Biorepository.” All biopsies were clinically indicated, and only extra tissue not required for diagnostic purposes was used for research purposes.
Author contributions
K. M., J. S. W, J. R., R. C. C., F. E. S., S. T., B. C., R. E. G., E. U. A., and K. N. C. performed the experiments, data acquisition, and analysis. K. M., J. S. W., L. K., E. U. A., J. C. H., and K. N. C. conceptualized and designed the studies. All authors participated in the drafting and critical appraisal of the manuscript. They all approve of the final version and agree to be accountable for all aspects of the work.
Acknowledgments
Confocal imaging was performed at the Institute for Systems Biomedicine in the Icahn School of Medicine at Mount Sinai. We acknowledge Kevin D. Costa for allowing the use of his live-cell microscopy system and Mufeng Hu and Vivian Au for technical assistance with image processing. KIBRA KO mice were provided by Dr. Richard Huganir (Howard Hughes Medical Institute, Johns Hopkins University). We acknowledge the Biorepository and Pathology Core and Microscopy Core at the Icahn School of Medicine at Mount Sinai for assistance with immunohistochemistry and image acquisition.
This work was supported by National Institutes of Health Grant R01 DK103022 (to K. N. C.) and an American Society of Nephrology Nephcure Kidney International–ASN Foundation for Kidney Research Career Development Award (to E. U. A.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- FSGS
- focal segmental glomerulosclerosis
- P-YAP
- phosphorylated YAP
- P-LATS
- phosphorylated LATS
- OE
- overexpression
- PAS
- periodic acid Schiff
- PS
- protamine sulfate
- SEM
- scanning electron microscopy
- TEM
- transmission electron microscopy
- HBSS
- Hanks' balanced salt solution
- FP
- foot process
- GBM
- glomerular basement membrane
- IHC
- immunohistochemistry
- PFA
- paraformaldehyde.
References
- 1. Greka A., and Mundel P. (2012) Cell biology and pathology of podocytes. Annu. Rev. Physiol. 74, 299–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Reiser J., Gupta V., and Kistler A. D. (2010) Toward the development of podocyte-specific drugs. Kidney Int. 77, 662–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Leeuwis J. W., Nguyen T. Q., Dendooven A., Kok R. J., and Goldschmeding R. (2010) Targeting podocyte-associated diseases. Adv. Drug Deliv. Rev. 62, 1325–1336 [DOI] [PubMed] [Google Scholar]
- 4. Saleem M. A., Zavadil J., Bailly M., McGee K., Witherden I. R., Pavenstadt H., Hsu H., Sanday J., Satchell S. C., Lennon R., Ni L., Bottinger E. P., Mundel P., and Mathieson P. W. (2008) The molecular and functional phenotype of glomerular podocytes reveals key features of contractile smooth muscle cells. Am. J. Physiol. Renal Physiol. 295, F959–F970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Saleem M. A., Ni L., Witherden I., Tryggvason K., Ruotsalainen V., Mundel P., and Mathieson P. W. (2002) Co-localization of nephrin, podocin, and the actin cytoskeleton: evidence for a role in podocyte foot process formation. Am. J. Pathol. 161, 1459–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Saleem M. A., O'Hare M. J., Reiser J., Coward R. J., Inward C. D., Farren T., Xing C. Y., Ni L., Mathieson P. W., and Mundel P. (2002) A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J. Am. Soc. Nephrol. 13, 630–638 [DOI] [PubMed] [Google Scholar]
- 7. Schiwek D., Endlich N., Holzman L., Holthöfer H., Kriz W., and Endlich K. (2004) Stable expression of nephrin and localization to cell-cell contacts in novel murine podocyte cell lines. Kidney Int. 66, 91–101 [DOI] [PubMed] [Google Scholar]
- 8. George B., Fan Q., Dlugos C. P., Soofi A. A., Zhang J., Verma R., Park T. J., Wong H., Curran T., Nihalani D., and Holzman L. B. (2014) Crk1/2 and CrkL form a hetero-oligomer and functionally complement each other during podocyte morphogenesis. Kidney Int. 85, 1382–1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schaldecker T., Kim S., Tarabanis C., Tian D., Hakroush S., Castonguay P., Ahn W., Wallentin H., Heid H., Hopkins C. R., Lindsley C. W., Riccio A., Buvall L., Weins A., and Greka A. (2013) Inhibition of the TRPC5 ion channel protects the kidney filter. J. Clin. Invest. 123, 5298–5309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Asanuma K., Kim K., Oh J., Giardino L., Chabanis S., Faul C., Reiser J., and Mundel P. (2005) Synaptopodin regulates the actin-bundling activity of α-actinin in an isoform-specific manner. J. Clin. Invest. 115, 1188–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yu J., Zheng Y., Dong J., Klusza S., Deng W. M., and Pan D. (2010) Kibra functions as a tumor suppressor protein that regulates Hippo signaling in conjunction with Merlin and Expanded. Dev. Cell 18, 288–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang K., Degerny C., Xu M., and Yang X. J. (2009) YAP, TAZ, and Yorkie: a conserved family of signal-responsive transcriptional coregulators in animal development and human disease. Biochem. Cell Biol. 87, 77–91 [DOI] [PubMed] [Google Scholar]
- 13. Kremerskothen J., Plaas C., Büther K., Finger I., Veltel S., Matanis T., Liedtke T., and Barnekow A. (2003) Characterization of KIBRA, a novel WW domain-containing protein. Biochem. Biophys. Res. Commun. 300, 862–867 [DOI] [PubMed] [Google Scholar]
- 14. Bork P., and Sudol M. (1994) The WW domain: a signalling site in dystrophin? Trends Biochem. Sci. 19, 531–533 [DOI] [PubMed] [Google Scholar]
- 15. Asanuma K., Campbell K. N., Kim K., Faul C., and Mundel P. (2007) Nuclear relocation of the nephrin and CD2AP-binding protein dendrin promotes apoptosis of podocytes. Proc. Natl. Acad. Sci. U.S.A. 104, 10134–10139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pan D. (2010) The hippo signaling pathway in development and cancer. Dev. Cell 19, 491–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moleirinho S., Chang N., Sims A. H., Tilston-Lünel A. M., Angus L., Steele A., Boswell V., Barnett S. C., Ormandy C., Faratian D., Gunn-Moore F. J., and Reynolds P. A. (2013) KIBRA exhibits MST-independent functional regulation of the Hippo signaling pathway in mammals. Oncogene 32, 1821–1830 [DOI] [PubMed] [Google Scholar]
- 18. Johnson R., and Halder G. (2014) The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat. Rev. Drug Discov. 13, 63–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sudol M., Shields D. C., and Farooq A. (2012) Structures of YAP protein domains reveal promising targets for development of new cancer drugs. Semin. Cell Dev. Biol. 23, 827–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stanger B. Z. (2012) Quit your YAPing: a new target for cancer therapy. Genes Dev. 26, 1263–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Campbell K. N., Wong J. S., Gupta R., Asanuma K., Sudol M., He J. C., and Mundel P. (2013) Yes-associated protein (YAP) promotes cell survival by inhibiting proapoptotic dendrin signaling. J. Biol. Chem. 288, 17057–17062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schwartzman M., Reginensi A., Wong J. S., Basgen J. M., Meliambro K., Nicholas S. B., D'Agati V., McNeill H., and Campbell K. N. (2016) Podocyte-specific deletion of Yes-associated protein causes FSGS and progressive renal failure. J. Am. Soc. Nephrol. 27, 216–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Szeto S. G., Narimatsu M., Lu M., He X., Sidiqi A. M., Tolosa M. F., Chan L., De Freitas K., Bialik J. F., Majumder S., Boo S., Hinz B., Dan Q., Advani A., John R., Wrana J. L., Kapus A., and Yuen D. A. (2016) YAP/TAZ are mechanoregulators of TGF-β-Smad signaling and renal fibrogenesis. J. Am. Soc. Nephrol. 27, 3117–3128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Duning K., Schurek E. M., Schlüter M., Bayer M., Reinhardt H. C., Schwab A., Schaefer L., Benzing T., Schermer B., Saleem M. A., Huber T. B., Bachmann S., Kremerskothen J., Weide T., and Pavenstädt H. (2008) KIBRA modulates directional migration of podocytes. J. Am. Soc. Nephrol. 19, 1891–1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kitiyakara C., Eggers P., and Kopp J. B. (2004) Twenty-one-year trend in ESRD due to focal segmental glomerulosclerosis in the United States. Am. J. Kidney Dis. 44, 815–825 [PubMed] [Google Scholar]
- 26. Polito M. G., de Moura L. A., and Kirsztajn G. M. (2010) An overview on frequency of renal biopsy diagnosis in Brazil: clinical and pathological patterns based on 9,617 native kidney biopsies. Nephrol. Dial. Transplant. 25, 490–496 [DOI] [PubMed] [Google Scholar]
- 27. Mohammadhoseiniakbari H., Rezaei N., Rezaei A., Roshan S. K., and Honarbakhsh Y. (2009) Pattern of glomerulonephritis in Iran: a preliminary study and brief review. Med. Sci. Monit. 15, PH109–PH114 [PubMed] [Google Scholar]
- 28. Wennmann D. O., Vollenbröker B., Eckart A. K., Bonse J., Erdmann F., Wolters D. A., Schenk L. K., Schulze U., Kremerskothen J., Weide T., and Pavenstädt H. (2014) The Hippo pathway is controlled by Angiotensin II signaling and its reactivation induces apoptosis in podocytes. Cell Death Dis. 5, e1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhao B., Wei X., Li W., Udan R. S., Yang Q., Kim J., Xie J., Ikenoue T., Yu J., Li L., Zheng P., Ye K., Chinnaiyan A., Halder G., Lai Z. C., and Guan K. L. (2007) Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 21, 2747–2761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hao Y., Chun A., Cheung K., Rashidi B., and Yang X. (2008) Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J. Biol. Chem. 283, 5496–5509 [DOI] [PubMed] [Google Scholar]
- 31. Wagrowska-Danilewicz M., and Danilewicz M. (2007) [Synaptopodin immunoexpression in steroid-responsive and steroid-resistant minimal change disease and focal segmental glomerulosclerosis]. Nefrologia 27, 710–715 [PubMed] [Google Scholar]
- 32. Chan E. H., Nousiainen M., Chalamalasetty R. B., Schäfer A., Nigg E. A., and Silljé H. H. (2005) The Ste20-like kinase Mst2 activates the human large tumor suppressor kinase Lats1. Oncogene 24, 2076–2086 [DOI] [PubMed] [Google Scholar]
- 33. Low B. C., Pan C. Q., Shivashankar G. V., Bershadsky A., Sudol M., and Sheetz M. (2014) YAP/TAZ as mechanosensors and mechanotransducers in regulating organ size and tumor growth. FEBS Lett. 588, 2663–2670 [DOI] [PubMed] [Google Scholar]
- 34. Makuch L., Volk L., Anggono V., Johnson R. C., Yu Y., Duning K., Kremerskothen J., Xia J., Takamiya K., and Huganir R. L. (2011) Regulation of AMPA receptor function by the human memory-associated gene KIBRA. Neuron 71, 1022–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kurihara H., Anderson J. M., Kerjaschki D., and Farquhar M. G. (1992) The altered glomerular filtration slits seen in puromycin aminonucleoside nephrosis and protamine sulfate-treated rats contain the tight junction protein ZO-1. Am. J. Pathol. 141, 805–816 [PMC free article] [PubMed] [Google Scholar]
- 36. Katschnig A. M., Kauer M. O., Schwentner R., Tomazou E. M., Mutz C. N., Linder M., Sibilia M., Alonso J., Aryee D. N. T., and Kovar H. (2017) EWS-FLI1 perturbs MRTFB/YAP-1/TEAD target gene regulation inhibiting cytoskeletal autoregulatory feedback in Ewing sarcoma. Oncogene 36, 5995–6005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Weins A., Wong J. S., Basgen J. M., Gupta R., Daehn I., Casagrande L., Lessman D., Schwartzman M., Meliambro K., Patrakka J., Shaw A., Tryggvason K., He J. C., Nicholas S. B., Mundel P., and Campbell K. N. (2015) Dendrin ablation prolongs life span by delaying kidney failure. Am. J. Pathol. 185, 2143–2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Griffin S. V., Olivier J. P., Pippin J. W., Roberts J. M., and Shankland S. J. (2006) Cyclin I protects podocytes from apoptosis. J. Biol. Chem. 281, 28048–28057 [DOI] [PubMed] [Google Scholar]
- 39. Wada T., Pippin J. W., Terada Y., and Shankland S. J. (2005) The cyclin-dependent kinase inhibitor p21 is required for TGF-β1-induced podocyte apoptosis. Kidney Int. 68, 1618–1629 [DOI] [PubMed] [Google Scholar]
- 40. Griffin S. V., Hiromura K., Pippin J., Petermann A. T., Blonski M. J., Krofft R., Takahashi S., Kulkarni A. B., and Shankland S. J. (2004) Cyclin-dependent kinase 5 is a regulator of podocyte differentiation, proliferation, and morphology. Am. J. Pathol. 165, 1175–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Marshall C. B., and Shankland S. J. (2007) Cell cycle regulatory proteins in podocyte health and disease. Nephron. Exp. Nephrol. 106, e51–e59 [DOI] [PubMed] [Google Scholar]
- 42. Liu-Chittenden Y., Huang B., Shim J. S., Chen Q., Lee S. J., Anders R. A., Liu J. O., and Pan D. (2012) Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 26, 1300–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li Z., Zhao B., Wang P., Chen F., Dong Z., Yang H., Guan K. L., and Xu Y. (2010) Structural insights into the YAP and TEAD complex. Genes Dev. 24, 235–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhao B., Ye X., Yu J., Li L., Li W., Li S., Yu J., Lin J. D., Wang C. Y., Chinnaiyan A. M., Lai Z. C., and Guan K. L. (2008) TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 22, 1962–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Diepenbruck M., Waldmeier L., Ivanek R., Berninger P., Arnold P., van Nimwegen E., and Christofori G. (2014) Tead2 expression levels control the subcellular distribution of Yap and Taz, zyxin expression and epithelial-mesenchymal transition. J. Cell Sci. 127, 1523–1536 [DOI] [PubMed] [Google Scholar]
- 46. Kanwar Y. S. (2015) Continuum of historical controversies regarding the structural-functional relationship of the glomerular ultrafiltration unit. Am. J. Physiol. Renal Physiol. 308, F420–F424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Azeloglu E. U., Hardy S. V., Eungdamrong N. J., Chen Y., Jayaraman G., Chuang P. Y., Fang W., Xiong H., Neves S. R., Jain M. R., Li H., Ma'ayan A., Gordon R. E., He J. C., and Iyengar R. (2014) Interconnected network motifs control podocyte morphology and kidney function. Sci. Signal. 7, ra12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Carpenter A. E., Jones T. R., Lamprecht M. R., Clarke C., Kang I. H., Friman O., Guertin D. A., Chang J. H., Lindquist R. A., Moffat J., Golland P., and Sabatini D. M. (2006) CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]