

Abstract
Significant progress has been made in understanding the principles underlying the development of liver fibrosis. This includes appreciating its dynamic nature, the importance of active fibrolysis in fibrosis regression, and the plasticity of cell populations endowing them with fibrogenic or fibrolytic properties. This is complemented by an increasing array of therapeutic targets with known roles in the progression or regression of fibrosis. With a key role for fibrosis in determining clinical outcomes and encouraging data from recently Food and Drug Administration-approved antifibrotics for pulmonary fibrosis, the development and validation of antifibrotic therapies has taken center stage in translational hepatology. In addition to summarizing the recent progress in antifibrotic therapies, the authors discuss some of the challenges ahead, such as achieving a better understanding of the interindividual heterogeneity of the fibrotic response, how to match interventions with the ideal patient population, and the development of better noninvasive methods to assess the dynamics of fibrogenesis and fibrolysis. Together, these advances will permit a better targeting and dose titration of individualized therapies. Finally, the authors discuss combination therapy with different antifibrotics as possibly the most potent approach for treating fibrosis in the liver.
Keywords: cirrhosis, collagen, fibroblast, fibrosis, liver, macrophage, myofibroblast, progression, regression, stellate cells, therapy
Our understanding of the mechanisms and specific components underlying the development and regression of liver fibrosis has matured toward clinical translation.1 Specialized cell types such as activated hepatic stellate cells (HSCs) and myofibroblasts (MFs)2,3 are central effectors of fibrogenesis (see “Origin and Function of Myofibroblasts in the Liver” by Wells and Schwabe in this issue), and other cells such as liver macrophages can promote either fibrogenesis or fibrolysis in a context-dependent manner (see “Resolution of Liver Fibrosis: Basic Mechanisms and Clinical Relevance” by Ramachandran, Iredale, and Fallowfield in this issue). Moreover, the underlying etiology of chronic liver damage determines both the mechanism and pattern of liver fibrosis, likely necessitating different approaches to antifibrotic therapy (see below). Instead of mere quantification of collagen and considering fibrosis as an endpoint, the dynamic processes of fibrogenesis and fibrolysis—the de novo formation and removal of connective tissue, respectively, that capture the dynamic nature of even advanced fibrosis— have taken center stage. Tissue injury is the most common stimulus for fibrogenesis, and immediately results in multiple coordinated processes aimed at initiating repair and regeneration, and at activating host defense.4 At early stages, initiating signals (DNA, adenosine triphosphate, other nucleotides and adenosine), responding cells (macrophages, platelets, liver sinusoidal endothelial cells [LSECs]), and soluble mediators (platelet-derived growth factor [PDGF], transforming growth factor-beta [TGF-β]) induce concomitant wound-healing responses, initiating repair, regeneration, and activation of host defense. With time, cells, cytokine responses, and matrix components become more specialized, but continue to have potent interactions with each other. Inflammation can either enhance the fibrogenic signal, for example, via secretion of soluble mediators (interleukin [IL] 1-β, IL-13, IL-17, and PDGF-BB), or induce fibrolysis (interferon- [IFN-] γ or IL-12). On the other hand, chronic inflammation is often regulated and dominated by the immunosuppressive TGF-β1, which is a highly potent fibrogenic factor. These interactions make inflammatory responses an attractive target, and focused anti-inflammatory approaches are expected to reduce tissue injury and fibrogenesis, without compromising liver regeneration, which is particularly attractive in inflammatory pathologies such as alcoholic and nonalcoholic hepatitis.
The differences between individuals that determine why some repair with a scar-free liver while others proceed to cirrhosis are determined by genetic and environmental factors (“second hits”), and the quantity of these different contributing factors appear to determine the outcome. Thus, the contribution of each cellular or signaling pathway may vary between groups of individuals. However, from a therapeutic perspective the situation seems manageable because the pathways that lead to fibrogenesis or induce fibrolysis are common between individuals, and only differ quantitatively. It also stresses the necessity of a personalized approach to treatment of fibrosis, using, for example, several biomarkers that quantify key fibrogenic or fibrolytic pathways. Notably, most of the pathways found for the liver are also central pathways in the development or regression of fibrosis in other organs and vice versa.1,5
It is important to recognize that fibrolysis is as complex and dynamic a process as fibrogenesis and provides additional therapeutic targets. Furthermore, cellular plasticity with economy of cellular populations is a common organizing principle. This is best demonstrated for liver macrophages that are key to the development of fibrogenesis as well as fibrolysis (see review by Ramachandran et al in this issue). This makes therapies that aim to delete cell populations deemed to be fibrogenic a blunt approach, which is likely to also limit fibrolysis.
Recognition of the full spectrum of changes associated with severe liver fibrosis is vital. In addition to quantitative and qualitative changes of the extracellular matrix (ECM), including increased ECM crosslinking and stiffness, liver fibrosis is associated with loss of hepatocytes, vascular remodeling, changes in cellular populations, and overall architectural distortion. The regenerative capacity of the liver is a great asset to all therapeutic strategies. However, therapies that aim to simply remove the ECM may not be effective against all the other pathological changes, and could even further impair liver function or increase the risk of liver cancer.
Principles of Antifibrotic Therapies
The recognition of heterogeneity in many aspects of fibrosis is a necessary step in therapeutic development. The fibrogenic pathways that are activated, and the relative amplitude of the inflammatory and fibrogenic responses vary significantly depending on the insult and its primary target cell, as exemplified by Schistosoma eggs (myeloid cells, lymphocytes), nonalcoholic steatohepatitis (NASH; hepatocytes, macrophages), or biliary obstruction (cholangiocytes). Inhibition of a single pathway upstream of the fibrogenic effector cells (HSCs and MFs) will likely generate a very different response in each of these conditions. Similarly, differences between early and advanced fibrosis will be extensive, as will be therapeutic responses. Further heterogeneity is present in the variable degree of fibrosis within the same liver. The very limited liver sampling possible by biopsy has entirely missed the variation that is present, and is now being revealed by noninvasive testing including elastography.
In view of this heterogeneity and the recent success of antiviral therapy, combination therapy for fibrosis is very attractive.6 The simplest approach in combination therapy is to target two vital but very different pathways to reduce upstream (chronic) inflammation and downstream ECM deposition. Combination therapy is also necessary because rapid, homogeneous, and monocausal fibrosis development in animal experimental data typically reveal single targets as being central to fibrogenesis, whereas modulation of such single molecules or pathways does not prove to be highly efficient in man. From the perspective of clinical drug development, the demonstration of antifibrotic efficacy in one organ makes the agent a candidate as an antifibrotic in other organs, and also a candidate for a second drug to be added as combination therapy.
Preclinical Testing
In Vitro and In Vivo Models
In vitro models are necessary for early drug discovery to advance our understanding of the molecular pathogenesis of liver fibrosis, and for high throughput testing once a target has been identified.7 These include culture-activated HSCs and HSC lines as well as other liver cells that are contributory to the fibrogenic or fibrolytic process. However, advanced preclinical proof of efficacy requires selected animal models, preferably mouse models that permit assessment of antifibrotic efficacy in the complex multicellular context and provide information on bioavailability, pharmacokinetics, pharmacodynamics, and toxicity. Because these models are only an approximation to the human scenario, there has been a tendency to omit a thorough in vivo preclinical validation before initiating larger phase 2 clinical studies. Examples are the 2-year studies of interferon-γ and the highly potent peroxisome proliferator activated receptor-γ (PPARγ) agonist Farglitazar in patients with advanced-stage hepatitis C, which yielded no effect by state-of-the-art biopsy-based fibrosis readouts.8,9
Animal models should reproduce the varied features of human liver fibrosis. These features include the degree and pattern of inflammation, biliary versus parenchymal damage, time course, and reversibility. Incorporation of the causative agent—hepatotropic virus, alcohol, or metabolic syndrome—is ideal, but can only be achieved for some disease or using humanized mice.10 Although no single model will perfectly represent even a given human etiology, useful predictions as to antifibrotic efficacy appear to be possible by using combinations. Thus, mice that lack the hepatocyte phospholipid flippase Mdr2 provide a model of spontaneous biliary fibrosis progression resembling primary sclerosing cholangitis, and discontinuation of toxin-administration in advanced toxin-induced fibrosis mimics advanced human parenchymal fibrosis with little tendency to reverse.11,12 Both models are characterized by only low-level inflammation and therefore show similarities to the target patients with advanced fibrosis of low-to-moderate inflammatory activity. Drugs that work in both models (inhibiting progression and inducing regression, respectively) may have a relatively high probability to be effective in man.
There has been significant progress in the development of rodent models of NASH. Earlier models produced components of NASH including steatosis and inflammation.13,14 Recently, diet-based models that use high-fat diets supplemented with cholesterol and fructose have captured central features of NASH including the metabolic syndrome, steatosis, inflammation, and fibrosis.15
An additional limitation is that the vast majority of studies are performed in a single strain of mice (typically C57BL/6), yet there are significant differences in fibrosis susceptibility between strains. Experiments are also typically done with young (6–12-week-old) mice, whereas liver fibrosis is usually a disease of older age, with older age as a risk factor for faster fibrosis progression.
Transgenic and Gene Deletion Models
Genetic models can confirm factors and mechanisms that drive fibrogenesis or fibrolysis in vivo, for example, transgenic mice with overexpression of PDGF-B, PDGF-C, or TGFβ1.16–18 However, these models do not reflect the multifaceted nature of human liver fibrosis, and lack chronic inflammatory liver injury, a key component in the development of fibrosis and long-term complications.19
Finally, in vivo models have to be done in an optimal and standardized quality, coupled with fibrosis readouts that accord to state of the art. This includes (1) group sizes of > 10 animals, (2) analysis of samples of sufficient size (5%–10% of the liver), and (3) use of complementary quantitative fibrosis and fibrolysis readouts. Notably, several past studies do not satisfy these criteria.7
Precision-Cut Tissue Slices
A criticism of animal studies is their unclear transferability to the humans, which may vary with the pharmacological target. Human precision-cut tissue slices (PCTS) that can be cultured for several days are ∼200-μm-thick punches of liver that partly reflect the multicellular human context.20,21 Precision-cut tissue slices can be obtained either from normal livers (resections, spontaneous fibrogenic activation ex vivo) or from cirrhotic explants. Multiple drugs can be tested in slices prepared from a small tissue block. This technology may serve as a preclinical bridge between animal models and the patient setting. However, more studies are needed for its validation.
One major obstacle is the species difference, with significant biological differences between rodents and humans.22 An approach to identify pathways that are important for fibrosis in humans is the concept of core pathways that are required for fibrosis in multiple organs and species.23 Increased testing of pathways in multiple organs in rodents is relatively straightforward, and able to provide a greater degree of certainty that the pathway will be important across different species. A second important issue is the high degree of homogeneity in experimental models. The test and control populations in experimental models are homogeneous across a wide range of parameters, including, age, sex, genetic background, diet, microbiome, etc. None of these will apply to the eventual human population, and it is relevant to ask if the efficacy of a compound as an antifibrotic is maintained if there is a controlled break in homogeneity in experimental models.
The Immune Response as an Antifibrotic Target
The immune response interacts with fibrogenesis and fibrolysis at multiple points, and is an attractive candidate for therapy.24 The healthy liver is notable for a very vigorous innate and subdued adaptive immune response.25 Among the innate cell population, liver macrophages have been most thoroughly investigated and have key functions in fibrogenesis and fibrolysis. The well-recognized resident macrophage population of the healthy liver (Kupffer cells [KCs]) are present at birth and are self-renewing.26 After injury, KCs initiate a fibrotic response via recruitment of additional innate immune cells, including large numbers of Ly6Chi inflammatory blood monocytes27 that quickly acquire the macrophage phenotype CD11b+ F4/80+ (Fig. 1).28–31 These infiltrating cells have the capacity to produce a wide range of cytokines, many of which have potent proinflammatory or direct profibrotic actions on HSCs and MFs, such as TNFα, IL-1β, TGF-β1, and PDGF-BB, respectively.32,33 They also express a range of chemokines like CCL-2, CCL-3, CCL-5, CCL-7, and CCL-8, which recruit MFs and other leukocytes.34 Targeting some of these molecules promises to be an effective antifibrotic strategy. To take TGF-β1 as an example, several strategies to block its activity have demonstrated efficacy in rodent models of liver fibrosis. These strategies include a fully humanized anti-TGF-β1 antibody (Lerdelimumab), soluble TGF-β1 receptors, blocking peptides, and a small molecule to block downstream activin receptor-like kinase activity (SB431542; NCT 00125385, 01665391, 01262001).35–39 Similarly, inhibition of several chemokines and their receptors demonstrated antifibrotic efficacy, including CCR5, CXCR4, and CXCR3 antagonists (NCT 00393120, 01413568).40,41 A shared concern is that these mediators affect different cell types and are involved in many processes including angiogenesis, and cellular proliferation and differentiation; their inhibition may have significant off-target effects as well.42 Some of these factors, especially chemokines, will also act differently if not in an opposite, fibrolytic way upon removal of the primary insult. Liver macrophage populations that have been vital for fibrogenesis undergo a major phenotypic switch, with enhanced production of e.g., matrix metalloproteinases (MMPs) to degrade the excess ECM and the release of proapoptotic ligands such as TRAIL, which can induce HSC and MF apoptosis.43–45 These proresolution macrophages have a distinct phenotype (CD11bhiF4/80intLY6Clow) and gene expression profile.32 For established fibrosis, enabling this phenotypic switch and enhancing the number of pro-resolution macrophages is an attractive antifibrotic approach.
Fig. 1.
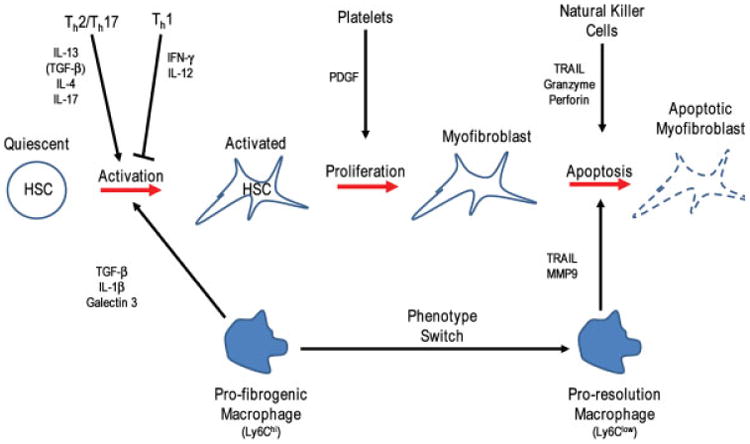
Multiple interactions between immune and profibrogenic cells. The progression of hepatic stellate cells (HSCs) from the quiescent to activated, to myofibroblasts, and eventually apoptosis is greatly influenced by paracrine signals from infiltrating blood monocytes which become tissue macrophages. At the initiation of injury, these tissue macrophages provide activation and proliferation signals, and during the resolution phase they provide apoptotic and reversion signals, but also actively digest and remove excess extracellular matrix. Additionally, innate (natural killer) and adaptive (Th1, Th2, and Th17) immune cells provide signals that can increase or decrease macrophage mediated fibrogenesis. Indirect cytokine production is shown in brackets. IL, interleukin; PDGF, platelet-derived growth factor; TGF, transforming growth factor.
The relative weight of the Th1 and Th2 T cell balance is an important determinant of fibrosis for innate immune and T cells.46,47 Thus the classically proinflammatory Th1 cytokines IFNγ and IL-12 are considered antifibrotic/fibrolytic, whereas the Th2 cytokines IL-4 and IL-13 are profibrogenic.47,48 The Th2 cytokines may be addressable by antibody-based therapies such as a bispecific antibody targeting IL-4 and IL-13.49 By analogy, macrophages can show a classical (M1) and an alternative (M2) polarization, which is induced by the same or similar cytokines that also induce Th1 versus Th2 polarization.50 However, there exist several subtypes of M2 macro-phages, with some of them possibly exhibiting antifibrotic effects, complicating simple Th1/M1 vs Th2/M2 polarizing approaches using cytokine (blocking) approaches.51,52 Therefore, skewing of this balance specifically toward Th1 (and M1) is more attractive than general inhibition of the Th2/M2 pathway, although such an approach needs to be balanced because it may enhance classical inflammation and tissue destruction.
The two related innate immune cell populations natural killer (NK) and natural killer T (NKT) cells have opposite effects. Natural killer cells have an important role in limiting fibrosis by inducing cell-cycle arrest and apoptosis of activated HSCs.53,54 Conversely, depletion and adoptive transfer experiments suggest that NKT cells can promote fibrogenesis, but the mechanism of their profibrotic action is not well characterized.55 More recently, type 2 innate lymphoid cells (ILC-2), which resemble Th2 T cells, have been demonstrated to be profibrogenic via secretion of IL13 and IL33, which directly activate HSCs.56
All antifibrotic therapies, particularly those that exert a regulatory activity, need to consider that the liver is never affected by fibrosis alone, but also by the underlying (usually inflammatory) disease. In this respect, fibrosis needs to be addressed in the context of the original disease. Antifibrotic therapies will affect many pathways. To increase efficacy and reduce side effects, therapies for specific fibrotic diseases will have to be well selected.
Regulating Platelet and Endothelial Function
Hepatic stellate cells are positioned adjacent to liver sinusoidal endothelial cells (LSECs), and the two have close functional interactions.57 After liver injury and the initiation of fibrosis are a loss of fenestrations in LSECs, increased expression of vasoconstrictors (ET-1 and angiotensin II), and decreased activity of vasodilators, most prominently nitric oxide (NO).58 In addition to these classic vascular changes, LSECs contribute to deposition of ECM (e.g., fibronectin and collagens I and IV), and cytokine production (e.g., TGF-β1 and PDGF-BB).59 Liver sinusoidal endothelial cells can also respond to changes in sinusoidal shear stress, with enhanced production of NO.60–62
Therapeutic targeting of LSECs in fibrosis has focused on their predominant role in regulating the dynamic part of intrahepatic portal hypertension, which is a major cause of morbidity and mortality in cirrhosis. Interventions have included broad spectrum kinase inhibitors such as sorafenib or sunitinib, and inhibitors of vascular endothelial growth factor and endothelial growth factor. Such interventions have resulted in changes that go beyond the hemodynamic to include reduction of fibrotic matrix.63,64 It is unclear how much of this reduction in fibrosis is due to regulation by LSECs, and how much of it is due to non-LSEC actions of these agents. However, as in inflammation, angiogenic mediators, while being profibrogenic during progression, can promote fibrolysis during regression.65 Liver sinusoidal endothelial cells also have a key role in regulating the relative response between liver regeneration and fibrosis. This is due to a stromal factor derived pathway, which can activate the chemokine receptors CXCR7 and CXCR4.66 After acute injury, activation of the CXCR7 pathway with recruitment of the downstream transcription factor Id1 results in a regenerative response. Chronic injury, however, results in a persistent activation of the FGF receptor 1 in LSEC that dampens the CXCR7-Id1 pathway, and activates a CXCR4 driven profibrotic pathway. Such pathways that regulate the switch between regeneration and fibrosis are excellent candidates for therapeutic intervention.67
Platelets are a rich source of profibrogenic factors, such as PDGF-BB and TGF-β1, but the role of platelets in fibrogenesis had been understudied.65,68,69 Recent reports have demonstrated that most if not all PDGF-BB in liver fibrosis derives from activated platelets and that its specific inhibition with a therapeutic antibody strongly attenuates fibrogenesis. Importantly, this effect is replicated with aspirin,8 a cheap and frequently used drug with an acceptable safety profile in early-to-moderate stages of liver disease. This finding demonstrates that we can expect marked (synergistic) antifibrotic effects by repurposing well-known drugs that are in use for other indications.
The ECM and Integrins as Antifibrotic Targets
A change in the composition and an increase in the amount of the ECM is the defining feature of all forms of fibrosis. In the normal liver, the extracellular matrix is composed predominantly of macromolecules including collagens (mainly the interstitial types I, III, V, VI, and the basement membrane types IV, XV, XVIII, and XIX), and a range of glycoproteins such as laminin isoforms and fibronectin, and several proteoglycans.70–72 During the development of rodent and human cirrhosis, there is a 5- to 10-fold increase in the content of collagens, particularly of fibril-forming types I and III, and an increase of elastin, laminins, and proteoglycans,73 which is accompanied by more highly crosslinked collagen fibers. The total amount of ECM is not only dependent on the rate of production, but also largely on the balance between the matrix degrading MMPs, and the inhibitors of metalloproteinases (TIMPs), especially TIMP-1.31 The MMPs are a family of endopeptidases that are produced by a wide range of cells, and taken together can degrade all the major constituents of the ECM.74 The TIMPs reduce MMP functionality by several mechanisms including stabilizing the proenzyme and also direct inhibition. Expression of TIMPs is more restricted than that of MMPs, and is high in activated HSCs. Several experiments have shown that alteration in either MMPs or TIMPs results in significant change in ECM deposition.
The ECM is not simply a downstream end product of the fibrotic cascade, but also directly feeds back onto it.71,75 An increase in the stiffness of the fibrotic matrix initially results in HSC and MF activation via receptor- (mainly integrin) mediated signal transduction from the altered ECM to the cellular cytoplasm and back to the ECM.76 Integrin receptors that (1) sense the collagen matrix and collagen-derived fragments, such as α1β1, α2β1, αvβ1, and αvβ3; (2) bind to fibronectin, such as αvβ3 and αvβ5; or (3) release active TGF-β1 (αvβ6 and αvβ8), which plays an important role in fibrogenesis.71,77,78 Taken together, these integrins and other ECM receptors mediate critical interactions between the ECM and hepatic cell populations, resulting in functional changes including adhesion, migration, proliferation, differentiation, and apoptosis, as well as modulation of cytokine, chemokine, and growth factor mediated signaling.71,79 Functional integrins are formed by noncovalent bonding of an α and a β subunit, with 24 known members in humans.80–82 In fibrosis, interest has focused on the role of αvβ6 and αvβ8 as activators of extracellular stored latent TGFβ1, which is proteolytically processed to active TGF-β1, for example, via MMP-14 mediated cleavage, upon cellular contraction and stretching.71,83–85 Latent TGFβ1 is tethered to αvβ6 or αvβ8 on activated cholangiocytes or HSCs/MFs, respectively via an arginine-glycine-aspartic acid motif.77,86,87 Integrin αvβ6 is virtually absent in the healthy liver and highly expressed after a range of insults.86,88,89 Therefore, the relative cellular specificity of the αv and especially TGF-β1 activating integrin αvβ6 permits selective inhibition of TGF-β activity in areas of mechanical stiffness and associated fibrogenesis. This is vital as total inhibition is known to result in unwanted proinflammatory changes.90 More generally, the family of αv integrins is expressed on many liver cell populations; genetic deletion or pharmacological inhibition of all αv integrins results in attenuated fibrogenesis,91 or in the abundant integrin αvβ3 (and αvβ5) that is mainly expressed on HSCs/ MF and macrophages.92,93
Collagens, the major ECM proteins in fibrosis, and elastin are stabilized via enzymatic crosslinking, which confers resistance to degradation, and thus may limit reversibility of established fibrosis.94 There has been a focus on the family of lysyl oxidases (LOX) that crosslink fibrillary collagen mainly at the nontriple helical ends (telopeptides) of the collagen molecules.11,95 LOX enzymes constitute a family of five members: LOX and LOX-like (LOXL) 1–4. They are secreted, copper-dependent amine oxidases with a variable N-terminal region and a conserved C-terminal domain that is necessary for catalytic activity. Expression of the LOX proteins is tightly controlled in a time- and organ-dependent manner during development, but aberrant expression and activity of these enzymes has been reported in a range of diseases associated with the ECM and in cancers,96,97 including an upregulation of LOX and LOXL2 in Wilson's disease, primary biliary and other etiologies of cirrhosis, and in pulmonary fibrosis.98–100 Hepatic stellate cells and portal MFs are major producers of LOX and LOXL2 in the liver.101 A humanized antibody (Simtuzumab) that blocks LOXL2 activity is currently being assessed in a large clinical study for liver fibrosis in patients with PSC or NASH (NCT01672853, NCT01672866, NCT01672879).99
Targeting Fibrosis Reversal
Recent animal studies have revealed that during experimental fibrosis regression up to half of the myofibroblasts undergo senescence and apoptosis, whereas the rest acquire a quiescent phenotype.102,103 The factors governing the inactivation of myofibroblasts are under investigation. For example, PPARγ plays a (limited) role in the re-establishment of the quiescent HSC phenotype,102 while matrix stiffness104 and crosslinking is currently addressed by LOXL2 inhibition (ClinicalTrials.gov, NCT01452308).99
Recruitment and activation of monocytes/macrophages is central to both fibrogenesis and fibrosis regression in rodents.105 Although targeting macrophage recruitment or polarization would be an attractive approach, the functional heterogeneity of macrophage subpopulations in humans has not yet been adequately characterized. Thus no clear links can be made yet from animal studies to human disease and the macrophage subsets may be dependent on the etiology of the liver disease. One rational attempt is the use of chemokine antagonists whose role in fibrogenesis seems to be preserved among species. Therefore, preventing the early recruitment of profibrotic mononuclear cells by CCL2 inhibition intrahepatic macrophages may be shifted toward the “restorative” subset, accelerating fibrosis regression.106
So Many Targets: Which Ones Are Attractive for Further Clinical Development?
Fig. 2 illustrates the complexity of cellular interactions and fibrogenic or fibrolytic signals exchanged between these cells. For the past 20 years there has been a steady addition to the number of molecules and pathways that are targets for antifibrotic therapy. TGFβ1 is one of the earliest such molecules and still occupies center stage. However, systemic inhibition of TGFβ1 results in increased inflammation.107 This spurred the targeting of specific steps in TGFβ1 activation, in a localized manner. Inhibition of integrin αvβ6, with reduction of TGFβ1 activation promises to be a highly effective and localized antifibrotic approach,86,88,89 and clinical trials using antibodies against avβ6 are underway.86 Connective tissue growth factor (CTGF) amplifies TGFβ1 signaling, and a monoclonal antibody targeting CTGF has shown promise in animal models of pulmonary fibrosis.108
Fig. 2.
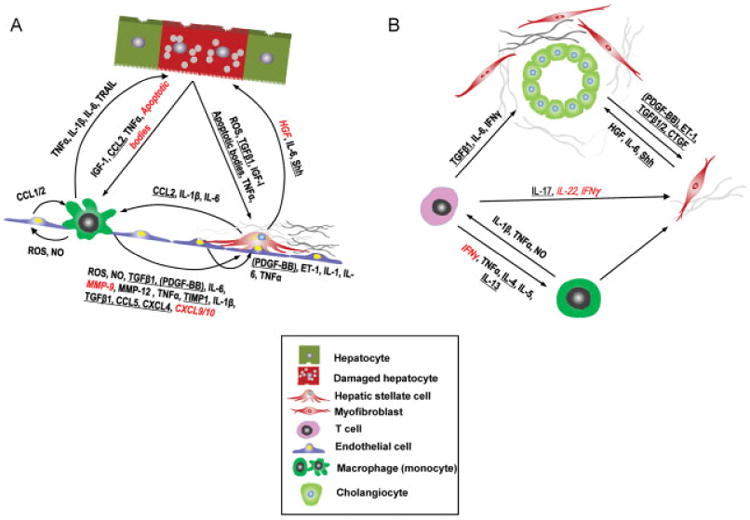
Multicellular context of fibrogenesis and fibrolysis: The postulated major cellular functional units and secreted factors that should be addressed in their complexity when designing effective antifibrotic strategies. (A) Vascular and (B) biliary unit. Profibrogenic targets are underlined, in contrast to putative fibrolysis-inducing targets in italics and red. Profibrogenic targets are underlined, in contrast to putative fibrolysis-inducing targets in italics. Modified from Schuppan and Kim.1 Baso, basophil; CCL, CC chemokine ligand; CTGF, connective tissue growth factor; CXCL, CXC chemokine ligand; ET-1, endothelin-1; HGF, hepatocyte growth factor; IFN, interferon; IGF, insulin-like growth factor; IL, interleukin; MMP, matrix metalloproteinase; NO, nitric oxide; PDGF-BB, platelet-derived growth factor with two subunits B (in parenthesis because a recent study indicates that most if not all PDGF-BB in liver fibrosis derives from activated platelets12; PMN, polymorphonuclear neutrophil; ROS, reactive oxygen species; TNFα, tumor necrosis factor α; Shh, sonic hedgehog; TGFβ1, transforming growth factor β1; Th, T helper cell; TIMP, tissue inhibitor of metalloproteinases; TRAIL, TNF-related apoptosis-inducing ligand; Treg, regulatory T cell.
Attenuating the activated phenotype of myofibroblasts is an attractive approach due to their key role in ECM deposition. Inhibition of the cannabinoid receptor 1 (CB1) reverses myofibroblast activation and attenuates experimental liver fibrosis.109 This has passed the proof of principle state, and peripheral-acting CB1 antagonists that may circumvent adverse side effects on the central nervous system like depression are being developed.110 In fibrotic NASH, progression is intimately linked to insulin resistance/type 2 diabetes, and the associated lipotoxic hepatocyte death and intestinal dysbiosis, providing rational targets for both anti-inflammatory and antifibrotic therapy in this condition.111,112 Therapeutic strategies include reducing oxidative stress, improving insulin signaling, activating the farnesoid × receptor receptor (e.g., with obeticholic acid), fibrosis-targeted inhibitors of hedgehog signaling, combined peroxisome proliferator activated receptor (PPAR)α/δ agonists,113–115 or manipulation of the altered gut microbiota using probiotics or microbiota transfer.112,116
Oxidative stress is an important cofactor in fibrosis, but the use of antioxidants has been disappointing.117 This may be due to differences between animal models and human disease, and the fibrosis stage and cell-specific regulation of oxidant and antioxidant pathways. Activation of NADPH oxidases (NOX1, NOX 2, and NOX4) induces HSC activation118–120 NOX4 can trigger apoptosis in hepatocytes.120 Inhibition of NOX1/NOX4 suppresses fibrogenesis in the CCl4 and bile duct ligation models, in pulmonary120–122 and in interstitial kidney fibrosis. A phase II trial is underway in diabetic kidney disease (ClinicalTrials.gov NCT02010242).
Tables 1 and 2 list relevant clinical drug trials using antifibrotic agents in liver fibrosis or other organ fibrosis with fibrosis as the primary or coprimary endpoint. What is remarkable is the diversity of agents that have been tested. They range from drugs with very broad or poorly characterized mechanism (e.g., omega-3 fats and vitamin D), to specific receptor inhibitors (losartan and liraglutide), broad but fairly low intensity anti-inflammatory and antiapoptotic effects (pentoxifylline and ursodeoxycholic acid), or multikinase inhibitors (nintedanib). This is a reflection of the wide range of biological processes that are involved in the development of liver fibrosis. Due to the obvious concerns of redundant pathways, and individual heterogeneity in active pathways that lead to fibrosis, there is a significant risk that many of the single agents listed may not have significant efficacy and/or display off-target side effects. However, the past and current studies are already providing a rich resource for designing effective treatments that would also exploit drug combinations in the near future. Notably, two antifibrotics (pirfenidone and nintedanib) have recently been approved by the Food and Drug Administration and the European Medicines Agency for the treatment of pulmonary fibrosis.
Table 1. Major studies with liver fibrosis as primary or coprimary endpoint (studies with at least 50 patients).
| Cause | Drug name (action), Treatment, Patients included (F,C, NR,SVR) | Efficacy | Year of completion/publication | Phase | No. of patients | NCTRef | |
|---|---|---|---|---|---|---|---|
| HCV (not exclusively antiviral agents) | Pentoxiphylline (anti-TNFα) vs. vit E; 1 y, r, db(F) | No results reported | 2006 | 3 | 100 | 00119119 | |
| IFNα-2b + R vs. IFNα-2b + R + Viusid (ascorbic acid, zinc, glycyrrhizic acid); 48 wk, r (F/NR) | Improved fibrosis score | 2007 | – | 100 | 124 | ||
| Farglitazar (PPARγ agonist); 52 wk, r, db (F/NR) | No effect | 2008/2010 | 2 | 225/265 | 002447519 | ||
| GS-9450 (pan-caspase inhibitor) vs. plac; 24 wk, nr, db (F/NR) | No results reported | 2010 | 2 | 307 | 00874796 | ||
| Irbesartan (AT1 R antagonist) vs. plac; 2 y, r, db (F/NR) | Pending | 2013 | 3 | 166 | 00265642 | ||
| Fuzheng Huayu (Chinese herbal drug) vs. plac, 48 wk, r, db (F) | Pending | 2014 | 2 | 100 | 00854087 | ||
| Pirfenidone (anti-inflammatory) vs. plac 2-year intervention | Pending | 2014 | 2–3 | 150 | 02161952 | ||
| HBV (not exclusively antiviral agents) | Salvianolic acid B (ingredient of Fuzheng Huayu) vs. IFNy; 6 mo, r, db (F) | No effect | 2002 | – | 60 | 125 | |
| Fuzheng Huayu) vs. plac; 6 mo, r, db (F); biopsy and serum fibrosis markers | Significant for fibrosis regression and fibrosis markers | 2005 | 226 | 126 | |||
| FG-3019 (anti-CTGF mAb) vs Entecavir vs. plac; 45 wk, r, db (F) | Pending | 2016 | 2 | 228 | 01217632 | ||
| Entecavir ± Fuzheng Huayu (Chinese herbal drug) vs. plac, 48 wk, r, db (C) | Pending | 2016 | 4 | 700 | 02241590 | ||
| HBV/HCV coinfected | Oltipraz (antiproliferative agent) vs. plac; 24 wk, r, db (F,C) | No effect | 2007/2011 | 2 | 83 | 00956098 | |
| PBC | UDCA (hydrophilic bile acid) vs. plac; 2 y, db (F,C) | No effect | 1991 | 3 | 146 | 127 | |
| UDCA vs. plac; 4 y, r, db (F,C) | Lower fibrosis progression rate; | 2000 | 4 | 103 | 128 | ||
| Obeticholic acid (FXR agonist) vs. plac; 12 mo-8 y, r, db (F); UE and serum fibrosis markers | Pending | 2023 | 3b | 350 | 02308111 | ||
| Alcoholic hepatitis | Candesartan (ACE inhibitor); 6 mo, r, db (F) | Histological improvement; 33.3% vs 11.6% (p = 0.020) | 2009/2012 | 1/2–2 | 85 | 00990639129 | |
| PSC | GS-6624 (anti-LOXL2 mAb) vs. plac; 96 wk, r, db(F) | Pending | 2015 | 2 | 225 | 01672853 | |
| NASH | Orlistat (pancreatic lipases inhibitor) vs. 1400 kcal diet (30% fat); 36 wk, r, ol (F) | No results reported | 2006 | 4 | 50 | 00160407 | |
| Pioglitazone (PPARy agonist) vs. plac; 6 mo, r, db | No effect | 2006 | 4 | 55 | 00227110130 | ||
| Pioglitazone vs. plac; 1 y, r, db (F) | Decreased fibrosis progression | 2008 | – | 74 | 131 | ||
| Pioglitazone vs. vit E vs. plac; 2 y, r, db (F) | Trend for decreased fibrosis progression for Pio groups | 2009/2010 | 3 | 247 | 00063622132 | ||
| Rosiglitazone (PPARγ agonist) vs. plac; 1 and 2 V, r (F) | No effect on fibrosis | 2010 | - | 53 | 133 | ||
| Pentoxifylline (anti-TNFα) vs. plac; 1 y, r, db (F) | Improved steatosis, lobular inflammation and fibrosis | 2010/2011 | 2 | 55 | 00590161134 | ||
| Rosiglitazone (Rosi) vs. Rosi + Metformin vs. Rosi + Losartan; 48 wk, r, ol (F) | No effect on fibrosis | 2011 | - | 137 | 135 | ||
| High-dose UDCA vs. plac, 1 y, r, db (F) | Significant reduction only of FibroTest | 2011 | 3 | 126 | 136 | ||
| Metformin (AMP kinase activator, antidiabetic); 1 y, r, db (F) | No results reported | 2012 | 4 | 80 | 00134303 | ||
| Metformin vs. insulin; 1 y, r, (C) | Pending | 2016 | 126 | NCT02234440 | |||
| Liraglutide (GLP-1 agonist) vs. plac; 48 wk, r, db(F) | No results reported | 2013 | 2 | 52 | 01237119 | ||
| Pentoxifylline + vit E vs. vit E; 3 mo (biopsy), r, db(F) | No results reported | 2013 | 3 | 120 | 01384578 | ||
| Losartan (AT1R antagonist) vs. plac; 2 y, r, db(F) | Pending | 2014 | 3 | 214 | 01051219 | ||
| Obeticholic acid (FXR agonist) vs. plac; 72 wk, r, db(F) | Significant for steatosis, lobular inflammation; marginally significant for fibrosis | 2014 | 2 | 280 | 01265498137 | ||
| Pioglitazone (PPARy agonist) vs. vit E vs. plac; 1.5 and 3 y, r, db (F) | Pending | 2014 | 4 | 90 | 00994682 | ||
| GS-6624 (anti-LOXL2 mAb; 75 mg vs. 125 mg) vs. plac; 100 wk, r, db (F) | Pending | 2015 | 2 | 225 | 01672866 | ||
| GS-6624 (200 mg vs. 700 mg) vs. plac; 100 wk, r, db (F,C) | Pending | 2015 | 2 | 225 | 01672879 | ||
| GFT505 (dual PPAR a/5 agonist); 52 wk, r, db (F) | Pending | 2015 | 2 | 270 | 01694849 | ||
| Pioglitazone (Pio) vs. vit E vs. vit E + Pio vs. plac; 1.5 and 3 y, r, db (F) | Pending | 2015 | 4 | 90 | 01002547 | ||
| Vit D vs. lifestyle counseling; 2 y, r, ol (F) | Pending | 2014 | 3 | 200 | 01623024 | ||
| Vit Dvs. plac; 48 wk, r, db (F) | Pending | 2015 | 2 | 60 | 01571063 | ||
| Omega-3 (fish oil) vs. plac; 1 y, r, db (F) | No results reported | 2010 | 2/3 | 64 | 00681408 | ||
| Omega-3 (fish oil); 18 mo, r, sb (F) | No results reported | 2013 | 2 | 100 | 00760513 | ||
| Docosahexaenoic acid; 2 y, r, db (F) | No results reported | 2011 | 1/2 | 60 | 00885313 | ||
| Eicosapentaenoic acid vs. plac; 1 y, r, db (F) | No results reported | 2012 | 2 | 243 | 01154985 | ||
| Diamel (dietary supplement) vs. plac vs. lifestyle counseling; 52 wk, r, db (F) | No results reported | 2012 | 3 | 158 | 00820651 | ||
| PolypiII (atorvastatin, valsartan); no biopsy (UE); 5 y, r, ol (F) | No results reported | 2018 | 3 | 1500 | 01245608 | ||
| NASH Surgery | Bariatric surgery (meta-analysis of 21 cohort studies) (F,C) | Variable effect | 2010 | – | 1643 | 138 | |
Abbreviations: ACE, angiotensin-converting enzyme; ATIR, angiotensin II receptor type 1; C, cirrhosis; CTGF, connective tissue growth factor; db, double-blind; F, fibrosis; FXR, farnesoid × receptor; GLP-1, glucagon-like peptide-1; IFN, interferon; IL, interleukin; LOXL2, lysyl oxidase-like 2; mAb, monoclonal antibody; NCT, number at ClinicalTrials.gov; nr, nonrandomized; NR, nonresponders; ol, open-label; plac, placebo; r, randomized; retro, retrospective analysis; TNFα, tumor necrosis factor α; UDCA, ursodeoxycholic acid; UE, ultrasound elastography; vit, vitamin.
Table 2. Studies in pulmonary and other fibrosis with fibrosis as primary or coprimary endpoint (studies with at least 50 patients).
| Fibrosis | Drug name/Treatment | Efficacy | Year of completion/publication | Phase | No. of patients | NCTRef. |
|---|---|---|---|---|---|---|
| Pulmonary | Etanercept (anti-TNFα) vs. plac; 48 wk, r, db | No effect | 2005/2008 | 2 | 88 | 00063869139 |
| N-acetylcystein (NAC, antioxidant) vs. plac; 1 y, r, db | Worsening of FVC and DLco in NAC-arm, no change in mortality | 2005 | 1/2 | 182 | 140 | |
| Bosentan (dual ET-1AR and ET-1 BR antagonist) vs. plac; 1 y, r, db Bosentan vs. plac; 12, 21 and 3 y (biopsy), r, db |
Worsening of PFT; decline in FVC, DLco and 02 saturation. No significant effect |
2005/2008 2010/2011 |
2/3 3 |
158 616 |
00071461 00391443 |
|
| Imatinib (kinase inhibitor) vs. plac; 92 wk, r, db | No effect | 2010 | 2/3 | 120 | 00131274141 | |
| Ambrisentan (ET-1AR antagonist) vs. plac; 92 wk, r, db | Terminated due to lack of efficacy | 2012 | 3 | 600 | 00768300142 | |
| Pirfenidone (anti-TGFβ, anti-TNFα, anti-IL-1β) vs. plac, 72 wk, r, db Pirfenidone vs. plac; 52 wk, r, db Pirfenidone vs. plac; 52 wk, r, db |
Study 004: reduced decline in FVC with high-dose pirfenidone | 2008 | 3 | 435 | 00287716143 | |
| Study 006: no difference in FVC Significant worsening of FVC Improved FVC, no difference in survival |
2008 2010 2014 |
3 3 3 |
344 275 555 |
00287729143–145 | ||
| BIBF1120 (Nintedanib, multi-RTK inhibitor) vs. plac; 1 y, r, db BIBF1120 vs. plac; 52 wk, r, db BIBF1120; 3 y, nr, ol |
Significantly reduced FVC decline and incidence of exacerbations Significantly reduced FVC decline Pending |
2011 2014 2015 |
2 2 |
432 1066 198 |
00514683146 01170065 |
|
| CNT0888 (anti-MCP1/CCL2 mAb) vs. plac; 74 wk, r, db | No results reported | 2012 | 2 | 126 | 00786201 | |
| QAX576 (anti-IL13 mAb); 4 wk, nr, ol | No results reported | 2009 | 2 | 52 | 00532233 | |
| FG-3019 (anti-CTGF mAb); 109 wk, ol | Pending | 2014 | 2 | 84 | 01262001 | |
| Myelofibrosis | GS-6624 (anti-LOXL2 mAb); 24 wk, r, ol | Pending | 2014 | 2 | 54 | 01369498 |
Abbreviations: ATIR, angiotensin II receptor type 1;CTGF, connective tissue growth factor; CXCR2, CXC chemokine receptor type 2; db, double-blind; DLCo, diffusing capacity of the lungs for carbon monoxide; ET-1A (B)R, endothelin-1 receptor type A(B); FVC, forced vital capacity; HMGCoAR, 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase; LOXL2, lysyl oxidase-like 2; mAb, monoclonal antibody; MCP1/CCL2, monocyte chemoattractant protein-1/CC chemokine ligand-2; mTOR, mammalian target of rapamycin; NCT, number at ClinicalTrials.gov; nr, nonrandomized; ol, open-label; PFT, pulmonary function test; plac, placebo; r, randomized; RTK, receptor tyrosine kinase; TNFα, tumor necrosis factor α.
The Patient Population to Be Studied
Recent reviews and guidelines highlight optimal patient selection and stratification for proof-of-concept clinical tri-als.1,5,123 Subjects should be stratified according to the major underlying etiology, gender, signs of the metabolic syndrome, alcohol use, concomitant medications, and routine surrogates of hepatic inflammation. They should be at an intermediate stage of fibrosis (e.g., Metavir stage 2–3) for highest probability to detect dynamic changes of progression or reversal. A noninvasive measure like transient elastography or acoustic radiation force imaging is helpful for preselection before biopsy is performed, which at present is still required as entry criterion and in follow-up. The inclusion of a genetic risk score for fibrosis progression, as validated for hepatitis C virus infection, is useful, but no such score has been validated for other etiologies. Although sampling variability of biopsy is high for viral hepatitis (25–30% for a one-stage difference), and even higher for fibrosis due to NASH and biliary diseases, high-quality antifibrotic drug trials that aim at biopsies of sufficient size (at least eight portal areas) and duration (2 years), and include ∼200 well-stratified patients have yielded reliable results.8,9 Current guidelines also suggest the inclusion of several biologically plausible surrogate markers of fibrosis or fibrosis progression, such as direct or indirect serum fibrosis markers, novel imaging technologies, or measurement of portal pressure in patients with advanced fibrosis. Many of these requirements have been fulfilled, such as in the currently largest trial testing the antifibrotic effect of a Loxl2-blocking antibody (Table 1). Finally, there is much activity to develop more sensitive and specific serological markers and imaging modalities for the assessment of fibrosis and especially fibrogenesis. Once validated in ongoing studies, such markers and technologies could dramatically reduce the time to validate a test drug or the number of patients needed.
Combination Therapies
Considering the expected synergies of modulating two or more fibrogenic (and/or fibrolytic) pathways and the potential to decrease or eliminate the side effects that may result from targeting a single mechanism, combinations of antifibrotic (anti-inflammatory) therapies hold great promise. Agents in such combinations can address the major cause of fibrosis, such as antivirals; derive from drugs with known safety profiles that are used for other cardiovascular or inflammatory indications in a process of drug repurposing; or be specifically targeted at fibrogenic or fibrolytic cells and pathways. Such drug combinations would factor in the multifactorial etiology of fibrosis and the quantitatively divergent fibrogenic pathways in each individual, which would likely also reduce potential side effects. However, despite its promise at present there is only scant data on the efficacy of combinations of potential antifibrotic agents due to the effort needed even at the preclinical stage.1 In man, clinical development of combination therapies that could guarantee thorough efficiency and low toxicity will only be possible with noninvasive tools that measure the effect of a given drug on its pharmacological target. In addition, we will need improved noninvasive biomarkers for the quantification of liver fibrosis, fibrogenesis, and liver function: surrogate markers for a personalized antifibrotic treatment that would permit titering of the given drugs and their combinations according to the individual antifibrotic response. The development and validation of such biomarkers has become a key focus of pharmaceutical and biotechnology companies with an interest in antifibrotic therapies.5,7,123
Acknowledgments
Studies related to this topic were supported by grants of the National Institutes of Health (NIH), German Research Foundation (DFG), German Ministry for Education and Research (BMBF), and European Union (EU) to DS, and of NIH and VA Merit Award to WZM. DS is the recipient of an Advanced Grant of the European Research Council related to liver fibrosis.
Abbreviations
- CB1
cannabinoid receptor 1
- CCl4
carbon tetrachloride
- CTGF
connective tissue growth factor
- ECM
extracellular matrix
- HSC
hepatic stellate cell
- IFN
interferon
- IL
interleukin
- KCs
Kupffer cells
- LOX
lysyl oxidase
- LSEC
liver sinusoidal endothelial cell
- MF
myofibroblast
- MMPs
matrix metalloproteinases
- NASH
nonalcoholic steatohepatitis
- NK
natural killer
- NKT
natural killer T
- NO
nitric oxide
- NOX
NADPH oxidase
- PCTS
precision-cut tissue slices
- PDGF
platelet-derived growth factor
- PPAR
peroxisome proliferator activated receptor
- TGF
transforming growth factor
- UDCA
ursodeoxycholic acid
References
- 1.Schuppan D, Kim YO. Evolving therapies for liver fibrosis. J Clin Invest. 2013;123(5):1887–1901. doi: 10.1172/JCI66028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mederacke I, Hsu CC, Troeger JS, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823. doi: 10.1038/ncomms3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wells RG. Portal fibroblasts in biliary fibrosis. Curr Pathobiol Rep. 2014;2(4):185–190. doi: 10.1007/s40139-014-0054-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luedde T, Kaplowitz N, Schwabe RF. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology. 2014;147(4):765–783. doi: 10.1053/j.gastro.2014.07.018. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedman SL, Sheppard D, Duffield JS, Violette S. Therapy for fibrotic diseases: nearing the starting line. Sci Transl Med. 2013;5(167):167s. doi: 10.1126/scitranslmed.3004700. r1. [DOI] [PubMed] [Google Scholar]
- 6.Marcellin P, Cooper C, Balart L, et al. Randomized controlled trial of danoprevir plus peginterferon alfa-2a and ribavirin in treatment-naïve patients with hepatitis C virus genotype 1 infection. Gastroenterology. 2013;145(4):790–800. doi: 10.1053/j.gastro.2013.06.051. e3. [DOI] [PubMed] [Google Scholar]
- 7.Popov Y, Schuppan D. Targeting liver fibrosis: strategies for development and validation of antifibrotic therapies. Hepatology. 2009;50(4):1294–1306. doi: 10.1002/hep.23123. [DOI] [PubMed] [Google Scholar]
- 8.Pockros PJ, Jeffers L, Afdhal N, et al. Final results of a double-blind, placebo-controlled trial of the antifibrotic efficacy of interferon-gamma1b in chronic hepatitis C patients with advanced fibrosis or cirrhosis. Hepatology. 2007;45(3):569–578. doi: 10.1002/hep.21561. [DOI] [PubMed] [Google Scholar]
- 9.McHutchison J, Goodman Z, Patel K, et al. Farglitizar Study Investigators. Farglitazar lacks antifibrotic activity in patients with chronic hepatitis C infection. Gastroenterology. 2010;138(4):1365–1373. 1373.e1–1373.e2. doi: 10.1053/j.gastro.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 10.Grompe M, Strom S. Mice with human livers. Gastroenterology. 2013;145(6):1209–1214. doi: 10.1053/j.gastro.2013.09.009. [DOI] [PubMed] [Google Scholar]
- 11.Popov Y, Sverdlov DY, Sharma AK, et al. Tissue transglutaminase does not affect fibrotic matrix stability or regression of liver fibrosis in mice. Gastroenterology. 2011;140(5):1642–1652. doi: 10.1053/j.gastro.2011.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshida S, Ikenaga N, Liu SB, et al. Extrahepatic platelet-derived growth factor-β, delivered by platelets, promotes activation of hepatic stellate cells and biliary fibrosis in mice. Gastroenterolo-gy. 2014;147(6):1378–1392. doi: 10.1053/j.gastro.2014.08.038. [DOI] [PubMed] [Google Scholar]
- 13.Takahashi Y, Soejima Y, Fukusato T. Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J Gastroenterol. 2012;18(19):2300–2308. doi: 10.3748/wjg.v18.i19.2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hebbard L, George J. Animal models of nonalcoholic fatty liver disease. Nature reviews Gastroenterol Hepatol. 2011;8:35–44. doi: 10.1038/nrgastro.2010.191. [DOI] [PubMed] [Google Scholar]
- 15.Charlton M, Krishnan A, Viker K, et al. Fast food diet mouse: novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am J Physiol Gastrointest Liver Physiol. 2011;301(5):G825–G834. doi: 10.1152/ajpgi.00145.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ueberham E, Löw R, Ueberham U, Schönig K, Bujard H, Gebhardt R. Conditional tetracycline-regulated expression of TGF-beta1 in liver of transgenic mice leads to reversible intermediary fibrosis. Hepatology. 2003;37(5):1067–1078. doi: 10.1053/jhep.2003.50196. [DOI] [PubMed] [Google Scholar]
- 17.Czochra P, Klopcic B, Meyer E, et al. Liver fibrosis induced by hepatic overexpression of PDGF-B in transgenic mice. J Hepatol. 2006;45(3):419–428. doi: 10.1016/j.jhep.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 18.Campbell JS, Hughes SD, Gilbertson DG, et al. Platelet-derived growth factor C induces liver fibrosis, steatosis, and hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2005;102(9):3389–3394. doi: 10.1073/pnas.0409722102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology. 2015;61(3):1066–1079. doi: 10.1002/hep.27332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olinga P, Schuppan D. Precision-cut liver slices: a tool to model the liver ex vivo. J Hepatol. 2013;58(6):1252–1253. doi: 10.1016/j.jhep.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 21.de Graaf IA, Olinga P, de Jager MH, et al. Preparation and incubation of precision-cut liver and intestinal slices for application in drug metabolism and toxicity studies. Nat Protoc. 2010;5(9):1540–1551. doi: 10.1038/nprot.2010.111. [DOI] [PubMed] [Google Scholar]
- 22.Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172(5):2731–2738. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 23.Mehal WZ, Iredale J, Friedman SL. Scraping fibrosis: expressway to the core of fibrosis. Nat Med. 2011;17(5):552–553. doi: 10.1038/nm0511-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol. 2014;14(3):181–194. doi: 10.1038/nri3623. [DOI] [PubMed] [Google Scholar]
- 25.Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology. 2012;143(5):1158–1172. doi: 10.1053/j.gastro.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 26.Yona S, Kim KW, Wolf Y, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38(1):79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol. 2014;60(5):1090–1096. doi: 10.1016/j.jhep.2013.12.025. [DOI] [PubMed] [Google Scholar]
- 28.Karlmark KR, Weiskirchen R, Zimmermann HW, et al. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology. 2009;50(1):261–274. doi: 10.1002/hep.22950. [DOI] [PubMed] [Google Scholar]
- 29.Hayashidani S, Tsutsui H, Shiomi T, et al. Anti-monocyte chemo-attractant protein-1 gene therapy attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation. 2003;108(17):2134–2140. doi: 10.1161/01.CIR.0000092890.29552.22. [DOI] [PubMed] [Google Scholar]
- 30.Lucas T, Waisman A, Ranjan R, et al. Differential roles of macro-phages in diverse phases of skin repair. J Immunol. 2010;184(7):3964–3977. doi: 10.4049/jimmunol.0903356. [DOI] [PubMed] [Google Scholar]
- 31.Lin SL, Castaño AP, Nowlin BT, Lupher ML, Jr, Duffield JS. Bone marrow Ly6Chigh monocytes are selectively recruited to injured kidney and differentiate into functionally distinct populations. J Immunol. 2009;183(10):6733–6743. doi: 10.4049/jimmunol.0901473. [DOI] [PubMed] [Google Scholar]
- 32.Ramachandran P, Pellicoro A, Vernon MA, et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci U S A. 2012;109(46):E3186–E3195. doi: 10.1073/pnas.1119964109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pradere JP, Kluwe J, De Minicis S, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology. 2013;58(4):1461–1473. doi: 10.1002/hep.26429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. 2010;30(3):245–257. doi: 10.1055/s-0030-1255354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thompson JE, Vaughan TJ, Williams AJ, et al. A fully human antibody neutralising biologically active human TGFbeta2 for use in therapy. J Immunol Methods. 1999;227(1-2):17–29. doi: 10.1016/s0022-1759(99)00060-5. [DOI] [PubMed] [Google Scholar]
- 36.Mori Y, Ishida W, Bhattacharyya S, Li Y, Platanias LC, Varga J. Selective inhibition of activin receptor-like kinase 5 signaling blocks profibrotic transforming growth factor beta responses in skin fibroblasts. Arthritis Rheum. 2004;50(12):4008–4021. doi: 10.1002/art.20658. [DOI] [PubMed] [Google Scholar]
- 37.Ueno H, Sakamoto T, Nakamura T, et al. A soluble transforming growth factor beta receptor expressed in muscle prevents liver fibrogenesis and dysfunction in rats. Hum Gene Ther. 2000;11(1):33–42. doi: 10.1089/10430340050016139. [DOI] [PubMed] [Google Scholar]
- 38.Yata Y, Gotwals P, Koteliansky V, Rockey DC. Dose-dependent inhibition of hepatic fibrosis in mice by a TGF-beta soluble receptor: implications for antifibrotic therapy. Hepatology. 2002;35(5):1022–1030. doi: 10.1053/jhep.2002.32673. [DOI] [PubMed] [Google Scholar]
- 39.Vogt J, Traynor R, Sapkota GP. The specificities of small molecule inhibitors of the TGFβ and BMP pathways. Cell Signal. 2011;23(11):1831–1842. doi: 10.1016/j.cellsig.2011.06.019. [DOI] [PubMed] [Google Scholar]
- 40.Gilliam BL, Riedel DJ, Redfield RR. Clinical use of CCR5 inhibitors in HIV and beyond. J Transl Med. 2011;9(Suppl 1):S9. doi: 10.1186/1479-5876-9-S1-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zaldivar MM, Pauels K, von Hundelshausen P, et al. CXC chemokine ligand 4 (Cxcl4) is a platelet-derived mediator of experimental liver fibrosis. Hepatology. 2010;51(4):1345–1353. doi: 10.1002/hep.23435. [DOI] [PubMed] [Google Scholar]
- 42.Hawinkels LJ, Ten Dijke P. Exploring anti-TGF-β therapies in cancer and fibrosis. Growth Factors. 2011;29(4):140–152. doi: 10.3109/08977194.2011.595411. [DOI] [PubMed] [Google Scholar]
- 43.Duffield JS, Forbes SJ, Constandinou CM, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115(1):56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pellicoro A, Aucott RL, Ramachandran P, et al. Elastin accumulation is regulated at the level of degradation by macrophage metalloelastase (MMP-12) during experimental liver fibrosis. Hepatology. 2012;55(6):1965–1975. doi: 10.1002/hep.25567. [DOI] [PubMed] [Google Scholar]
- 45.Popov Y, Sverdlov DY, Bhaskar KR, et al. Macrophage-mediated phagocytosis of apoptotic cholangiocytes contributes to reversal of experimental biliary fibrosis. Am J Physiol Gastrointest Liver Physiol. 2010;298(3):G323–G334. doi: 10.1152/ajpgi.00394.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wynn TA, Cheever AW, Jankovic D, et al. An IL-12-based vaccination method for preventing fibrosis induced by schistosome infection. Nature. 1995;376(6541):594–596. doi: 10.1038/376594a0. [DOI] [PubMed] [Google Scholar]
- 47.Shi Z, Wakil AE, Rockey DC. Strain-specific differences in mouse hepatic wound healing are mediated by divergent T helper cytokine responses. Proc Natl Acad Sci U S A. 1997;94(20):10663–10668. doi: 10.1073/pnas.94.20.10663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chiaramonte MG, Donaldson DD, Cheever AW, Wynn TA. AnIL-13 inhibitor blocks the development of hepatic fibrosis during a T-helper type 2-dominated inflammatory response. J Clin Invest. 1999;104(6):777–785. doi: 10.1172/JCI7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.May C, Sapra P, Gerber HP. Advances in bispecific biotherapeutics for the treatment of cancer. Biochem Pharmacol. 2012;84(9):1105–1112. doi: 10.1016/j.bcp.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 50.Wan J, Benkdane M, Teixeira-Clerc F, et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology. 2014;59(1):130–142. doi: 10.1002/hep.26607. [DOI] [PubMed] [Google Scholar]
- 51.Barron L, Wynn TA. Fibrosis is regulated by Th2 and Th17 responses and by dynamic interactions between fibroblasts and macrophages. Am J Physiol Gastrointest Liver Physiol. 2011;300(5):G723–G728. doi: 10.1152/ajpgi.00414.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Duffield JS, Lupher M, Thannickal VJ, Wynn TA. Host responses in tissue repair and fibrosis. Annu Rev Pathol. 2013;8:241–276. doi: 10.1146/annurev-pathol-020712-163930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Muhanna N, Abu Tair L, Doron S, et al. Amelioration of hepatic fibrosis by NK cell activation. Gut. 2011;60(1):90–98. doi: 10.1136/gut.2010.211136. [DOI] [PubMed] [Google Scholar]
- 54.Radaeva S, Sun R, Jaruga B, Nguyen VT, Tian Z, Gao B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology. 2006;130(2):435–452. doi: 10.1053/j.gastro.2005.10.055. [DOI] [PubMed] [Google Scholar]
- 55.Gao B, Radaeva S. Natural killer and natural killer T cells in liver fibrosis. Biochim Biophys Acta. 2013;1832(7):1061–1069. doi: 10.1016/j.bbadis.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McHedlidze T, Waldner M, Zopf S, et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity. 2013;39(2):357–371. doi: 10.1016/j.immuni.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Iwakiri Y, Shah V, Rockey DC. Vascular pathobiology in chronic liver disease and cirrhosis - current status and future directions. J Hepatol. 2014;61(4):912–924. doi: 10.1016/j.jhep.2014.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bhunchet E, Fujieda K. Capillarization and venularization of hepatic sinusoids in porcine serum-induced rat liver fibrosis: a mechanism to maintain liver blood flow. Hepatology. 1993;18(6):1450–1458. [PubMed] [Google Scholar]
- 59.Herbst H, Frey A, Heinrichs O, et al. Heterogeneity of liver cells expressing procollagen types I and IV in vivo. Histochem Cell Biol. 1997;107(5):399–409. doi: 10.1007/s004180050126. [DOI] [PubMed] [Google Scholar]
- 60.Shah V, Haddad FG, Garcia-Cardena G, et al. Liver sinusoidal endothelial cells are responsible for nitric oxide modulation of resistance in the hepatic sinusoids. J Clin Invest. 1997;100(11):2923–2930. doi: 10.1172/JCI119842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Langer DA, Das A, Semela D, et al. Nitric oxide promotes caspase-independent hepatic stellate cell apoptosis through the generation of reactive oxygen species. Hepatology. 2008;47(6):1983–1993. doi: 10.1002/hep.22285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Deleve LD, Wang X, Guo Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology. 2008;48(3):920–930. doi: 10.1002/hep.22351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tugues S, Fernandez-Varo G, Muñoz-Luque J, et al. Antiangiogenic treatment with sunitinib ameliorates inflammatory infiltrate, fibrosis, and portal pressure in cirrhotic rats. Hepatology. 2007;46(6):1919–1926. doi: 10.1002/hep.21921. [DOI] [PubMed] [Google Scholar]
- 64.Thabut D, Routray C, Lomberk G, et al. Complementary vascular and matrix regulatory pathways underlie the beneficial mechanism of action of sorafenib in liver fibrosis. Hepatology. 2011;54(2):573–585. doi: 10.1002/hep.24427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang L, Kwon J, Popov Y, et al. Vascular endothelial growth factor promotes fibrosis resolution and repair in mice. Gastroenterology. 2014;146(5):1339–50. doi: 10.1053/j.gastro.2014.01.061. e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ding BS, Cao Z, Lis R, et al. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature. 2014;505(7481):97–102. doi: 10.1038/nature12681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marra F, Tacke F. Roles for chemokines in liver disease. Gastroenterology. 2014;147(3):577–594. doi: 10.1053/j.gastro.2014.06.043. e1. [DOI] [PubMed] [Google Scholar]
- 68.Kinnman N, Francoz C, Barbu V, et al. The myofibroblastic conversion of peribiliary fibrogenic cells distinct from hepatic stellate cells is stimulated by platelet-derived growth factor during liver fibrogenesis. Lab Invest. 2003;83(2):163–173. doi: 10.1097/01.lab.0000054178.01162.e4. [DOI] [PubMed] [Google Scholar]
- 69.Nocito A, Georgiev P, Dahm F, et al. Platelets and platelet-derived serotonin promote tissue repair after normothermic hepatic ischemia in mice. Hepatology. 2007;45(2):369–376. doi: 10.1002/hep.21516. [DOI] [PubMed] [Google Scholar]
- 70.Schuppan D, Cramer T, Bauer M, Strefeld T, Hahn EG, Herbst H. Hepatocytes as a source of collagen type XVIII endostatin. Lancet. 1998;352(9131):879–880. doi: 10.1016/S0140-6736(05)60006-2. [DOI] [PubMed] [Google Scholar]
- 71.Schuppan D, Ruehl M, Somasundaram R, Hahn EG. Matrix as a modulator of hepatic fibrogenesis. Semin Liver Dis. 2001;21(3):351–372. doi: 10.1055/s-2001-17556. [DOI] [PubMed] [Google Scholar]
- 72.Myers JC, Li D, Bageris A, Abraham V, Dion AS, Amenta PS. Biochemical and immunohistochemical characterization of human type XIX defines a novel class of basement membrane zone collagens. Am J Pathol. 1997;151(6):1729–1740. [PMC free article] [PubMed] [Google Scholar]
- 73.Schuppan D. Structure of the extracellular matrix in normal and fibrotic liver: collagens and glycoproteins. Semin Liver Dis. 1990;10(1):1–10. doi: 10.1055/s-2008-1040452. [DOI] [PubMed] [Google Scholar]
- 74.Arthur MJ, Iredale JP, Mann DA. Tissue inhibitors of metalloproteinases: role in liver fibrosis and alcoholic liver disease. Alcohol Clin Exp Res. 1999;23(5):940–943. doi: 10.1111/j.1530-0277.1999.tb04208.x. [DOI] [PubMed] [Google Scholar]
- 75.Iredale JP, Thompson A, Henderson NC. Extracellular matrix degradation in liver fibrosis: biochemistry and regulation. Biochim Biophys Acta. 2013;1832(7):876–883. doi: 10.1016/j.bbadis.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 76.Janmey PA, Wells RG, Assoian RK, McCulloch CA. From tissue mechanics to transcription factors. Differentiation. 2013;86(3):112–120. doi: 10.1016/j.diff.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sheppard D. Integrin-mediated activation of latent transforming growth factor beta. Cancer Metastasis Rev. 2005;24(3):395–402. doi: 10.1007/s10555-005-5131-6. [DOI] [PubMed] [Google Scholar]
- 78.Aluwihare P, Mu Z, Zhao Z, et al. Mice that lack activity of alphavbeta6- and alphavbeta8-integrins reproduce the abnormalities of Tgfb1- and Tgfb3-null mice. J Cell Sci. 2009;122(Pt 2):227–232. doi: 10.1242/jcs.035246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Henderson NC, Sheppard D. Integrin-mediated regulation of TGFβ in fibrosis. Biochim Biophys Acta. 2013;1832(7):891–896. doi: 10.1016/j.bbadis.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110(6):673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 81.Huttenlocher A, Horwitz AR. Integrins in cell migration. Cold Spring Harb Perspect Biol. 2011;3(9):a005074. doi: 10.1101/cshperspect.a005074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Campbell ID, Humphries MJ. Integrin structure, activation, and interactions. Cold Spring Harb Perspect Biol. 2011;3(3):4994. doi: 10.1101/cshperspect.a004994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Munger JS, Huang X, Kawakatsu H, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96(3):319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 84.Mu D, Cambier S, Fjellbirkeland L, et al. The integrin alpha(v) beta8 mediates epithelial homeostasis through MT1-MMP-de-pendent activation of TGF-beta1. J Cell Biol. 2002;157(3):493–507. doi: 10.1083/jcb.200109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol. 2007;179(6):1311–1323. doi: 10.1083/jcb.200704042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Patsenker E, Popov Y, Stickel F, Jonczyk A, Goodman SL, Schuppan D. Inhibition of integrin alphavbeta6 on cholangiocytes blocks transforming growth factor-beta activation and retards biliary fibrosis progression. Gastroenterology. 2008;135(2):660–670. doi: 10.1053/j.gastro.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kitamura H, Cambier S, Somanath S, et al. Mouse and human lung fibroblasts regulate dendritic cell trafficking, airway inflammation, and fibrosis through integrin αvβ8-mediated activation of TGF-β. J Clin Invest. 2011;121(7):2863–2875. doi: 10.1172/JCI45589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Popov Y, Patsenker E, Stickel F, et al. Integrin alphavbeta6 is a marker of the progression of biliary and portal liver fibrosis and a novel target for antifibrotic therapies. J Hepatol. 2008;48(3):453–464. doi: 10.1016/j.jhep.2007.11.021. [DOI] [PubMed] [Google Scholar]
- 89.Wang B, Dolinski BM, Kikuchi N, et al. Role of alphavbeta6 integrin in acute biliary fibrosis. Hepatology. 2007;46(5):1404–1412. doi: 10.1002/hep.21849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shull MM, Ormsby I, Kier AB, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multi-focal inflammatory disease. Nature. 1992;359(6397):693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Henderson NC, Arnold TD, Katamura Y, et al. Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med. 2013;19(12):1617–1624. doi: 10.1038/nm.3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhou X, Murphy FR, Gehdu N, Zhang J, Iredale JP, Benyon RC. Engagement of alphavbeta3 integrin regulates proliferation and apoptosis of hepatic stellate cells. J Biol Chem. 2004;279(23):23996–24006. doi: 10.1074/jbc.M311668200. [DOI] [PubMed] [Google Scholar]
- 93.Patsenker E, Popov Y, Stickel F, et al. Pharmacological inhibition of integrin alphavbeta3 aggravates experimental liver fibrosis and suppresses hepatic angiogenesis. Hepatology. 2009;50(5):1501–1511. doi: 10.1002/hep.23144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Issa R, Zhou X, Constandinou CM, et al. Spontaneous recovery from micronodular cirrhosis: evidence for incomplete resolution associated with matrix cross-linking. Gastroenterology. 2004;126(7):1795–1808. doi: 10.1053/j.gastro.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 95.Lucero HA, Kagan HM. Lysyl oxidase: an oxidative enzyme and effector of cell function. Cell Mol Life Sci. 2006;63(19-20):2304–2316. doi: 10.1007/s00018-006-6149-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kagan HM, Li W. Lysyl oxidase: properties, specificity, and biological roles inside and outside of the cell. J Cell Biochem. 2003;88(4):660–672. doi: 10.1002/jcb.10413. [DOI] [PubMed] [Google Scholar]
- 97.Barker HE, Cox TR, Erler JT. The rationale for targeting the LOX family in cancer. Nat Rev Cancer. 2012;12(8):540–552. doi: 10.1038/nrc3319. [DOI] [PubMed] [Google Scholar]
- 98.Kagan HM. Lysyl oxidase: mechanism, regulation and relationship to liver fibrosis. Pathol Res Pract. 1994;190(9-10):910–919. doi: 10.1016/S0344-0338(11)80995-7. [DOI] [PubMed] [Google Scholar]
- 99.Barry-Hamilton V, Spangler R, Marshall D, et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat Med. 2010;16(9):1009–1017. doi: 10.1038/nm.2208. [DOI] [PubMed] [Google Scholar]
- 100.Vadasz Z, Kessler O, Akiri G, et al. Abnormal depositionofcollagen around hepatocytes in Wilson's disease is associated with hepatocyte specific expression of lysyl oxidase and lysyl oxidase like protein-2. J Hepatol. 2005;43(3):499–507. doi: 10.1016/j.jhep.2005.02.052. [DOI] [PubMed] [Google Scholar]
- 101.Perepelyuk M, Terajima M, Wang AY, et al. Hepatic stellate cells and portal fibroblasts are the major cellular sources of collagens and lysyl oxidases in normal liver and early after injury. Am J Physiol Gastrointest Liver Physiol. 2013;304(6):G605–G614. doi: 10.1152/ajpgi.00222.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kisseleva T, Cong M, Paik Y, et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci U S A. 2012;109(24):9448–9453. doi: 10.1073/pnas.1201840109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Troeger JS, Mederacke I, Gwak GY, et al. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology. 2012;143(4):1073–83. doi: 10.1053/j.gastro.2012.06.036. e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Olsen AL, Bloomer SA, Chan EP, et al. Hepatic stellate cells require a stiff environment for myofibroblastic differentiation. Am J Physiol Gastrointest Liver Physiol. 2011;301(1):G110–G118. doi: 10.1152/ajpgi.00412.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18(7):1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Baeck C, Wei X, Bartneck M, et al. Pharmacological inhibition of the chemokine C-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing Ly-6C(+) macrophage infiltration in mice. Hepatology. 2014;59(3):1060–1072. doi: 10.1002/hep.26783. [DOI] [PubMed] [Google Scholar]
- 107.Samarakoon R, Overstreet JM, Higgins PJ. TGF-β signaling in tissue fibrosis: redox controls, target genes and therapeutic opportunities. Cell Signal. 2013;25(1):264–268. doi: 10.1016/j.cellsig.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang Q, Usinger W, Nichols B, et al. Cooperative interaction of CTGF and TGF-βinanimal models of fibrotic disease. Fibrogenesis Tissue Repair. 2011;4(1):4. doi: 10.1186/1755-1536-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Muñoz-Luque J, Ros J, Fernández-Varo G, et al. Regression of fibrosis after chronic stimulation of cannabinoid CB2 receptor in cirrhotic rats. J Pharmacol Exp Ther. 2008;324(2):475–483. doi: 10.1124/jpet.107.131896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tam J, Cinar R, Liu J, et al. Peripheral cannabinoid-1 receptor inverse agonism reduces obesity by reversing leptin resistance. Cell Metab. 2012;16(2):167–179. doi: 10.1016/j.cmet.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Henao-Mejia J, Elinav E, Jin C, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482(7384):179–185. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mehal WZ. The Gordian Knot of dysbiosis, obesity and NAFLD. Nat Rev Gastroenterol Hepatol. 2013;10(11):637–644. doi: 10.1038/nrgastro.2013.146. [DOI] [PubMed] [Google Scholar]
- 113.Bohinc BN, Diehl AM. Mechanisms of disease progression in NASH: new paradigms. Clin Liver Dis. 2012;16(3):549–565. doi: 10.1016/j.cld.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 114.Mudaliar S, Henry RR, Sanyal AJ, et al. Efficacy and safety of the farnesoid × receptor agonist obeticholic acid inpatients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology. 2013;145(3):574–82.e1. doi: 10.1053/j.gastro.2013.05.042. [DOI] [PubMed] [Google Scholar]
- 115.Guy CD, Suzuki A, Zdanowicz M, et al. NASH CRN. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology. 2012;55(6):1711–1721. doi: 10.1002/hep.25559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wood NJ. Microbiota: dysbiosis driven by inflammasome deficiency exacerbates hepatic steatosis and governs rate of NAFLD progression. Nat Rev Gastroenterol Hepatol. 2012;9(3):123. doi: 10.1038/nrgastro.2012.21. [DOI] [PubMed] [Google Scholar]
- 117.Phung N, Pera N, Farrell G, Leclercq I, Hou JY, George J. Pro-oxidant-mediated hepatic fibrosis and effects of antioxidant intervention in murine dietary steatohepatitis. Int J Mol Med. 2009;24(2):171–180. doi: 10.3892/ijmm_00000220. [DOI] [PubMed] [Google Scholar]
- 118.Paik YH, Iwaisako K, Seki E, et al. The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91(phox) mediate hepatic fibrosis in mice. Hepatology. 2011;53(5):1730–1741. doi: 10.1002/hep.24281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jiang JX, Venugopal S, Serizawa N, et al. Reduced nicotinamide adenine dinucleotide phosphate oxidase 2 plays a key role in stellate cell activation and liver fibrogenesis in vivo. Gastroenterology. 2010;139(4):1375–1384. doi: 10.1053/j.gastro.2010.05.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jiang JX, Chen X, Serizawa N, et al. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic Biol Med. 2012;53(2):289–296. doi: 10.1016/j.freeradbiomed.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Thannickal VJ. Mechanisms of pulmonary fibrosis: role of activated myofibroblasts and NADPH oxidase. Fibrogenesis Tissue Repair. 2012;5(Suppl 1) doi: 10.1186/1755-1536-5-S1-S23. Proceedings of Fibroproliferative disorders: from biochemical analysis to targeted therapies Petro E Petrides and David Brenner)S23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Aoyama T, Paik YH, Watanabe S, et al. Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology. 2012;56(6):2316–2327. doi: 10.1002/hep.25938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Torok NJ, Dranoff JA, Schuppan D, et al. Strategies and endpoints of antifibrotic drug trials Summary and Recommendations from the AASLD Emerging Trends Conference 2014; Hepatology; 2015. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Vilar Gomez E, Gra Oramas B, Soler E, Llanio Navarro R, Ruenes Domech C. Viusid, a nutritional supplement, in combination with interferon alpha-2b and ribavirin in patients with chronic hepatitis C. Liver Int. 2007;27(2):247–259. doi: 10.1111/j.1478-3231.2006.01411.x. [DOI] [PubMed] [Google Scholar]
- 125.Liu P, Hu YY, Liu C, et al. Clinical observation of salvianolic acid B in treatment of liver fibrosis in chronic hepatitis B. World J Gastroenterol. 2002;8(4):679–685. doi: 10.3748/wjg.v8.i4.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Liu P, Hu YY, Liu C, et al. Multicenter clinical study on Fuzhenghuayu capsule against liver fibrosis due to chronic hepatitis B. World J Gastroenterol. 2005;11(19):2892–2899. doi: 10.3748/wjg.v11.i19.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Poupon RE, Poupon R, Balkau B The UDCA-PBC Study Group. Ursodiol for the long-term treatment of primary biliary cirrhosis. N Engl J Med. 1994;330(19):1342–1347. doi: 10.1056/NEJM199405123301903. [DOI] [PubMed] [Google Scholar]
- 128.Corpechot C, Carrat F, Bonnand AM, Poupon RE, Poupon R. The effect of ursodeoxycholic acid therapy on liver fibrosis progression in primary biliary cirrhosis. Hepatology. 2000;32(6):1196–1199. doi: 10.1053/jhep.2000.20240. [DOI] [PubMed] [Google Scholar]
- 129.Kim MY, Cho MY, Baik SK, et al. Beneficial effects of candesartan, an angiotensin-blocking agent, on compensated alcoholic liver fibrosis - a randomized open-label controlled study. Liver Int. 2012;32(6):977–987. doi: 10.1111/j.1478-3231.2012.02774.x. [DOI] [PubMed] [Google Scholar]
- 130.Belfort R, Harrison SA, Brown K, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med. 2006;355(22):2297–2307. doi: 10.1056/NEJMoa060326. [DOI] [PubMed] [Google Scholar]
- 131.Aithal GP, Thomas JA, Kaye PV, et al. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology. 2008;135(4):1176–1184. doi: 10.1053/j.gastro.2008.06.047. [DOI] [PubMed] [Google Scholar]
- 132.Sanyal AJ, Chalasani N, Kowdley KV, et al. NASH CRN. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362(18):1675–1685. doi: 10.1056/NEJMoa0907929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ratziu V, Charlotte F, Bernhardt C, et al. LIDO Study Group. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology. 2010;51(2):445–453. doi: 10.1002/hep.23270. [DOI] [PubMed] [Google Scholar]
- 134.Zein CO, Yerian LM, Gogate P, et al. Pentoxifylline improves nonalcoholic steatohepatitis: a randomized placebo-controlled trial. Hepatology. 2011;54(5):1610–1619. doi: 10.1002/hep.24544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Torres DM, Jones FJ, Shaw JC, Williams CD, Ward JA, Harrison SA. Rosiglitazone versus rosiglitazone and metformin versus rosiglitazone and losartan in the treatment of nonalcoholic steatohepatitis in humans: a 12-month randomized, prospective, open-label trial. Hepatology. 2011;54(5):1631–1639. doi: 10.1002/hep.24558. [DOI] [PubMed] [Google Scholar]
- 136.Ratziu V, de Ledinghen V, Oberti F, et al. FRESGUN. A randomized controlled trial of high-dose ursodesoxycholic acid for nonalcoholic steatohepatitis. J Hepatol. 2011;54(5):1011–1019. doi: 10.1016/j.jhep.2010.08.030. [DOI] [PubMed] [Google Scholar]
- 137.Neuschwander-Tetri BA, Loomba R, Sanyal AJ, et al. for the NASH Clinical Research Network. Farnesoid × nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 14:2014. 61934. doi: 10.1016/S0140-6736(14)61933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Chavez-Tapia NC, Tellez-Avila FI, Barrientos-Gutierrez T, Mendez-Sanchez N, Lizardi-Cervera J, Uribe M. Bariatric surgery for non-alcoholic steatohepatitis in obese patients. Cochrane Database Syst Rev. 2010;1(1):CD007340. doi: 10.1002/14651858.CD007340.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Raghu G, Brown KK, Costabel U, et al. Treatment of idiopathic pulmonary fibrosis with etanercept: an exploratory, placebo-controlled trial. Am J Respir Crit Care Med. 2008;178(9):948–955. doi: 10.1164/rccm.200709-1446OC. [DOI] [PubMed] [Google Scholar]
- 140.Demedts M, Behr J, Buhl R, et al. IFIGENIA Study Group. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. 2005;353(21):2229–2242. doi: 10.1056/NEJMoa042976. [DOI] [PubMed] [Google Scholar]
- 141.Daniels CE, Lasky JA, Limper AH, Mieras K, Gabor E, Schroeder DR Imatinib-IPF Study Investigators. Imatinib treatment for idiopathic pulmonary fibrosis: Randomized placebo-controlled trial results. Am J Respir Crit Care Med. 2010;181(6):604–610. doi: 10.1164/rccm.200906-0964OC. [DOI] [PubMed] [Google Scholar]
- 142.Raghu G, Behr J, Brown KK, et al. ARTEMIS-IPF Investigators*. Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial. Ann Intern Med. 2013;158(9):641–649. doi: 10.7326/0003-4819-158-9-201305070-00003. [DOI] [PubMed] [Google Scholar]
- 143.Noble PW, Albera C, Bradford WZ, et al. CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377(9779):1760–1769. doi: 10.1016/S0140-6736(11)60405-4. [DOI] [PubMed] [Google Scholar]
- 144.Taniguchi H, Ebina M, Kondoh Y, et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur Resp J. 2010;35(4):821–829. doi: 10.1183/09031936.00005209. [DOI] [PubMed] [Google Scholar]
- 145.King TE, Jr, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–2092. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 146.Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–2082. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]