

Abstract
Tumor cells display on their surface several molecular chaperones that normally reside in the endoplasmic reticulum. Because this display is unique to cancer cells, these chaperones are attractive targets for drug development. Previous epitope-mapping of autoantibodies (AutoAbs) from prostate cancer patients identified the 78-kDa glucose-regulated protein (GRP78) as one such target. Although we previously showed that anti-GRP78 AutoAbs increase tissue factor (TF) procoagulant activity on the surface of tumor cells, the direct effect of TF activation on tumor growth was not examined. In this study, we explore the interplay between the AutoAbs against cell surface–associated GRP78, TF expression/activity, and prostate cancer progression. First, we show that tumor GRP78 expression correlates with disease stage and that anti-GRP78 AutoAb levels parallel prostate-specific antigen concentrations in patient-derived serum samples. Second, we demonstrate that these anti-GRP78 AutoAbs target cell-surface GRP78, activating the unfolded protein response and inducing tumor cell proliferation through a TF-dependent mechanism, a specific effect reversed by neutralization or immunodepletion of the AutoAb pool. Finally, these AutoAbs enhance tumor growth in mice bearing human prostate cancer xenografts, and heparin derivatives specifically abrogate this effect by blocking AutoAb binding to cell-surface GRP78 and decreasing TF expression/activity. Together, these results establish a molecular mechanism in which AutoAbs against cell-surface GRP78 drive TF-mediated tumor progression in an experimental model of prostate cancer. Heparin derivatives counteract this mechanism and, as such, represent potentially appealing compounds to be evaluated in well-designed translational clinical trials.
Keywords: GRP78, prostate cancer, tissue factor, tumor cell biology, tumor promoter, autoantibodies, targeting
Introduction
Prostate cancer is the most frequent malignant tumor in men, whose age-adjusted incidence rates have dramatically increased with the introduction of serum PSA2 screening and early detection (1). Although patients with organ-confined disease may have a more favorable prognosis, metastatic disease remains incurable. Secondary thrombotic complications can increase morbidity and mortality in cancer patients. Several lines of evidence demonstrate that such thrombogenic events occur at the surface of tumor cells and involve an enhanced expression and procoagulant activity of TF (2). In addition to its critical role in hemostasis and blood clot formation, a growing body of genetic and pharmacological investigation indicates that tumor cell-expressed TF contributes to tumor growth via an array of proangiogenic and immunomodulating cytokines, chemokines, and growth factors (3).
GRP78 has long been established as an endoplasmic reticulum (ER)-resident stress-response chaperone that facilitates the correct folding and assembly of newly synthesized proteins (4). However, over the past decade, several studies demonstrated that, when present on the surface of cancer cells, GRP78 also functions as a signaling receptor (5); for instance, the binding of α2-macroglobulin to prostate cancer cells through csGRP78 triggers insulin-like responses (6) and regulates transcriptional activation of c-MYC target genes (7) to promote tumor proliferation and survival (8). In summary, the atypical presence of functional GRP78 “moonlighting” (9) at the tumor cell surface has unambiguously been confirmed by many independent reports as part of the UPR (reviewed in Ref. 10). Since the first report of GRP78 translocation on the external side of the cell (11) and the presence of circulating GRP78-targeting AutoAbs (12, 13), csGRP78-targeted theranostic applications have been validated in a wide variety of human tumors, including prostate (14, 15), breast (16, 17), ovarian (18), and brain (19) cancer, along with melanoma (20), leukemias, and lymphomas (21, 49).
In previous collective work, by epitope-mapping (“fingerprinting”) the circulating repertoire of AutoAbs in prostate cancer patients, we identified the consensus motif CNVSDKSC (12) as a specifically recognized epitope that corresponds to an N-terminal domain (Leu98–Leu115) of GRP78 (22). This immunogenic portion of GRP78 is also a functional binding site for α2-macroglobulin at the cell surface (23, 24). In addition, we elucidated the mechanism whereby AutoAbs against cell-surface GRP78 modulate TF activity via release of Ca2+ from the ER into the cytosol (25). All of these data indicate that binding of the AutoAbs to GRP78 at the cell surface elicits multiple signal transduction responses with a functional role in cancer. Consistent with these observations, we have shown that men with prostate cancer may have >12-fold higher serum levels of anti-GRP78 AutoAbs than age-matched cancer-free men, which correlates with metastatic disease and, ultimately, with a decreased overall survival (22).
Here we have used a xenograft mouse model of human prostate cancer to investigate the dependence on TF of tumor growth potentiation by anti-GRP78 AutoAbs. Given that these AutoAbs recognize an epitope on csGRP78 that overlaps with its heparin-binding domain (22), we also evaluate and validate a low-molecular weight heparin (LMWH), enoxaparin, as a potential inhibitor of tumor progression. Together, the results presented in this study show that the humoral anti-GRP78 AutoAb response is a viable therapeutic target in experimental models, with perspective potential application in patients with prostate cancer.
Results
GRP78 levels correlate with prostate cancer grade
Previous studies by Pootrakul et al. (26) and Daneshmand et al. (27) demonstrated a link between GRP78 expression and disease stage in a large patient cohort diagnosed with prostate cancer. To confirm GRP78 expression during prostate cancer progression, we have exploited the Human Protein Atlas (28), a tissue-based immunohistochemical (IHC) map of the human proteome. The reported samples can be pathologically classified following the Gleason scoring system (29) as follows: not otherwise specified = grade 1; low grade = grade 2; high grade = grade 4. Fig. 1A (top) shows low-level staining for GRP78 in grade 1 prostate cancer. In grade 2 prostate cancer (middle), GRP78 expression increases in most tumor cells. Finally, in grade 4 prostate cancer (bottom), there is a strong immunostaining for GRP78. Both cytoplasmic and membranous positivity for GRP78 are reported for these index samples in the Human Protein Atlas. These findings suggest that increased amounts of total GRP78, paralleled by cell-surface relocation of the protein (9), correlate with the progression of human prostate cancer, consistent with our previous report (15).
Figure 1.

Prostate cancer grade correlates with GRP78 expression and anti-GRP78 AutoAb titers. A, specimens from prostate cancer patients stained for GRP78 were graded pathologically. Scale bar, 100 μm. Top, grade 1: limited numbers of GRP78-positive cells. Middle, grade 2: increased numbers of GRP78-positive cells. Bottom, grade 4: high positivity for GRP78. Expression of GRP78 is accompanied by protein “moonlighting” and presence on the tumor cell surface (representative cells are indicated by red arrows). B, anti-GRP78 AutoAb titers correlate with serum PSA concentrations in patients with prostate cancer, as evaluated by Pearson correlation (p < 0.05, r = 38.4%, n = 48). C, patients grouped in low (1–2 ng/ml, n = 12), medium PSA (3–7 ng/ml, n = 14), high (8–19 ng/ml, n = 13), or very high (≥20 ng/ml, n = 8) PSA levels show progressively increasing levels of anti-GRP78 AutoAb titers. Controls were age-matched individuals (n = 9) not diagnosed with prostate cancer. Values are shown as mean ± S.E. (error bars); *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant. D, anti-GRP78 AutoAb titers are significantly lower in prostate cancer patients after (24 weeks postoperative) versus before prostatectomy (90.88 μg/ml versus 124.2 μg/ml; ***, p < 0.0001).
AutoAb titers against csGRP78 increase with serum PSA concentration in patients with prostate cancer
Having shown that GRP78 expression increases with grade in human prostate cancer (Fig. 1A), we next evaluated the circulating pool of anti-GRP78 AutoAbs in a panel of serum samples (n = 48) from prostate cancer patients (Ontario Tumor Bank, Ontario, Canada). Utilizing an established ELISA protocol (25), we showed that anti-GRP78 AutoAb titers increase with serum concentrations of PSA, a marker that has been proposed as a surrogate for prostate cancer grade (30) (Fig. 1B, Pearson correlation test; p < 0.05, r = 0.38). Although the correlation was considered modest, it is statistically significant for this small number of samples. Thus, a larger cohort of patients will be further investigated. Based on this, we classified the patient-derived samples based on their serum PSA concentrations as low (1–2 ng/ml), medium (3–7 ng/ml), high (8–19 ng/ml), and very high (≥20 ng/ml), following the D'Amico clinical standard in prostate cancer risk categories (31). Age-matched, prostate cancer–free men served as negative controls. This analysis demonstrated that the anti-GRP78 AutoAb response is progressively amplified with the increase in serum PSA concentrations, with the patient cohort having markedly higher anti-GRP78 AutoAb titers than the control cohort (Fig. 1C).
Because the half-life of circulating antibodies is generally less than 3 weeks, a sustained and consistent antigen presentation is required to achieve functional levels of antigen-specific IgG-type responses (32). To evaluate whether a similar mechanism takes place in the case of anti-GRP78 AutoAbs, we evaluated a selected cohort of prostate cancer patients. Two patient cohorts treated with radical prostatectomy were examined preoperatively (cohort 1) and 24 weeks postoperatively (cohort 2) for their anti-GRP78 AutoAbs titers (Fig. 1D). Consistently with this working hypothesis, we found a significant decrease in anti-GRP78 AutoAb titers in patients after surgical resection of the primary tumor (cohort 1) compared with preoperative levels (cohort 2) (t test, p < 0.0001). To date, these postoperative patients (cohort 2) have undetectable serum PSA concentrations (PSA < 1 ng/ml; data not shown) and are in clinical remission.
Binding of anti-GRP78 AutoAbs to csGRP78 activates the UPR
Recently, we showed that binding of anti-GRP78 AutoAbs to csGRP78 results in Ca2+ efflux from the ER and UPR activation, a pro-survival pathway initiated by ER stress (25). Here, we first confirmed the presence of csGRP78 in the prostate cancer cell line DU145 by flow cytometry (Fig. 2A). Treatment of DU145 cells with pathological (i.e. found in prostate cancer patient sera) concentrations (60 μg/ml) of anti-GRP78 AutoAbs induced expression of UPR markers at both the protein (Fig. 2, B and C) and RNA levels (Fig. 2D). This included increased expression of the protein-disulfide isomerase (PDI), inositol-requiring enzyme-1α (IRE1α), phosphorylation of the initiation factor eIF2α (p-eIF2α), and spliced X-box-binding protein-1 (sXBP1). In contrast, expression of CCAAT/enhancer-binding protein (C/EBP) homologous protein (CHOP), an apoptosis marker, did not change in response to anti-GRP78 AutoAbs (Fig. 2, B and C), data indicative of response specificity. Remarkably, mRNA levels of GRP78 itself are increased >5-fold upon cell treatment with the AutoAbs (Fig. 2C), suggesting a positive activation feedback loop in cancer cells. As a control, PC-3 cells were treated with a pathological concentration of anti-GRP78 autoantibodies (60 μg/ml). Different from the DU145 cell line, the PC-3 cell line expresses a negligible amount of csGRP78 (25, 22). Unlike thapsigargin and tunicamycin, anti-GRP78 autoantibodies fail to increase GRP78 and TF expression in the PC-3 cell line (Fig. 2E). Collectively, these findings suggest that UPR induction is a reliable surrogate marker to establish a link between anti-GRP78 AutoAbs and csGRP78 in human prostate cancer cells.
Figure 2.
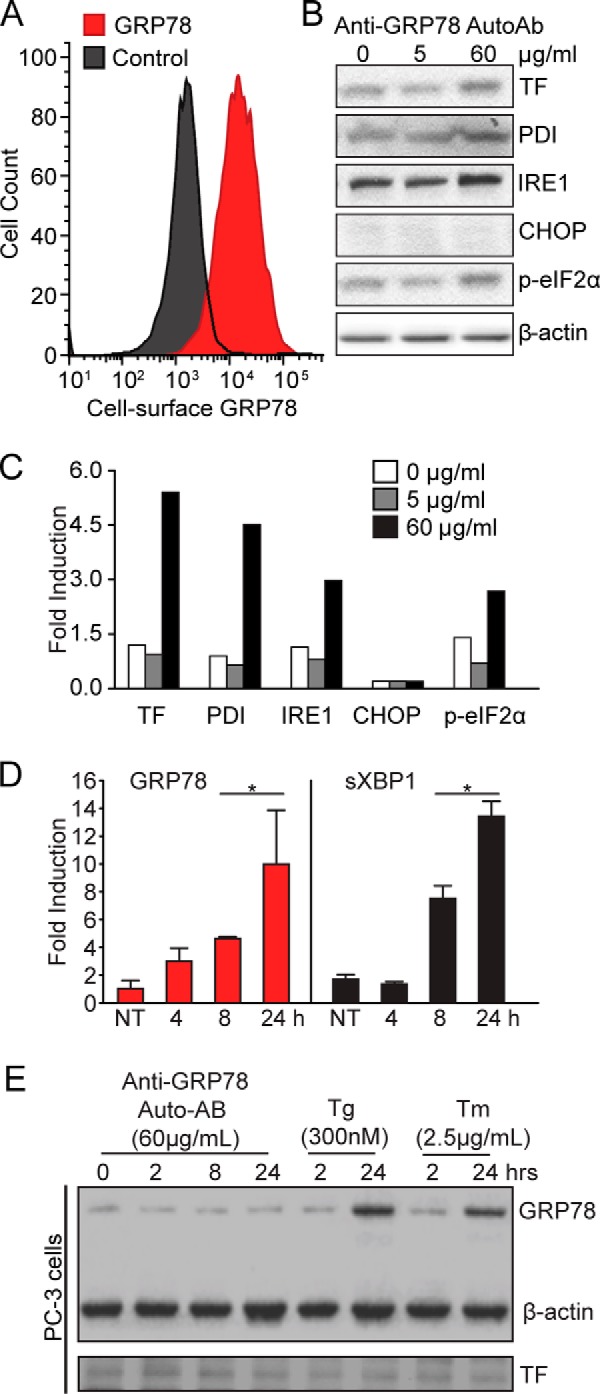
Treatment with anti-GRP78 AutoAbs increases the expression of TF and UPR markers in DU145 human prostate cancer cells. A, flow cytometry analysis of csGRP78 amounts in DU145 cells. B, pathological doses of anti-GRP78 AutoAbs (60 μg/ml) increase protein expression of both TF and markers of UPR activation (PDI, IRE1, and phospho-eIF2α), compared with non-treated (0 μg/ml) cells or cells treated with a normal dose of anti-GRP78 AutoAbs (5 μg/ml). β-Actin was used as a loading control. C, protein bands of the immunoblot in B were quantified with ImageJ, and values were normalized to β-actin. Error bars are ± S.D. p-eIF2α, phosphorylation of the initiation factor eIF2 α. D, quantitative real-time PCR analysis of GRP78 and spliced XBP1 mRNA expression in DU145 cells treated with a pathological dose of anti-GRP78 AutoAbs (60 μg/ml). Results are expressed as -fold induction over non-treated (NT) cells (*, p < 0.05; n = 3). E, pathological doses of anti-GRP78 AutoAbs (60 μg/ml) do not increase GRP78 expression in PC-3 cells; thapsigargin (Tg; 300 nm) or tunicamycin (Tm; 2.5 μg/ml) was used as a control UPR inducer. β-Actin was used as a loading control.
Tumor growth induced by anti-GRP78 AutoAbs requires TF
In previous work, de Ridder et al. (33) established that the humoral response against csGRP78 promotes tumor growth and proliferation. Further, we and others have demonstrated that treating human prostate cancer cells with anti-GRP78 AutoAbs increases TF expression and activation, thereby promoting cell proliferation (25, 34). Given these foundational reports, we evaluated whether anti-GRP78 AutoAbs would stimulate tumor growth in vivo. DU145 prostate cancer cells were subcutaneously implanted into the flank of non-obese diabetic/severe combined immunodeficiency (NOD/SCID) mice. Animals were administered at the site of the tumor with (i) vehicle only (PBS), (ii) negative control antibodies (human IgG), or (iii) anti-GRP78 AutoAbs. Six weeks post-administration, animals treated with anti-GRP78 AutoAbs showed markedly larger tumors compared with both vehicle– and human IgG control–treated groups (Fig. 3A). This accelerated tumor growth became even more pronounced at week 8, when the experiment was terminated. In parallel, tumor expression of TF protein increased upon treatment with anti-GRP78 AutoAbs, compared with human IgG treatment (Fig. 3B), consistent with a similar increase in TF mRNA levels (Fig. 3C). As observed in vitro for DU145 prostate cancer cells, tumor xenografts from mice treated with anti-GRP78 AutoAbs expressed substantially higher levels of GRP78 and other UPR markers, such as activating transcription factor-4 (ATF4) and sXBP1, along with the proliferation marker Ki67 and the angiogenic marker vascular endothelial growth factor receptor-2 (VEGFR2), compared with control-treated mice (Fig. 3C). Consistent with these findings, gene expression analysis by using NanoString® technology identified numerous UPR genes in addition to GRP78, including the protein kinase RNA-like endoplasmic reticulum kinase (PERK), calnexin, calreticulin, ATF4, ATF6, and total XBP1 that were clearly up-regulated in xenografts from mice treated with anti-GRP78 AutoAbs (Fig. 3D and supplemental Fig. 1). We observed a 48% increase in proliferating (Ki67-positive) cells in xenografts exposed to anti-GRP78 AutoAbs (n = 1309, 72.54-pixel2 average cell area) compared with those exposed to human IgG control (n = 676, 60.09-pixel2 average cell area) (Fig. 3E).
Figure 3.
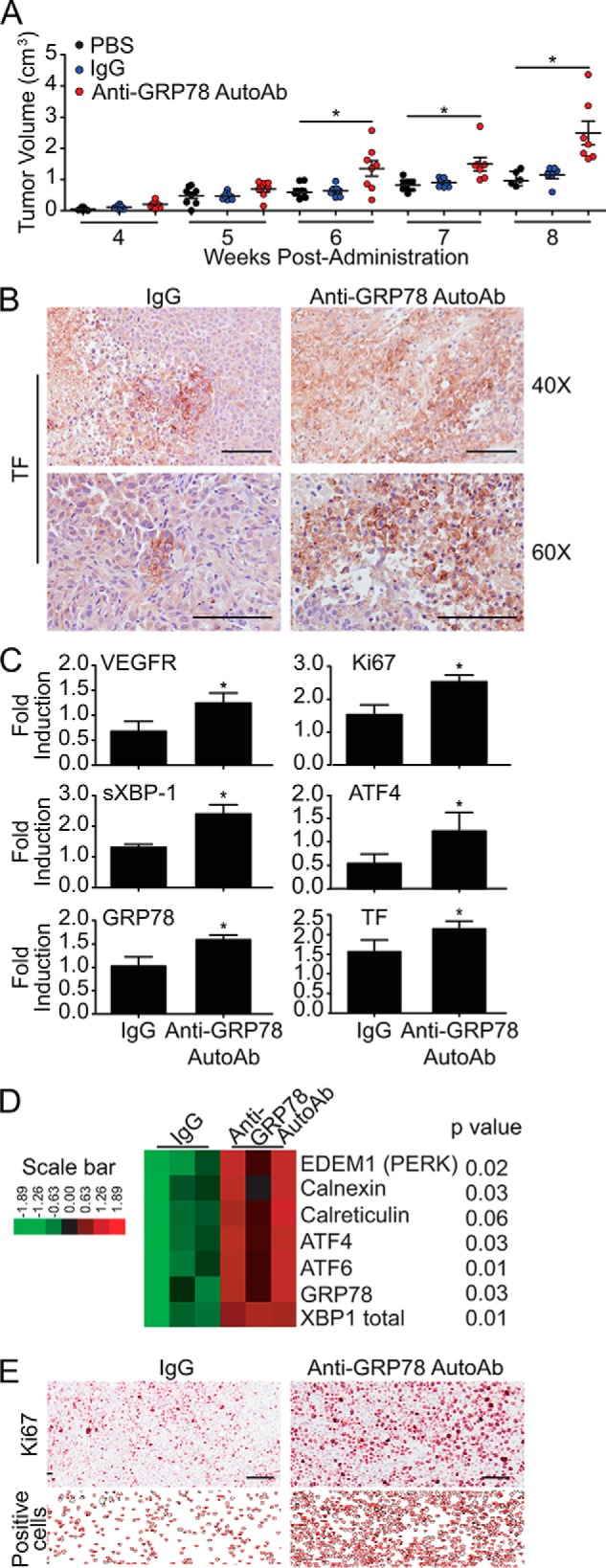
Anti-GRP78 AutoAbs accelerate tumor growth in mice and increase protein expression of TF and UPR markers. A, treatment of NOD/SCID mice bearing DU145 xenografts with anti-GRP78 AutoAbs (60 μg/ml) significantly increases tumor growth compared with PBS-treated or human IgG-treated (60 μg/ml) mice (*, p < 0.05; **, p < 0.001 versus IgG and PBS treatment; n = 8/group). Mean tumor volume ± S.E. (error bars) is shown. B, IHC analysis of tumors from mice treated with anti-GRP78 AutoAbs demonstrates an increase in TF expression compared with human IgG treatment. Scale bar, 100 μm. C, quantitative real-time PCR analysis of tumors from mice treated with anti-GRP78 AutoAbs demonstrates increased mRNA expression of TF, VEGR, Ki67, spliced XBP1, ATF4, and GRP78 (*, p < 0.05 versus IgG treatment; n = 3). D, gene expression levels of seven UPR markers (derived by NanoString®) following treatment with control human IgG (60 μg/ml) or anti-GRP78 AutoAbs (60 μg/ml). p values are indicated for each gene (n = 3/treatment). E, IHC analysis of the proliferation marker Ki67 demonstrates an increase in positive cells in the anti-GRP78 AutoAb-treated group compared with control human IgG. Representative images of five sections are shown. ImageJ was used to count the number of Ki67-positive cells. Scale bar, 100 μm.
We next evaluated whether TF would directly mediate the effect of anti-GRP78 AutoAbs on increased tumor progression by transducing DU145 prostate cancer cells with TF-targeted shRNA lentiviral vectors. A lentiviral vector encoding the GFP was used to produce the control DU145GFP cells. Western blot analysis of four stable antibiotic-resistant (TF-silenced) clones revealed variable decreases in TF protein expression (Fig. 4A), with clone 4 showing undetectable levels of endogenous TF (henceforth designated as DU145 knockdown, DU145KD). Treatment with the anti-GRP78 AutoAbs increased TF activity in DU145GFP and DU145 clone 1 (DU145#1; TF expression reduced but still detectable), but, as expected due to the absence of TF protein, not in in DU145KD cells (Fig. 4B). A concentration-dependent response to GRP78 AutoAbs was noted when comparing DU145GFP (high TF response), DU145 clone 1 (intermediate TF response), and DU145KD (undetectable TF response), a result again indicative of specificity (Fig. 4B).
Figure 4.

TF downmodulation in DU145 cells inhibits anti-GRP78 AutoAb-mediated TF activity and tumor progression. A, Western blot analysis of TF expression in cells transduced with lentiviral particles encoding TF shRNA. Control cells were transduced with lentiviral particles encoding GFP (DU145GFP). β-Actin was used as a loading control. Clone 4 demonstrates high efficiency of TF knockdown (DU145KD). B, TF procoagulant (PCA) activity is induced by the anti-GRP78 AutoAbs and is proportional to TF expression levels (*, p < 0.05 AutoAbs (+) versus non-treated (−) cells; n = 5). C, in vivo, DU145KD xenografts grow more slowly than DU145GFP xenografts (*, p < 0.05). D, IHC analysis demonstrates lower TF expression in DU145KD xenografts, compared with DU145GFP (left); TF total protein amounts were lower in the DU145 TFKD animal group versus control (Western blotting; right). Scale bar, 100 μm. Mean tumor volume ± S.D. (error bars) is shown. E, anti-GRP78 AutoAb treatment (60 μg/ml) does not alter the rate of tumor growth in DU145KD xenografts. Human IgG (60 μg/ml) treatment was used as a control (n = 5/group). Mean tumor volumes ± S.D. are shown. ns, not significant. F, treatment of DU145 cells with Tg (5 μm), anti-GRP78 AutoAbs (60 μg/ml), or patient sera increases TF activity, whereas immunodepletion of anti-GRP78 AutoAbs prevents TF activity (*, p < 0.05 versus NT cells; #, p < 0.05 versus AutoAb treatment; n = 5/group). G, pretreatment of sera from prostate cancer patients with human recombinant GRP78 (hrGRP78) (60 μg/ml) prevents the activation of TF in DU145 cells (*, p < 0.05 versus NT cells; #, p < 0.05 versus corresponding native serum; n = 5/group).
We next examined the contribution of TF to tumor progression in xenografts obtained in NOD/SCID mice by implant of either DU145GFP or DU145KD cells (5 × 105 cells/animal). In this experiment, we used a smaller number of cells to allow for longer monitoring period of these tumors. Six weeks post-administration, DU145KD tumors were significantly smaller than control DU145GFP tumors (Fig. 4C). Immunostaining showed decreased TF expression in DU145KD tumors compared with DU145GFP tumors (Fig. 4D) and confirmed that TF presence and activity are necessary for tumor growth. To define the contribution of TF to tumor growth mediated by anti-GRP78 AutoAbs, DU145KD cells were implanted in NOD/SCID mice. Because these xenografts grow relatively slowly (Fig. 4C), in these experiments, we implanted higher numbers of human prostate cancer cells (1 × 106/animal). This step was implemented to maintain consistency with in vivo experiments by using parental DU145 cells. Animals were randomized in two cohorts that received a weekly administration of either anti-GRP78 AutoAbs or human IgG control at the tumor site. We observed no differences in tumor growth between the anti-GRP78 AutoAb-treated cohort and the human IgG control cohort (Fig. 4E). These findings demonstrate that the tumor growth induction triggered by anti-GRP78 AutoAbs (Fig. 4C) is TF-dependent.
Immunodepletion or neutralization of anti-GRP78 AutoAbs inhibits TF activity
Because binding of purified anti-GRP78 AutoAbs to csGRP78 increases TF expression (Fig. 3, B and C) and activity (Fig. 5B), we next evaluated the specificity of this mechanism in serum samples from prostate cancer patients. TF procoagulant activity was substantially increased in cultured DU145 cells treated with either purified anti-GRP78 AutoAbs (60 μg/ml) or undiluted patient sera containing 60 μg/ml of anti-GRP78 AutoAbs (Fig. 4F), but not in sera that was immunodepleted of anti-GRP78 AutoAbs as described (25). Thapsigargin was used as a positive control (25) due to its inherent ability to increase cytosolic Ca2+ concentration and to activate TF (Fig. 4F). In addition, neutralization of the anti-GRP78 AutoAb pool by pretreatment of patient sera with human recombinant GRP78 (hrGRP78, 60 μg/ml) clearly reduced TF activation, data indicative of specificity. Together, these findings demonstrate that circulating anti-GRP78 AutoAbs are predominant activators of TF in serum samples of prostate cancer patients, which seem to contain no additional factors which can enhance TF activity.
Figure 5.

Heparin and enoxaparin prevent the binding of anti-GRP78 AutoAbs to DU145 cells and inhibit tumor growth. A, heparin pretreatment (50 IU/ml) inhibits the effect of anti-GRP78 AutoAbs on TF activation, whereas it does not affect TF activity on non-treated (NT) or Tg-induced (5 μm) DU145 cells (*, p < 0.05 versus NT; #, p < 0.05 versus treatment with anti-GRP78 AutoAbs; n = 5/group). B, heparin (50–100 IU/ml) and enoxaparin (5–40 mg/ml) prevent anti-GRP78 AutoAb-mediated activation of TF in DU145 cells (*, p < 0.05 versus NT cells; #, p < 0.05 versus AutoAb treatment; n = 5/group). C, GRP78 binding and immunostaining (green) by anti-GRP78 AutoAbs on intact DU145 cells (left image) is abolished by pretreating cells with heparin (10 IU/ml; middle image) and enoxaparin (50 μg/ml; right image). DAPI was used to stain the nuclei (blue). Scale bar, 20 μm. D, NOD/SCID mice bearing DU145 xenografts were treated with either anti-GRP78 AutoAbs (60 μg/ml) or human IgG (60 μg/ml, control), followed by a secondary treatment with either enoxaparin (6 mg/kg/week) or PBS (control). The observed potentiation of tumor growth by the anti-GRP78 AutoAbs (**, p < 0.001 versus IgG, n = 6/group, left) is abrogated by a secondary treatment with enoxaparin (n = 6/group, right). At week 6, all mice receiving anti-GRP78 AutoAb/PBS treatment reached the end point. ns, not significant. E, gene expression levels of seven UPR markers (derived by NanoString®) following treatment with control human IgG (60 μg/ml) or anti-GRP78 AutoAbs (60 μg/ml), followed by enoxaparin (6 mg/kg/week) treatment. p values are indicated for each gene; n = 3/treatment.
Unfractionated heparin and LMWH block the binding of anti-GRP78 AutoAbs to csGRP78 and reduce tumor growth in vivo
Previous studies have shown that GRP78 possesses a heparin-binding domain spanning residues Leu98–Thr102 (35) that is coincident with the epitope recognized by the anti-GRP78 AutoAbs (12). We therefore examined whether heparin could compete for the binding of anti-GRP78 AutoAbs to csGRP78. Pretreatment of DU145 cells with heparin (50 IU/ml) prevented activation of TF by the anti-GRP78 AutoAbs but did not affect thapsigargin-induced TF activity (Fig. 5A). In addition to standard (unfractionated) heparin, we also examined a LMWH, enoxaparin, confirming a similar inhibition of anti-GRP78 AutoAb-induced TF activation (Fig. 5B). Furthermore, DU145 cells pretreated with enoxaparin showed markedly lower binding of anti-GRP78 AutoAbs to csGRP78, as demonstrated by fluorescence imaging, compared with untreated cells (Fig. 5C). Based on these observations, we examined the ability of enoxaparin to counteract the tumor progression effects of anti-GRP78 AutoAbs in vivo. Immunodeficient mice were xenografted with DU145 prostate cancer cells, followed by weekly administration of either anti-GRP78 AutoAbs or control human IgG at the tumor site. One week after the first AutoAb treatment, mice received either enoxaparin (6 mg/kg/week) or PBS (control). As expected, animals treated with anti-GRP78 AutoAbs exhibited significantly larger tumors compared with control IgG-treated animals (Fig. 5D, left). In contrast, mice in the enoxaparin-treated group were unresponsive to the anti-GRP78 AutoAbs; the observed, slow tumor growth was not different in PBS-treated mice (Fig. 5D, right). We concluded that enoxaparin treatment blocks the portion of tumor growth elicited by anti-GRP78 AutoAb, whereas it does not possess inherent anti-tumor properties in vivo, at least in this experimental xenograft mouse model.
Finally, gene expression was assessed with NanoString® in tumors from mice treated with either anti-GRP78 AutoAbs or human IgG control followed by enoxaparin. This analysis showed no consistent difference in the induction of UPR genes (Fig. 5E and supplemental Fig. 1). These findings indicate that enoxaparin abolishes the effect of anti-GRP78 AutoAbs on UPR activation mediated via csGRP78 (compare with Fig. 3D and supplemental Fig. 1).
Discussion
In this study, we present data that support an intriguing and unique role for anti-GRP78 AutoAbs as potentiators of human prostate cancer cell proliferation in vitro and tumor growth in vivo, using the DU145 model cell line. By using experimental cell culture, mouse models, and clinical samples, we consistently demonstrate a functional role of anti-GRP78 AutoAbs as a determinant of prostate cancer progression. Mechanistically, anti-GRP78 AutoAbs purified from serum samples of prostate cancer patients target GRP78 on tumor cell surfaces, triggering an increased expression and activation of TF, a mediator of tumor proliferation. This effect is evidently specific, as either immunodepleting or neutralizing these AutoAbs revealed no other factor in the serum of prostate cancer patients capable of inducing TF expression or activity. Moreover, we demonstrate for the first time that anti-GRP78 AutoAbs directed (largely or exclusively) against the Leu98–Leu115 epitope within the N-terminal domain of GRP78 markedly increase TF-dependent tumor growth. Perhaps most strikingly, the LMWH enoxaparin reverts this anti-GRP78 AutoAb-induced potentiation of tumor growth but otherwise has no anti-tumor effect. Taken together, the results presented here support a working model (Fig. 6) whereby TF activation via binding of anti-GRP78 AutoAbs to GRP78 on the cancer cell surface represents a critical mechanistic switch for tumor growth and, therefore, a potential target pathway for therapeutic intervention.
Figure 6.

Summary model. Anti-GRP78 AutoAbs increase tumor growth by binding to cell-surface GRP78 and activating TF. This interaction can be interrupted by treatment with heparin or enoxaparin. Binding of anti-GRP78 AutoAbs to cell-surface GRP78 induces elevated cytosolic Ca2+ concentrations that can activate the pro-survival UPR pathway and increase TF activity. Overall, this interaction leads to an increased rate of tumor growth, improved angiogenesis, and, potentially, enhanced survival via UPR activation.
GRP78 has long been recognized as a stress-response molecular chaperone that is instrumental in the folding and assembly of newly synthesized proteins within the ER (36). In addition, GRP78 redirects misfolded proteins for ER-associated degradation, regulates ionized Ca2+ homeostasis, and controls the activation of the UPR following ER stress. However, a wide range of human tumor cell lines show that GRP78 can function in a manner distinct from its well-established chaperone activity. In fact, a large body of work has recently revealed that GRP78 may associate with the cell surface (“protein moonlighting”) (9) under stress conditions, such as cancer, to function as a modulator of signal transduction in cell proliferation, tumor invasion, and immunity (5, 10). A humoral response against csGRP78 correlates with poor prognosis in patients with prostate cancer (12), and anti-GRP78 AutoAbs from prostate cancer patients exhibit mitogenic potential on various human prostate cancer cell lines, including 1-LN and DU145 (22). These findings imply that the AutoAb response directed against csGRP78 represents not only a candidate marker of disease progression but also a functional element for tumor cell proliferation and prostate cancer growth. Although recent studies suggest that immunity against GRP78 accelerates tumor growth in vivo (33), the direct contribution of these particular anti-GRP78 AutoAbs, as well as the underlying mechanism(s) of this enhanced tumorigenicity, have not as yet been fully defined.
By using archived tumor tissue and serum samples from an annotated patient cohort diagnosed with different stages of prostate cancer, we demonstrated a substantial increase in GRP78 in more advanced disease. Further, there was a positive correlation between serum PSA concentration levels and anti-GRP78 AutoAb titers in a subset of prostate cancer patients having advanced disease before receiving surgical treatment. Circulating anti-GRP78 AutoAbs have also been reported in other cancers, including colorectal (37), hepatocellular (38), and ovarian carcinomas (18, 39); however, the humoral immune response against csGRP78 in human prostate cancer appears to be notably striking compared with other malignant solid tumors (12). Thus, the presence of circulating anti-GRP78 AutoAbs in the blood of cancer patients should now be considered as a candidate serological marker for early prostate cancer detection (12, 40).
It is well-documented that an increase in antigen presentation on the surface of cancer cells can drive antibody production (32). Thus, we compared anti-GRP78 AutoAb titers in the pre-prostatectomy setting (patients with active prostate cancer) with the post-prostatectomy setting (prostate cancer patients deemed to have no evidence of disease). Postoperative patients demonstrated much lower levels of anti-GRP78 AutoAbs compared with preoperative patients. This finding supports the concept that the expression of GRP78 on the surface of prostate cancer cells induces an anti-GRP78 AutoAb response. Whereas postoperative patients had significantly lower anti-GRP78 AutoAb titers compared with patients with active disease, these titers were not reduced to the levels observed in the control cohort of non-prostate cancer patients. We are currently investigating whether anti-GRP78 AutoAb titers would eventually return to normal levels over time in patients undergoing radical prostatectomies as primary surgical management of prostate cancer with curative intent; if so, one might speculate that the anti-GRP78 circulating titers could serve as an early humoral warning of disease progression and/or tumor recurrence.
The correlation of anti-GRP78 AutoAbs with prostate cancer grade suggests a potential contributory role in mediating disease progression. We examined the mechanisms by which anti-GRP78 AutoAbs enhance prostate cancer cell survival and tumor growth. We have previously shown that the binding of anti-GRP78 AutoAbs to csGRP78 depleted ER Ca2+ stores (25). This process is necessary to activate the UPR pro-survival pathway (24). Interestingly, the high concentrations of anti-GRP78 AutoAbs observed in patients with prostate cancer induced several UPR markers at both the protein and RNA levels. However, there was no change in the expression of the pro-apoptotic marker CHOP, a bZIP transcription factor induced by the ATF6 and PERK pathways (41), an observation internally consistent with the system specificity. UPR activation enables cells to either manage current stress conditions or activate apoptosis. Mild activation of the UPR leads to adaptation to cellular stresses by means of increased chaperone expression, suggesting a link with the selective intrinsic instabilities of apoptotic proteins, such as CHOP, to create a strong pro-survival bias (41). GRP78 expression is a hallmark of the UPR and has recently been linked to chemo-resistance (42); therefore, we are currently investigating whether UPR activation following the specific binding of anti-GRP78 AutoAbs to csGRP78 might indeed confer protection against apoptosis and contribute to tumor growth and metastasis.
We have previously elucidated the mechanism by which binding of anti-GRP78 AutoAbs to csGRP78 increases TF expression and activity (25). It is worth noting that, although TF and GRP78 are expressed on the cell surface, the interaction between the two proteins is casual at best (6), supporting an indirect mechanism of TF induction. Further, it is well-established that TF expression and activation correlate with increased tumor progression (3). In addition to the risk of thrombosis among cancer patients (43), TF has long been recognized to increase inflammation, angiogenesis, and tumor proliferative capacity (3). Based on this background, we monitored tumor growth in an in vivo model system, the immunodeficient NOD/SCID mice, that allows the growth of human tumor xenografts in the presence of a complete repertoire of coagulation factors (44). Studies showed that tumors derived from wild-type DU145 prostate cancer cells, but not DU145 cells with silenced TF expression, responded with enhanced tumor growth upon anti-GRP78 AutoAb treatment. Consistently, AutoAb-treated tumors showed UPR activation and increased levels of Ki67 and VEGFR2. These findings indicate that other pathways that influence tumor and stromal cell proliferation and viability are activated following binding of anti-GRP78 AutoAbs to target csGRP78.
Impaired TF expression and/or function can potentially limit tumor progression; however, direct modification of TF expression is challenging, given the inherent risk of coagulopathy induction in clinical settings (45). Instead, here we have explored alternative approaches to block the interaction of anti-GRP78 AutoAbs with csGRP78 on prostate cancer cells. Previous studies reported that GRP78 contains a heparin-binding site consisting of Leu98–Thr102 (35), which resides within the N-terminal region of GRP78 known to contain the epitope for anti-GRP78 AutoAb binding (22). Although heparin and enoxaparin could function as anti-cancer agents, the underlying molecular mechanisms have not been fully characterized (46). The in vitro data presented here show that heparin and LMWH are sufficient to disrupt the binding of anti-GRP78 AutoAbs to csGRP78 and to reduce TF activity commensurately. Heparin treatment did not affect the induction of TF activity by thapsigargin (data not shown), indicating that heparin does not inhibit the release of ER Ca2+ that contributes to TF activation (25). In addition, we have demonstrated that enoxaparin pretreatment inhibited anti-GRP78 AutoAbs binding to the surface of DU145 cells. Together, these data indicate that heparin/enoxaparin can successfully inhibit TF activity by counteracting the effect of anti-GRP78 AutoAbs. These findings also provide an innovative mechanism by which heparin reduces local anticoagulant effects independent of its ability to inactivate thrombin and Xa via antithrombin.
Finally, we designed an interventional study to begin to assess the ability of enoxaparin to block the growth of DU145 xenografts in the presence or absence of anti-GRP78 AutoAbs and in an animal model (NOD/SCID mice) in which a complete coagulation system is present. Treatment with enoxaparin abolished the effect of anti-GRP78 AutoAbs on accelerated prostate cancer growth in these tumor-bearing mice. Consistently, analysis of tumors treated with anti-GRP78 AutoAbs and enoxaparin demonstrated no UPR activation, compared with tumors treated only with anti-GRP78 AutoAbs. Our studies suggest that LMWH agents such as enoxaparin, which can interfere with AutoAb targeting of csGRP78, might well comprise a unique therapeutic strategy to treat prostate cancer and perhaps other human tumors that induce anti-GRP78 humoral responses. Of course, the potential benefits of enoxaparin or heparin as a therapeutic intervention in this setting must be carefully balanced against the bleeding risk in translational clinical trials.
In summary, the results presented here uncover an unrecognized molecular mechanism by which anti-GRP78 AutoAbs drive TF-mediated tumor progression in the setting of prostate cancer. Furthermore, these findings also suggest a translational approach to treat prostate cancer with either heparin or heparin-like molecules in prostate cancer patients with elevated levels of circulating anti-GRP78 AutoAbs.
Experimental procedures
Cell culture and recombinant proteins
Tumor cell lines were purchased from the American Type Culture Collection (Manassas, VA). The human prostate cancer cell line, DU145, was cultured in minimum essential medium plus GLUTAMAXTM (Invitrogen). Media were supplemented with 10% FBS (Sigma-Aldrich) containing 100 units/ml penicillin and 100 μg/ml streptomycin (Invitrogen). Cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2. Thapsigargin (Sigma-Aldrich, Oakville, Canada) was used as a positive control for the TF activity assay. Stock solutions were prepared in DMSO and diluted in physiological buffer to achieve a final concentration of 5 μm. Human recombinant GRP78 was resuspended at 60 μg/ml in PBS and used to neutralize anti-GRP78 AutoAbs.
Human Protein Atlas
Representative images of prostate adenocarcinoma, not otherwise specified (M-81403, patient ID 3040), low-grade (M-814031, patient ID 3957), and high-grade (M-814033, patient ID 3978) were downloaded from the publicly available database of the Human Protein Atlas (http://www.proteinatlas.org)3 and were analyzed for total and surface-associated GRP78. Prostate cancer was graded in these samples by pathologists, and a grade from 1 to 5 was assigned based on the microscopic resemblance between the cancerous cells and normal prostate tissue.
Serum PSA concentration and anti-GRP78 AutoAb titers
Human specimens were obtained from the Ontario Tumor Bank, Ontario Institute for Cancer Research. Blood samples were taken from a cohort of patients diagnosed with prostate cancer before treatment administration. Serum PSA concentrations were provided by the Ontario Tumor Bank. Anti-GRP78 AutoAb titers were measured with an established ELISA protocol as described (25).
Isolation of anti-GRP78 AutoAbs from patient samples
A study by Mintz et al. (12) has identified the CNVSDKSC peptide that resembles the N-terminal domain of GRP78. Positive serum reactivity to the CNVSDKSC peptide was specifically linked to disease progression and shorter survival in a large patient population (12). Blood samples and corresponding sera from patients (n = 24) with prostate cancer were obtained from the Department of Urology, St. Joseph's Healthcare Hamilton (Hamilton, Canada). Written informed consent was obtained from each patient and approved by the Research Ethics Board of St. Joseph's Healthcare Hamilton (approval 08-3047). Anti-GRP78 AutoAbs were purified by affinity against the N-terminal region of GRP78 (Leu98–Leu115) (25). Briefly, a conformational peptide, CNVSDKSC, mimicking the 3D domain of Leu98–Leu115 of GRP78 was synthesized (CanPeptide Inc., Pointe-Claire, Canada) and immobilized on a Sepharose A column (SulfoLink protein kit, catalog no. 44995, ThermoFisher). Patient sera were incubated with the immobilized conformational peptide columns and eluted as per the manufacturer's instructions. Eluted fractions were dialyzed with PBS, concentrated using the Amicon Ultra 15 centrifugal filters, stored 4 °C, and admixed to produce a pooled source of anti-GRP78 AutoAbs used for treatment. It is worth noting that the established 60 μg/ml pathological dose of anti-GRP78 AutoAbs was first reported by Gonzalez-Gronow et al. (22), in which anti-GRP78 AutoAb titers were analyzed in a prostate cancer patient cohort in comparison with cancer-free, age-matched control patients. This study reported an average anti-GRP78 AutoAb titer of 60 μg/ml in prostate cancer patients compared with 1 order of magnitude less (i.e. 5–7 μg/ml) in the control group.
Immunoblotting
Tumor cells were lysed in denaturing sample buffer, and total proteins were separated on a 10% SDS-polyacrylamide gel under reducing conditions. Following separation, proteins were transferred to nitrocellulose membranes (Bio-Rad) with a Trans-Blot semi-dry apparatus (Bio-Rad). Membranes were blocked overnight in Tris-buffered saline plus 0.1% Tween 20 (TBST) containing 5% skim milk and subsequently incubated with the following primary antibodies: anti-GRP78/Bip (catalog no. 610979, BD Transduction), anti-phospho-eIF2α (catalog no. 9721S, BD Transduction), anti-VEGF pathway sampler kit (catalog no. 8696, Cell Signaling, Danvers, MA), anti-TF (catalog no. 4502, American Diagnostica, Stamford, CT), anti-CHOP (catalog no. B3, Santa Cruz Biotechnology, Inc., Dallas, TX) followed by the appropriate HRP-conjugated secondary antibodies (Dako, Carpentaria, CA) diluted in TBST containing 1% skim milk. Signals were visualized with the Western Lighting chemiluminescence reagent (PerkinElmer Life Sciences) and Kodak X-OMAT Blue XB-1 film (PerkinElmer Life Sciences) in a Kodak X-OMAT 1000A processor (Mississauga, Canada). To control for protein loading, immunoblots were stripped and reprobed with anti-β-actin antibody (catalog no. A544, Sigma-Aldrich). ImageJ was used for densitometric band quantification using the watershed algorithm as described on the National Institutes of Health website (27).
Continuous measurement of cell-surface TF procoagulant activity
We have previously developed an assay for the measurement of TF procoagulant activity on intact tumor cells (47). Briefly, cells were seeded into a 96-well tissue culture plate (10,000 cells/well) the day before the experiment. The culture media were removed, and cells were washed once in TBS. A mixture containing 1 nm human FVIIa, 30 nm human FX, 10 mm CaCl2, and 0.4 mm chromogenic substrate S-2765 (Diapharma, West Chester, OH) in TBS was added to each well. Following the addition of the test agent diluted in TBS, the absorbance at 405 nm was measured every 2 min for 3 h at 37 °C. A standard curve was generated in which 100 units of TF activity was defined as the activity of 450 μg (0.3 μl) of human recombinant TF (as determined by the American Diagnostica ELISA). Vmax was calculated with SoftMax Pro and used to determine the amount of FXa generated per 10,000 cells (units/10,000 cells).
RNA isolation and real-time PCR
RNA was isolated with the Qiagen RNeasy Mini kit (Qiagen), and cDNA was synthesized with the Applied Biosystems high-capacity cDNA reverse transcription kit (Applied Biosystems). Real-time PCRs were carried out based on standard protocols with the SYBR Green MasterMix (Applied Biosystems) using gene-specific forward and reverse primers (supplemental Table 1).
TF-silenced prostate cancer cells
DU145 cells grown to 50% confluence in a 12-well plate were infected with lentiviral particles carrying a human TF-specific shRNA sequence (Santa Cruz Biotechnology). After 48 h, colonies were selected in media containing 5 μg/ml puromycin (Santa Cruz Biotechnology) and assessed for TF expression (Western blotting) and activity (continuous assay) (47). As a control, DU145 cells were infected with lentiviral particles containing the GFP sequence only. Colonies of lentivirus-infected cells were selected by incubation in puromycin-containing media.
Mouse xenograft models of human prostate cancer
All experiments were approved and performed following the guidelines of the McMaster University Research Ethics Board as outlined in the Animal Utilization Protocol (Hamilton, Canada). Mice had access to food and water ad libitum. To obtain xenografts of human prostate cancer cells, DU145GFP or DU145KD cells were included in 0.1 ml of MatrigelTM and administered subcutaneously into the flanks of 7-week-old NOD/SCID male mice (Jackson Laboratory); all mice were implanted with 1 × 106 cells, except for in Fig. 4C, where mice were implanted with 5 × 105 cells to allow for a longer monitoring period. Tumor formation was monitored daily and measured weekly. When tumors reached 5% of body weight, mice were sacrificed and tumors were collected. Tumor volume was determined based on the following equation: volume = length × width2 × 0.52.
Anti-GRP78 AutoAb treatment in vivo
Tumor-bearing mice were weight-matched and randomized into treatment cohorts for weekly injections. Control human IgG or anti-GRP78 AutoAb concentrations were calculated based on a total blood volume of 2.2 ml as reported for this mouse strain by Jackson Laboratory: 168 μg [IgG or anti-GRP78 AutoAb]/2.2 ml [blood] = 76 μg/ml. The animal study was organized as follows: (i) PBS (0.1 ml), (ii) control human IgG, or (iii) anti-GRP78 AutoAbs (each 168 μg in 0.1 ml of PBS). Tumor formation was monitored daily and measured weekly. When tumors reached 5% of normal body weight, mice were sacrificed, and tumors were collected and processed for mRNA analysis. For the enoxaparin intervention study, the intervention cohort (n = 5) received an additional injection of enoxaparin sodium (Lovenox®; Sanofi Aventis) diluted in 0.9% sterile saline at a concentration of 1 mg/ml and administered intratumorally at 6 mg/kg/week. The control cohort (n = 5) received 100 μl of vehicle only (PBS).
RNA isolation and NanoString® analysis
Total RNA was isolated from flash-frozen mouse tumor tissues with the RNeasy lipid tissue minikit (catalog no. 74804, Qiagen). For quality assurance, only RNA samples with an RNA integrity number greater than 5 were used for NanoString® analysis. Data were normalized against seven housekeeping genes, namely IPO8, GUSB, TBP, YWHAZ, ACTB, GAPDH, and RPLP2. p values were corrected for multiple comparisons with the Benjamin–Hochberg procedure by nSolverTM software (NanoString Technologies, Seattle, WA), and heat maps were produced with Java Treeview software (Seattle, WA). Hierarchical gene clustering was performed by Euclidean distance and complete linkage analysis.
GRP78 immunofluorescence and analysis of csGRP78 by flow cytometry
DU145 cells were grown in chamber slides. Upon 50% confluence, cells were left untreated or treated with 50 μg/ml enoxaparin or 10 IU/ml of heparin for 4 h. Live cells were incubated with either anti-GRP78 AutoAbs (60 μg/ml) or PBS for 2 h. Cells were fixed in 4% paraformaldehyde, and anti-GRP78 AutoAbs were detected with the anti-human Alexa488 secondary antibody (Molecular Probes). Nuclei were visualized by DAPI (Sigma). Slides were imaged with an Olympus DSU microscope. For flow cytometry analysis, DU145 cells were harvested in Accutase (Sigma) and incubated with minimum essential medium supplemented with 30% FBS and 10 μg/ml purified human IgG (catalog no. I2511, Sigma) for 20 min at 4 °C. Cells were washed with ice-cold PBS, and 1 × 106-cell aliquots were incubated with 1 μg of phycoerythrin-conjugated human anti-GRP78 AutoAbs or phycoerythrin-conjugated isotype control for 40 min at 4 °C in 100 μl of PBS containing 0.5% BSA. After two washes, cells were resuspended in ice-cold PBS for flow cytometry analysis (AccuriC6, BD Biosciences). Data were analyzed by FlowJo version 10.0.8 (FlowJo LLC).
Immunohistochemical staining
Tissues from tumor-bearing mice were fixed in formalin and paraffin-embedded. Four-micrometer sections were deparaffinized and blocked with 5% normal goat (for Ki67 staining) or rabbit serum (for TF staining). Sections were incubated with primary antibody for 1 h at room temperature, followed by either goat anti-rabbit or rabbit anti-goat biotinylated secondary antibody (Vector Laboratories, Burlingame, CA) diluted 1:500 in 0.05 m Tris buffer, pH 7.5, for 30 min and subsequently streptavidin-HRP (Zymed Laboratories Inc.) diluted 1:20 for 5 min. Sections were developed in Nova Red HRP substrate (Vector Laboratories) and counterstained with hematoxylin. ImageJ software was used to determine the number of Ki67-positive cells as described (48).
Statistics
GraphPad software (Prism version 6.07) was used to analyze quantitative data. Correlation studies of clinical data were analyzed based on the Pearson correlation test. Significance of differences between controls and treatments was determined by analysis of variance. Upon finding significance with analysis of variance, an unpaired Student's t test was performed. p ≤ 0.05 was considered significant.
Author contributions
A. A. A. designed research studies, conducted experiments, acquired data, analyzed data, and wrote the manuscript. P. L. acquired data (in vivo). F. M. conducted experiments and acquired data (in vitro and in vivo). E. P. designed the experimental protocol. S. L. conducted experiments (immunohistochemistry). C. A. F. C. conducted experiments (immunofluorescence). J. H. P. provided reagents. M. G.-G. provided reagents and analyzed data. J. H. provided reagents. S. V. P., M. C., and A. K. provided reagents and analyzed data. J. R. wrote the manuscript and analyzed data. G. G. wrote the manuscript. S. D. conducted experiments (flow cytometry). S. M. wrote the manuscript. R. P. designed research studies, analyzing data, and wrote the manuscript. W. A. designed research studies, analyzed data, and wrote the manuscript. B. S. provided reagents, analyzed data, and wrote the manuscript. R. C. A. designed research studies, analyzed data, and wrote the manuscript.
Supplementary Material
Acknowledgments
Biological Materials were provided by the Ontario Tumor Bank, which is funded by the Ontario Institute for Cancer Research. We thank Life Science Editors for editing assistance.
This work was supported in part by Prostate Cancer Canada (PCC) Discovery Grants 2010-590 and D2017-1949 (to R. C. A.), McMaster Surgical Associate Grant 176725 (to B. S.), and research awards from the Gillson–Longenbaugh Foundation (to R. P. and W. A.). This work was also supported by St. Joseph's Healthcare Hamilton. R. Pasqualini and W. Arap have equity in Alvos Therapeutics (Arrowhead Research Corp.), which is subject to certain restrictions under university policy. The University of New Mexico Health Sciences Center manages and monitors the terms of these arrangements in accordance with its conflict-of-interest policy.
This article contains supplemental Table 1 and Fig. 1.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- PSA
- prostate-specific antigen
- ATF4 and ATF6
- activating transcription factors 4 and 6, respectively
- AutoAb
- autoantibody
- C/EBP
- CCAAT/enhancer-binding protein
- CHOP
- C/EBP homologous protein
- csGRP78
- cell-surface GRP78
- ER
- endoplasmic reticulum
- GRP78
- 78-kDa glucose-regulated protein
- IRE1α
- inositol-requiring enzyme-1α
- LMWH
- low-molecular weight heparin
- PERK
- protein kinase RNA-like endoplasmic reticulum kinase
- TF
- tissue factor
- Tg
- thapsigargin
- VEGFR2
- vascular endothelial growth factor receptor-2
- sXBP1
- spliced X-box–binding protein-1
- UPR
- unfolded protein response
- IHC
- immunohistochemical
- PDI
- protein-disulfide isomerase
- NOD/SCID
- non-obese diabetic/severe combined immunodeficiency.
References
- 1. Hoffman R. M., Meisner A. L., Arap W., Barry M., Shah S. K., Zeliadt S. B., and Wiggins C. L. (2016) Trends in United States prostate cancer incidence rates by age and stage, 1995–2012. Cancer Epidemiol. Biomarkers Prev. 25, 259–263 [DOI] [PubMed] [Google Scholar]
- 2. Abdulkadir S. A., Carvalhal G. F., Kaleem Z., Kisiel W., Humphrey P. A., Catalona W. J., and Milbrandt J. (2000) Tissue factor expression and angiogenesis in human prostate carcinoma. Hum. Pathol. 31, 443–447 [DOI] [PubMed] [Google Scholar]
- 3. Rak J., Milsom C., May L., Klement P., and Yu J. (2006) Tissue factor in cancer and angiogenesis: the molecular link between genetic tumor progression, tumor neovascularization, and cancer coagulopathy. Semin. Thromb. Hemost. 32, 54–70 [DOI] [PubMed] [Google Scholar]
- 4. Ni M., and Lee A. S. (2007) ER chaperones in mammalian development and human diseases. FEBS Lett. 581, 3641–3651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gonzalez-Gronow M., Selim M. A., Papalas J., and Pizzo S. V. (2009) GRP78: a multifunctional receptor on the cell surface. Antioxid. Redox Signal. 11, 2299–2306 [DOI] [PubMed] [Google Scholar]
- 6. Misra U. K., and Pizzo S. V. (2015) Activated α2-macroglobulin binding to human prostate cancer cells triggers insulin-like responses. J. Biol. Chem. 290, 9571–9587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gopal U., Gonzalez-Gronow M., and Pizzo S. V. (2016) Activated α2-macroglobulin regulates transcriptional activation of c-MYC target genes through cell surface GRP78 protein. J. Biol. Chem. 291, 10904–10915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Misra U. K., Payne S., and Pizzo S. V. (2011) Ligation of prostate cancer cell surface GRP78 activates a proproliferative and antiapoptotic feedback loop: a role for secreted prostate-specific antigen. J. Biol. Chem. 286, 1248–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jeffery C. J. (1999) Moonlighting proteins. Trends Biochem. Sci. 24, 8–11 [DOI] [PubMed] [Google Scholar]
- 10. Lee A. S. (2014) Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 14, 263–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Delpino A., Piselli P., Vismara D., Vendetti S., and Colizzi V. (1998) Cell surface localization of the 78 kD glucose regulated protein (GRP 78) induced by thapsigargin. Mol. Membr. Biol. 15, 21–26 [DOI] [PubMed] [Google Scholar]
- 12. Mintz P. J., Kim J., Do K. A., Wang X., Zinner R. G., Cristofanilli M., Arap M. A., Hong W. K., Troncoso P., Logothetis C. J., Pasqualini R., and Arap W. (2003) Fingerprinting the circulating repertoire of antibodies from cancer patients. Nat. Biotechnol. 21, 57–63 [DOI] [PubMed] [Google Scholar]
- 13. Cohen M., and Petignat P. (2011) Purified autoantibodies against glucose-regulated protein 78 (GRP78) promote apoptosis and decrease invasiveness of ovarian cancer cells. Cancer Lett. 309, 104–109 [DOI] [PubMed] [Google Scholar]
- 14. Arap M. A., Lahdenranta J., Mintz P. J., Hajitou A., Sarkis A. S., Arap W., and Pasqualini R. (2004) Cell surface expression of the stress response chaperone GRP78 enables tumor targeting by circulating ligands. Cancer Cell 6, 275–284 [DOI] [PubMed] [Google Scholar]
- 15. Ferrara F., Staquicini D. I., Driessen W. H., D'Angelo S., Dobroff A. S., Barry M., Lomo L. C., Staquicini F. I., Cardo-Vila M., Soghomonyan S., Alauddin M. M., Flores L. G. 2nd, Arap M. A., Lauer R. C., Mathew P., et al. (2016) Targeted molecular-genetic imaging and ligand-directed therapy in aggressive variant prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 10.1073/pnas.16154001135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Miao Y. R., Eckhardt B. L., Cao Y., Pasqualini R., Argani P., Arap W., Ramsay R. G., and Anderson R. L. (2013) Inhibition of established micrometastases by targeted drug delivery via cell surface-associated GRP78. Clin. Cancer Res. 19, 2107–2116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dobroff A. S., D'Angelo S., Eckhardt B. L., Ferrara F., Staquicini D. I., Cardo-Vila M., Staquicini F. I., Nunes D. N., Kim K., Driessen W. H., Hajitou A., Lomo L. C., Barry M., Krishnamurthy S., Sahin A., et al. (2016) Towards a transcriptome-based theranostic platform for unfavorable breast cancer phenotypes. Proc. Natl. Acad. Sci. U.S.A. 113, 12786–12791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Delie F., Petignat P., and Cohen M. (2012) GRP78 protein expression in ovarian cancer patients and perspectives for a drug-targeting approach. J. Oncol. 2012, 468615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Virrey J. J., Dong D., Stiles C., Patterson J. B., Pen L., Ni M., Schönthal A. H., Chen T. C., Hofman F. M., and Lee A. S. (2008) Stress chaperone GRP78/BiP confers chemoresistance to tumor-associated endothelial cells. Mol. Cancer Res. 6, 1268–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Papalas J. A., Vollmer R. T., Gonzalez-Gronow M., Pizzo S. V., Burchette J., Youens K. E., Johnson K. B., and Selim M. A. (2010) Patterns of GRP78 and MTJ1 expression in primary cutaneous malignant melanoma. Mod. Pathol. 23, 134–143 [DOI] [PubMed] [Google Scholar]
- 21. Rasche L., Duell J., Castro I. C., Dubljevic V., Chatterjee M., Knop S., Hensel F., Rosenwald A., Einsele H., Topp M. S., and Brändlein S. (2015) GRP78-directed immunotherapy in relapsed or refractory multiple myeloma: results from a phase 1 trial with the monoclonal immunoglobulin M antibody PAT-SM6. Haematologica 100, 377–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gonzalez-Gronow M., Cuchacovich M., Llanos C., Urzua C., Gawdi G., and Pizzo S. V. (2006) Prostate cancer cell proliferation in vitro is modulated by antibodies against glucose-regulated protein 78 isolated from patient serum. Cancer Res. 66, 11424–11431 [DOI] [PubMed] [Google Scholar]
- 23. Misra U. K., Gonzalez-Gronow M., Gawdi G., Hart J. P., Johnson C. E., and Pizzo S. V. (2002) The role of Grp 78 in α2-macroglobulin-induced signal transduction. Evidence from RNA interference that the low density lipoprotein receptor-related protein is associated with, but not necessary for, GRP 78-mediated signal transduction. J. Biol. Chem. 277, 42082–42087 [DOI] [PubMed] [Google Scholar]
- 24. Misra U. K., Gonzalez-Gronow M., Gawdi G., Wang F., and Pizzo S. V. (2004) A novel receptor function for the heat shock protein Grp78: silencing of Grp78 gene expression attenuates α2M*-induced signalling. Cell Signal. 16, 929–938 [DOI] [PubMed] [Google Scholar]
- 25. Al-Hashimi A. A., Caldwell J., Gonzalez-Gronow M., Pizzo S. V., Aboumrad D., Pozza L., Al-Bayati H., Weitz J. I., Stafford A., Chan H., Kapoor A., Jacobsen D. W., Dickhout J. G., and Austin R. C. (2010) Binding of anti-GRP78 autoantibodies to cell surface GRP78 increases tissue factor procoagulant activity via the release of calcium from endoplasmic reticulum stores. J. Biol. Chem. 285, 28912–28923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pootrakul L., Datar R. H., Shi S. R., Cai J., Hawes D., Groshen S. G., Lee A. S., and Cote R. J. (2006) Expression of stress response protein Grp78 is associated with the development of castration-resistant prostate cancer. Clin. Cancer Res. 12, 5987–5993 [DOI] [PubMed] [Google Scholar]
- 27. Daneshmand S., Quek M. L., Lin E., Lee C., Cote R. J., Hawes D., Cai J., Groshen S., Lieskovsky G., Skinner D. G., Lee A. S., and Pinski J. (2007) Glucose-regulated protein GRP78 is up-regulated in prostate cancer and correlates with recurrence and survival. Hum. Pathol. 38, 1547–1552 [DOI] [PubMed] [Google Scholar]
- 28. Uhlén M., Fagerberg L., Hallström B. M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A., Olsson I., Edlund K., Lundberg E., Navani S., Szigyarto C. A., et al. (2015) Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- 29. Epstein J. I., Amin M. B., Reuter V. E., and Humphrey P. A. (2017) Contemporary Gleason grading of prostatic carcinoma: an update with discussion on practical issues to implement the 2014 International Society of Urological Pathology (ISUP) Consensus Conference on Gleason Grading of Prostatic Carcinoma. Am. J. Surg. Pathol. 41, e1–e7 [DOI] [PubMed] [Google Scholar]
- 30. Teeter A. E., Presti J. C. Jr, Aronson W. J., Terris M. K., Kane C. J., Amling C. L., and Freedland S. J. (2013) Do nomograms designed to predict biochemical recurrence (BCR) do a better job of predicting more clinically relevant prostate cancer outcomes than BCR? A report from the SEARCH database group. Urology 82, 53–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. D'Amico A. V., Whittington R., Malkowicz S. B., Schultz D., Blank K., Broderick G. A., Tomaszewski J. E., Renshaw A. A., Kaplan I., Beard C. J., and Wein A. (1998) Biochemical outcome after radical prostatectomy, external beam radiation therapy, or interstitial radiation therapy for clinically localized prostate cancer. JAMA 280, 969–974 [DOI] [PubMed] [Google Scholar]
- 32. Vieira P., and Rajewsky K. (1988) The half-lives of serum immunoglobulins in adult mice. Eur. J. Immunol. 18, 313–316 [DOI] [PubMed] [Google Scholar]
- 33. de Ridder G. G., Gonzalez-Gronow M., Ray R., and Pizzo S. V. (2011) Autoantibodies against cell surface GRP78 promote tumor growth in a murine model of melanoma. Melanoma Res. 21, 35–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Milsom C., and Rak J. (2008) Tissue factor and cancer. Pathophysiol. Haemost. Thromb. 36, 160–176 [DOI] [PubMed] [Google Scholar]
- 35. Hansen L. K., O'Leary J. J., Skubitz A. P., Furcht L. T., and McCarthy J. B. (1995) Identification of a homologous heparin binding peptide sequence present in fibronectin and the 70 kDa family of heat-shock proteins. Biochim. Biophys. Acta 1252, 135–145 [DOI] [PubMed] [Google Scholar]
- 36. Ron D., and Walter P. (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529 [DOI] [PubMed] [Google Scholar]
- 37. Raiter A., Vilkin A., Gingold R., Levi Z., Halpern M., Niv Y., and Hardy B. (2014) The presence of anti-GRP78 antibodies in the serum of patients with colorectal carcinoma: a potential biomarker for early cancer detection. Int. J. Biol. Markers 29, e431–e435 [DOI] [PubMed] [Google Scholar]
- 38. Shao Q., Ren P., Li Y., Peng B., Dai L., Lei N., Yao W., Zhao G., Li L., and Zhang J. (2012) Autoantibodies against glucose-regulated protein 78 as serological diagnostic biomarkers in hepatocellular carcinoma. Int. J. Oncol. 41, 1061–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chinni S. R., Falchetto R., Gercel-Taylor C., Shabanowitz J., Hunt D. F., and Taylor D. D. (1997) Humoral immune responses to cathepsin D and glucose-regulated protein 78 in ovarian cancer patients. Clin. Cancer Res. 3, 1557–1564 [PubMed] [Google Scholar]
- 40. Quinones Q. J., de Ridder G. G., and Pizzo S. V. (2008) GRP78: a chaperone with diverse roles beyond the endoplasmic reticulum. Histol. Histopathol. 23, 1409–1416 [DOI] [PubMed] [Google Scholar]
- 41. Rutkowski D. T., Arnold S. M., Miller C. N., Wu J., Li J., Gunnison K. M., Mori K., Sadighi Akha A. A., Raden D., and Kaufman R. J. (2006) Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLos Biol. 4, e374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gifford J. B., Huang W., Zeleniak A. E., Hindoyan A., Wu H., Donahue T. R., and Hill R. (2016) Expression of GRP78, master regulator of the unfolded protein response, increases chemoresistance in pancreatic ductal adenocarcinoma. Mol. Cancer Ther. 15, 1043–1052 [DOI] [PubMed] [Google Scholar]
- 43. Van Hemelrijck M., Adolfsson J., Garmo H., Bill-Axelson A., Bratt O., Ingelsson E., Lambe M., Stattin P., and Holmberg L. (2010) Risk of thromboembolic diseases in men with prostate cancer: results from the population-based PCBaSe Sweden. Lancet Oncol. 11, 450–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fang C. H., Lin T. C., Guha A., Nemerson Y., and Konigsberg W. H. (1996) Activation of factor X by factor VIIa complexed with human-mouse tissue factor chimeras requires human exon 3. Thromb. Haemost. 76, 361–368 [PubMed] [Google Scholar]
- 45. Khorana A. A. (2009) Cancer and thrombosis: implications of published guidelines for clinical practice. Ann. Oncol. 20, 1619–1630 [DOI] [PubMed] [Google Scholar]
- 46. Mousa S. A., Linhardt R., Francis J. L., and Amirkhosravi A. (2006) Anti-metastatic effect of a non-anticoagulant low-molecular-weight heparin versus the standard low-molecular-weight heparin, enoxaparin. Thromb. Haemost. 96, 816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Caldwell J. A., Dickhout J. G., Al-Hashimi A. A., and Austin R. C. (2010) Development of a continuous assay for the measurement of tissue factor procoagulant activity on intact cells. Lab. Invest. 90, 953–962 [DOI] [PubMed] [Google Scholar]
- 48. Schneider C. A., Rasband W. S., and Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Staquicini D. I., D'Angelo S., Ferrara F., Karjalainen K., Sharma G., Smith T. L., Tarleton C. A., Jaalouk D. E., Kuniyasu A., Baze W. B., Chaffee B. K., Barnhart K. F., Koivunen E., Marchiò S., Sidman R. L., et al. (2017) Therapeutic targeting of membrane-associated GRP78 in leukemia and lymphoma: preclinical efficacy in vitro and formal toxicity study of BMTP-78 in rodents and nonhuman primates. Pharmacogenomics J., 10.1038/tpj.2017.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.