

Summary
Double strand breaks (DSBs) of the DNA in eukaryotic cells are predominantly repaired by non-homologous end joining (NHEJ). The histone chaperone, anti-silencing factor 1a (ASF1a) interacts with MDC1 and is recruited to sites of DSB to facilitate the interaction of phospho-ATM with MDC1 and the phosphorylation of MDC1, which is required for the recruitment of RNF8/RNF168 histone ubiquitin ligases. Thus, ASF1a deficiency reduces histone ubiquitination at DSBs, decreasing the recruitment of 53BP1 and decreases NHEJ, rendering cells more sensitive to DSBs. This role of ASF1a in DSB repair cannot be provided by the closely related ASF1b, and does not require its histone chaperone activity. Homozygous deletion of ASF1a is seen in 10-15% of certain cancers, suggesting that loss of NHEJ may be selected in some malignancies, and that the deletion can be used as a molecular biomarker for cancers susceptible to radiotherapy or to DSB-inducing chemotherapy.
Keywords: ASF1a, non-homologous end joining (NHEJ), homologous recombination (HR), ubiquitination, E3 ligase, histone, bleomycin, RNF8, MDC1, BRCA1, RNF168, 53BP1, ATM
Graphical abstract
DNA double stranded breaks (DSB) are harmful lesions that contribute to genomic instability, with failure to repair these breaks leading to abnormal development, pre-mature aging, and tumorigenesis (Helleday et al., 2007; Jackson and Bartek, 2009; Li et al., 2007; McKinnon, 2009; Moynahan and Jasin, 2010). Non-homologous end joining (NHEJ) and homologous recombination (HR) are the primary repair pathways used to resolve DSBs (Ceccaldi et al., 2016). While HR repair is restricted to S-G2 phases of the cell-cycle due to the availability of complementary homologous sequences, NHEJ repair occurs throughout the cell cycle with a preference for the G0 and G1phases (Heyer et al., 2010). Although NHEJ repair is more error-prone than HR, it is predominantly utilized in vertebrates presumably because most cells in the body spend more time in G0 and G1 (Kass and Jasin, 2010; Lieber, 2010).
Ubiquitination of histones adjacent to DSB sites is an important branch point that promotes NHEJ over HR (Doil et al., 2009; Huen et al., 2007; Kolas et al., 2007; Mailand et al., 2007). 53BP1 binding to DSBs is dependent on ubiquitination of histones H2A and H2AX (shortened to H2A/X) at lys15 and also on dimethylation of H4 at lysine 20 (Botuyan et al., 2006; Fradet-Turcotte et al., 2013; Zgheib et al., 2009). Recruitment of 53BP1-Rif1 complex to the DSBs promotes NHEJ repair and prevents the HR-promoting BRCA1-CtIP complex from binding to DSB sites (Chapman et al., 2013; Di Virgilio et al., 2013; Feng et al., 2013; Zimmermann et al., 2013), even though the BRCA1-RAP80-Abraxas-BRCC36 complex (non-functional for HR) is recruited to the ubiquitinylated histones (Kim et al., 2007a; Sobhian et al., 2007; Wang et al., 2007). Recruitment of BRCA1-Rap80 is a second way by which HR repair is inhibited by histone ubiquitination by depleting the pool of BRCA1 available for promoting HR by associating with CtIP, Bach1 and Rad51 (Hu et al., 2011). Thus, ubiquitination at DSBs is a crucial feature in DNA Damage Repair for determining whether a DSB is repaired by NHEJ or HR.
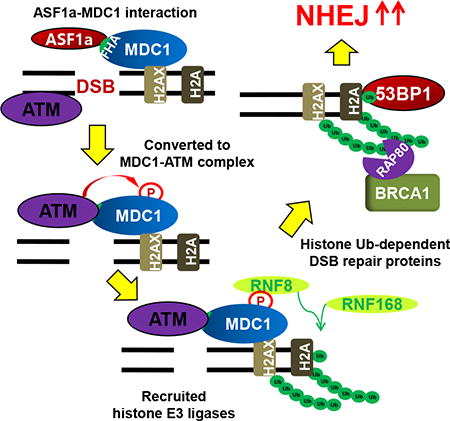
The first step in the repair of DSBs is the MRE11-RAD50-NBS1 (MRN) complex mediated recruitment and activation of ATM kinase triggered by DSBs (Paull, 2015). ATM phosphorylates H2AX at Serine 139, which facilitates phospho-H2AX interaction with MDC1 (Lou et al., 2006; Stewart et al., 2003; Stucki et al., 2005). Although the initial localization of ATM to DSBs requires MRN, the interaction of autophosphorylated ATM with MDC1 is required for the prolonged association of phospho-ATM to DSBs (So et al., 2009) and for ATM dependent phosphorylation of MDC1, which promotes interaction of phospho-MDC1 with RNF8 E3 ligase (Huen et al., 2007; Mailand et al., 2007). RNF8 recruits RNF168 (Doil et al., 2009; Stewart et al., 2009; Thorslund et al., 2015), which initiates ubiquitination on H2A/X (Mattiroli et al., 2012).
ASF1 (anti-silencing factor 1) is a histone chaperone that interacts with newly synthesized H3-H4 heterodimers and hands over the histones to the histone chaperone CAF-1 for nucleosome assembly after replication or repair (Mello et al., 2002; Tyler et al., 1999; Tyler et al., 2001; Winkler et al., 2012). Higher eukaryotes contain two paralogs of yeast ASF1, ASF1a and ASF1b with significant difference in sequence in their C-terminal parts (De Koning et al., 2007; Munakata et al., 2000; Sillje and Nigg, 2001). ASF1a promotes H3 acetylation at lys56 by the CBP/p300 acetyltransferase, which is required for nucleosome reassembly after DNA repair (Das et al., 2009; Groth et al., 2007). ASF1 is required for checkpoint recovery and the return to normal cell cycle after DNA damage repair (Tsabar et al., 2016), but there is no report that shows ASF1 is directly involved in DSB repair. We have discovered that ASF1a is specifically required to promote NHEJ and thus suppress HR. Specifically, ASF1a is required for the phosphorylated ATM to interact with and phosphorylate MDC1, which is essential for the recruitment of RNF8/RNF168 and histone ubiquitination, steps that are themselves essential for 53BP1 recruitment and NHEJ.
Results
ASF1a is required for NHEJ repair and resistance to DSB
NHEJ/DsRed293B cells (Golding et al., 2009; Mueller et al., 2013) contain a mutant DsRed gene stably integrated in the genome that is activated when two specific DSB sites are cut by I-SceI endonuclease and repaired by NHEJ to produce a functional DsRed gene. ASF1a knockdown with two different siRNAs of ASF1a (siASF1a-147 (Groth et al., 2005) and -355 (Groth et al., 2007)), reduced NHEJ without decreasing the expression of I-SceI (Figure 1A and 1B). As expected, knockdown of 53BP1 decreased NHEJ while knockdown of BRCA1 had no effect on NHEJ.
Figure 1. ASF1a is required for NHEJ and resistance to DSBs.
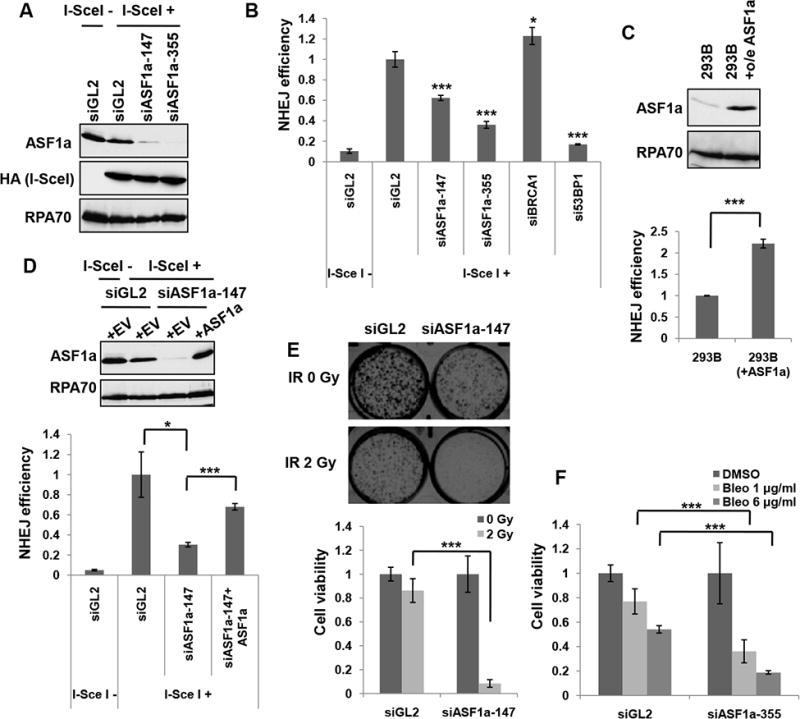
(A) Immunoblots of the NHEJ/DsRed293B lysates transfected with two different ASF1a targeting siRNAs, 48 hr after transfection of HA-I-SceI plasmids. HA-I-SceI was detected by anti-HA antibody. (B) ASF1a knockdown reduces NHEJ efficiency. NHEJ efficiency is measured as described in the METHOD DETAILS and represented as mean ± S.D. of triplicates. ***, P< 0.005; *, P< 0.05. (C) ASF1a overexpression increases NHEJ efficiency. 293B having stable overexpression (o/e) of ASF1a was compared with wild-type 293B for ASF1a expression level in the immunoblot (top) and NHEJ efficiency (bottom). Mean ± S.D. from triplicate measurements. (D) Rescue of NHEJ in siASF1a-transfected 293B cells by expression of siRNA-resistant ASF1a. Empty (+EV) or ASF1a expressing vector resistant to siASF1a (+ASF1a) was co-transfected with HA-I-SceI. Immunoblots (top) and quantitation of NHEJ efficiency (bottom). Mean ± S.D. of triplicates. (E) Depletion of ASF1a renders cells sensitive to ionizing radiation (IR). Cell viability was quantified and presented as mean ± S.D. from triplicate measurements (lower panel). Representative images (upper panel). (F) Dose-dependent sensitivity to bleomycin of ASF1a depleted cells. The indicated dose of bleomycin was treated for 24 hr after 48 hr from first siRNA transfection. Mean ± S.D. from triplicates.
In contrast, overexpression of ASF1a stimulated NHEJ (Figure 1C). Expression of a siRNA-resistant ASF1a ameliorated the reduction in NHEJ repair seen upon siASF1a transfection, indicating that the decrease in NHEJ is specific to ASF1a decrease and not due to any off-target activity of the siRNA (Figure 1D). Furthermore, depletion of ASF1a renders the cells more sensitive to ionizing radiation and bleomycin, agents that induce DSBs that are mostly repaired by NHEJ (Figure 1E and 1F). Overall, these results suggest that ASF1a is required for NHEJ repair.
ASF1a knockout reduces NHEJ and increases HR repair
To confirm a role of ASF1a in NHEJ repair, we generated CRISPR/CAS9 mediated deletions of the ASF1a in NHEJ/DsRed293B cells (Figure 2A). PCR using primers across the sgRNA targeted sites verified the genomic deletion of both ASF1a alleles (example in Figure 2B), and immunoblotting showed a corresponding loss of ASF1a protein (Figure 2C). The gene targeting did not affect the protein level of MCM9, another DSB repair gene that overlaps with the ASF1a gene (Fig. 2C). Transfection of I-SceI expressing plasmids into these clonal cell lines confirmed that NHEJ efficiency was reduced in ASF1a knockout cells (Figure 2D), and this was rescued by re-expression of ASF1a (Figure 2E and 2F), indicating that the suppression of NHEJ was specifically due to the loss of ASF1a. Furthermore we found that disappearance of γH2AX after a transient DSB induced by a pulse of bleomycin was significantly retarded in ASF1a knockout compared to wild type (Figure 2G and S1A). This too suggests that NHEJ mediated repair of DSB is impaired in ASF1a depleted cells.
Figure 2. Knockout of ASF1a reduces NHEJ and promotes HR.
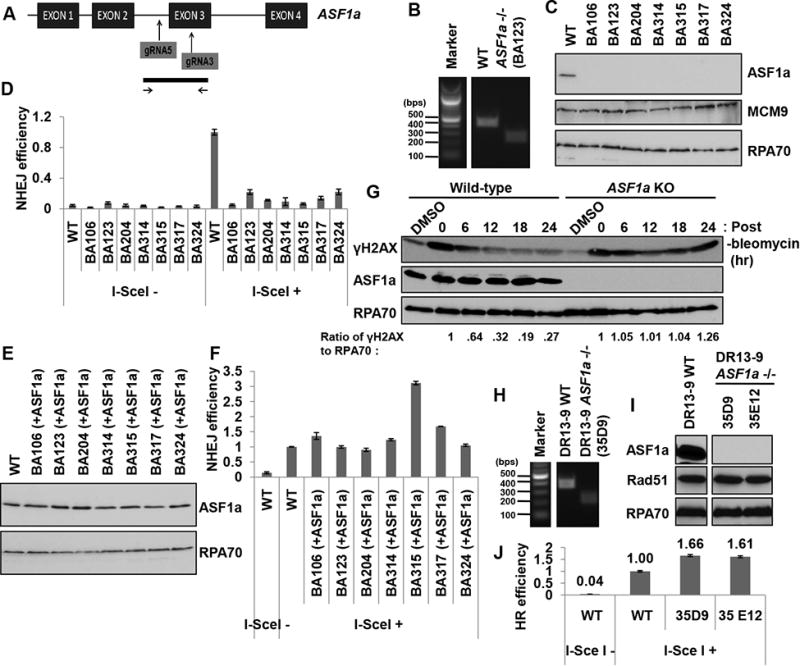
(A) A schematic of the targeting strategy for ASF1a knockout in 293B or HeLa DR13-9 cells using the CRSPR/CAS9 system. The sgRNAs targeting the ASF1a gene (top) and the region interrogated to identify the deletion (bottom) are shown. (B) A representative image of the PCR product from the 293B clones: Wild type and BA123 (with a homozygous deletion of the ASF1a gene). (C) A western blot showing ASF1a protein level in 293B wild-type and ASF1a null clones. (D) Knocking-out ASF1a suppresses NHEJ efficiency. The percentage of DsRed-positive cells in each cell-line was normalized to that of wild-type cells transfected with HA-I-SceI. Mean ± S.D. from triplicates. (E and F) ASF1a expression in ASF1a knockout cells rescues NHEJ efficiency. ASF1a was stably expressed in ASF1a knockout cell lines using retroviral infection. Immunoblots of those lysates (E) and NHEJ assay (F). Mean ± S.D. from triplicates. (G) Decrease of γH2AX after DSB is slowed in ASF1a knockout cells. The ratio of γH2AX to RPA70 signal was quantitated at each time point and normalized to the ratio at the 0 hr point. See also Figure S1A. (H) Representative image of PCR on genomic DNA from HeLa DR13-9 clones: wild type and 35D9. (I) A western blot showing ASF1a protein level in wild-type and ASF1a null clones of HeLa DR13-9. (J) Knocking-out ASF1a promotes HR repair. HR was measured as described in the METHOD DETAILS. Mean ± S.D. from triplicates. See also Figure S1B to S1D.
NHEJ and HR are competing DNA repair pathways (Lieber, 2010; Sonoda et al., 2006). The HeLa DR13-9 cell line has two mutated partial GFP gene fragments that is able to produce functional GFP signal only when an I-SceI induced DSB on the GFP gene is repaired by HR (Ransburgh et al., 2010). HeLa DR13-9 ASF1a-/- cells were generated using the same sgRNAs as in Figure 2A (Figure 2H and 2I). The ASF1a knockout lines showed an increase of HR efficiency compared to wild-type cells (Figure 2J). We ruled out a trivial explanation that the increase in HR was due to an increase in Rad51 by immunoblotting for the latter (Figure 2I). We also obtained HeLa DR13-9 ASF1a knockout cells from a single sgRNA targeting exon3 of ASF1a gene followed by NHEJ repair causing a frame-shift. In these clones, too, the ASF1a knockout up-regulated HR efficiency (Figure S1B and S1C). We clarified that the effect on HR efficiency was not from a cell cycle defect in the ASF1a knockout cells (Figure S1D). Therefore, the complete absence of ASF1a decreases NHEJ and lead to a compensatory increase of HR.
ASF1a is required for the recruitment of 53BP1 and RAP80 and inhibiting the recruitment of CtIP at DSBs
As described in the introduction, the choice between NHEJ and HR for DSB repair depends on the antagonistic recruitment of 53BP1-Rif1 (for NHEJ) and BRCA1-CtIP (for HR) (Panier and Boulton, 2014). Indeed, bleomycin induced 53BP1 foci were significantly decreased in ASF1a-depleted cells as well as in ASF1a knockout cells (Figure 3A to C and S2). Depletion of RNF8, the E3 ligase, also decreased 53BP1 foci (positive control). However, 53BP1 focus formation in ASF1a depleted cells was restored by siRNA-resistant ASF1a (Figure 3D).
Figure 3. ASF1a is required for the recruitment of 53BP1 and RAP80 at DSBs.
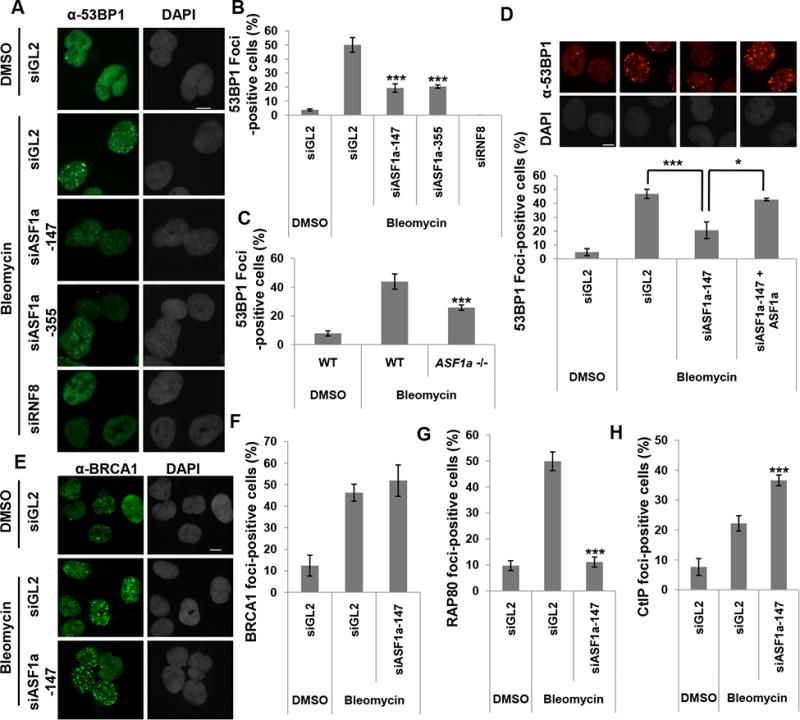
(A to C) Decrease of 53BP1 foci upon depletion of ASF1a in U2OS cells or in ASF1a -/-cells after treatment with bleomycin for 1 hr before fixation. Representative images (A) and quantitation (B and C). Cells with >20 foci of 53BP1 were counted. Scale bar, 10 μm. Mean ± S.D. of triplicates. ***, P < 0.005. Scale bar, 10 μm. See also Figure S2. (D) Rescue of 53BP1 foci in ASF1a-depleted U2OS cells by siRNA-resistant ASF1a. Representative images (top) and Mean ± S.D. of triplicates. ***, P< 0.005; *, P< 0.05 (bottom). (E and F) No change of BRCA1 foci upon depletion of ASF1a. Representative images (E) and quantitation (F). (G) RAP80 foci are decreased in ASF1a depleted U2OS cells. (H) CtIP foci are increased in ASF1a depleted U2OS cells. The quantitations in (F-G) is the same as in (B), and cells having over 5 foci of CtIP were counted in (H).
Turning to HR, we noted that BRCA1 focus formation was not affected by the depletion of ASF1a (Figure 3E and 3F). However, BRCA1 promotes or suppresses HR by forming complexes with different proteins (Coleman and Greenberg, 2011; Hu et al., 2011; Li and Greenberg, 2012). We, therefore, measured focus-formation by RAP80, which suppresses HR when associated with BRCA1, and CtIP, which promotes HR when co-recruited with BRCA1 (Figure 3G and 3H). RAP80 focus-formation was decreased, whereas CtIP focus-formation was increased upon ASF1a knockdown. Thus, ASF1a selectively promotes recruitment of the HR-suppressing BRCA1-RAP80 and inhibits the binding of the HR-promoting BRCA1-CtIP complex. These results suggest that ASF1a promotes NHEJ repair over HR by facilitating the recruitment of 53BP1 and BRCA1-RAP80 while inhibiting the recruitment of BRCA1-CtIP complex.
ASF1a is required for the proper localization of ATM and phosphorylation of MDC1 at DSB by facilitating the ATM-MDC1 interaction
To understand why 53BP1 recruitment was inhibited by loss of ASF1a, we investigated the cascade of protein recruitment at DSBs beginning with ATM, the kinase at the apex of this cascade (Shiloh and Ziv, 2013). ATM is activated by autophosphorylation at Ser-1981, which is required for its prolonged retention on DSBs through its interaction with MDC1 (So et al., 2009). Following DNA damage, auto-phosphorylation of ATM at Ser-1981 (pS1981-ATM) and phosphorylation of ATM substrates such as CHK2, NBS1 and H2AX, were not decreased in ASF1a-depleted cells (Figure 4A), suggesting that the MRN dependent activation of ATM did not require ASF1a. However knockdown of ASF1a decreased pS1981-ATM foci in bleomycin treated cells (Figure 4B and 4C), while ectopic expression of siRNA-resistant ASF1a restored the pS1981-ATM foci (Figure 4D). To examine the kinetics of localization of activated ATM to DSB, the pS1981-ATM foci-positive cells were counted at different time points after pulse-treatment of bleomycin (Figure 4E). Although initial increase of the foci-positive cells was seen even in ASF1a knockout cells like wild type, the increase was rapidly diminished in the knockout cell from 1 hr post-treatment, whereas the p-ATM foci persisted in the wild type cells (Figure 4E). These results suggest that ASF1a is required not for the initial recruitment and activation of ATM, but for the prolonged localization of activated ATM at DSBs.
Figure 4. ASF1a promotes the localization of ATM and phosphorylation of MDC1 at DSB by facilitating the ATM-MDC1 interaction.
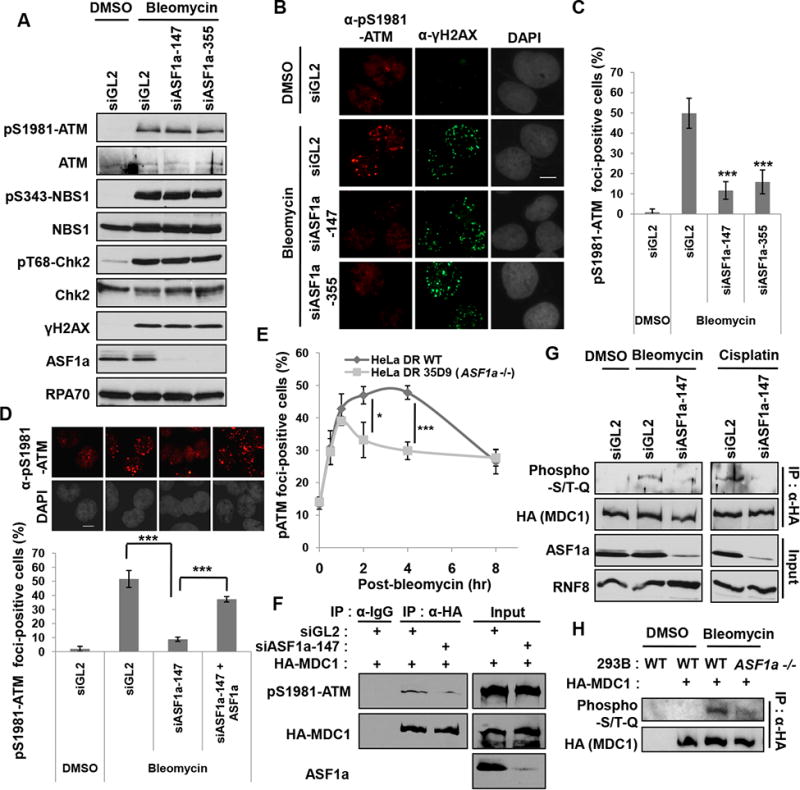
(A) Immunoblots with indicated antibodies show no decrease in DNA damage induced autophosphorylation of ATM or in ATM phosphorylation of NBS1, Chk2 and H2AX after depletion of ASF1a. (B and C) Decreased pS1981-ATM foci after depletion of ASF1a. Representative images of immunostaining (B) and quantification of foci-positive cells (C). Scale bar, 10 μm. Mean ± S.D. of triplicates. ***, P < 0.005. (D) siRNA-resistant ASF1a rescues pS1981-ATM foci-formation. Representative images (top) and quantification of cells >10 pS1981-ATM foci (bottom). Mean ± S.D. of triplicates. (E) A rapid disappearance of pS1981-ATM foci-formation upon ASF1a knockout. The cells having >5 pS1981-ATM foci in wild type or ASF1a -/- HeLa DR13-9 were counted as positive cells at indicated time points after pulse-treatment of 40 μgml-1 bleomycin for 20 min. Mean ± S.D. of triplicates. *, P < 0.05; ***, P < 0.005. (F) Defective interaction of MDC1 with phospho-ATM after ASF1a knockdown. (G and H) ASF1a is required for ATM phosphorylation on MDC1. HEK293T cells transfected with indicated siRNAs (G), 293B wild type and ASF1a -/- cells (H) were transfected by HA-MDC1 plasmid and DMSO, bleomycin or cisplatin was added for 14 hr before harvest.
The stable localization of phospho-ATM to DSB is dependent on its interaction with MDC1. Immunoprecipitated MDC1 normally co-precipitated phospho-ATM upon DNA damage, but this was decreased by ASF1a knockdown, indicating that ASF1a is required for the stable interaction between MDC1 and phospho-ATM (Figure 4F). Phosphorylation of MDC1 by ATM as measured with anti-phospho-S/TQ antibody, which recognizes sites phosphorylated by ATM, showed that the phosphorylation of MDC1 after DNA damage by bleomycin or cisplatin was markedly decreased upon ASF1a depletion or ASF1a knockout (Figure 4G and 4H). Thus ASF1a facilitates the stable interaction of phospho-ATM with MDC1 to promote DSB localization of ATM and the phosphorylation of MDC1.
ASF1a interacts with MDC1, and the FHA domain of MDC1 is required for ASF1a localization to DSBs
Since our data suggest that ASF1a has a role for DSB localization of ATM required for MDC1 phosphorylation, we investigated whether there was any physical interaction between ASF1a and MDC1. HA-ASF1a immunoprecipitates contained endogenous MDC1 protein even before cells see any DNA damage (Figure 5A). Although the ASF1a V94R mutant did not interact with H3, its interaction with endogenous MDC1 indicates that ASF1a-MDC1 interaction is independent of ASF1a's histone chaperone function (Figure 5B). The enrichment of ASF1a ChIP signal at the I-SceI site after induction of a DSB shows that ASF1a is itself recruited to the DSB (Figure 5C). The localization of ASF1a to the I-SceI cut site is reduced when MDC1 is depleted (Figure 5C). MDC1-depleted DNA damaged cells showed less ASF1a immunofluorescence after pre-extraction of nuclei, suggesting that MDC1 helps recruit ASF1a to a more insoluble chromatin-associated fraction (Figure 5D). In contrast, RNF8 depletion did not affect the recruitment of ASF1a to the insoluble fraction (Figure 5D). To check whether ASF1a and MDC1 were colocalized at DSB sites, we isolated MDC1-bound DNA by ChIP of MDC1, then used the eluate from the first precipitate for a second ChIP with anti-ASF1a antibody followed by PCR for the DNA at the I-SceI cut site. The serial ChIP experiment revealed that ASF1a is co-localized with MDC1 at the I-SceI cut sites (Figure 5E, +I-SceI, siGL2). The signal from the serial ChIP experiment decreased after MDC1 depletion, confirming the specificity of the first ChIP with anti-MDC1 antibody. In addition bleomycin-induced ASF1a foci overlapped with MDC1 foci (Figure 5F). Thus MDC1 helps recruit ASF1a to DSBs.
Figure 5. ASF1a interacts with MDC1, and the FHA domain of MDC1 is required for ASF1a localization to DSBs.
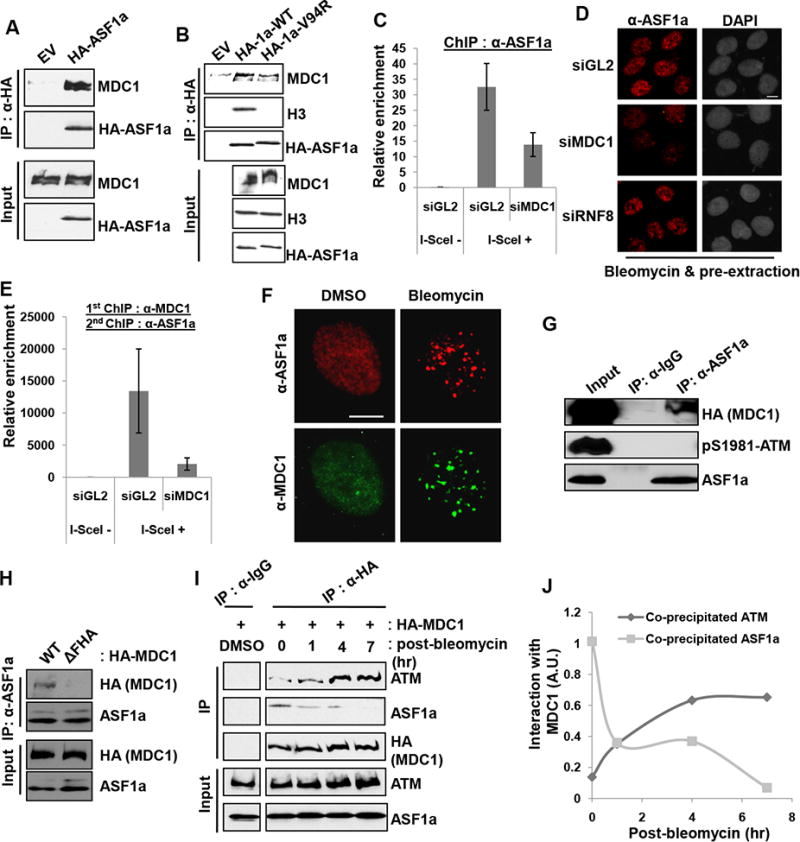
(A) HA-ASF1a precipitates contain endogenous MDC1. Immunoblots of immunoprecipitates or input lysate. (B) Histone binding by ASF1a not required for the interaction with MDC1. Wild-type or Val94Arg mutant of HA-ASF1a was immunoprecipitated with anti-HA antibody. (C) MDC1 is required for ASF1a localization at I-SceI cut site. ChIP assay was performed as described in the METHOD DETAILS. (D) MDC1 dependent association of ASF1a with chromatin after DNA damage. U2OS cells treated with bleomycin for 1 hr followed by pre-extraction, to remove soluble proteins, were fixed and immunostained. Scale bar, 10 μm. (E) Co-localization of MDC1 and ASF1a to I-SceI cut site. The eluate from anti-MDC1 ChIP was applied to an anti-ASF1a ChIP. (F) Co-localization of ASF1a- and MDC1-foci at DSBs. (G) Untagged ASF1a co-immunoprecipitates MDC1 but not phospho-ATM. (H) ASF1a-MDC1 interaction needs the FHA domain of MDC1. HA-MDC1 wild type or FHA deletion mutant (∆55-124 a.a.) was overexpressed in HEK293T cells followed by bleomycin treatment for 14 hr. (I and J) MDC1 interaction with ASF1a decreases as its interaction with ATM increases after DNA damage. The immunoblots with indicated antibodies (I) and quantification of co-precipitated ATM or ASF1a with HA-MDC1 after pulse-treatment of bleomycin (J).
Immunoprecipitation of untagged ASF1a also co-precipitated MDC1, but not phospho-ATM (Figure 5G). Although the MDC1 immunoprecipitate contains phospho-ATM (Figure 4F), the ASF1a immunoprecipitate, which contains MDC1, does not contain phospho-ATM (Figure 5G, lane 3), suggesting that MDC1 may form independent complexes with ASF1a and p-ATM. Interestingly, the deletion of the FHA domain on MDC1 decreased its interaction with ASF1a (Figure 5H), suggesting that ASF1a, like phospho-ATM (Lou et al., 2006), interacts with the FHA domain of MDC1. To examine the interaction kinetics of MDC1 with ATM or ASF1a upon DNA damage, we performed anti-HA-MDC1 immunoprecipitation assay at different time points after pulse-treatment of bleomycin (Figure 5I and 5J). ASF1a interacts with MDC1 at early time points and the interaction decreased as the ATM-MDC1 interaction increased after DNA damage (Figure 5I and 5J). Thus ASF1a interacts with the FHA domain of MDC1 to facilitate the stable interaction of phospho-ATM with MDC1 and is required for the subsequent phosphorylation of MDC1 by ATM (Figure 4G), but is excluded from the phospho-ATM-MDC1 complex.
ASF1a is required for RNF8, but not MDC1, recruitment at DSB
MDC1 phosphorylation by ATM promotes the recruitment of E3 ligases such as RNF8 and RNF168 to the DSB through the MDC1-RNF8 interaction (reviewed in (Nakada, 2016)). Therefore, we next asked whether ASF1a is required for the recruitment of MDC1, RNF8 or RNF168 at DSBs (Figure 6). MDC1 is co-localized with RNF8 upon bleomycin treatment (Figure S3). Knockout of ASF1a did not decrease bleomycin-induced MDC1 focus-formation, but decreased the number of RNF8 foci in the nucleus (Figure 6A to 6C). Furthermore, the MDC1-RNF8 interaction measured by co-immunoprecipitation from bleomycin treated cells was diminished in ASF1a -/- cells (Figure 6D). Chromatin immunoprecipitation assay (ChIP) of HeLa DR13-9 cells with a single I-SceI cut site showed the enrichment of RNF8, MDC1 and RNF168 at the DSB induced by I-SceI (Figure 6E to 6I). Knockdown of ASF1a diminished RNF8 and RNF168, but not MDC1, recruitment at the I-SceI cut site (Figure 6E to 6G). We confirmed that RNF8 recruitment, but not MDC1 recruitment, depended on ASF1a in the ASF1a knockout cells (Figure 6H and 6I). Therefore, ASF1a is required for the MDC1-RNF8 interaction at a step after MDC1 recruitment but before RNF8-RNF168 recruitment, consistent with its requirement for MDC1 phosphorylation.
Figure 6. ASF1a is required for RNF8, but not MDC1, recruitment at DSBs.
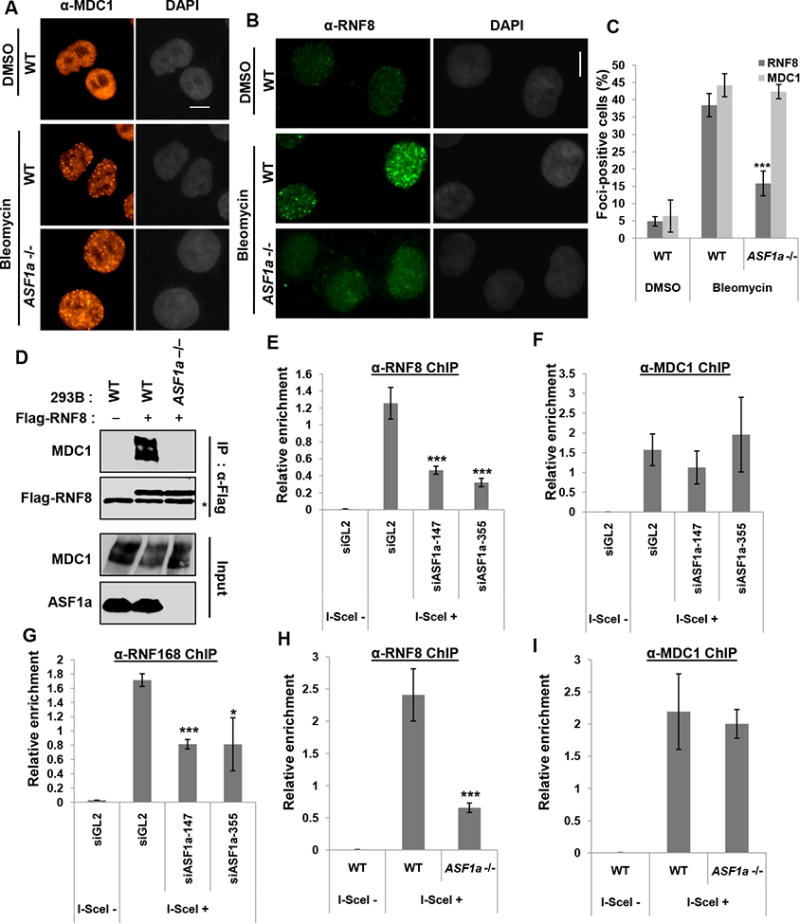
(A to C) Decrease of bleomycin-induced RNF8 foci, but not MDC1 foci in ASF1a knockout cells. HeLa DR13-9 wild type or ASF1a -/- cells were incubated with 20 μgml-1 bleomycin for 40 min followed by fixation. Representative images of immunostaining with indicated antibodies (A and B), and quantification of cells with >5 foci of RNF8 or MDC1 (C). See colocalization of RNF8 and MDC1 foci in Figure S3. Scale bar, 10 μm. Mean ± S.D. of triplicates. ***, P < 0.005. (D) Decreased interaction of MDC1 with RNF8 after DNA damage in ASF1a knockout cells. Plasmid expressing Flag-RNF8 was transfected in 293B wild type and ASF1a -/- cells followed by bleomycin treatment before harvest. Asterisk indicates immunoglobulin heavy chain. (E to I) ASF1a is required for RNF8/168 localization, not MDC1, at I-SceI cut site. HeLa DR13-9 cells transfected with indicated siRNAs (E to G), wild type and ASF1a -/- cells (H and I) were applied to ChIP assay using anti-RNF8 (E and H), -MDC1 (F and I) and -RNF168 (G) antibodies. Mean ± SD of triplicates. ***, P < 0.005; *, P< 0.05.
RNF8 recruitment was not decreased in ASF1b depleted cells, indicating that ASF1a is the only ASF1 paralog required for the recruitment of RNF8 to the DSB (Figure S4A and S4B). The E3 ligase RNF2/RING1B, which is responsible for H2A/X ubiquitination at lys119-120 during DNA Damage Repair (Pan et al., 2011; Wu et al., 2011), continued to be recruited to the DSB in ASF1a-depleted cells (Figure S4C), indicating that ASF1a, like MDC1, is specifically required for the recruitment to DSBs of RNF8 and RNF168, but not RNF2/RING1B.
Histone ubiquitination on H2A/X and formation of FK2 foci require a histone chaperone independent function of ASF1a
Since RNF8-RNF168 recruitment to the DSB is impaired by ASF1a deficiency, we expected that ASF1a would be required for some specific types of histone ubiquitination at the DSB. Bleomycin increases phosphorylation of H2AX at Ser-139 to form H2AX (Figure 4A) and mono-ubiquitination of H2A/X as detected by the upshifted H2A or H2AX signal on immunoblots (Figure 7A). Depletion or knockout of ASF1a down-regulated the mono-ubiquitination of H2AX and of H2A (Figure 7A-C). Mono-ubiquitination of the histones that is detected by the shifted band was restored by expression of siASF1a-resistant ASF1a gene (Figure 7B). However, H2A ubiquitination specifically on K119, carried out by RNF2/RING1B, is not decreased by siASF1a (Figure S4D) or by knockout of ASF1a (Figure 7C). A better known target of RNF8, H1.2 (Thorslund et al., 2015), also showed decrease in ubiquitination in ASF1a knockout cells treated with bleomycin (Figure 7C). These results suggest that ASF1a is required for RNF8-RNF168 dependent ubiquitination on histones after DNA damage response. Consistent with this, bleomycin- or IR-induced FK2 foci, a marker for the poly-ubiquitin chains at DSB, were also decreased upon ASF1a knockdown (Figure 7D-F). siRNF8 and siRNF168 were used to knockdown factors known to be required for FK2 foci formation to provide a positive control for the experiment (Figure 7D and 7E). Thus, our results indicate that ASF1a is required for ubiquitination of histones by RNF8/RNF168 on specific sites at DSB sites, but not for PRC1 dependent ubiquitination of H2A on K119.
Figure 7. DSB-dependent ubiquitination on histones and FK2 focus formation require ASF1a.
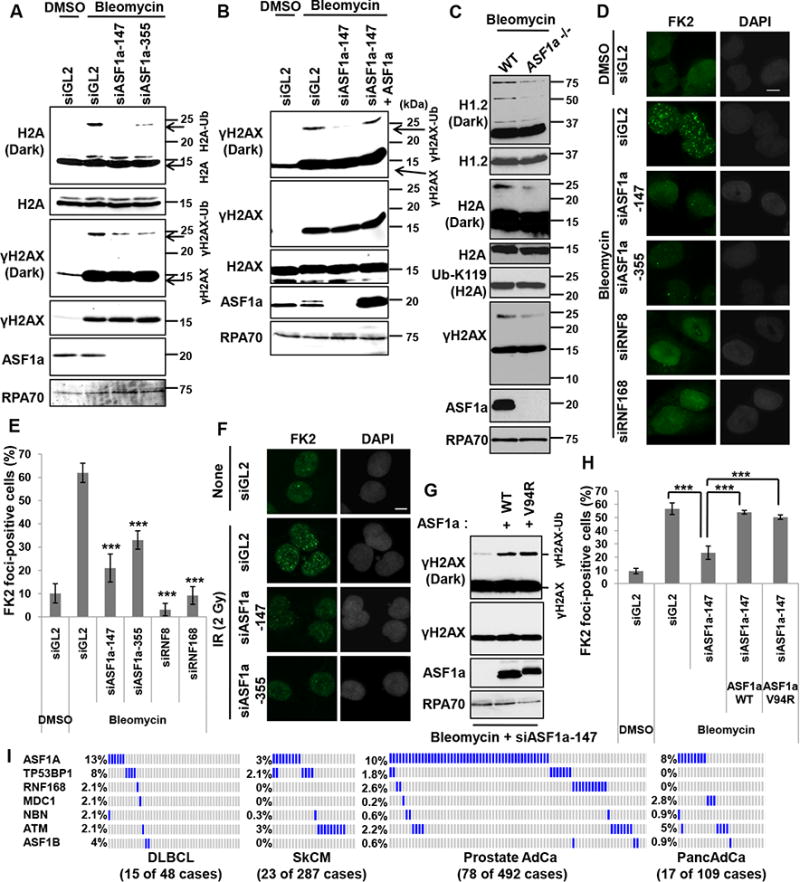
(A) DSB-induced H2A or H2AX mono-ubiquitination is dependent on ASF1a. HEK293T cells were transfected with indicated siRNAs and treated with bleomycin. (B) Expression of siRNA-resistant ASF1a restores γH2AX mono-ubiquitination in cells depleted of endogenous ASF1a. Indicated siRNAs were transfected into either wild type U2OS or U2OS cells with stable overexpression of siRNA-resistant ASF1a. (C) Decrease of RNF8/168-dependent ubiquitination of H1.2, H2A and γH2AX, but not PRC1 dependent ubiquitination of H2A K119, in ASF1a knockout cells. HeLa DR13-9 wild type or ASF1a -/- cells were incubated with 20 μgml-1 bleomycin for 80 min. (D, E and F) Immunostaining with anti-FK2 antibody in ASF1a depleted cells. U2OS cells were transfected with indicated siRNA and immunostained following treatment with 5 μgml-1 bleomycin for 1 hr (D and E) or at 90 min post-IR (2 Gy) (F). Representative images (D and F) and quantification of (D) in (E). Mean ± SD of triplicates. ***, P < 0.005. (G) Rescue of mono-ubiquitination of H2AX by wild-type or Val94Arg mutant ASF1a in ASF1a-depleted U2OS cells. (H) Rescue of FK2 foci by wild-type or Val94Arg mutant ASF1a in ASF1a-depleted U2OS cells. Indicated siRNAs were transfected into either wild type U2OS or U2OS cells with stable overexpression of siRNA-resistant ASF1a wild type or V94R mutant. Cells were immunostained after treatment of 10 μgml-1 bleomycin for 1 hr. Mean ± SD of triplicates. See also Figure S5. (I) High frequency of homozygous deletion of ASF1a gene in cancers. Cbioportal (http://cbioportal.org) was analyzed and each blue or gray bar represents a patient with or without a homozygous deletion in the indicated gene, respectively. To save space a large number of gray bars were omitted from the rows in SkCM, Prostate AdCa and PancAdCa. The number of patients whose tumors had homozygous deletion of at least one of these repair genes (excluding ASF1B) is shown below in each cancer.
Since, ASF1a has a role in chaperoning the H3-H4 heterodimer (Mello et al., 2002; Tyler et al., 1999; Tyler et al., 2001), we examined whether the V94 residue of ASF1a, essential for interacting with histones, was essential for histone-ubiquitination. Exogenous V94R mutant-ASF1a and wt-ASF1a restored mono-ubiquitination and FK2 foci formation equally well when expressed in cells depleted of endogenous ASF1a by siRNA (Figure 7G and 7H). Furthermore expression of V94R mutant-ASF1a in ASF1a knockout cells suppressed HR, promoted NHEJ and 53BP1 foci formation like wt-ASF1a (Figure S5A to S5C). In the • H2AX disappearance assay shown in Figure 2G, the overexpression of V94R in ASF1a knockout cells restored the rapid disappearance of • H2AX as well as overexpression of wild type ASF1a (Figure S5D). All these results indicate that V94R mutant of ASF1a has no defect in supporting NHEJ and, thus ASF1a promotes NHEJ at DSB independent of its histone chaperone function.
ASF1a deletion is seen in a significant fraction of certain malignancies
The genomic region containing the ASF1a gene, 6q22.31 is sometimes deleted in human cancers (Braggio et al., 2012; Kim et al., 2007b; Sung et al., 2011). We analyzed copy-number or sequencing data of cancer patients at CBioportal, to discover that homozygous deletion of ASF1a, unlike ASF1b, is seen in a significant fraction of several common malignancies, such as Diffuse Large B Cell Lymphoma (DLBCL), Skin Melanoma (SkCM), Prostate adenocarcinoma (Prostate AdCa) and Pancreatic Adenocarcinoma (PancAdCa) (Figure 7I). Genes important for DSB repair, ASF1a, TP53BP1, RNF168, MDC1, NBN and ATM, are inactivated in 27% of DLBCL and 15% of Prostate AdCa. ASF1a deletion is the mode of repair inactivation in a large fraction of those patients (13% and 10%, respectively). The high frequency of ASF1a deletion is comparable to, or exceeds, that of known master genes of DSB repair like TP53BP1 and ATM. It is interesting to note that in most cases where a deletion in genes of this pathway are seen, there is exclusive deletion of only one member of the pathway suggesting that the tumor may be selecting for the inactivation of DSB repair by mutating at least one gene in the pathway. It is also important to note that the related gene, ASF1b, is not deleted at anywhere near the frequency seen for ASF1a, highlighting the difference between ASF1a and ASF1b in DSB repair.
Given that ASF1a depletion renders cells more sensitive to radiation or to DSB-inducing agents (Figure 1E and 1F), these results also suggest that molecular screening for ASF1a deletion could become a biomarker for identifying patients who will benefit particularly from radiotherapy or DSB-inducing chemotherapy.
Discussion
The initial recruitment of either 53BP1 or BRCA1-CtIP to DSBs is a major determinant in selecting which pathway will resolve a DSB: HR or NHEJ (Bothmer et al., 2010; Bouwman et al., 2010; Bunting et al., 2010; Difilippantonio et al., 2008; Dimitrova et al., 2008). Our results surprisingly implicate a protein formerly thought to be only a histone chaperone, ASF1a, in promoting NHEJ repair while limiting HR, by facilitating the recruitment of 53BP1 (and BRCA1-RAP80) at the expense of BRCA1-CtIP.
We also pinpoint the step where NHEJ is impaired in the absence of ASF1a. It is already known that the recruitment of 53BP1 and of RAP80 is dependent on histone ubiquitination by RNF8 and RNF168 (reviewed in (Nakada, 2016)). The novel finding here is that although ATM phosphorylation of H2AX, Chk2 and NBS1, and MDC1 recruitment at DSBs does not require ASF1a, the latter is required for phosphorylation of MDC1 on S/TQ sites by ATM kinase. Phospho-MDC1 is necessary for interaction with the FHA domain of RNF8 to recruit the ubiquitin ligase to the DSB (Huen et al., 2007; Kolas et al., 2007). This explains how ASF1a facilitates the recruitment of RNF8 and RNF168 to the DSB, the subsequent ubiquitination of H2A/X, and the recruitment of 53BP1 and RAP80 to promote NHEJ. Although we see a good correlation between restoration of 53BP1 focus formation and NHEJ repair, there is report that suggests NHEJ, as measured in Class Switch Recombination may not require 53BP1 focus formation (Minter-Dykhouse et al., 2008). Therefore we cannot exclude the possibility that ASF1a has additional functions in NHEJ independent of 53BP1 focus formation.
The FHA domain of MDC1 has been suggested to interact with repair proteins such as ATM, Chk2, MRN complex and Rad51 (Goldberg et al., 2003; Lou et al., 2006; Lou et al., 2003b; So et al., 2009; Xu and Stern, 2003; Zhang et al., 2005a). The FHA domain of MDC1 interacts with ATM and is important for the stabilization of activated ATM at DSBs. The same FHA domain is also used for interaction with ASF1a, which has a role in promoting ATM binding to MDC1. There is no tripartite complex composed of ASF1a, MDC1 and ATM, and so the ASF1a-MDC1 interaction is necessary prior to the stable binding of phospho-ATM with MDC1. In fact MDC1 separately interacts with ATM and ASF1a upon DNA damage response. The initial binding of MDC1 to ASF1a switches to ATM within a few hours of DNA damage. This converse interaction of MDC1 towards ATM and ASF1a is consistent with a model that there is a handoff of MDC1 from ASF1a to phospho-ATM. Considering the interaction of the FHA domain on MDC1 with multiple DDR factors, it is also possible that the ASF1a-MDC1 interaction is indirect and mediated by other factors. Therefore it will be worthwhile to later explore how the binding of multiple repair proteins like Chk2, MRN or Rad51 to the FHA domain of MDC1 is controlled in a spatial- and temporal manner by ASF1a and conversely whether the interaction of these proteins with MDC1 is necessary for the MDC1-ASF1a interaction.
Unlike yeast, higher eukaryotes possess two paralogs of ASF1, ASF1a and ASF1b. The ubiquitination of H2A/X, interaction with MDC1, NHEJ efficiency or DSB localization of 53BP1 does not require the residue essential for ASF1a's histone chaperone activity. The independence from histone interaction is consistent with the observation that ASF1a's sister histone chaperone, ASF1b, cannot support RNF8 recruitment or NHEJ when ASF1a is absent. The histone chaperone activities of ASF1a and ASF1b derive from the conserved N-terminal domain of the two proteins, while the C-terminal domain is not conserved. Thus the DSB repair function that is unique to ASF1a may be mediated by unique C-terminal region. This difference in function of ASF1a and ASF1b is supported by other reports showing differences between the two paralogs. Unlike ASF1b, which is expressed primarily in proliferating cells, ASF1a is expressed in proliferating and quiescent cells (Corpet et al., 2011). ASF1a has specialized roles even as a histone chaperone: (a) it is the main donor to the H3.3-H4 histone chaperone HIRA (Tagami et al., 2004; Tang et al., 2006; Zhang et al., 2005b) and (b) ASF1a alone participates in H3 acetylation at Lys56 by CBP/p300 after DNA damage (Battu et al., 2011; Das et al., 2009). It is interesting that depletion of ASF1a but not ASF1b increased HR efficiency in an I-SceI based gene conversion assay (Duro et al., 2010). This, too, is consistent with our results, but the increase in HR could be because ASF1a depletion decreases NHEJ repair.
Recently, in a control experiment Li and Tyler found that human ASF1a did not affect the kinetics of DSB repair when I-PpoI was used to induce a DSB (Li and Tyler, 2016) but this could be due to incomplete depletion of ASF1a by shRNA. In our experiments, we transfected siRNAs twice at 24 hr-interval to effect the maximum depletion of ASF1a. Indeed a single transfection of siRNA was insufficient to affect NHEJ (data not shown), presumably because residual ASF1a is sufficient for NHEJ. This is also why we generated ASF1a knockout cell lines by CRISPR/CAS system to confirm that ASF1a is required to promote NHEJ and suppress HR (Figure 2).
Finally, the implication of ASF1a in promoting NHEJ may be of clinical significance. As shown in Fig. 7I, the Cancer Genome Atlas project reports that 13% of Diffuse Large B Cell Lymphoma, 3% of Skin Melanoma, 10% of Prostate adenocarcinoma and 8% of Pancreatic Adenocarcinoma suffer homozygous deletion of ASF1a gene. In addition, although there are six genes in this pathway that are deleted in these cancers, it is interesting that the usually only one gene in the pathway is deleted, suggesting that the tumors select for inactivation of the pathway, which can be effected by deleting any one gene in the pathway. Our results suggest that such cancers may be unable to complete NHEJ, leading to an increased sensitivity to chemotherapy that causes DSBs and to radiotherapy. This is a subject that needs to be carefully investigated because of the implication for personalized therapy of tumors.
Star*Methods
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ASF1a | Cell Signaling | Cat#2990 |
| ASF1b | Cell Signaling | Cat#2769 |
| Phospho-S343-NBS1 | Cell Signaling | Cat#3001 |
| Phospho-T68-Chk2 | Cell Signaling | Cat#2661 |
| γH2AX (S139) | Cell Signaling | Cat#2577 |
| H2AX | Cell Signaling | Cat#2595 |
| Ubiquityl-H2A (K119) | Cell Signaling | Cat#8240 |
| H2A | Cell Signaling | Cat#2578 |
| HA | Santa Cruz | Cat#sc-805 |
| Rad51 | Santa Cruz | Cat#sc-8349 |
| BRCA1 | Santa Cruz | Cat#sc-6954 |
| RNF8 | Santa Cruz | Cat#sc-271462 |
| RNF168 | Santa Cruz | Cat#sc-101125 |
| ATM | Santa Cruz | Cat#sc-7230 |
| Chk2 | Santa Cruz | Cat#sc-9064 |
| 53BP1 | Santa Cruz | Cat#sc-22760 |
| RPA70 | Calbiochem | Cat#NA13 |
| MCM9 | (Lee et al., 2015) | N/A |
| 53BP1 | BD Biosciences | Cat#6122522 |
| RAP80 | Bethyl Laboratories | Cat#A300-763A |
| CtIP | Novus Biological | Cat#NB100-79810 |
| NBS1 | Novus Biological | Cat#NB100-143 |
| CAF1 (p60) | Novus Biological | Cat#NB100-57523 |
| Anti-ubiquitinylated protein antibody (clone FK2) | EMD Millipore | Cat#04-263 |
| Phospho-S1981-ATM | Abcam | Cat#ab81292 |
| ATM | Abcam | Cat#ab78 |
| H1.2 | Abcam | Cat#ab17677 |
| H3 | Abcam | Cat#ab1791 |
| MDC1 | Abcam | Cat#ab11171 |
| MDC1 | Abcam | Cat#ab50003 |
| RNF2/RING1B | Abcam | Cat#ab3832 |
| FLAG | Sigma | Cat#F1804 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Bleomycin Sulfate | Enzo Life Sciences | BML-AP302-0010 |
| Cisplatin | Sigma | Cat#P4394 |
| Lipofectamine RNAiMAX Transfection Reagent | Invitrogen | Cat#13778150 |
| Lipofectamine 2000 Transfection Reagent | Invitrogen | Cat#11668019 |
| QuickExractIM DNA Extraction Solution | Epicentre | Cat#QE0905T |
| Ez-View Red Anti-HA Affinity Beads | Sigma | Cat#E6779 |
| Critical Commercial Assays | ||
| In-Fusion HD Cloning kit | Takara | Cat#638910 |
| Deposited Data | ||
| Raw imaging data | This paper | http://dx.doi.org/10.17632/3tjvrx6nn9.1 |
| Experimental Models: Cell Lines | ||
| NHEJ/DsRed293B | (Golding et al., 2009; Mueller et al., 2013) | N/A |
| NHEJ/DsRed293B ASF1a knockout cell lines | This paper | N/A |
| NHEJ/DsRed293B ASF1a knockout cell lines stably expressing ASF1a wildtype | This paper | N/A |
| NHEJ/DsRed293B ASF1a knockout cell lines stably expressing ASF1a V94R mutant | This paper | N/A |
| HeLa DR13-9 | (Ransburgh et al., 2010) | N/A |
| HeLa DR13-9 ASF1a knockout cell lines | This paper | N/A |
| HEK 293T | ATCC | CRL-3216 |
| U2OS | ATCC | HTB-96 |
| U2OS stably expressing ASF1a wildtype | This paper | N/A |
| U2OS stably expressing ASF1a V94R | This paper | N/A |
| U2OS pCW-ASF1a (TetOn) | This paper | N/A |
| Oligonucleotides | ||
| siASF1a-147, 5′-AAGUGAAGAAUACGAUCAAGU-3′ | (Groth et al., 2005) | N/A |
| siASF1a-355, 5′-GAGACAGAAUUAAGGGAAA-3′ | (Groth et al., 2007) | N/A |
| siASF1b, 5′-AACAACGAGUACCUCAACCCU-3′ | (Das et al., 2009) | N/A |
| siBRCA1, 5′-CCUGUCUCCACAAAGUGUG-3′ | (Zhu and Dutta, 2006) | N/A |
| si53BP1, 5′-GCCAGGUUCUAGAGGAUGA-3′ | (Wang et al., 2002) | N/A |
| siRNF8, 5′-GGAGAUAGCCCAAGGAGAA-3′ | (Nakada et al., 2012) | N/A |
| siRNF168, 5′-GGCGAAGAGCGAUGGAGGA-3′ | (Mosbech et al.) | N/A |
| siMDC1, 5′-UCCAGUGAAUCCUUGAGGU-3′ | (Lou et al., 2003a) | N/A |
| hASF1a targeting gRNA3 (CTAATTACTTGTACCTATCG AGG) | This paper | N/A |
| hASF1a targeting gRNA5 (TTACCTAGAACCATCAGTT GAGG) | This paper | N/A |
| PCR primers to validate ASF1a KO: forward, AGTCATG CTTTCAAGTATCAAGGGTCC; reverse, GTCTGGTTTT ACTGGTGGATTTTCCC | This paper | N/A |
| qPCR primers for ChIP (I-SceI cut site: forward, TACGG CAAGCTGACCCTGAA; reverse, GAAGTCGTGCTGCT TCATGT), (2 kb upstream of cut site (control): forward, GCCCATATATGG AGTTCCGC; reverse, CCCTATTGGCGTTACTATGG) | (Lee et al., 2015) | N/A |
| Primers for siASF1a-147-resistant ASF1a: forward, GTG GGCTCTGCAGAAAGCGAAGAATATGATCAAGTTTTA GACTC; reverse, GAGTCTAAAACTTGATCATATTCTTC GCTTTCTGCAGAGCCCAC | This paper | N/A |
| Primers for ASF1a V94R mutantion: forward, GATGCAG TAGGCGTAACTAGGGTGCTAATTACTTGTACC; reverse, GGTACAAGTAATTAGCACCCTAGTTACGCCT ACTGCATC | This paper | N/A |
| Recombinant DNA | ||
| HA-ASF1a WT (siASF1a-147 resistant) | This paper | N/A |
| HA-ASF1a V94R (siASF1a-147 resistant) | This paper | N/A |
| FLAG-RNF8 | This paper | N/A |
| Untagged ASF1a WT (siASF1a-147 resistant) | This paper | N/A |
| Untagged ASF1a V94R (siASF1a-147 resistant) | This paper | N/A |
| HA-MDC1 WT | (Wu et al., 2008) | N/A |
| HA-MDC1 ∆FHA | (Wu et al., 2008) | N/A |
| pCβASce (I-SceI) | Addgene | Cat#26477 |
| ASF1a gene targeting gRNA | This paper | N/A |
| Cas9 nuclease | Addgene | Cat#41815 N/A |
| pLKO.1-blast | This paper | N/A |
| Software and Algorithms | ||
| ZEN 2 (blue edition) | Zeiss, | N/A |
| Photoshop 7.0 | Adobe | N/A |
| ImageJ | NIH | N/A |
| CellQuest™ Pro | BD Biosciences | N/A |
| Flowing software Ver. 2.5 | http://flowingsoftwar e.btk.fi/ | N/A |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Anindya Dutta (ad8q@virginia.edu).
Experimental Model and Subject Details
Cell culture
U2OS, NHEJ/DsRed293B, HeLa DR13-9 and HEK293T were maintained in Dulbecco's modified Eagle's medium (Thermo Scientific) containing 10% fetal bovine serum (FBS; Sigma-Aldrich) and penicillin/streptomycin (1%, Gibco) at 37°C. NHEJ/DsRed293B cells were obtained from the Dr. J. Larner (University of Virginia) and K Valerie (Virginia Commonwealth University), and HeLa DR13-9 cells were a gift from JD Parvin (Ohio State University).
Method Details
Generation of stable expressing cell lines or knockout cell lines of ASF1a
To generate a cell line stably expressing siRNA-resistant ASF1a wild type or V94R mutant, the corresponding genes were inserted into retroviral vector pLHCX (Clontech), which were cotransfected with retroviral packaging vector into HEK293T cells using Lipofectamin 2000 (Invitrogen) according to the manufacturer's instruction. The supernatant of the culture medium was filtered by a 0.45-μm filter, and added to U2OS or NHEJ/DsRed293B (either wild type or ASF1a KO). The infected cells were selected by the medium containing 200 μgml-1 hygromycin B (Sigma-Aldrich) over 7 days. Alternatively, ASF1a gene was inserted into a doxycycline inducible lentiviral vector pCW, and U2OS cell lines generated by lentiviral infection followed by puromycin selection. 1 μgml-1 doxycycline was added to express the ASF1a protein for 48 hr.
gRNAs were cloned into pCR-Blunt II-TOPO backbone vector, gRNF_GFP-T2 (Addgene 41820) using In-Fusion HD cloning kit (Takara). gRNA target sequences were as follows. gRNA3: CTAATTACTTGTACCTATCGAGG gRNA5: TTACCTAGAACCATCAGTTGAGG. Human codon optimized Cas9 nuclease (hCas9) expression vector was obtained from Addgene (41815). Cell culture 293T cells were co-transfected with Cas9, two gRNAs, and vector containing blasticidin-S resistance gene (pLKO.1-blast) using Lipofectamine 2000 according to the manufacturer's protocol. pLKO.1-blast was cloned using pLKO.1-puro vector (Addgene 8453). Blasticidin S was added 2 days after transfection until untransfected cells were killed. Blasticidin S selected cells were diluted and plated into 96 well plates for single cell clone isolation. Genomic DNA from each single cell clone was obtained by the QuickExtract™ DNA Extraction Solution (Epicentre) and subjected to conventional PCR using the primers, forward, 5′-AGTCATGCTTTCAAGTATCAAGGGTCC-3′, and reverse, 5′-GTCTGGTTTTACTGGTGGATTTTCCC-3′ to check the genomic deletion. Together with this, the immunoblot for anti-ASF1a was performed to verify absence of the ASF1a protein. Cell cycle distribution of HeLa DR13-9 wild type or ASF1a KO cells in figure S1D was analyzed using a BD FACS Calibur and Flowing software (ver. 2.5).
NHEJ and HR assays
For NHEJ assay, 2 or 4 μg I-SceI expression vector pCβASce was transfected using Lipofectamine2000 (Invitrogen) into either siRNA-transfected or ASF1a knockout NHEJ/DsRed293B cells plated on 6 well dish at 50% confluency. For HR assay, 2 μg pCβASce was transfected into HeLa DR13-9 wild type or ASF1a KO cell lines. After 48 hr, the DsRed- and GFP-expressing cells were counted by the FL2 and FL1 channels in flow cytometric analysis (BD FACS Calibur and CellQuest™ Pro), respectively. The efficiency was calculated by normalizing the DsRed or GFP positive cells (%) to that of the siGL2 transfected or ASF1a wild type cell line also transfected with pCβASce. The DsRed signal in each knockout cell line untransfected with I-SceI was taken as background. H2AX disappearance after pulse-treatment of bleomycin was monitored to examine endogenous NHEJ efficiency. 293B wild type or ASF1a KO cells were incubated with 10 μgml-1 bleomycin for 1 hr followed by washing out, then harvested at different time points. Whole cell extracts were used for western blotting.
Immunoprecipitation, immunoblotting and antibodies
For immunoprecipitation, HEK293T or NHEJ/DsRed293B cells were lysed by lysis buffer containing 20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1% NP-40, 0.5 mM EDTA, 0.5 mM EGTA, 5 mM MgCl2, 10 mM NaF, 0.1 mM sodium vanadate, and protease inhibitors. After brief sonication followed by centrifugation at 15,000 rpm for 30 min, 1-2 mg of lysates were incubated with the indicated antibodies or 8 μl of EZview Red Anti-HA Affinity beads (E6779; Sigma) for 5 hr or overnight. The immuno-complex by was pulled down with protein A-conjugated agarose beads (GE healthcare). The beads were washed 5 times with the lysis buffer. For the interaction of HA-MDC1 with pS1981-ATM, a plasmid expressing HA-MDC1 was transfected in HEK293T cells with either control siRNA (siGL2) or siASF1a-147 followed by bleomycin treatment for 14 hr. For the interaction of HA-ASF1a with MDC1, IP was performed with anti-HA antibody from undamaged HEK293T expressing HA-ASF1a.For the interaction of HA-MDC1 with ATM and ASF1a in Fig. 5I and 5J, HA-MDC1 transfected HEK293T cells were harvested at indicated time points after pulse-treatment of 10 μgml-1 bleomycin for 1 hr, then immunoprecipitated with anti-HA antibody. The IPed protein amount was measured by imageJ software and normalized to pull-downed HA-MDC1 at each time point. For immunoblotting, total cell extract was subjected to SDS-polyacrylamide gel electrophoresis. Bleomycin or cisplatin was usually added for 14 hr before cell harvest, except where indicated. Antibodies for this study were as follows: anti-ASF1a (2990S), anti-ASF1b (2769S), anti-pS343-NBS1 (3001S), anti-pT68-Chk2 (2661S) anti-γ-H2AX (S139) (2577S), anti-H2AX (2595S), anti-Ubiquityl-H2A (K119)-H2A (8240S) and anti-H2A (2578S) (Cell Signaling Technology); anti-HA (sc-805), anti-Rad51 (sc-8349), anti-BRCA1 (sc-6954), anti-RNF8 (sc-271462), anti-RNF168 (sc-101125), anti-ATM (sc-7230), anti-Chk2 (sc-9064) and anti-53BP1 (sc-22760) (Santa Cruz Biotechnology); anti-RPA70 (NA13; Calbiochem); anti-MCM9 (home-made; raised in rabbits against the C-terminal 100 a.a of human MCM9 protein); anti-53BP1 (6122522; BD Biosciences); anti-RAP80 (A300-763A; Bethyl laboratories); anti-CtIP (NB100-79810), anti-NBS1 (NB100-143) and anti-CAF-1 (p60) (NB100-57523) (Novus Biological); anti-ubiquitinylated proteins antibody, clone FK2 (04-263) (EMD Millipore); anti-pS1981-ATM (ab81292), anti-ATM (ab78), anti-H1.2 (ab17677), anti-H3 (ab1791), anti-MDC1 (ab11171), anti-MDC1 (ab50003) and anti-RNF2/RING1B (ab3832) (Abcam); anti-FLAG (F1804; Sigma).
siRNAs and mutagenesis
The sequences of the siRNAs used in this paper are as follows: siASF1a-147, 5′-AAGUGAAGAAUACGAUCAAGU-3′; siASF1a-355, 5′-GAGACAGAAUUAAGGGAAA-3′; siASF1b, 5′-AACAACGAGUACCUCAACCCU-3′; siBRCA1, 5′-CCUGUCUCCACAAAGUGUG-3′; si53BP1, 5′-GCCAGGUUCUAGAGGAUGA-3′; siRNF8, 5′-GGAGAUAGCCCAAGGAGAA-3′; siRNF168, 5′-GGCGAAGAGCGAUGGAGGA-3′; siMDC1, 5′-UCCAGUGAAUCCUUGAGGU-3′. Each siRNA was transfected into the cells by Lipofectamine RNAi MAX (Invitrogen). siRNAs for ASF1a depletion were transfected twice at a 24 hr-interval to maximize the knockdown efficiency.
siRNA-resistant WT or V94R mutant ASF1a was made using the following primers: siASF1a-147 resistant, forward, 5′-GTGGGCTCTGCAGAAAGCGAAGAATATGATCAAGTTTTAGACTC-3′ and, reverse, 5′-GAGTCTAAAACTTGATCATATTCTTCGCTTTCTGCAGAGCCCAC-3′; V94R, forward, 5′-GATGCAGTAGGCGTAACTAGGGTGCTAATTACTTGTACC-3′ and, reverse, 5′-GGTACAAGTAATTAGCACCCTAGTTACGCCTACTGCATC-3′.
Cell survival assay
The clonogenic assay for cell viability was performed using siASF1a-transfected U2OS cells. After 2nd transfection of siRNA at second day, 2000 cells were plated on 6 well dishes. After 48 hr from 1st transfection of siASF1a, the indicated amount of bleomycin was incubated with cells for 24 hr, or cells were irradiated with 2 Gy of ionizing radiation (IR). Cell viability was measured by staining with crystal violet at 7 days from the treatment.
Immunofluorescence
The immunofluorescence was performed as previously described (Lee et al., 2015). U2OS or HeLa DR13-9 cells were grown on coverslips, washed with PBS twice, and fixed with 4% paraformaldehyde with 0.1 % Triton X-100 for 10 min. 0.5% Triton X-100 containing PBS was used to permeabilize the fixed cells followed by blocking with 10% FBS in PBST (0.1% Triton X-100 in PBS) overnight at 4°C. Endogenous protein was labeled with the primary antibodies for 2h, then incubated with Alexa Fluor 555 anti-rabbit (A21429; Life Technologies) or Alexa Fluor 488 anti-mouse (A11029; Life Technologies) immunoglobulin G secondary antibody in the same blocking buffer for 40 min at room temperature. The nuclei of cells were stained with 4′, 6′-diamidino-2-phenylindole (Vector Laboratories, Inc) and the stained cells were imaged by Zeiss AXIO observer A-1 equipped with Zeiss EC Plan-Apochromax 63X/1.4 oil and Zeiss AXIOCAM MRC. Acquired images were analyzed using Axiovision software and brightness and contrast of obtained images were adjusted using ZEN 2 (Zeiss, blue edition) and Photoshop 7.0 (Adobe). For colocalization of ASF1a and MDC1 foci, we used U2OS stable cells having the doxycycline inducible lentiviral ASF1a to overexpress ASF1a protein by treatment of 1 μgml-1 doxycycline for 48 hr.
Chromatin immunoprecipitation (ChIP) assay
The ChIP assay was performed as previously described with slight modifications (Negishi et al., 2010). HeLa DR13-9 cells were fixed with 1% formaldehyde for 20 minutes at RT followed by additional incubation with 0.125 M Glycine for 5 minutes. The cells were washed with PBS and lysed by SDS lysis buffer (50 mM Tris-HCl (pH 8.0), 10 mM EDTA (pH 8.0), 0.1% SDS and protease inhibitor cocktail (Roche)) on ice for 15 min followed by sonication (30 s-on / 30 s-off, 8 times at 10% amplitude) using Sonic Dismembrator model 500 (Fisher Scientific)). Lysates were diluted (1:10) using dilution buffer (50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1% Triton X-100, 0.1% SDS and protease inhibitor cocktail (Roche)) and incubated overnight with Protein G conjugated Dynabeads (Invitrogen), which were bound with indicated antibodies. The beads were sequentially washed with RIPA-150 (50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% SDS and 0.1% sodium deoxycholate), LiCl buffer (10 mM Tris-HCl (pH 8.0), 0.15 M LiCl, 1 mM EDTA, 0.5% NP-40, 0.5% sodium deoxycholate) and Tris-EDTA (TE) buffer and the beads were incubated in elution buffer (10 mM Tris-HCl (pH 8.0), 300 mM NaCl, 5 mM EDTA, and 0.5% SDS) at 65°C overnight. The DNA recovered by phenol:chloroform extraction was analyzed by qPCR using ABI 7300 real time PCR system (Applied Biosystems). Primer sequences used for qPCR were as follows: I-SceI cut site (F1, 5′-TACGGCAAGCTGACCCTGAA-3′; R1, 5′-GAAGTCGTGCTGCTTCATGT-3′) and 2 kb upstream of cut site (control) (F2, 5′-GCCCATATATGGAGTTCCGC-3′; R2, 5′-CCCTATTGGCGTTACTATGG-3′). The relative enrichment of each protein at I-SceI cut site was calculated by fold signal at I-SceI cut site relative to control site 2 kb upstream of cut site in each sample. For ChIP-reChIP, the beads from 1st immunoprecipitation were washed and incubated for 30 min at 37°C in resuspension buffer (10 mM DTT in TE). The supernatant was diluted 10 times with dilution buffer and mixed with the beads conjugated with next antibody overnight.
Homozygous deletion of ASF1a gene in cancers
Copy-number variation or sequencing data collated in Cbioportal (http://cbioportal.org) was analyzed to look for homozygous deletions in the indicated genes of DSB repair pathway, with ASF1B as a negative control.
Quantification and Statistical Analysis
In all the statistical analyse with a p value, students' t test was used. In each case, * and *** stands for p < 0.05 and p < 0.005, respectively. All the error bars used in the figures were obtained from three independent experiments as indicated in the relevant legends, and data were represented as mean ± standard deviation (SD).
Data and Software Availability
The raw data files for images are available at Mendeley (http://dx.doi.org/10.17632/3tjvrx6nn9.1).
Supplementary Material
Highlights.
-ASF1a, which can be homozygously deleted in cancers, promotes NHEJ and suppresses HR
-ASF1a interacts with MDC1 to facilitate the latter's interaction with activated ATM
-ASF1a thus promotes phosphorylation of MDC1 and the recruitment of RNF8 to DSBs
-ASF1a is essential for histone ubiquitination and 53BP1 recruitment at DSB and NHEJ
Acknowledgments
We thank Dr. Zhenkun Lou for supplying the plasmids for wild-type and deletion mutant of MDC1. Also, we thank members of the Dutta laboratory for helpful discussions and Dr. Michael Ortega for his careful reading and revision of the manuscript. This work was supported by the Farrow Fellowship, the NCI Cancer Center Support Grant P30 CA44579 (to K.Y.L.), R01 CA60499 and CA166054 (to A.D.).
Footnotes
Authors Contribution: K.Y.L. designed the study, executed most of the experiments and wrote the first draft of the paper. J.S.I. performed experiments implicating ASF1a in DSB repair. E.S. generated the knockout cell lines and some of plasmids. A.D. designed the study and edited the paper. K.Y.L. and A.D. analyzed and discussed results.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Battu A, Ray A, Wani AA. ASF1A and ATM regulate H3K56-mediated cell-cycle checkpoint recovery in response to UV irradiation. Nucleic Acids Res. 2011;39:7931–7945. doi: 10.1093/nar/gkr523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothmer A, Robbiani DF, Feldhahn N, Gazumyan A, Nussenzweig A, Nussenzweig MC. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J Exp Med. 2010;207:855–865. doi: 10.1084/jem.20100244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17:688–695. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braggio E, Dogan A, Keats JJ, Chng WJ, Huang G, Matthews JM, Maurer MJ, Law ME, Bosler DS, Barrett M, et al. Genomic analysis of marginal zone and lymphoplasmacytic lymphomas identified common and disease-specific abnormalities. Mod Pathol. 2012;25:651–660. doi: 10.1038/modpathol.2011.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Rondinelli B, D'Andrea AD. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016;26:52–64. doi: 10.1016/j.tcb.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, Sartori AA, Adams IR, Batista FD, Boulton SJ. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell. 2013;49:858–871. doi: 10.1016/j.molcel.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman KA, Greenberg RA. The BRCA1-RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. J Biol Chem. 2011;286:13669–13680. doi: 10.1074/jbc.M110.213728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corpet A, De Koning L, Toedling J, Savignoni A, Berger F, Lemaitre C, O'Sullivan RJ, Karlseder J, Barillot E, Asselain B, et al. Asf1b, the necessary Asf1 isoform for proliferation, is predictive of outcome in breast cancer. EMBO J. 2011;30:480–493. doi: 10.1038/emboj.2010.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das C, Lucia MS, Hansen KC, Tyler JK. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature. 2009;459:113–117. doi: 10.1038/nature07861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Koning L, Corpet A, Haber JE, Almouzni G. Histone chaperones: an escort network regulating histone traffic. Nat Struct Mol Biol. 2007;14:997–1007. doi: 10.1038/nsmb1318. [DOI] [PubMed] [Google Scholar]
- Di Virgilio M, Callen E, Yamane A, Zhang W, Jankovic M, Gitlin AD, Feldhahn N, Resch W, Oliveira TY, Chait BT, et al. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science. 2013;339:711–715. doi: 10.1126/science.1230624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Difilippantonio S, Gapud E, Wong N, Huang CY, Mahowald G, Chen HT, Kruhlak MJ, Callen E, Livak F, Nussenzweig MC, et al. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature. 2008;456:529–533. doi: 10.1038/nature07476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova N, Chen YC, Spector DL, de Lange T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature. 2008;456:524–528. doi: 10.1038/nature07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, Ellenberg J, Panier S, Durocher D, Bartek J, et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009;136:435–446. doi: 10.1016/j.cell.2008.12.041. [DOI] [PubMed] [Google Scholar]
- Duro E, Lundin C, Ask K, Sanchez-Pulido L, MacArtney TJ, Toth R, Ponting CP, Groth A, Helleday T, Rouse J. Identification of the MMS22L-TONSL complex that promotes homologous recombination. Mol Cell. 2010;40:632–644. doi: 10.1016/j.molcel.2010.10.023. [DOI] [PubMed] [Google Scholar]
- Feng L, Fong KW, Wang J, Wang W, Chen J. RIF1 counteracts BRCA1-mediated end resection during DNA repair. J Biol Chem. 2013;288:11135–11143. doi: 10.1074/jbc.M113.457440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fradet-Turcotte A, Canny MD, Escribano-Diaz C, Orthwein A, Leung CC, Huang H, Landry MC, Kitevski-LeBlanc J, Noordermeer SM, Sicheri F, Durocher D. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature. 2013;499:50–54. doi: 10.1038/nature12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg M, Stucki M, Falck J, D'Amours D, Rahman D, Pappin D, Bartek J, Jackson SP. MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature. 2003;421:952–956. doi: 10.1038/nature01445. [DOI] [PubMed] [Google Scholar]
- Golding SE, Morgan RN, Adams BR, Hawkins AJ, Povirk LF, Valerie K. Pro-survival AKT and ERK signaling from EGFR and mutant EGFRvIII enhances DNA double-strand break repair in human glioma cells. Cancer Biol Ther. 2009;8:730–738. doi: 10.4161/cbt.8.8.7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth A, Corpet A, Cook AJ, Roche D, Bartek J, Lukas J, Almouzni G. Regulation of replication fork progression through histone supply and demand. Science. 2007;318:1928–1931. doi: 10.1126/science.1148992. [DOI] [PubMed] [Google Scholar]
- Groth A, Ray-Gallet D, Quivy JP, Lukas J, Bartek J, Almouzni G. Human Asf1 regulates the flow of S phase histones during replicational stress. Mol Cell. 2005;17:301–311. doi: 10.1016/j.molcel.2004.12.018. [DOI] [PubMed] [Google Scholar]
- Helleday T, Lo J, van Gent DC, Engelward BP. DNA double-strand break repair: from mechanistic understanding to cancer treatment. DNA Repair (Amst) 2007;6:923–935. doi: 10.1016/j.dnarep.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Heyer WD, Ehmsen KT, Liu J. Regulation of homologous recombination in eukaryotes. Annu Rev Genet. 2010;44:113–139. doi: 10.1146/annurev-genet-051710-150955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Scully R, Sobhian B, Xie A, Shestakova E, Livingston DM. RAP80-directed tuning of BRCA1 homologous recombination function at ionizing radiation-induced nuclear foci. Genes Dev. 2011;25:685–700. doi: 10.1101/gad.2011011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, Chen J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007;131:901–914. doi: 10.1016/j.cell.2007.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass EM, Jasin M. Collaboration and competition between DNA double-strand break repair pathways. FEBS Lett. 2010;584:3703–3708. doi: 10.1016/j.febslet.2010.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Chen J, Yu X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science. 2007a;316:1202–1205. doi: 10.1126/science.1139621. [DOI] [PubMed] [Google Scholar]
- Kim JH, Dhanasekaran SM, Mehra R, Tomlins SA, Gu W, Yu J, Kumar-Sinha C, Cao X, Dash A, Wang L, et al. Integrative analysis of genomic aberrations associated with prostate cancer progression. Cancer Res. 2007b;67:8229–8239. doi: 10.1158/0008-5472.CAN-07-1297. [DOI] [PubMed] [Google Scholar]
- Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, Panier S, Mendez M, Wildenhain J, Thomson TM, et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 2007;318:1637–1640. doi: 10.1126/science.1150034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KY, Im JS, Shibata E, Park J, Handa N, Kowalczykowski SC, Dutta A. MCM8-9 complex promotes resection of double-strand break ends by MRE11-RAD50-NBS1 complex. Nat Commun. 2015;6:7744. doi: 10.1038/ncomms8744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Vogel H, Holcomb VB, Gu Y, Hasty P. Deletion of Ku70, Ku80, or both causes early aging without substantially increased cancer. Mol Cell Biol. 2007;27:8205–8214. doi: 10.1128/MCB.00785-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ML, Greenberg RA. Links between genome integrity and BRCA1 tumor suppression. Trends Biochem Sci. 2012;37:418–424. doi: 10.1016/j.tibs.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Tyler JK. Nucleosome disassembly during human non-homologous end joining followed by concerted HIRA- and CAF-1-dependent reassembly. Elife. 2016;5 doi: 10.7554/eLife.15129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou Z, Chini CC, Minter-Dykhouse K, Chen J. Mediator of DNA damage checkpoint protein 1 regulates BRCA1 localization and phosphorylation in DNA damage checkpoint control. J Biol Chem. 2003a;278:13599–13602. doi: 10.1074/jbc.C300060200. [DOI] [PubMed] [Google Scholar]
- Lou Z, Minter-Dykhouse K, Franco S, Gostissa M, Rivera MA, Celeste A, Manis JP, van Deursen J, Nussenzweig A, Paull TT, et al. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol Cell. 2006;21:187–200. doi: 10.1016/j.molcel.2005.11.025. [DOI] [PubMed] [Google Scholar]
- Lou Z, Minter-Dykhouse K, Wu X, Chen J. MDC1 is coupled to activated CHK2 in mammalian DNA damage response pathways. Nature. 2003b;421:957–961. doi: 10.1038/nature01447. [DOI] [PubMed] [Google Scholar]
- Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007;131:887–900. doi: 10.1016/j.cell.2007.09.040. [DOI] [PubMed] [Google Scholar]
- Mattiroli F, Vissers JH, van Dijk WJ, Ikpa P, Citterio E, Vermeulen W, Marteijn JA, Sixma TK. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell. 2012;150:1182–1195. doi: 10.1016/j.cell.2012.08.005. [DOI] [PubMed] [Google Scholar]
- McKinnon PJ. DNA repair deficiency and neurological disease. Nat Rev Neurosci. 2009;10:100–112. doi: 10.1038/nrn2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello JA, Sillje HH, Roche DM, Kirschner DB, Nigg EA, Almouzni G. Human Asf1 and CAF-1 interact and synergize in a repair-coupled nucleosome assembly pathway. EMBO Rep. 2002;3:329–334. doi: 10.1093/embo-reports/kvf068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minter-Dykhouse K, Ward I, Huen MS, Chen J, Lou Z. Distinct versus overlapping functions of MDC1 and 53BP1 in DNA damage response and tumorigenesis. J Cell Biol. 2008;181:727–735. doi: 10.1083/jcb.200801083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosbech A, Lukas C, Bekker-Jensen S, Mailand N. The deubiquitylating enzyme USP44 counteracts the DNA double-strand break response mediated by the RNF8 and RNF168 ubiquitin ligases. J Biol Chem. 288:16579–16587. doi: 10.1074/jbc.M113.459917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol. 2010;11:196–207. doi: 10.1038/nrm2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller AC, Sun D, Dutta A. The miR-99 family regulates the DNA damage response through its target SNF2H. Oncogene. 2013;32:1164–1172. doi: 10.1038/onc.2012.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munakata T, Adachi N, Yokoyama N, Kuzuhara T, Horikoshi M. A human homologue of yeast anti-silencing factor has histone chaperone activity. Genes Cells. 2000;5:221–233. doi: 10.1046/j.1365-2443.2000.00319.x. [DOI] [PubMed] [Google Scholar]
- Nakada S. Opposing roles of RNF8/RNF168 and deubiquitinating enzymes in ubiquitination-dependent DNA double-strand break response signaling and DNA-repair pathway choice. J Radiat Res. 2016;57(1):i33–i40. doi: 10.1093/jrr/rrw027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada S, Yonamine RM, Matsuo K. RNF8 regulates assembly of RAD51 at DNA double-strand breaks in the absence of BRCA1 and 53BP1. Cancer Res. 2012;72:4974–4983. doi: 10.1158/0008-5472.CAN-12-1057. [DOI] [PubMed] [Google Scholar]
- Negishi M, Saraya A, Mochizuki S, Helin K, Koseki H, Iwama A. A novel zinc finger protein Zfp277 mediates transcriptional repression of the Ink4a/arf locus through polycomb repressive complex 1. PLoS One. 2010;5:e12373. doi: 10.1371/journal.pone.0012373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan MR, Peng G, Hung WC, Lin SY. Monoubiquitination of H2AX protein regulates DNA damage response signaling. J Biol Chem. 2011;286:28599–28607. doi: 10.1074/jbc.M111.256297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol. 2014;15:7–18. doi: 10.1038/nrm3719. [DOI] [PubMed] [Google Scholar]
- Paull TT. Mechanisms of ATM Activation. Annu Rev Biochem. 2015;84:711–738. doi: 10.1146/annurev-biochem-060614-034335. [DOI] [PubMed] [Google Scholar]
- Ransburgh DJ, Chiba N, Ishioka C, Toland AE, Parvin JD. Identification of breast tumor mutations in BRCA1 that abolish its function in homologous DNA recombination. Cancer Res. 2010;70:988–995. doi: 10.1158/0008-5472.CAN-09-2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013;14:197–210. [PubMed] [Google Scholar]
- Sillje HH, Nigg EA. Identification of human Asf1 chromatin assembly factors as substrates of Tousled-like kinases. Curr Biol. 2001;11:1068–1073. doi: 10.1016/s0960-9822(01)00298-6. [DOI] [PubMed] [Google Scholar]
- So S, Davis AJ, Chen DJ. Autophosphorylation at serine 1981 stabilizes ATM at DNA damage sites. J Cell Biol. 2009;187:977–990. doi: 10.1083/jcb.200906064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, Livingston DM, Greenberg RA. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science. 2007;316:1198–1202. doi: 10.1126/science.1139516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda E, Hochegger H, Saberi A, Taniguchi Y, Takeda S. Differential usage of non-homologous end-joining and homologous recombination in double strand break repair. DNA Repair (Amst) 2006;5:1021–1029. doi: 10.1016/j.dnarep.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Stewart GS, Panier S, Townsend K, Al-Hakim AK, Kolas NK, Miller ES, Nakada S, Ylanko J, Olivarius S, Mendez M, et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 2009;136:420–434. doi: 10.1016/j.cell.2008.12.042. [DOI] [PubMed] [Google Scholar]
- Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421:961–966. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123:1213–1226. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- Sung CO, Kim SC, Karnan S, Karube K, Shin HJ, Nam DH, Suh YL, Kim SH, Kim JY, Kim SJ, et al. Genomic profiling combined with gene expression profiling in primary central nervous system lymphoma. Blood. 2011;117:1291–1300. doi: 10.1182/blood-2010-07-297861. [DOI] [PubMed] [Google Scholar]
- Tagami H, Ray-Gallet D, Almouzni G, Nakatani Y. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell. 2004;116:51–61. doi: 10.1016/s0092-8674(03)01064-x. [DOI] [PubMed] [Google Scholar]
- Tang Y, Poustovoitov MV, Zhao K, Garfinkel M, Canutescu A, Dunbrack R, Adams PD, Marmorstein R. Structure of a human ASF1a-HIRA complex and insights into specificity of histone chaperone complex assembly. Nat Struct Mol Biol. 2006;13:921–929. doi: 10.1038/nsmb1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorslund T, Ripplinger A, Hoffmann S, Wild T, Uckelmann M, Villumsen B, Narita T, Sixma TK, Choudhary C, Bekker-Jensen S, Mailand N. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature. 2015;527:389–393. doi: 10.1038/nature15401. [DOI] [PubMed] [Google Scholar]
- Tsabar M, Waterman DP, Aguilar F, Katsnelson L, Eapen VV, Memisoglu G, Haber JE. Asf1 facilitates dephosphorylation of Rad53 after DNA double-strand break repair. Genes Dev. 2016;30:1211–1224. doi: 10.1101/gad.280685.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler JK, Adams CR, Chen SR, Kobayashi R, Kamakaka RT, Kadonaga JT. The RCAF complex mediates chromatin assembly during DNA replication and repair. Nature. 1999;402:555–560. doi: 10.1038/990147. [DOI] [PubMed] [Google Scholar]
- Tyler JK, Collins KA, Prasad-Sinha J, Amiott E, Bulger M, Harte PJ, Kobayashi R, Kadonaga JT. Interaction between the Drosophila CAF-1 and ASF1 chromatin assembly factors. Mol Cell Biol. 2001;21:6574–6584. doi: 10.1128/MCB.21.19.6574-6584.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, Elledge SJ. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007;316:1194–1198. doi: 10.1126/science.1139476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Matsuoka S, Carpenter PB, Elledge SJ. 53BP1, a mediator of the DNA damage checkpoint. Science. 2002;298:1435–1438. doi: 10.1126/science.1076182. [DOI] [PubMed] [Google Scholar]
- Winkler DD, Zhou H, Dar MA, Zhang Z, Luger K. Yeast CAF-1 assembles histone (H3-H4)2 tetramers prior to DNA deposition. Nucleic Acids Res. 2012;40:10139–10149. doi: 10.1093/nar/gks812. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wu CY, Kang HY, Yang WL, Wu J, Jeong YS, Wang J, Chan CH, Lee SW, Zhang X, Lamothe B, et al. Critical role of monoubiquitination of histone H2AX protein in histone H2AX phosphorylation and DNA damage response. J Biol Chem. 2011;286:30806–30815. doi: 10.1074/jbc.M111.257469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Luo K, Lou Z, Chen J. MDC1 regulates intra-S-phase checkpoint by targeting NBS1 to DNA double-strand breaks. Proc Natl Acad Sci U S A. 2008;105:11200–11205. doi: 10.1073/pnas.0802885105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Stern DF. NFBD1/MDC1 regulates ionizing radiation-induced focus formation by DNA checkpoint signaling and repair factors. FASEB J. 2003;17:1842–1848. doi: 10.1096/fj.03-0310com. [DOI] [PubMed] [Google Scholar]
- Zgheib O, Pataky K, Brugger J, Halazonetis TD. An oligomerized 53BP1 tudor domain suffices for recognition of DNA double-strand breaks. Mol Cell Biol. 2009;29:1050–1058. doi: 10.1128/MCB.01011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ma Z, Treszezamsky A, Powell SN. MDC1 interacts with Rad51 and facilitates homologous recombination. Nat Struct Mol Biol. 2005a;12:902–909. doi: 10.1038/nsmb991. [DOI] [PubMed] [Google Scholar]
- Zhang R, Poustovoitov MV, Ye X, Santos HA, Chen W, Daganzo SM, Erzberger JP, Serebriiskii IG, Canutescu AA, Dunbrack RL, et al. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev Cell. 2005b;8:19–30. doi: 10.1016/j.devcel.2004.10.019. [DOI] [PubMed] [Google Scholar]
- Zhu W, Dutta A. An ATR- and BRCA1-mediated Fanconi anemia pathway is required for activating the G2/M checkpoint and DNA damage repair upon rereplication. Mol Cell Biol. 2006;26:4601–4611. doi: 10.1128/MCB.02141-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 53BP1 regulates DSB repair using Rif1 to control 5' end resection. Science. 2013;339:700–704. doi: 10.1126/science.1231573. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.