

Abstract
Objectives
Paragangliomas of the head and neck and cranial base are typically benign, slow‐growing tumors arising within the jugular foramen, middle ear, carotid bifurcation, or vagus nerve proper. The objective of this study was to provide a comprehensive characterization of our institutional experience with clinical management of these tumors and posit an algorithm for diagnostic evaluation and treatment.
Methods
This was a retrospective cohort study of patients undergoing treatment for paragangliomas of the head and neck and cranial base at our institution from 2000–2017. Data on tumor location, catecholamine levels, and specific imaging modalities employed in diagnostic work‐up, pre‐treatment cranial nerve palsy, treatment modality, utilization of preoperative angiographic embolization, complications of treatment, tumor control and recurrence, and hereditary status (ie, succinate dehydrogenase mutations) were collected and summarized.
Results
The mean (SD) age of our cohort was 51.8 (±16.1) years with 123 (63.4%) female patients and 71 (36.6%) male patients. Catecholamine‐secreting lesions were found in nine (4.6%) patients. Fifty‐one patients underwent genetic testing, with mutations identified in 43 (20 SDHD, 13 SDHB, 7 SDHD, 1 SDHA, SDHAF2, and NF1). Observation with serial imaging, surgical extirpation, radiation, and stereotactic radiosurgery were variably employed as treatment approaches across anatomic subsites.
Conclusion
An algorithmic approach to clinical management of these tumors, derived from our longitudinal institutional experience and current empiric evidence, may assist otolaryngologists, radiation oncologists, and geneticists in the care of these complex neoplasms.
Level of Evidence
4
Keywords: Paraganglioma, glomus, succinate dehydrogenase
INTRODUCTION
Paragangliomas are rare, hypervascular neoplasms arising from neural crest‐derived cell clusters in the head and neck/cranial base, mediastinum, abdomen, and pelvis.1 With an overall incidence of 1 in 30,000–100,000, head and neck paragangliomas (HNPGLs) arise, in order of decreasing frequency, from the carotid body (carotid body paragangliomas, CBP), jugular bulb (JP), vagus nerve (cranial nerve [CN] X; VP), the tympanic branch of the glossopharyngeal (CN IX) or auricular branch of CN X (TP), and the cervical sympathetic chain (SCP).2
As many as 40% of HNPGLs are now known to arise in patients with a hereditary predisposition. Most commonly implicated are germline mutations in one of the four subunits (A‐D) of the succinate dehydrogenase complex of the mitochondrial electron transport chain and its flavination co‐factor (SDHA‐D, SDHAF2, collectively referred to as SDHx).3, 4 Screening for pathologic SDHx mutations in all patients with incident HNPGL diagnosis is now the standard of care.5 Though HNPGLs are infrequent secretors (< 3% of tumors) of catecholamines, laboratory evaluation of plasma and/or urine catecholamine metabolites should be included in the diagnostic workup to identify the rare secreting HNPGL and, far more importantly, a concomitant pheochromocytoma.6 Contrast‐enhanced cross‐sectional imaging by computer axial tomography (CT) or magnetic resonance imaging (MRI) of the head and neck/cranial base are essential tools to delineate the extent of disease, including great vessel involvement, expected CN anatomy, temporal bone or intracranial extension of HNPGL, and to exclude the presence of multifocal tumors.7, 8
The principal treatment approaches for HNPGL include surgery, fractionated radiation, and stereotactic radiosurgery, although observation with serial imaging may also be considered.9 Recommendations for HNPGL treatment depend on a number of factors. First, HNPGLs exhibit an indolent growth pattern, with an average tumor‐doubling time of 4.2–5.5 years,10, 11 and are seldom malignant.12 Second, fractionated radiation therapy (eg, 45–50 Gy), stereotactic radiosurgery (SRS), and surgery offer comparable 10‐year control rates, HNPGL‐specific survival, and distant‐metastasis free survival, all in excess of 97%.13, 14 As operative morbidity to CNs VII–XII is often unavoidable, radiation or SRS may offer improved functional outcomes with regard to speech, swallowing, and facial nerve and shoulder function. As such, a steadily‐increasing proportion of patients with HNPGL are being treated with non‐curative or debulking surgery with preservation of CNs followed by adjuvant radiation, or exclusively by non‐surgical therapies.15, 16 Finally, patient‐related factors, including age and goals of care, medical comorbidities, and genetic status also serve as critical factors to consider before embarking on any specific treatment path.
Ultimately, clinical management of these tumors remains challenging owing to heterogeneity in clinical presentation, existence of multiple treatment alternatives, and potential to cause serious detriment to critical functions of the head and neck.17 Herein, we provide a report of our institutional experience with 194 patients with HNPGLs from 2000–2017, with an emphasis on our approach to clinical evaluation, treatment, and genetic investigation of CBP, JP, VP, TP, SCP, and multi‐focal HNPGL patient cohorts. Finally, we posit an evidence‐based algorithm for multi‐disciplinary clinical evaluation and management of these diverse neoplasms.
MATERIALS AND METHODS
Following approval by the University of Michigan Institutional Review Board (HUM00120115), a retrospective medical record review was conducted of 194 patients with 233 paragangliomas (228 HNPGL, five concomitant mediastinal/abdominal paragangliomas or pheochromocytoma) who underwent treatment at our institution from 2000–2017. DataDirect18 and EMERSE19 search functions were utilized to identify patients from the electronic health record for inclusion in our study. Data on patient demographics, tumor characteristics, initial CN deficits and signs and symptoms at presentation, diagnostic evaluation, treatment, follow‐up, and results of genetic evaluations were collected.
HNPGL size was determined by anatomic pathology or head and neck MRI, in cases of non‐surgical treatment approaches. Tumors were considered functional (ie, catecholamine‐secreting) when patients had plasma or urine metanephrine levels elevated ≥1.5 times our laboratory's reference range. Duration of clinical follow‐up was defined as the time interval from the date of treatment (ie, surgery or final dose of radiation) or first patient encounter (for patients observed) to the last clinical evaluation or patient death.
Descriptive statistics and student's t‐test (two‐tailed, α = 0.05) were performed using SPSS (Version 22.0, Chicago, Illinois, U.S.A.).
RESULTS
Patient Demographics & Paraganglioma Localization
The mean (SD) age of our cohort was 51.8 (±16.1) years with a range of 13.7 to 85.2 years. Of 194 patients, 123 (63.4%) were female and 71 (36.6%) were male. The distribution of HNPGL sub‐sites is depicted in Figure 1, panel A. A patient index and detailed localization of multi‐focal and/or metastatic paragangliomas is presented in Figure 1, panel B.
Figure 1.

Localization of paraganglioma (s) of the head and neck in 194 patients at our institution from 2000–2017. Panel A: Frequency histogram depicting total patients and patients with confirmed mutations in SDHx gene by paraganglioma location (Other paraganglioma: includes parotid gland/CN VII paraganglioma, infratemporal‐pterygopalatine paraganglioma, and gangliocytic paraganglioma of skull base. Panel B: Index and localization of 24 patients with multi‐focal or metastatic paragangliomas. bilat = bilateral; CBP = carotid body paragangliomal; CNM = cervical nodal metastases; FLM = frontal lobe metastases; GPP = glossopharyngeal paraganglioma; JP = jugular paraganglioma; L = left‐sided; MP = mediastinal paraganglioma; PAP = para‐aortic paraganglioma; Pheo = pheochromocytoma; PM = pulmonary metastases; R = right‐sided; SCP = sympathetic chain paraganglioma; SGP = supraglottic paraganglioma; SLNP = superior laryngeal nerve paraganglioma; TP = tympanic paraganglioma; VM = vertebral body metastases; VP = vagal paraganglioma. † Paragangliomas developed sequentially. ‡ Metastatic disease
Genetic Testing for Hereditary Paraganglioma‐Pheochromocytoma Syndromes
Genetic testing for SDHx variants become widely adopted only after 2008. In our entire cohort of HNPGL patients, 51 (26.3%) underwent genetic counseling and testing, 44 of which were diagnosed with HNPGL in 2008 or later. Of the 51 patients undergoing genetic testing, 43 (84.3%) were confirmed to carry a pathogenic mutation in a known susceptibility gene. Mutations in SDHD were most common (20/43, 46.5%), followed by SDHB (13/43, 30.2%), and SDHC (7/43, 16.3%). Pathogenic mutations in SDHA, SDHAF2, and NF1 were identified in the final three patients with unilateral CBP, unilateral CBP, and SCP with concomitant bilateral pheochromocytoma, respectively. The highest frequency of SDHx mutations was seen in patients with SCP, multi‐focal or metastatic HNPGL, and bilateral CBP (100.0% of patients tested within each cohort).
Carotid Body Paraganglioma (Unilateral)
Depicted in Figure 2 is a flow diagram detailing patient demographics, presenting signs and symptoms, diagnostic evaluation, tumor characteristics, treatment, and follow‐up for our cohort of patients with solitary unilateral CBP. The most common presentation of unilateral CBP was painless, enlarging neck mass (30 patients, 48.4%). Pre‐treatment CN palsy was rare, with only three patients (4.8%) presenting with cranial nerve X or XII weakness on initial physical exam. Similarly, functional CBP were rare, with notable metanephrine elevations in only four of 24 patients in whom metanephrines were assessed pre‐treatment. During the study period, a single patient was diagnosed with a malignant, unilateral CBP based on aggressive soft‐tissue extension into the brachial plexus and cervical nerve roots; this patient was treated with sub‐total resection and adjuvant radiation.
Figure 2.
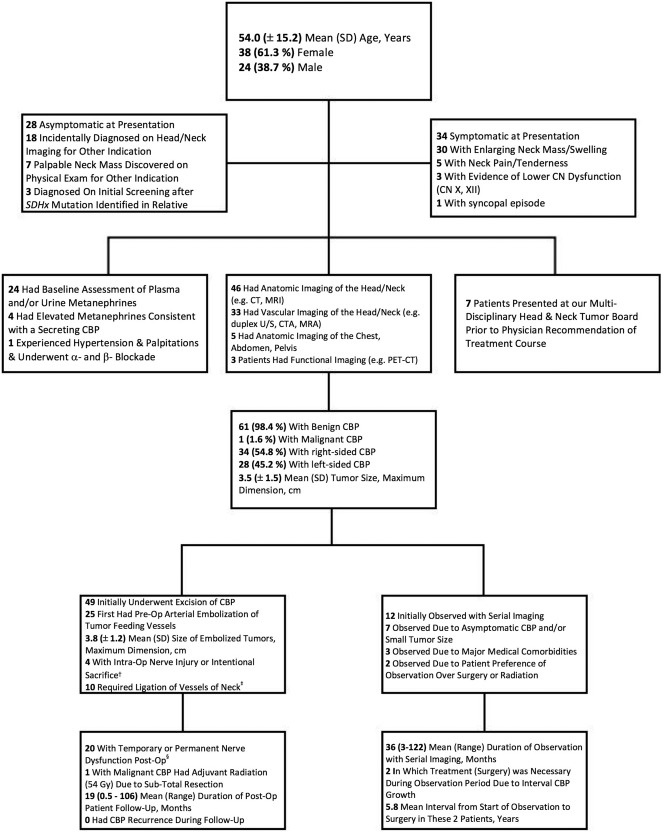
Diagnostic evaluation & treatment of patients (n = 62) with solitary, unilateral carotid body tumors (CBP). † CN IX, X, XII. ‡ External carotid artery, lingual artery, superior thyroid artery, thyrocervical trunk, superior thyroid artery, and internal jugular vein. § CN VII, marginal mandibular branch: 4 patients, CN IX: 3 patients, CN X: 10 patients, CN XI: 2 patients, CN XII: 8 patients, cervical sympathetic trunk: 3 patients
Among 49 patients with CBP treated initially with surgery, 25 patients first underwent preoperative angiographic embolization. There was no difference in CBP size between patients undergoing preoperative embolization versus those that did not (3.8 ± 1.2 cm vs. 3.4 ± 1.6 cm, p = 0.337). Preoperative tumor embolization was complicated by new CN weakness in two patients (CN X, XII, respectively) and a right, fronto‐parietal ischemic stroke in one patient, precluding definitive treatment of CBP. Among the 12 patients with CBP initially undergoing a “wait and scan” approach, 2 (16.7%) eventually underwent extirpation due to CBP growth, with a mean time interval to treatment of 5.8 years.
Carotid Body Paraganglioma (Bilateral)
The mean (SD) age of the bilateral CBP patient cohort (14 patients) was 49.0 (±11.9) years, with a gender distribution of 8 (57.1%) females and 6 (42.9%) males. No patients with bilateral CBP exhibited CN dysfunction at presentation, and all tumors were non‐secreting, benign paragangliomas. A staged surgical excision of bilateral CBP was performed in 8/14 patients with a mean (range) time interval of 11 (0.1–72) months between operations. Seven of these staged excisions began with the larger of the bilateral CBP. Staged surgical excisions of bilateral CBP resulted in development of new CN palsies in two patients (unilateral CN X weakness, unilateral CN XII weakness and bilateral Horner's syndrome, respectively). Additionally, 5/8 patients (62.5%) were treated for new‐onset hypertension due to baroreceptor dysfunction in the postoperative period.
Surgical excision of the larger CBP and observation with serial imaging of the smaller, contralateral tumor was the treatment strategy for four of 14 bilateral CBP patients. The mean (range) duration of observation for the smaller of bilateral CBP was 73 (6–140) months with no patients requiring additional treatment for CBP growth or new symptoms during the observation period. The final two patients were diagnosed with small, asymptomatic bilateral CBP within the last three years and have been observed during this time frame.
Jugular Paraganglioma
The flow diagram for the diagnostic evaluation and treatment of 41 patients with solitary JP is depicted in Figure 3. The highest frequency of pre‐treatment CN deficits was seen in the JP cohort, with 28/41 (68.3%) patients presenting with dysfunction of one or more CNs (IX, X, XI, XII). Primary treatment modalities for JP varied; 16 (39.0%) underwent surgical excision of JP, 11 (26.8%) received conventional fractionated radiation, four (9.8%) underwent SRS, and 10 (24.4%) were followed with serial imaging. Like the CBP cohort, tumor size was similar between embolized and non‐embolized JP (3.5 ± 2.0 cm vs. 3.0 ± 1.7 cm, p = 0.675). Embolization was complicated in two patients by new CN IX–X deficits and bilateral punctate thalamic strokes, respectively. In almost all cases, our surgeons opted for a sub‐total resection with CN preservation (14/16 operations), with adjuvant radiation to the jugular foramen in 8/14 patients.
Figure 3.

Diagnostic evaluation & treatment of patients (n = 41) with solitary, unilateral jugular paraganglioma (JP). † 2 patients with facial paralysis or weakness, 3 with neck/occiput pain, 1 with dysarthria, and 1 with altered vision and papilledema. ‡ External carotid artery, occipital artery, and internal jugular vein
Vagal Paraganglioma
Figure 4 depicts the flow diagram for the diagnostic evaluation and treatment of 21 patients with solitary VP. Evidence of CN X palsy and unilateral vocal cord dysfunction was observed in seven (33.3%) patients at presentation. VP were the largest of any HNPGL in our cohort, with a mean (SD) tumor size of 5.3 (±1.9) cm. One patient was diagnosed with malignant VP, again due to aggressive cervical soft‐tissue discovered on post‐surgical pathology. Surgical excision with preoperative embolization of VP was the most common treatment path (11/2, 52.4%). Surgery resulted in complete unilateral CN X palsy in all patients, with additional postoperative deficits including CN IX, XII, and the cervical sympathetic chain in two patients.
Figure 4.
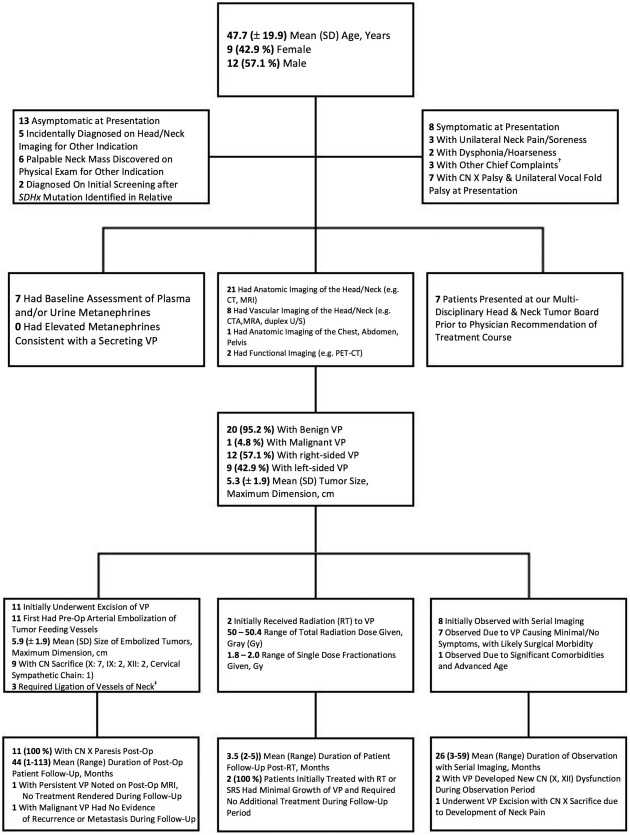
Diagnostic evaluation & treatment of patients (n = 21) with solitary, unilateral vagal paraganglioma (VP). † 1 patient with painless neck swelling/fullness, 1 with pulsatile tinnitus, 1 with dysphagia. ‡ Ascending pharyngeal artery, facial artery, internal carotid artery, external carotid artery
Tympanic Paraganglioma
The mean (SD) age of the TP cohort (n = 22) was 60.3 (±13.8) years, with a gender distribution of 19 (86.4%) females and 3 (13.6%) males. Unilateral pulsatile tinnitus (13/22, 59.1%) and conductive hearing loss (7/22, 31.8%) were the most common presenting symptoms of TP. All TP were benign tumors, with a mean (SD) tumor size of 0.7 (±0.6) cm, the smallest HNPGL in our entire cohort. Of the 19 patients with TP undergoing surgical excision, none experienced postoperative complications or developed recurrence during the follow‐up period (mean 12.1 months). The remaining three patients with TP were observed, with none requiring definitive TP treatment during a mean observation period of 24 months.
Sympathetic Chain Paraganglioma
The seven patients with solitary SCP were comparatively younger than other HNPGL groups, with a mean (SD) patient age of 32.7 (±10.6) years. Of six patients with symptomatic SCP at presentation, four (57.1%) had evidence of unilateral Horner's syndrome and two (28.6%) were diagnosed during workup for new‐onset hypertension caused by catecholamine‐secreting lesions. The final patient with SCP was diagnosed incidentally on initial screening after an SDHx mutation was identified in a family member. The mean (SD) size of tumors in this cohort, all benign lesions, was 4.3 (±2.1) cm.
Six patients were treated primarily with angiographic embolization and excision of SCP, leading to unilateral Horner's syndrome in five of six (83.3%) and additional CN palsies (X, XII) in four of six (66.7%). The remaining SCP patient was observed and did not require definitive treatment during 18 months of follow‐up.
Multi‐Focal/Metastatic HNPGL
The mean (SD) age of patients with multi‐focal or metastatic HNPGL was 46.5 (±13.8) years. Referring to Figure 1, panel B, SDHD mutations were confirmed in five patients (patients 2, 4, 6, 17, 18), SDHB mutations in two (patients 14, 22), SDHC mutations in four (patients 8, 13, 15, 22), and NF1 mutations in one (patient 16). Four patients (patients 18, 22–24) in this cohort were diagnosed with malignant HNPGL due to the presence of metastatic disease.
DISCUSSION
We present a relatively large cohort of HNPGL patients, with a strong representation of particularly rare HNPGL sub‐sites (eg, VP, TP, SCP), and evident diversity in genetic perturbations, diagnostic evaluation, and treatment protocols. Consistent with previous reports, there was a strong female predilection of HNPGL, a broad range of patient age at diagnosis, and a substantial proportion of patients with a hereditary predisposition to HNPGL (84.3% among those tested).20, 21 Similarly, the relative prevalence of HNPGL subsites and SDHx mutations in our cohort (unilateral CBP and SDHD most common, SCP and SDHA least common, respectively) closely matches frequency distributions seen in other HNPGL patient cohorts.22
Across all HNPGL subsites at our institution, screening for functional paragangliomas with plasma or urine metanephrines was performed in a minority of HNPGL cases (Figs. 2, 3, 4), often in patients who self‐reported a history of hypertension, anxiety, or palpitations or in patients with suspected or confirmed SDHx‐related HNPGL. In total, we identified hypersecreting HNPGL in nine (4.6%) patients, a proportion skewed upwards slightly by two patients with paragangliomas derived from sympathetic, rather than parasympathetic, paraganglia. Elevation of metanephrines in one patient presenting with CBP (patient 14, Fig. 1, panel B) led to a diagnosis of concomitant, hypersecreting para‐aortic paraganglioma. Plasma metanephrines have a marginal sensitivity of roughly 22% for HNPGL, though their utility lies in their near 100% sensitivity for diagnosing a concomitant catecholamine‐secreting paragangliomas at other sites in SDHx mutation carriers that would demand prioritized treatment.23, 24 Thus, a baseline assessment of plasma metanephrines should be performed in all patients with HNPGL.
Multimodal imaging techniques were often employed (Figs. 2, 3, 4) in our cohort. Angiography commonly supplemented initial contrast‐enhanced CT and MRI in all HNPGL subsites except for TP. Angiography allowed for mapping of dominant tumor feeding vessels (eg, ascending pharyngeal artery) in preparation for embolization and was useful in identifying important collateral vessels from the internal carotid and vertebral arteries requiring preservation during surgical extirpation.8 Baseline and post‐treatment audiometry was uniformly employed in JP and TP cases to assess long‐term hearing outcomes in these patients.25
In our entire HNPGL cohort, observation with serial imaging was a popular approach for patients with small, asymptomatic HNPGL (Figs. 2, 3, 4) to define tumor growth pattern and avoid operative morbidity to lower cranial nerves. A “watchful waiting” protocol was employed in 40/194 patients (20.6%) with HNPGL over a range of 24–73 months of follow‐up. A switch to active treatment planning occurred in seven (17.5%) patients due to radiologic growth of HNPGL (two CBP, one JP), pain (one VP), or new cranial nerve dysfunction (two VP, one SCP). A dearth of retrospective evidence supports “watchful waiting” as a viable initial management in appropriately selected HNPGL patients due to indolent growth patterns and exceedingly low rates of malignant degeneration or death due to HNPGL progression.26, 27, 28, 29
Iatrogenic damage to lower cranial nerves is the foremost risk in surgical extirpation of HNPGL.30, 31 New cranial nerve dysfunction after surgery was seen in 20/49 (40.8%) patients with unilateral CBP and 6/16 (37.5%) patients with JP. The former is a considerably higher number than in other published reports likely due to a referral bias favoring a greater number of Shamblin III tumors treated at our institution.32 Permanent CN X palsy and unilateral Horner's syndrome was virtually universal in VP (11/11 patients) and SCP (5/6 patients) cohorts undergoing surgery. Nevertheless, cure rates afforded by surgery were excellent across all HNPGL subsites. In the JP cohort, function‐preserving, sub‐total resection was followed by adjuvant radiation in 8/14 (51.7%) patients (Fig. 3). In five of these cases, adjuvant radiation was administered expeditiously after resection. Conversely, radiation was prompted by tumor recurrence/regrowth in three cases (roughly one in five JP patients undergoing sub‐total resection) validating rates of recurrence/regrowth seen in other JP cohorts.33 Increased utilization of this function‐preserving approach to JP treatment may therefore necessitate patient counseling regarding likelihood of adjuvant treatment and/or timely adjuvant radiation therapy to preempt tumor recurrence/regrowth.
At our institution, unilateral and bilateral CBP, JP, VP, and SCP frequently undergo angiographic embolization 24–72 hours prior to surgery, though our data suggest larger size does not reliably influence surgeon preference for embolization. At present, there remains a paucity of strong data on which HNPGL characteristics (eg, size, subsite) predict greater benefit of preoperative embolization on outcomes.34, 35 Therefore, a best practices approach to HNPGL embolization should involve a case‐based consideration of institutional preferences, level of interventional/vascular radiology expertise, and tumor‐specific factors such as size, subsite, and proximity to lower CNs.
Fractioned radiation (RT) and stereotactic radiosurgery (SRS) produce long‐term, durable HNPGL control and were most commonly employed in our JP and VP cohorts, providing excellent tumor control over 2–119 months of follow‐up and causing no treatment complications (Figs. 3, 4). These therapies are clearly highly‐efficacious, safe alternatives to surgery and are best suited for patients with large CBP, JP, VP, or multi‐focal HNPGL who have a high likelihood of operative morbidity to lower cranial nerves or contraindications to surgery (eg, medical comorbidities).13
Genetic testing for hereditary predisposition to HNPGL is crucial for active treatment planning, screening for co‐occurring pheochromocytoma and multifocal tumors, evaluation of at‐risk relatives, and in determining protocols for life‐long surveillance.3 Some have posited a step‐by‐step strategy for genetic testing based on clinical features of HNPGL (eg, begin with SDHD or SDHB testing in multi‐focal or malignant HNPGL, respectively).36 However, although some genotype‐phenotype correlations in specific SDHx‐related HNPGL are evident,37 there is a significant clinical overlap of syndromes caused by different SDHx mutations. For this reason, and as genetic testing becomes more accessible and affordable,38 initial screening for known pathogenic mutations in all 12 of the known hereditary susceptibility genes likely improves sensitivity and patient care in this population.
CONCLUSIONS
Management of each HNPGL case should be individualized and tailored to specific patient‐, tumor‐, and genetic‐factors. However, an algorithmic approach to management, as we posit (Figure 5) based on our extensive institutional expertise, can provide a general framework for best practices of current HNPGL evaluation and management.
Figure 5.
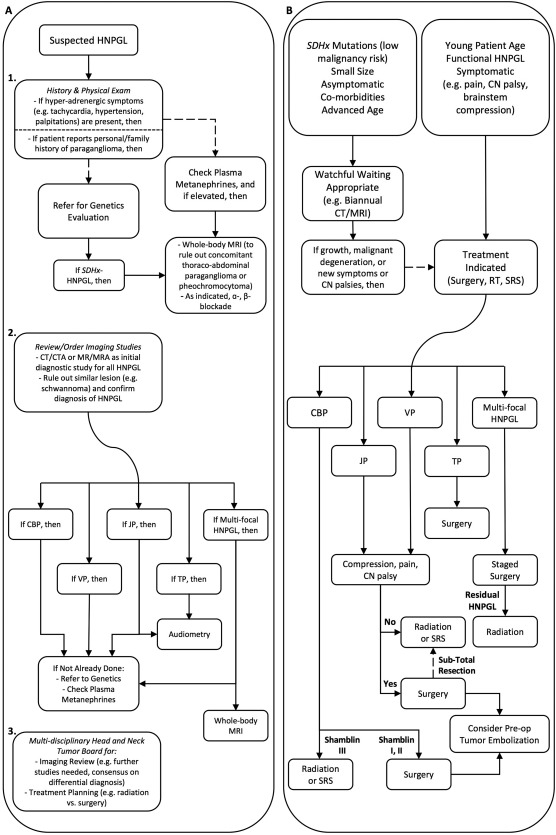
Proposed clinical algorithm for diagnostic evaluation & treatment of HNPGL. Panel A: Beginning with suspected HNPGL, diagnostic evaluation should commence with patient history and physical exam (1), followed by head and neck imaging (2) to confirm diagnosis of HNPGL and case presentation at Tumor Board (3). Panel B: Following comprehensive diagnostic evaluation as in panel A, providers should consider a number of tumor‐ and patient‐related factors when choosing between observation and protocols for active treatment of HNPGL
AUTHOR CONTRIBUTIONS
Conception or Design of Study: JDS, TE, GJB. Acquisition, Analysis, or Interpretation of Data: JDS, RNH, OAD, TE, GJB. Preparation and Revision of Manuscript: JDS, RNH, OAD, MEP, CRB, GTW, TE, GJB. Approval of the Manuscript: JDS, RNH, OAD, MEP, CRB, GTW, TE, GJB.
ACKNOWLEDGMENTS
The authors would like to thank the diverse faculty and staff of the Departments of Otolaryngology–Head and Neck Surgery, Radiology, Radiation Oncology, and Genetics at the University of Michigan who see and care for these patients. JDS received funding support from an NIH T32 Training Grant (T32 DC 5356‐15).
Financial Disclosure: The authors have no financial relationships to disclose relevant to this manuscript.
Conflicts of Interest: The authors have no conflicts of interest to disclose relevant to this manuscript.
BIBLIOGRAPHY
- 1. Fishbein L. Pheochromocytoma and paraganglioma: Genetics, diagnosis, and treatment. Hematol Oncol Clin North Am 2016;30:135–150. [DOI] [PubMed] [Google Scholar]
- 2. Pellitteri PK, Rinaldo A, Myssiorec D, et al. Paragangliomas of the head and neck. Oral Oncol 2004;40:563–575. [DOI] [PubMed] [Google Scholar]
- 3. Dahia PLM. Pheochromocytoma and paraganglioma pathogenesis: Learning from genetic heterogeneity. Nat Rev Cancer 2014;14:108–119. [DOI] [PubMed] [Google Scholar]
- 4. PIccini V, Rapizzi E, Bacca A, et al. Head and neck paragangliomas: Genetic spectrum and clinical variability in 79 consecutive patients. Endocr Relat Cancer 2012;19(2):149–155. [DOI] [PubMed] [Google Scholar]
- 5. Boedeker CC, Hensen EF, Neumann HPH, et al. Genetics of hereditary head and neck paragangliomas. Head Neck 2014;36;907–916. [DOI] [PubMed] [Google Scholar]
- 6. Van duinen N, Corssmit EP, de Jong WH, Brookman D, Kema IP, Romijn JA. Plasma levels of free metanephrines and 3‐methoxytyramine indicate a higher number of biochemically active HNPGL than 24‐h urinary excretion rates of catecholamines and metabolites. Eur J Endocrinol 2013;169(3):377–382. [DOI] [PubMed] [Google Scholar]
- 7. Corrales CE, Fischbein N, Jackler RK. Imaging innovations in temporal bone disorders. Otolaryngol Clin North Am 2015;48(2):263–280. [DOI] [PubMed] [Google Scholar]
- 8. Woolen S, Gemmete JJ. Paragangliomas of the head and neck. Neuroimag Clin N Am 2016;26:259–278. [DOI] [PubMed] [Google Scholar]
- 9. Mendenhall WM, Amdur RJ, Vaysberg M, et al. Head and neck paragangliomas. Head Neck 2011;33:1530–1534. [DOI] [PubMed] [Google Scholar]
- 10. Jansen JC, van den Berg R, Kuiper A, van der Mey AG, Zqinderman AH, Cornelisse CJ. Estimation of growth rate in patients with head and neck paragangliomas influences the treatment proposal. Cancer 2000;88:2811–2816. [PubMed] [Google Scholar]
- 11. Michalowska I, Cwikla JB, Michalski W, et al. Growth rate of paragangliomas related to germline mutations of the SDHX genes. Endocr Pract 2017;23(3):342–352. [DOI] [PubMed] [Google Scholar]
- 12. Timmers HJ, Gimenez‐Roqueplo AP, Mannelli M, Pacak K. Clinical aspects of SDHx‐related pheochromocytoma and paraganglioma. Endocr Relat Cancer 2009;16(2):391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gilbo P, Morris CG, Amdur RJ, et al. Radiotherapy for benign head and neck paragangliomas: A 45‐year experience. Cancer 2014;120:3738–3743. [DOI] [PubMed] [Google Scholar]
- 14. Anttila T, Hayry V, Nicoli T, et al. A two‐decade experience of head and neck paragangliomas in a whole population‐based single centre cohort. Eur Arch Otorhinolaryngol 2015;272:2045–2053. [DOI] [PubMed] [Google Scholar]
- 15. Ngwenya LB, Chiocca EA. Treatment for paragangliomas: Passing the test of time. World Neurosurg 2012;77:639–641. [DOI] [PubMed] [Google Scholar]
- 16. Kollert M, Minovi AA, Draft W, Bockmuhl U. Cervical paragangliomas‐tumor control and long‐term functional results after surgery. Skull Base 2006;16(4):185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hu K, Persky MS. Treatment of head and neck paragangliomas. Cancer Control 2016;23(3):228–241. [DOI] [PubMed] [Google Scholar]
- 18.University of Michigan Medical School Office of Research. DataDirect. https://datadirect.med.umich.edu. Accessed June 3, 2017.
- 19. Hanauer DA. EMERSE: The Electronic Medical Record Search Engine. AMIA Annu Symp Proc 2006:941. [PMC free article] [PubMed] [Google Scholar]
- 20. Papaspyrou K, Mewes T, Rossmann H, et al. Head and neck paragangliomas: Report of 175 patients (1989–2010). Head Neck 2012;34:632–637. [DOI] [PubMed] [Google Scholar]
- 21. Erickson D, Kudva YC, Ebersold MJ, et al. Benign paragangliomas: clinical presentation and treatment outcomes in 236 patients. J Clin Endocrinol Metab 2001;86(11):5210–5216. [DOI] [PubMed] [Google Scholar]
- 22. Taieb D, Kaliski A, Boedeker CC, et al. Current approaches and recent developments in the management of head and neck paragangliomas. Endocr Rev 2014;35:795–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boot C, Toole B, Johnson SJ, Ball S, Neely D. Single‐centre study of the diagnostic performance of plasma metanephrines with seated sampling for the diagnosis of phaeochromocytoma/paraganglioma. Ann Clin Biochem 2017;54(1):143–148. [DOI] [PubMed] [Google Scholar]
- 24. Rao D, Peitzsch M, Prejbisz A, et al. Plasma methoxytyramine: clinical utility with metanephrines for diagnosis of pheochromocytoma and paraganglioma. Eur J Endocrinol 2017;177(2):103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Medina M, Prasad SC, Patnaik U, et al. The effects of tympanomastoid paragangliomas on hearing and the audiological outcomes after surgery over a long‐term follow‐up. Audiol Neurootol 2014;19(5):342–350. [DOI] [PubMed] [Google Scholar]
- 26. Carlson ML, Sweeney AD, Wanna GB, Netterville JL, Haynes DS. Natural history of glomus jugulare: a review of 16 tumors managed with primary observation. Otolaryngol Head Neck Surg 2015;152(1):98–105. [DOI] [PubMed] [Google Scholar]
- 27. Langerman A, Athavale SM, Rangarajan SV, Sinard RJ, Netterville JL. Natural history of cervical paragangliomas: Outcomes of observation of 43 patients. Arch Otolaryngol Head Neck Surg 2012;138(4):341–345. [DOI] [PubMed] [Google Scholar]
- 28. Jansen TTG, Timmers HJLM, Marres HAM, et al. Feasibility of a wait‐and‐scan period as initial management strategy for head and neck paraganglioma. Head Neck 2017;39(10):2088–2094. [DOI] [PubMed] [Google Scholar]
- 29. Prasad SC, Mimoune HA, D'Orazio F, et al. The role of wait‐and‐scan and the efficacy of radiotherapy in the treatment of temporal bone paragangliomas. Otol Neurotol 2014;35(5):922–931. [DOI] [PubMed] [Google Scholar]
- 30. Bacciu A, Medina M, Ait Mimoune H, et al. Lower cranial nerves function after surgical treatment of Fisch Class C and D tympanojugular paragangliomas. Eur Arch Otorhinolaryngol 2015;272(2):311–319. [DOI] [PubMed] [Google Scholar]
- 31. Lope Ahmad RA, Sivalingam S, Konishi M, et al. Oncologic outcome in surgical management of jugular paraganglioma and factors influencing outcomes. Head Neck 2013;35(4):527–534. [DOI] [PubMed] [Google Scholar]
- 32. Anand VK, Alemar GO, Sanders TS. Management of the internal carotid artery during carotid body tumor surgery. Laryngoscope 1995;105:231–235. [DOI] [PubMed] [Google Scholar]
- 33. Li D, Zeng XJ, Hao SY, et al. Less‐aggressive surgical management and long‐term outcomes of jugular foramen paragangliomas: a neurosurgical perspective. J Neurosurg 2016;125(5):1143–1154. [DOI] [PubMed] [Google Scholar]
- 34. Power AH, Bower TC, Kasperbauer J, et al. Impact of preoperative embolization on outcomes of carotid body tumor resections. J Vasc Surg 2012;56:979–989. [DOI] [PubMed] [Google Scholar]
- 35. Jackson RS, Myhill JA, Padhya TA, McCaffrey JC, McCaffrey TV, Mhaskar RS. The effects of preoperative embolization on carotid body paraganglioma surgery: A systematic review and meta‐analysis. Otolaryngol Head Neck Surg 2015;153(6):943–950. [DOI] [PubMed] [Google Scholar]
- 36. Neumann HPH, Erlic Z, Boedeker CC, et al. Clinical predictors for germline mutations in head and neck paraganglioma patients: cost reduction strategy in genetic diagnostic process as fall‐out. Cancer Res 2009;69:3650–3656. [DOI] [PubMed] [Google Scholar]
- 37. Ricketts CJ, Forman JR, Rattenberry E, et al. Tumor risks and genotype‐phenotype‐proteotype analysis in 358 patients with germline mutations in SDHB and SDHD . Hum Mutat 2010;31:41–51. [DOI] [PubMed] [Google Scholar]
- 38. Uhlmann WR, Schwalm K, Raymond VM. Development of a streamlined work flow for handling patients’ genetic testing insurance authorizations. J Genet Couns 2017;26(4):657–668. [DOI] [PubMed] [Google Scholar]