

Abstract
Protein chaperones play a critical role in proteostasis. The activity of the major endoplasmic reticulum chaperone BiP (GRP78) is regulated by Fic-mediated AMPylation during resting states. By contrast, during times of stress, BiP is deAMPylated. Here, we show that excessive AMPylation by a constitutively active FicE247G mutant is lethal in Drosophila. This lethality is cell-autonomous, as directed expression of the mutant FicE247G to the fly eye does not kill the fly but rather results in a rough and reduced eye. Lethality and eye phenotypes are rescued by the deAMPylation activity of wild-type Fic. Consistent with Fic acting as a deAMPylation enzyme, its activity was both time- and concentration-dependent. Furthermore, Fic deAMPylation activity was sufficient to suppress the AMPylation activity mediated by the constitutively active FicE247G mutant in Drosophila S2 lysates. Further, we show that the dual enzymatic activity of Fic is, in part, regulated by Fic dimerization, as loss of this dimerization increases AMPylation and reduces deAMPylation of BiP.
Keywords: dimerization, Drosophila, endoplasmic reticulum stress (ER stress), GRP78, unfolded protein response (UPR), AMPylation, BiP, Fic, deAMPylation
Introduction
AMPylation is a post-translational modification involving the covalent attachment of AMP to the hydroxyl group of a threonine, serine, or tyrosine residue. This modification was first discovered by Stadtman and colleagues (1) studying the regulation of glutamine synthetase by a post-translational modification with AMP. Interestingly, this modification can be reversed by a repeated catalytic site in the second domain of glutamine synthetase adenyltransferase (2, 3). Five decades later, AMPylation was rediscovered in studies with a bacterial virulence factor called VopS that encodes a Fic (filamentation-induced by cAMP) domain-containing protein (4). These identified AMPylators contain a Fic domain with a conserved HPFX(D/E)GN(G/K)R motif required for catalytic activity (5). Bacterial genomes appear to encode multiple Fic domain proteins with varied transferase activities (6, 7), whereas metazoan genomes encode only a single Fic domain–containing protein with a conserved topology and function.
In previous studies, we identified a Drosophila Fic domain-containing protein, Fic, that includes a single transmembrane domain upstream of conserved tetratricopeptide repeats (TPRs)2 and a Fic domain. These domains are highly conserved in metazoan Fic domain proteins and are oriented with the TPR and Fic domains located within the ER lumen (8). Loss of Drosophila Fic results in defects in synaptic transmission in the visual system (8). We observed that the ER-localized Drosophila Fic is responsible for AMPylation of the chaperone BiP (GRP78) (9). During the unfolded protein response (UPR), BiP is both deAMPylated and transcriptionally up-regulated to help cells cope with ER stress (10). We found that BiP is AMPylated by Fic in resting, unstressed cells and deAMPylated by Fic in cells responding to ER stress. Since these initial studies, other groups have observed AMPylation of BiP by conserved metazoan Fic proteins, known as FicD or HYPE in mammals and Fic-1 in Caenorhabditis elegans (11–13). Thus, BiP, like other chaperones, is regulated by ATP and substrate binding but additionally by transcription and the conserved post-translational modification AMPylation (9).
One key factor to understanding the regulation of AMPylation by Fic is the role of a conserved autoinhibitory helix found in most Fic domain proteins. This inhibitory helix contains a conserved glutamate residue that is critical for the regulation of AMPylation activity of Fic domain proteins (9, 14, 15). This glutamate typically forms a salt bridge with a conserved arginine in the Fic domain active site. The salt bridge between these glutamate and arginine residues blocks the coordination of the γ phosphate of ATP in the active site, thus inhibiting the AMPylation activity of the Fic domain (14). Mutation of this conserved glutamate in Fic (FicE247G in Drosophila) and in other eukaryotic Fic proteins results in constitutively active AMPylating enzymes with apparent reduced substrate specificity (9, 12, 16). However, it is unclear how the inhibitory salt bridge is regulated in vivo to permit AMPylation of substrates, including BiP. Structural studies have suggested that a dimerization event might be involved in the regulation of Fic-mediated AMPylation activity (12, 15). In humans and C. elegans, mutations in the predicted dimer interface of FicD and Fic-1 resulted in decreased auto-AMPylation, indicating dimerization may enhance AMPylation activity (12, 15).
To further investigate the regulation of Fic activity in vivo, we generated transgenes for the expression of wild-type and mutant Fic proteins. We found that expression of constitutively active FicE247G has no detectable phenotype in flies, as long as endogenous Fic is also present. By contrast, FicE247G overexpression in a fic null fly is lethal. Similarly, eye-specific expression of FicE247G caused a rough eye phenotype, but only in the absence of endogenous Fic. We hypothesize that the FicE247G toxicity in fic null flies is due to constitutive AMPylation without corresponding deAMPylation activity mediated by wild-type Fic. Supporting this hypothesis, we observed deAMPylation activity by wild-type Fic and found that this deAMPylation activity by wild-type Fic is active in the presence of the constitutive AMPylating activity of FicE247G. Furthermore, we found that dimerization of Fic was not essential for its deAMPylation activity, as a single mutation (I271D) in Fic that disrupts dimerization retains the ability to deAMPylate endogenous BiP. Interestingly, we found that disruption of Fic dimerization leads to increased AMPylation of recombinant BiP, indicating that dimerization could play a regulatory role in this enzymatic activity. In summary, we demonstrate that the balance of AMPylation and deAMPylation by Fic is essential for cell survival and is regulated, in part, by the dimerization state of Fic.
Results
Endogenous Fic expression is protective against the constitutive AMPylating activity of FicE247G
To study the effect of Fic in Drosophila, we used the CRISPR-Cas9 system to generate a fic null mutant, referred to as fic30C. The CRISPR-Cas9–induced fic30C indel introduced a 101-base pair deletion resulting in a frameshift and early stop codon after leucine 155 of the coding sequence, creating a null fic allele (Fig. 1A and supplemental Fig. S1). As observed previously with other fic mutants, homozygous fic30C flies are viable with no externally visible phenotypes (8). Next, to assess the effect of Fic expression, we generated transgenic flies with C-terminal V5 and His6-tagged Fic proteins under control of the Gal4/UAS system. All UAS-Fic transgenes were inserted at the same locus on chromosome 3. These flies, in contrast to flies previously generated with randomly inserted UAS-Fic transgenes (8), exhibit undistinguishable expression levels for all three Fic proteins (Fig. 1B).
Figure 1.

Recessive toxicity of FicE247G expression. A, structure of the Drosophila fic gene and protein with the regulatory Glu-247, dimerization Ile-271, and catalytic His-375 residues. The CRISPR/Cas9-induced fic30C mutation induces a 101-base pair deletion, generating an early stop codon N-terminal to the conserved TPR domain. B, a representative Western blot shows expression of UAS-Fic transgenes (wild type, H375A, and E247G) under control of the ubiquitous Da-Gal4 driver in a wild-type background. Quantification of Western blots from three separate experiments showed no significant difference in expression of the UAS-Fic transgenes. C, Da-Gal4 homozygous females were crossed to the indicated UAS-Fic/TM6b, Hu males either in a wild-type or fic30C background. The graph represents percentage of progeny inheriting the indicated UAS-Fic transgene together with the Gal4 driver. The expected Mendelian percentage is 50%. Bars, mean of three independent crosses; dots, values for independent crosses; error bars, S.D. The total number of flies scored for each genotype was at least 150. D, representative SEM images of eyes expressing the indicated UAS-Fic transgene with LongGMR-Gal4 in a wild-type or fic30C background. Scale bar, 100 μm. E, representative SEM images of eyes expressing the indicated UAS-Fic transgene along with LongGMR-Gal4, UAS-FicE247G in a fic30C background. Scale bar, 100 μm.
We initially tested the effect of Fic transgene overexpression on the viability of wild-type flies. Flies homozygous for the ubiquitous Da-Gal4 driver on chromosome 3 (Da-Gal4/Da-Gal4) were crossed to flies containing the chromosome 3 balancer TM6B, Hu and one copy of a UAS-Fic transgene encoding either wild-type Fic, constitutively active FicE247G, or catalytically inactive FicH375A. From this cross, 50% of progeny were expected to be flies ubiquitously expressing the transgene. Viability of flies was determined by scoring adult flies for the presence or absence of a visible dominant chromosome 3 marker (TM6B, Hu). In these experiments, no difference was observed between the expected and observed numbers of flies expressing the wild-type UAS-Fic transgene compared with those with the balancer chromosome (Fig. 1C). Likewise, expression of either UAS-FicE247G or UAS-FicH375A transgenes under control of the Da-Gal4 driver did not affect viability of flies in a fic wild-type background.
Next, we assessed the effect of Fic transgene overexpression in fic30C null flies. For this purpose, fic30C/30C; Da-Gal4/Da-Gal4 flies were crossed to fic30C/30C; UAS-Fic/TM6B flies, fic30C/30C; UAS-FicH375A/TM6B flies, or fic30C/30C; UAS-FicE247G/TM6B flies. From these crosses, 50% of progeny were expected to be flies ubiquitously expressing the transgene. As observed in a fic wild-type background, we found no difference between the expected and observed viability of fic30C flies expressing the UAS-Fic or UAS-FicH375A transgenes under Da-Gal4 control (Fig. 1C). However, expression of the UAS-FicE247G transgene under the ubiquitous Da-Gal4 promoter was lethal in the fic30C null background. These data indicate that expression of the wild-type Fic protein from the endogenous fic locus is sufficient to suppress lethality caused by FicE247G overexpression.
Tissue-specific expression of FicE247G exhibits cell autonomous and recessive toxicity
To compare the effect of tissue-specific expression of the overactive FicE247G with wild-type Fic, we used the photoreceptor-specific LongGMR-Gal4 promoter for specific expression in the eye. This allowed us to detect eye-specific phenotypes in the surviving adult flies. Consistent with the results from ubiquitous expression, LongGMR-Gal4–driven expression of either wild-type Fic or FicE247G transgenes resulted in no externally visible defects in a fic wild-type background (Fig. 1D). Expression of wild-type UAS-Fic in the fic30C null flies did not interfere with normal eye development. However, in fic30C null flies, expression of the overactive AMPylating FicE247G mutant resulted in flies with a severe eye phenotype characterized by smaller eyes with disturbed ommatidial structures. To confirm that this rough eye defect was caused by the loss of wild-type Fic in the fic30C null flies, we tested the effect of LongGMR-Gal4, UAS-FicE247G expression in hemizygous fic30C/Df(2L)BSC296 flies (supplemental Fig. S1). As observed in fic30C null flies, overexpression of UAS-FicE247G in the eye resulted in a rough eye phenotype.
Because the presence of endogenous Fic in flies appears to counterbalance the lethality and eye morphology defects of UAS-FicE247G expression, we next asked whether overexpression of UAS-Fic or UAS-FicH375A transgenes would rescue the rough eye phenotype of fic30C; LongGMR-Gal4, UAS-FicE247G flies (Fig. 1E). As in flies expressing endogenous Fic, overexpression of the wild-type UAS-Fic transgene results in normal eye morphology. However, overexpression of the catalytically dead UAS-FicH375A transgene failed to rescue the rough eye phenotype.
We hypothesized that the lethality and rough eye phenotypes of UAS-FicE247G are caused by toxicity of this enzyme. To test this, TUNEL assays were performed on late third instar larval eye discs of wild-type and fic30C null flies expressing UAS-Fic or UAS-FicE247G transgenes under the control of LongGMR-Gal4 (supplemental Fig. S2). As predicted, eye discs from fic30C animals expressing UAS-FicE247G were heavily positive for TUNEL staining, indicating cell death. However, eye discs from wild-type and fic30C animals in which wild-type UAS-Fic was expressed show no significant TUNEL staining.
These experiments indicate that expression of the overactive AMPylating FicE247G in the absence of wild-type Fic is toxic to cells. We have observed in previous in vitro studies that FicE247G is an enzyme with hyperactivity and reduced specificity (9). Indeed, other in vitro and structural studies have also found that mutations to the autoinhibitory helix of eukaryotic Fic enzymes result in overactive and promiscuous AMPylators (11, 12). Furthermore, the nature of the developmental eye defects under control of LongGMR-Gal4 suggests that the toxicity of dFicE247G is cell-autonomous. Taken together, these data indicate that the catalytic activity of wild-type Fic protein counterbalances the unregulated AMPylating activity of the FicE247G mutant.
Recombinant Fic mediates deAMPylation of in vitro AMPylated BiP
Recently, it was found that FicD, the mammalian homolog of Fic, could both AMPylate and deAMPylate BiP and that disruption of the autoinhibitory α helix of FicD could inhibit the deAMPylation activity of FicD (17). Based on these recent findings and our fly studies, we hypothesized that both the AMPylation and deAMPylation activity of Fic are conserved functions for this enzyme and are essential for proper regulation and survival of a cell.
To address this hypothesis, we investigated whether an AMPylated BiP substrate could be deAMPylated by Fic. For these assays, recombinant [32P]AMP-BiPT229A was generated by in vitro AMPylation of purified His6-BiPT229A, a mutant with diminished ATP hydrolysis activity (18). We compared the in vitro deAMPylation activity of recombinant wild-type Fic and the catalytically dead FicH375A using [32P]AMP-BiPT229A as a substrate (Fig. 2A). The addition of recombinant wild-type Fic resulted in a concentration-dependent reduction of the [32P]AMP signal from [32P]AMP-BiPT229A protein (Fig. 2A, lanes 2–5), whereas the addition of the catalytically dead FicH375A did not reduce this signal (Fig. 2A, lanes 6–9). This indicates that wild-type Fic, but not FicH375A, is able to deAMPylate recombinant AMPylated BiP.
Figure 2.

Fic-mediated deAMPylation of BiP. A, Fic-mediated deAMPylation of in vitro AMPylated BiPT229A ([α-32P]AMP) was assayed by autoradiograph. A representative Coomassie Blue gel and autoradiograph of concentration-dependent deAMPylation activity of HIS-FicΔ70 (lanes 2–5) and HIS-FicΔ70H375A (lanes 6–9) are shown. B, Fic-mediated deAMPylation of endogenous BiP from S2 lysates was assayed by Western blotting. Shown is a representative Western blot of S2 lysates untreated (lanes 1–4), treated with CHX (lanes 5–8), or treated with DTT (lanes 9–12) for 4 h. S2 lysates were incubated with recombinant enzymes: GST-FicΔ70 (lanes 2, 6, and 10), GST-FicΔ70E247G (lanes 3, 7, and 11), or GST-FicΔ70H375A (lanes 4, 8, and 12). The asterisk indicates a nonspecific band. Blots were probed with anti-AMP-threonine, anti-BiP, and anti-GST antibodies. C, concentration-dependent deAMPylation activity of GST-FicΔ70 (lanes 2–8) was assayed by Western blotting. The asterisk indicates a nonspecific band. Blots were probed with anti-AMP-threonine, anti-BiP, and anti-GST antibodies. D, time-dependent deAMPylation activity of 50 nm GST-FicΔ70 (lanes 1–7) was assayed by Western blotting. Blots were probed with anti-AMP-threonine, anti-BiP, and anti-GST antibodies. E, representative Western blot of GST-FicΔ70-mediated deAMPylation of endogenous BiP in the presence of 250 nm ATP and GST-FicΔ70 (lane 2), GST-FicΔ70E247G (lane 3), or equimolar GST-FicΔ70 and GST-FicΔ70E247G (lane 4). Blots were probed with anti-AMP-threonine, anti-BiP, and anti-dFic antibodies.
Recombinant Fic mediates deAMPylation of AMPylated BiP in cell lysates
Next, we asked whether the addition of recombinant Fic could deAMPylate endogenous BiP from Drosophila Schneider 2 (S2) lysates. AMPylation of endogenous BiP was observed in S2 lysates by Western blot analysis using the anti-AMP-threonine antibody (19) (Fig. 2B). As observed previously, treating cells with cyclohexamide (CHX) increased levels of BiP AMPylation in S2 cells due to the fact that this compound reduces ER stress by halting protein translation (Fig. 2B, lanes 5–8) (9). By contrast, treatment of S2 cells with DTT, which reduces disulfide bonds, causes up-regulation of the UPR, resulting in the loss of BiP AMPylation (Fig. 2B, lanes 9–12) (9).
To validate our Western blot detection of AMPylation and to determine the site(s) on which endogenous BiP was AMPylated in S2 cells, we purified endogenous BiP using concanavalin A beads and performed LC-MS/MS analysis to identify AMPylated residues (supplemental Fig. S3). From this analysis, we found that endogenous BiP was AMPylated at a single site, Thr-518, a conserved threonine in the substrate-binding domain. This site has previously been identified in mammalian cells (13). In earlier studies, we identified Thr-366, a conserved residue in the ATPase domain of BiP, as a site of AMPylation (9); however, under our current conditions, only Thr-518 AMPylation was observed on endogenous BiP in these lysates. Of note, the Thr-518 AMPylation was detected in untreated and CHX-treated S2 cells but not found in DTT-treated S2 cells, consistent with our Western blot analysis of BiP AMPylation (9) (Fig. 2B).
To determine whether Fic has deAMPylation activity, we tested the ability of recombinant protein to remove AMPylation from endogenous BiP in Drosophila (S2) lysates. Recombinant proteins were added to solubilized organelle fractions of lysates and incubated for 1 h at 30 °C. In both untreated and CHX-treated cells, the addition of wild-type recombinant Fic resulted in the loss of BiP AMPylation, as indicated by Western blot analysis using the anti-AMP-threonine antibody (Fig. 2B, lanes 2 and 6). No difference was observed in DTT-treated samples, as no AMPylated BiP was present to be modified (Fig. 2B, lane 10). The addition of recombinant overactive AMPylating FicE247G or the catalytically dead FicH375A mutant proteins did not alter the steady-state levels of BiP AMPylation in these lysates (Fig. 2B, lanes 3, 7, and 11 and lanes 4, 8, and 12, respectively). We also tested the ability of recombinant Fic to deAMPylate BiP from lysates treated with tunicamycin, a UPR-inducing drug, and 4-phenylbutyrate, an ER stress inhibitor (supplemental Fig. S4). We found that the addition of recombinant wild-type Fic to lysates treated with 4-phenylbutyrate did result in loss of BiP AMPylation, whereas tunicamycin-treated lysates contained no observed AMPylated BiP to modify (supplemental Fig. S4).
Of note, the recombinant FicE247G remained auto-AMPylated when added to the lysate, indicating that endogenous Fic did not recognize FicE247G as a substrate for deAMPylation (Fig. 2B, lanes 3, 7, and 11). Another interesting observation is that recombinant Fic is able to efficiently deAMPylate BiP from CHX-treated lysates. Under CHX conditions, endogenous Fic becomes an AMPylator of BiP. This suggests that in vivo some other factor(s) control the enzymatic activity of Fic and the balance of BiP AMPylation. In vitro, the additional factor(s) may not be present or may be unable to efficiently regulate the recombinant Fic added to these lysates.
Fic-mediated deAMPylation is enzymatic
The fact that the catalytic histidine His-375 is required for deAMPylation of BiP strongly suggests that the deAMPylation of BiP is an enzymatic process rather than a block of Fic AMPylation activity. To determine whether the deAMPylation activity of Fic is enzymatic, deAMPylation of endogenous BiP was assayed over a range of concentrations and times. The levels of observed endogenous BiP AMPylation decreased with the addition of higher concentrations of recombinant Fic (Fig. 2C). Furthermore, when a moderate concentration of Fic (50 nm) was added to the lysate and incubated over an extended period of time, the levels of BiP AMPylation decreased in a time-dependent manner (Fig. 2D). These data show a time and concentration dependence of the deAMPylation activity mediated by Fic, thereby supporting our proposal that Fic is an enzyme that deAMPylates the modified BiP substrate.
Fic deAMPylation can compete with unregulated FicE247G AMPylation in cell lysates
Interestingly, no increased BiP AMPylation was observed in any of the lysates incubated with FicE247G (Fig. 2B, lanes 3, 7, and 11). We predict that the lack of additional AMPylation could be explained by the lack of additional sources of ATP and the presence of low levels of deAMPylation activity by endogenous Fic. This would be consistent with our observations in wild-type flies, where the activity of overexpressed exogenous FicE247G is overcome by low levels of endogenous wild-type Fic (Fig. 1). Because these lysates contain very low levels of Fic (9), a small amount of Fic could indeed counterbalance the unregulated AMPylating activity of FicE247G without altering the appropriate levels of BiP AMPylation in the cell, as observed in flies overexpressing the UAS-FicE247G transgene.
We predicted that the addition of an exogenous ATP source may alter the balance of AMPylation and deAMPylation activity when FicE247G is added to S2 lysate. Consistent with this hypothesis, when ATP was added to S2 lysates, the addition of recombinant FicE247G resulted in an observed increase in BiP AMPylation (Fig. 2E, lane 3), whereas the addition of recombinant Fic was still able to efficiently deAMPylate BiP (Fig. 2E, lane 2). To determine whether recombinant Fic could also compete with the AMPylation activity of recombinant FicE247G, equal amounts of recombinant wild-type Fic and FicE247G were incubated in S2 lysates supplemented with ATP (Fig. 2E, lane 4). The addition of Fic together with FicE247G to the S2 lysate resulted in deAMPylation of endogenous BiP, indicating that recombinant wild-type Fic can outcompete FicE247G. Because FicE247G retains high AMPylating activity, the ability of Fic to counterbalance FicE247G enzymatic activity indicates that the deAMPylation activity of Fic is also quite high in S2 lysates. Furthermore, the apparent increased efficacy of endogenous Fic also suggests that additional factors or modifications to endogenous Fic must occur in the ER to control the activity of this bifunctional enzyme.
We also investigated whether competition between wild-type Fic and constitutively active FicE247G occurs during in vitro AMPylation of a substrate. For these assays, purified recombinant BiP ATPase domain, His6-BiP(27–407), was used as a substrate for Fic-mediated AMPylation, as this was previously shown to be modified (9). AMPylation assays were performed by incubating FicE247G with BiP(27–407) in AMPylation buffer with ATP at 30 °C for 1 h. After incubation with 100 nm FicE247G, AMPylation of 4 μm BiP(27–407) was detected by Western blot analysis using the anti-AMP-Thr antibody (supplemental Fig. S5 (A and B), lane 3). To validate our Western blot detection of BiP(27–407) AMPylation, we performed LC-MS/MS analysis on BiP(27–407) from these AMPylation assays. LC-MS/MS analysis confirmed that BiP(27–407) is predominantly AMPylated at Ser-365/Thr-366 (supplemental Fig. S5C) with other, less abundant AMPylation sites also identified in the sample (supplemental Table S1). This is consistent with our previously published observations on Fic in vitro AMPylation of BiP (9).
To determine whether recombinant Fic could compete with FicE247G for BiP(27–407) in vitro, increasing concentrations (from 25 to 800 nm) of Fic were added to AMPylation reactions containing 100 nm FicE247G and 4 μm BiP(27–407) (supplemental Fig. S5A, lanes 4–9). The addition of increasing amounts of Fic to the reaction resulted in decreasing BiP(27–407) AMPylation (supplemental Fig. S5A, lanes 4–9). As a control, we observed no effect on the in vitro AMPylation of BiP(27–407) treated with increasing concentrations of FicH375A (supplemental Fig. S5B, lanes 4–9).
Appreciably more Fic was required to completely counteract the AMPylation of BiP(27–407) by FicE247G in vitro. One possible explanation for this is that FicE247G is a more efficient enzyme than wild-type Fic in vitro. This somewhat contradicts our observation that endogenous Fic can counterbalance the toxic effects of transgenic expression of FicE247G in vivo. Of note, in previous studies, we observed that the truncated form of BiP is a better in vitro substrate for AMPylation than full-length BiP (9). Therefore, deAMPylation of this truncated form of BiP could be somewhat compromised. Alternatively, Fic deAMPylation activity may be enhanced by other factors in vivo, such as changes in post-translational modifications or effects on the dimerization state of the Fic enzyme that are not present in this in vitro reaction.
Fic AMPylation of BiP is attenuated by Fic dimerization
Because the AMPylation activity of human FicD and C. elegans Fic-1 was shown to be reduced in dimerization mutants (12, 15), we wanted to know whether the AMPylation and deAMPylation activity of Drosophila Fic was also affected by dimerization. To test this, we mutated an isoleucine (Ile-271) in the predicted dimer interface of Fic shown to be critical for dimerization in C. elegans Fic-1 and human FicD (12, 15). To prevent dimerization artifacts from the GST tag, we utilized an N-terminal His6-tagged Fic expression system. As predicted, purified Fic and FicI271D eluted by size exclusion chromatography as a dimer and monomer, respectively (Fig. 3A). This was also true for purified FicE247G and FicE247G,I271D. Furthermore, native gel migration of purified FicI271D and FicE247G,I271D was significantly faster than Fic, FicE247G, and FicH375A, consistent with a change in the oligomeric state of these mutants (Fig. 3B).
Figure 3.

Fic AMPylation of full-length BiP is inhibited by dimerization. A, representative traces of FPLC elution of His6-FicΔ70, His6-FicΔ70I271D, His6-FicΔ70E247G, and His6-FicΔ70E247G,I271D. Molecular mass standards alcohol dehydrogenase (150 kDa), conalbumin (75 kDa), ovalbumin (43 kDa), and ribonuclease (13.7 kDa) are labeled in gray. B, representative immunoblots of His6-FicΔ70, His6-FicΔ70I271D, His6-FicΔ70E247G, His6-FicΔ70E247G,I271D, and His6-FicΔ70H375A from 6% native polyacrylamide gel and 8% SDS-polyacrylamide gel. Blots were probed with anti-dFic antibody. C, representative immunoblots of auto-AMPylation of His6-FicΔ70E247G (EG, lane 1) and His6-FicΔ70E247G,I271D (EGID, lane 2). Blots were probed with anti-AMP-threonine and anti-dFic antibodies. D, representative Coomassie Blue gel and autoradiographs of AMPylation assays in which 4 μm His6-BiPT229A was used as a substrate for varying concentrations of His6-FicΔ70 (lanes 2–4), His6-FicΔ70I271D (lanes 5–7), His6-FicΔ70E247G (lanes 8–10), and His6-FicΔ70E247G,I271D (lanes 11–13). Short and long exposures of autoradiographs are shown to display dynamic range of each enzyme activity.
Initially, we tested the AMPylation activity of our dimer mutant. We hypothesized that, as observed in human and C. elegans, disruption of Fic dimerization would lead to decreased AMPylation activity of the enzyme. Because wild-type Fic exhibits very low AMPylation activity, we compared the AMPylation activity of the constitutively active AMPylating mutant FicE247G with its corresponding dimerization mutant FicE247G,I271D. We observed that auto-AMPylation levels of recombinant FicE247G,I271D were markedly lower than recombinant FicE247G by Western blot analysis (Fig. 3C). To determine whether dimerization of Fic alters its AMPylation activity for the substrate BiP, AMPylation assays were performed in the presence of [α-32P]ATP. Several concentrations (100–400 nm) of recombinant Fic, FicI271D, FicE247G, and FicE247G,I271D were added to 4 μm His6-BiPT229A and incubated for 30 min at 30 °C (Fig. 3D). Incubation of BiP without Fic enzyme or with wild-type Fic showed no [32P]BiP AMPylation signal by autoradiograph (Fig. 3D, lanes 1–4). However, incubation of BiP with FicE247G showed a clear [32P]BiP AMPylation signal (Fig. 3D, lanes 8–10), indicating that FicE247G is an active AMPylating enzyme. Strikingly, the addition of FicI271D to BiP also resulted in [32P]BiP AMPylation signal (Fig. 3D, lanes 5–7), indicating that FicI271D is also an active AMPylating enzyme. Furthermore, the addition of the FicE247G,I271D mutant resulted in a substantially stronger [32P]BiP signal than either FicE247G or FicI271D (Fig. 3D, lanes 11–13), suggesting that the AMPylating activity of the double mutant is significantly higher than that of either mutant alone. This also suggests that the regulatory effects of the autoinhibitory helix salt bridge and the dimerization interface have independent but synergistic effects on Fic AMPylation activity.
Fic deAMPylation activity does not require dimerization
Next, we tested the deAMPylation activity of our dimer mutant on AMPylated BiP. Because both enzymatic activities of Fic use the same active site, we predicted that, like AMPylation, the deAMPylation activity of Fic would also be altered by the disruption of dimerization. To determine whether dimerization is required for deAMPylation activity, we performed in vitro deAMPylation assays using recombinant [32P]AMP-BiPT229A as a substrate as described above. Recombinant Fic, FicI271D, FicE247G, FicE247G,I271D, and FicH375A were each added to [32P]AMP-BiPT229A and incubated for 60 min in the absence of ATP (Fig. 4A). In the samples treated with wild-type Fic or the monomeric FicI271D, the [32P]AMP signal from [32P]AMP-BiPT229A was greatly reduced compared with the no-enzyme control (Fig. 4A, lanes 1–3), whereas the FicE247G-, FicE247G,I271D-, and FicH375A-treated samples showed no reduction in [32P]AMP signal of BiPT229A (Fig. 4A, lanes 4–6). Both wild-type Fic and monomeric FicI271D were further tested in a concentration-dependent manner, and we observed that the addition of these enzymes resulted in a concentration-dependent reduction of the [32P]AMP signal from [32P]AMP-BiPT229A protein (Fig. 4B). However, the enzymatic activity of monomeric FicI271D appeared to be slightly compromised, as more enzyme was required to fully deAMPylate the substrate (Fig. 4B, lanes 6–9) compared with wild-type Fic (Fig. 4B, lanes 2–5).
Figure 4.
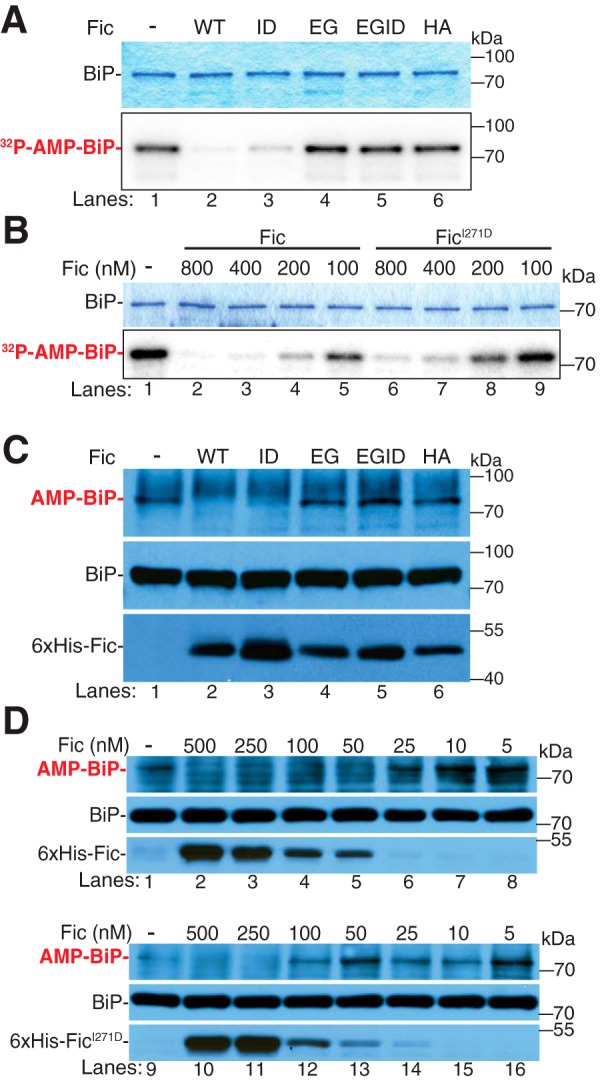
Fic deAMPylation is independent of dimerization. A, representative Coomassie Blue gel and autoradiographs of deAMPylation assays in which in vitro AMPylated BiP ([α-32P]AMP) was incubated with recombinant enzymes: His6-FicΔ70 (WT, lane 2), His6-FicΔ70I271D (ID, lane 3), His6-FicΔ70E247G (EG, lane 4), His6-FicΔ70E247G,I271D (EGID, lane 5), or GST-FicΔ70H365A (HA, lane 6). B, representative Coomassie Blue gel and autoradiographs of deAMPylation assays in which in vitro AMPylated BiP ([α-32P]AMP) was used as a substrate for varying concentrations of His6-FicΔ70 (lanes 2–5) and His6-FicΔ70I271D (lanes 6–9). C, representative Western blot of S2 lysates incubated with recombinant enzymes: His6-FicΔ70 (WT, lane 2), His6-FicΔ70I271D (ID, lane 3), His6-FicΔ70E247G (EG, lane 4), His6-FicΔ70E247G,I271D (EGID, lane 5), or GST-FicΔ70H365A (HA, lane 6). Blots were probed with anti-AMP-threonine, anti-BiP, and anti-dFic antibodies. D, concentration-dependent deAMPylation activities of His6-FicΔ70 (lanes 2–8) and His6-FicΔ70I271D (lanes 10–16) were assayed by Western blotting. Blots were probed with anti-AMP-threonine, anti-BiP, and anti-dFic antibodies.
To determine whether dimerization is required for deAMPylation of endogenous BiP, recombinant His6-Fic proteins were incubated with organelle fractions of Drosophila S2 cell lysates for 1 h at 30 °C (Fig. 4C). Consistent with the observed deAMPylation activity toward recombinant BiP, both wild-type Fic and the monomeric FicI271D proteins were able to deAMPylate endogenous BiP, whereas FicE247G, FicE247G,I271D, and FicH375A were not able to deAMPylate BiP. Again, compared with wild-type Fic, the deAMPylation activity of FicI271D is slightly reduced, as higher concentrations of FicI271D enzyme are required to completely deAMPylate endogenous BiP (Fig. 4D). When a limiting concentration of FicI271D (50 nm) is added to the lysate and incubated for 90 min, the levels of BiP AMPylation decreased in a time-dependent manner (supplemental Fig. S6, lanes 8–14); however, deAMPylation of BiP is not taken to completion as was observed with wild-type Fic (supplemental Fig. S6, lanes 1–7). Taken together, these data indicate that FicI271D retains its deAMPylation activity despite also having increased AMPylation activity.
Fic dimerization is not required to rescue FicE247G toxicity
Because FicE247G and FicE247G,I271D both display increased AMPylation activity, we wanted to know the effects of these mutant proteins in vivo. We generated UAS-FicI271D and UAS-FicE247G,I271D transgenic flies with C-terminal V5 and His6 tags as described above. As with UAS-Fic, UAS-FicE247G, and UAS-FicH375A, Western analysis of the monomeric UAS-FicI271D and UAS-FicE247G,I271D proteins indicates stable expression of these transgenes in flies (Fig. 5A).
Figure 5.
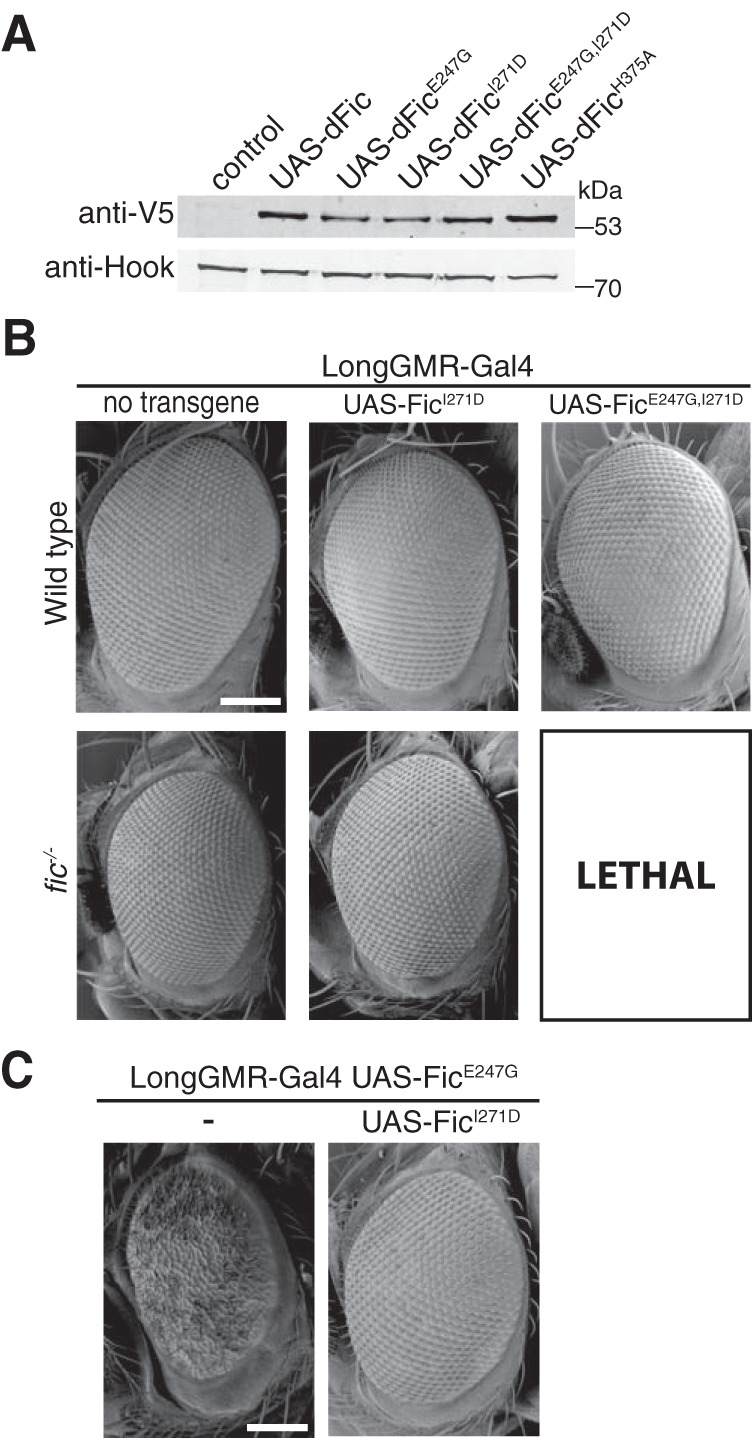
Fic dimerization is not required to rescue FicE247G toxicity. A, a representative Western blot shows expression of UAS-Fic transgenes (Fic, FicE247G, FicI271D, FicE247G,271D, FicH375A) under control of the ubiquitous Da-Gal4 driver in a wild-type background. B, representative SEM images of eyes expressing the indicated UAS-Fic transgene with LongGMR-Gal4 in a wild-type or fic30C background. Scale bar, 100 μm. C, representative SEM images of eyes expressing the indicated UAS-Fic transgene along with LongGMR-Gal4, UAS-FicE247G in a fic30C background. Scale bar, 100 μm.
Next, we assessed the effect of Fic transgene overexpression in fic30C null fly eyes. Consistent with the results from previous experiments, LongGMR-Gal4–driven expression of either FicE247G or FicE247G,I271D transgenes resulted in no externally visible defects in a fic wild-type background (Fig. 5B). Furthermore, expression of the monomeric FicI271D in fic30C null flies did not interfere with normal eye development. However, expression of the UAS-FicE247G,I271D transgene under the LongGMR-Gal4 promoter was lethal in the fic30C null background. Overexpression of toxic proteins under the LongGMR-Gal4 promoter has previously been shown to cause lethality, most likely due to LongGMR-Gal4 expression in larval and pupal neurons outside of the visual system (20). The lethality of UAS-FicE247G,I271D under the LongGMR-Gal4 promoter suggests that the observed increase in in vitro AMPylation is retained in vivo because corresponding UAS-FicE247G flies are viable in the same genetic background. Furthermore, these data indicate that expression of the wild-type Fic protein from the endogenous fic locus is sufficient to suppress lethality caused by FicE247G,I271D overexpression, supporting the hypothesis that both deAMPylation and AMPylation are required for cell survival.
Next, we asked whether overexpression of the UAS-FicI271D transgene would rescue the rough eye phenotype of fic30C; LongGMR-Gal4, UAS-FicE247G flies (Fig. 5C). Again, as with the UAS-Fic transgene (Fig. 1E), overexpression of the monomeric UAS-FicI271D transgene suppressed the UAS-FicE247G–induced eye phenotype. Taken together, these data suggest that the remaining deAMPylation activity of FicI271D is sufficient to counterbalance the enhanced AMPylation activity of this enzyme in vivo.
Discussion
Overall, our studies have shown that uncontrolled Fic-mediated AMPylation is lethal, and endogenous wild-type Fic can rescue flies from this lethality. Additionally, we have demonstrated that deAMPylation is likely to be the default activity of wild-type Fic and that this enzyme must change in a way so that net AMPylation of a substrate can occur. In these studies, we have also found that Fic dimerization attenuates the AMPylation activity of Fic toward its substrate BiP but is not critical for the deAMPylation activity of Fic.
Wild-type Fic appears to principally exhibit deAMPylation activity. During low ER stress, the activity of Fic appears as an AMPylator, thereby promoting the AMPylation of a portion of the available BiP and generating a subpopulation of inactive BiP that is held in reserve in the ER. When ER stress is elevated, Fic-mediated deAMPylation of BiP during ER stress would then increase the pool of active BiP, allowing for a more rapid and more acute response to stress. It is important to note that BiP resides in multiple conformational states (21) that are regulated by ER stress, which may also impact its accessibility as a Fic substrate.
Exactly how this activity is switched in vivo has yet to be identified; however, there are two independent mutations in Fic that alter its AMPylation and deAMPylation activity on BiP (Fig. 6). The first mutation, FicE247G, is a well-characterized overactive AMPylating mutant. Mutation of this conserved glutamate results in the disruption of an inhibitory salt bridge in the active site that promotes the proper coordination of ATP in the binding pocket and AMPylation activity (9, 14, 15). Importantly, disruption of this salt bridge also completely disrupts deAMPylation activity of the enzyme. The second mutation, FicI271D, has been observed in this study and by others to disrupt homodimerization of Fic (12, 15). Disruption of Fic dimerization increases AMPylation activity for its substrate BiP while reducing Fic auto-AMPylation and without significantly attenuating Fic's deAMPylation activity. Interestingly, despite the increased AMPylation activity of FicI271D, flies overexpressing this mutant do not display the toxicity associated with FicE247G. This observation further supports the hypothesis that both deAMPylation and AMPylation are required for cell survival. Indeed, these two mutations appear to work by distinct molecular mechanisms, as combining these two mutations resulted in a synergistic effect on the AMPylation activity of FicE247G,I71D.
Figure 6.
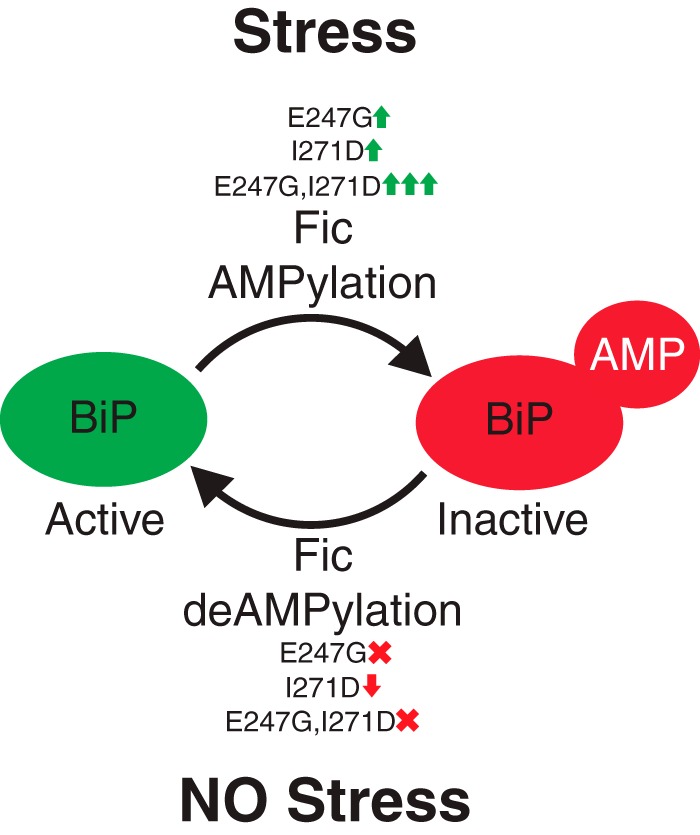
Model of Fic AMPylation and deAMPylation activity. See “Discussion” for details.
It is intriguing that dimerization would inhibit the AMPylation activity of Fic but not significantly alter the deAMPylation activity of this enzyme. It is tempting to speculate that the dimerization state of Fic could be a key regulatory factor working with another unknown component or modification in vivo. Future studies on how the bimodal activities of Fic are regulated within the ER will be critical to understanding how Fic regulation of BiP is controlled in the ER.
Experimental procedures
Cell culture and fly work
Drosophila melanogaster Schneider 2 (S2) cells were cultured with standard protocols and grown at 27 °C in Schneider's medium supplemented with 10% fetal bovine serum and antibiotics (22). Flies were maintained using standard conditions. The Bloomington Stock Center provided Da-Gal4 (BS 55850) and LongGMR-Gal4 (BS 8121) driver lines as well as the fic deficiency stock Df(fic) = Df(2L)BSC296 and OreR.
Fly stocks
OreR, fic30C, Df(2L)BSC296 (Bloomington Stock #25720), w;; Da-Gal4 (Bloomington Stock #55850), w;; LongGMR-Gal4 (Bloomington Stock #8121), w;; UAS-V5-FicWT_attP-B3/TM6B,hu, w;; UAS-V5-FicE247G_attP-B3/TM6B,hu, w;; UAS-V5-FicH375A_attP-B3/TM6B,hu, w;; UAS-V5-FicI271D_attP-B3/TM6B,hu, w;; UAS-V5-FicE247G,I271D_attP-B3/TM6B,hu, w; fic30C; Da-Gal4,w; fic30C; LongGMR-Gal4, w;fic30C; UAS-V5-FicWT_attP-B3/TM6B,hu, w; fic30C; UAS-V5-FicE247G_attP-B3/TM6B,hu, w; fic30C; UAS-V5-FicH375A_attP-B3/TM6B,hu, w; fic30C; UAS-V5-FicI271D_attP-B3/TM6B,hu, w; fic30C/CyO; UAS-V5-FicE247G,I271D_attP-B3/TM6B,hu, w;; LongGMR-Gal4,UAS-V5-FicWT_attP-B3/TM6B,hu, w;; LongGMR-Gal4,UAS-V5-FicE247G_attP-B3/TM6B,hu, w; fic30C; LongGMR-Gal4,UAS-V5-FicWT_attP-B3/TM6B,hu, w; fic30C; LongGMR-Gal4,UAS-V5-FicE247G_attP-B3/TM6B,hu, w; fic30C/Df(2L)BSC296; LongGMR-Gal4,UAS-V5-FicE247G_attP-B3/TM6B,hu.
Generation of fic30C null mutation
CRISPR-mediated mutagenesis (23) was performed with tools and the protocol available from the O'Connor-Giles, Wildonger, and Harrison laboratories (23). Mutagenesis was performed in ywv;;Nanos-Cas9 using two gRNAs targeting fic-Exon1 (gRNA1, CCGGAAGTCCTACTGCGCTA; gRNA2, ACCGTCAGCGCACGGCAGA). Injections were performed by Rainbow Transgenic Services (Camarillo, CA). The surviving F1 progenies were crossed to balancer stocks, and subsequently, potential founder F2 males were individually crossed to the balancer stock and screened for CRISPR-induced indels. Mutations were detected by PCR amplification of the target region, reannealing, T7 endonuclease treatment, and polyacrylamide gel electrophoresis analysis of potential cleavage products. Sanger sequencing of PCR products detected the precise sequence of candidate deletions.
Generation of UAS-V5-Fic constructs
Fic cDNA sequences (FicWT, FicE247G, and FicH375A) were subcloned into a pUASt vector modified by insertion of an AttB sequence, sequence-verified, and injected into embryos (BestGene, Chino Hills, CA) at the 62E1 landing site (24). To generate the FicI271D and FicE247G,I271D mutants, gBlocks (IDT, Coralville, IA) containing the respective mutant sequences were synthesized, subcloned into the pUASt-V5-Fic-AttB vector via the HiFi Assembly Kit (New England Biolabs, Ipswich, MA), and injected into embryos at the same site. Da-Gal4–driven expression levels of all constructs were determined by Western blotting. In brief, five flies were homogenized in 200 μl of lysis buffer (10% SDS, 6 m urea, and 50 mm Tris-HCl, pH 6.8, plus 10% DTT), boiled for 2 min, and spun for 10 min at 20,000 × g to remove debris. 10 μl were separated by SDS-PAGE and transferred to nitrocellulose membranes. Blots were probed with anti-V5 (1:3000; R960-25 Invitrogen) and anti-Hook (1:8000 (25)) and detected using fluorescence-labeled antibodies and an Odyssey scanner (LI-COR Biosciences, Lincoln, NE).
Survival analysis of flies expressing FicE247G
To assess the lethality of unregulated Fic, we expressed C-terminally V5-His6-tagged UAS-Fic, UAS-FicH375A, or UAS-FicE247G via the ubiquitous Da-Gal4 driver in either wild-type or fic30C null flies. Offspring were scored, and the number of adults with both Da-Gal4 and the UAS-Fic variant were compared with the number of heterozygous sibling controls. Percent of expected was calculated from the ratio of recovered flies of the relevant genotypes compared with half the total number (number observed/(total number/2)). Crosses were repeated three times. The total number of flies scored was at least 150 for all crosses.
Electron microscopy
SEM of fly eyes were obtained as described previously (26, 27). In short, eyes were fixed in 2% paraformaldehyde, 2% glutaraldehyde, 0.2% Tween 20, and 0.1 m cacodylate buffer, pH 7.4, for 2 h. Samples were washed four times with increasing ethanol (25–100%) for 12 h each followed by a series of hexamethyldisilazane washes (25–100% in ethanol) for 1 h each. Flies were air-dried overnight and mounted on SEM stubs, and the bodies were coated in fast-drying silver paint. Flies were sputter-coated with a gold/pallidum mixture for 60 s and imaged at ×900 magnification, with extra high tension set at 3.0 kV on a scanning electron microscope (SIGMA, Zeiss). Stereoscopic light images were acquired on a V12 stereoscope (Zeiss) at ×100 zoom. Stacks of ∼30 × 10-μm optical sections were merged and filtered with CZFocus software (Zeiss).
TUNEL staining
Cell death was detected with the TUNEL method as described previously (28). Briefly, eye discs from wandering third instar larvae were dissected into PBS, fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 in PBS (PBST), and incubated in TUNEL reaction mixture (In Situ Cell Death Detection Kit-TMR Red, Roche Applied Science) for 2 h. Tissues were mounted in Vectashield with DAPI (Vectorlabs, Burlingame, CA). Stacks of 20 × 1-μm confocal sections were acquired with an LSM 710 confocal microscope (Carl Zeiss) and equally processed for brightness and contrast using ImageJ (National Institutes of Health).
Plasmid construction
Plasmids for bacterial expression of His6-Δ26BiP, His6-BiP(27–407), GST-FicΔ70, GST-FicΔ70E247G, and GST-FicΔ70H375A were generated previously (9). Plasmids for bacterial expression of His6-FicΔ70, His6-FicΔ70E247G, and His6-FicΔ70H375A were generated by subcloning the Fic genes from GST-FicΔ70, GST-FicΔ70E247G, and GST-FicΔ70H375A, respectively, into the pet28A vector using BamHI and NotI sites. Plasmids for bacterial expression of His6-FicΔ70I271D and His6-FicΔ70E247G,I271D were generated by PCR amplification and ligation of His6-FicΔ70 and His6-FicΔ70E247G using primers 5′-GACGGCAAATCCGATGACGAGCACAACGAGATCTTGGGCATGGATCTGGC-3′ and 5′-TACGGCCATGCGGGTCTCTAGG-3′. Plasmids for bacterial expression of His6-Δ26BiPT229A were generated by PCR amplification and ligation of His6-Δ26BiP using primers 5′-GCCTTCGATGTCTCCCTGCTGACC-3′ and 5′-GCCGCCGCCCAAATCGAAC-3′.
Purification of recombinant proteins
For protein purification, plasmids for expression of His6-Δ26BiPT229A, His6-BiP(27–407), His6-FicΔ70, His6-FicΔ70E247G, His6-FicΔ70H375A, His6-FicΔ70I271D, and His6-FicΔ70E247G,I271D were transformed into Rosetta cells, grown to mid-log phase (A600 ∼0.5), and induced with 400 μm isopropyl β-d-1-thiogalactopyranoside at 23 °C for 4 h. Plasmids for expression of GST, GST-FicΔ70, GST-FicΔ70E247G, and GST-FicΔ70H375A were transformed into Rosetta cells, grown to mid-log phase (A600 ∼0.5), and induced with 400 μm isopropyl β-d-1-thiogalactopyranoside at 37 °C for 2 h. Cells were lysed using a cell disrupter (Emulsiflex-C3), and proteins were purified using standard nickel-affinity (Thermo Scientific) and GST-affinity (Thermo Scientific) purification protocols. FPLC of His6-FicΔ70, His6-FicΔ70E247G, His6-FicΔ70I271D, and His6-FicΔ70E247G,I271D was performed on a Superdex 200 10/300 column (GE Healthcare).
AMPylation and deAMPylation of BiP ATPase domain
In vitro AMPylation and deAMPylation assays of purified His6-BiP(27–407) were performed as described previously (9). Recombinant His6-BiP(27–407) proteins and recombinant GST-FicΔ70, GST-FicΔ70E247G, and GST-FicΔ70H375A were used as substrates and enzymes, respectively. Reactions were performed in AMPylation buffer (20 mm Tris, pH 7.5, 100 mm NaCl, 10 mm MgCl2, 5 mm MnCl2, 5 mm CaCl2, 250 μm ATP) at 30 °C for 1 h. Reactions were stopped by the addition of SDS sample buffer. Samples were analyzed by Western blot analysis using the anti-AMP-Thr antibody (19) and anti-BiP antibody (Abcam MAC143). Detection was performed by standard HRP methods.
In vitro AMPylation and deAMPylation of endogenous BiP
In vitro deAMPylation of endogenous BiP was performed on ER/mitochondrial fractions of S2 cells. To induce ER stress, S2 cells were treated with 5 mm DTT for 4 h. To reduce ER stress, S2 cells were treated with 100 μg/ml CHX for 4 h. S2 cells were harvested with cell scrapers, suspended in a HNMEK lysis buffer (20 mm HEPES, pH 7.4, 50 mm NaCl, 2 mm MgCl2, 2 mm EDTA, 10 mm KCl, 20 mm NaF, 50 nm EGTA, protease inhibitors), and lysed using a Dounce homogenizer. Lysates were centrifuged at 500 × g for 10 min at 4 °C to remove nuclei and cellular debris. The supernatant was collected and centrifuged at 10,000 × g for 10 min to pellet ER and mitochondrial membranes. ER/mitochondrial fractions were washed once in HNMEK lysis buffer and centrifuged at 10,000 × g for 10 min. ER/mitochondrial pellets were suspended in deAMPylation buffer (50 mm HEPES, pH 7.4, 5 mm MgCl, 100 mm KCl, 1 mm CaCl2, 0.1% Triton). Recombinant GST-FicΔ70, GST-FicΔ70E247G, GST-FicΔ70H375A, His6-FicΔ70, His6-FicΔ70E247G, His6-FicΔ70H375A, His6-FicΔ70I271D, and His6-FicΔ70E247G,I271Dwere used as enzymes. Reactions were performed at a final concentration of ∼4 mg/ml of total cellular protein at 30 °C for 1 h. For in vitro AMPylation of endogenous BiP, reactions were performed in deAMPylation buffer supplemented with 250 μm ATP.
In vitro AMPylation and deAMPylation of recombinant BiP
In vitro AMPylation of recombinant His6-Δ26BiPT229A was performed as follows. Recombinant His6-Δ26BiPT229A was used as a substrate for the enzymes His6-FicΔ70, His6-FicΔ70E247G, His6-FicΔ70H375A, His6-FicΔ70I271D, and His6-FicΔ70E247G,I271D. Reactions were performed in deAMPylation buffer supplemented with 250 mm cold ATP and ∼0.5 μCi of [α-32P]ATP at 30 °C for 30 min. Reactions were stopped by the addition of SDS sample buffer. Samples were analyzed by SDS-PAGE stained with Coomassie Brilliant Blue, and 32P radioactive signal was detected with a Typhoon FLA 7000 (GE Healthcare) after ∼18-h exposure to storage phosphor screens.
For in vitro deAMPylation of recombinant [32P]AMP-BiPT229A, His6-Δ26BiPT229A was first in vitro AMPylated and repurified as described previously (17). Briefly, 10 μg of recombinant His6-Δ26BiPT229A preincubated with 15 μm cold ATP was added to 1 μg of GST-FicΔ70E247G and 20 μCi of [α-32P]ATP in deAMPylation buffer and incubated at room temperature for 1 h. Reactions were stopped by the addition of 400 μl of deAMPylation buffer supplemented with 500 mm KCl. The AMPylated His6-Δ26BiPT229A was repurified using a standard nickel-affinity (Thermo Scientific) and GST-affinity (Thermo Scientific) purification protocols and buffer-exchanged in an Amicon Ultra 0.5-ml 30-kDa centrifugal filter (Millipore Sigma). For in vitro deAMPylation reactions, recombinant His6-FicΔ70, His6-FicΔ70E247G, His6-FicΔ70H375A, His6-FicΔ70I271D, and His6-FicΔ70E247G,I271D were used as enzymes. Reactions were performed at a final substrate concentration of 800 nm cold AMP-BiPT229A and 80 nm radiolabeled [32P]AMP-BiPT229A in each reaction. Reactions were performed in deAMPylation buffer at room temperature for 1 h. Samples were analyzed by SDS-PAGE stained with Coomassie Blue, and 32P radioactive signal was detected with a Typhoon FLA 7000 (GE Healthcare) after ∼18-h exposure to storage phosphor screens.
Mass spectrometry analysis
Samples were run on SDS-polyacrylamide gels and stained with Coomassie Blue dye to isolate proteins before analysis by mass spectrometry. Sample preparation included reduction and alkylation of proteins using DTT and iodoacetamide, overnight digestion with trypsin, and desalting via solid-phase extraction. LC-MS/MS experiments were carried out on a Thermo Scientific EASY-nLC 1200 liquid chromatography system coupled to a Thermo Scientific Orbitrap Fusion Lumos mass spectrometer. MS1 spectra were acquired in the Orbitrap (resolution = 120,000). Peptide fragmentation subsequently took place via high-energy collision-induced dissociation, and MS2 spectra were acquired in the ion trap. Resulting MS/MS spectral data were searched using the Mascot search engine (Matrix Science) for peptide identification. Additionally, AMPylation PTM assignments were manually verified from fragmentation spectra. The spectral count totals displayed in supplemental Table S1 refer to the number of MS/MS spectra identified with a Mascot score above a threshold of 15.
Concanavalin A purification of BiP for mass spectrometry
Concanavalin A pulldowns were performed on ER/mitochondrial fractions of S2 cells prepared as described above. ER/mitochondrial fractions were suspended in RIPA buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 5 mm MgCl2, 5 mm MgCl2, 5 mm CaCl2, 1 mm PMSF, protease inhibitor mixture) for 30 min on ice and centrifuged at 10,000 × g for 10 min at 4 °C. The soluble lysate was incubated with equilibrated concanavalin A beads overnight at 4 °C. The beads were washed three times with RIPA buffer. To elute endogenous BiP from the concanavalin A beads, the pulldowns were incubated with RIPA buffer + 5 mm ATP at room temperature for 15 min. Eluate was transferred to a new tube, TCA-precipitated, and suspended in SDS loading buffer for SDS-PAGE and Western blot analysis.
Author contributions
A. K. C. conceived and executed experiments, analyzed data, and drafted and revised the manuscript. A. T. M. conceived and executed experiments, analyzed data, and revised the manuscript. J. Z. and K. A. S. performed mass spectrometric analysis of protein samples and revised the manuscript. H. K. and K. O. conceived experiments, analyzed data, and revised the manuscript.
Supplementary Material
Acknowledgments
We thank the members of the Orth and Krämer laboratories for discussion and technical assistance. We thank the Bloomington Stock Center (National Institutes of Health Grant P40OD018537) for flies, Lauren Tyra for performing the SEM experiments, and the Molecular and Cellular Imaging Facility at the University of Texas Southwestern Medical Center for help with electron microscopy.
This work was funded by National Institutes of Health (NIH) Grant R01 GM115188, Welch Foundation Grant I-1561, and Once Upon a Time … Foundation (to K. O.), NIH Grants EY010199 and EY021922 (to H. K.), and National Science Foundation Graduate Research Fellowship 1000176311 (to A. T. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Table S1 and Figs. S1–S6.
- TPR
- tetratricopeptide repeat
- ER
- endoplasmic reticulum
- UPR
- unfolded protein response
- CHX
- cyclohexamide
- SEM
- scanning electron microscopy
- RIPA
- radioimmune precipitation assay
- UAS
- upstream activating sequence.
References
- 1. Kingdon H. S., Shapiro B. M., and Stadtman E. R. (1967) Regulation of glutamine synthetase, VIII. ATP: glutamine synthetase adenylyltransferase, an enzyme that catalyzes alterations in the regulatory properties of glutamine synthetase. Proc. Natl. Acad. Sci. U.S.A. 58, 1703–1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Anderson W. B., and Stadtman E. R. (1970) Glutamine synthetase deadenylylation: a phosphorolytic reaction yielding ADP as nucleotide product. Biochem. Biophys. Res. Commun. 41, 704–709 [DOI] [PubMed] [Google Scholar]
- 3. Brown M. S., Segal A., and Stadtman E. R. (1971) Modulation of glutamine synthetase adenylylation and deadenylylation is mediated by metabolic transformation of the P11-regulatory protein. Proc. Natl. Acad. Sci. U.S.A. 68, 2949–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yarbrough M. L., Li Y., Kinch L. N., Grishin N. V., Ball H. L., and Orth K. (2009) AMPylation of Rho GTPases by Vibrio VopS disrupts effector binding and downstream signaling. Science 323, 269–272 [DOI] [PubMed] [Google Scholar]
- 5. Harms A., Stanger F. V., and Dehio C. (2016) Biological diversity and molecular plasticity of FIC domain proteins. Annu. Rev. Microbiol. 70, 341–360 [DOI] [PubMed] [Google Scholar]
- 6. Garcia-Pino A., Zenkin N., and Loris R. (2014) The many faces of Fic: structural and functional aspects of Fic enzymes. Trends Biochem. Sci. 39, 121–129 [DOI] [PubMed] [Google Scholar]
- 7. Roy C. R., and Cherfils J. (2015) Structure and function of Fic proteins. Nat. Rev. Microbiol. 13, 631–640 [DOI] [PubMed] [Google Scholar]
- 8. Rahman M., Ham H., Liu X., Sugiura Y., Orth K., and Krämer H. (2012) Visual neurotransmission in Drosophila requires expression of Fic in glial capitate projections. Nat. Neurosci. 15, 871–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ham H., Woolery A. R., Tracy C., Stenesen D., Krämer H., and Orth K. (2014) Unfolded protein response-regulated Drosophila Fic (dFic) protein reversibly AMPylates BiP chaperone during endoplasmic reticulum homeostasis. J. Biol. Chem. 289, 36059–36069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kozutsumi Y., Segal M., Normington K., Gething M. J., and Sambrook J. (1988) The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 332, 462–464 [DOI] [PubMed] [Google Scholar]
- 11. Sanyal A., Chen A. J., Nakayasu E. S., Lazar C. S., Zbornik E. A., Worby C. A., Koller A., and Mattoo S. (2015) A novel link between Fic (filamentation induced by cAMP)- mediated adenylylation/AMPylation and the unfolded protein response. J. Biol. Chem. 290, 8482–8499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Truttmann M. C., Cruz V. E., Guo X., Engert C., Schwartz T. U., and Ploegh H. L. (2016) The Caenorhabditis elegans protein FIC-1 is an AMPylase that covalently modifies heat-shock 70 family proteins, translation elongation factors and histones. PLoS Genet. 12, e1006023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Preissler S., Rato C., Chen R., Antrobus R., Ding S., Fearnley I. M., and Ron D. (2015) AMPylation matches BiP activity to client protein load in the endoplasmic reticulum. Elife 4, e12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Engel P., Goepfert A., Stanger F. V., Harms A., Schmidt A., Schirmer T., and Dehio C. (2012) Adenylylation control by intra- or intermolecular active-site obstruction in Fic proteins. Nature 482, 107–110 [DOI] [PubMed] [Google Scholar]
- 15. Bunney T. D., Cole A. R., Broncel M., Esposito D., Tate E. W., and Katan M. (2014) Crystal structure of the human, FIC-domain containing protein HYPE and implications for its functions. Structure 22, 1831–1843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Truttmann M. C., Zheng X., Hanke L., Damon J. R., Grootveld M., Krakowiak J., Pincus D., and Ploegh H. L. (2017) Unrestrained AMPylation targets cytosolic chaperones and activates the heat shock response. Proc. Natl. Acad. Sci. U.S.A. 114, E152–E160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Preissler S., Rato C., Perera L., Saudek V., and Ron D. (2017) FICD acts bifunctionally to AMPylate and de-AMPylate the endoplasmic reticulum chaperone BiP. Nat. Struct. Mol. Biol. 24, 23–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gaut J. R., and Hendershot L. M. (1993) Mutations within the nucleotide binding site of immunoglobulinbinding protein inhibit ATPase activity and interfere with release of immunoglobulin heavy chain. J. Biol. Chem. 268, 7248–7255 [PubMed] [Google Scholar]
- 19. Hao Y. H., Chuang T., Ball H. L., Luong P., Li Y., Flores-Saaib R. D., and Orth K. (2011) Characterization of a rabbit polyclonal antibody against threonine-AMPylation. J. Biotechnol. 151, 251–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ray M., and Lakhotia S. C. (2015) The commonly used eye-specific sev-GAL4 and GMR-GAL4 drivers in Drosophila melanogaster are expressed in tissues other than eyes also. J. Genet. 94, 407–416 [DOI] [PubMed] [Google Scholar]
- 21. Preissler S., Chambers J. E., Crespillo-Casado A., Avezov E., Miranda E., Perez J., Hendershot L. M., Harding H. P., and Ron D. (2015) Physiological modulation of BiP activity by trans-protomer engagement of the interdomain linker. Elife 4, e08961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ceriani M. F. (2007) Basic protocols for Drosophila S2 cell line: maintenance and transfection. Methods Mol. Biol. 362, 415–422 [DOI] [PubMed] [Google Scholar]
- 23. Gratz S. J., Cummings A. M., Nguyen J. N., Hamm D. C., Donohue L. K., Harrison M. M., Wildonger J., and O'Connor-Giles K. M. (2013) Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics 194, 1029–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Venken K. J. T., He Y., Hoskins R. A., and Bellen H. J. (2006) P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science 314, 1747–1751 [DOI] [PubMed] [Google Scholar]
- 25. Krämer H., and Phistry M. (1996) Mutations in the Drosophila hook gene inhibit endocytosis of the boss transmembrane ligand into multivesicular bodies. J. Cell Biol. 133, 1205–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wolff T. (2011) Preparation of Drosophila eye specimens for scanning electron microscopy. Cold Spring Harb. Protoc. 2011, 1383–1385 [DOI] [PubMed] [Google Scholar]
- 27. Nandi N., Tyra L. K., Stenesen D., and Krämer H. (2014) Acinus integrates AKT1 and subapoptotic caspase activities to regulate basal autophagy. J. Cell Biol. 207, 253–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vasudevan D., and Ryoo H. D. (2016) Detection of cell death in Drosophila tissues. Methods Mol. Biol. 1419, 131–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.