

Abstract
Gaussian accelerated molecular dynamics (GaMD) is a recently developed enhanced sampling technique that provides efficient free energy calculations of biomolecules. Like the previous accelerated molecular dynamics (aMD), GaMD allows for “unconstrained” enhanced sampling without the need to set predefined collective variables and so is useful for studying complex biomolecular conformational changes such as protein folding and ligand binding. Furthermore, because the boost potential is constructed using a harmonic function that follows Gaussian distribution in GaMD, cumulant expansion to the second order can be applied to recover the original free energy profiles of proteins and other large biomolecules, which solves a long-standing energetic reweighting problem of the previous aMD method. Taken together, GaMD offers major advantages for both unconstrained enhanced sampling and free energy calculations of large biomolecules. Here, we have implemented GaMD in the NAMD package on top of the existing aMD feature and validated it on three model systems: alanine dipeptide, the chignolin fast-folding protein, and the M3 muscarinic G protein-coupled receptor (GPCR). For alanine dipeptide, while conventional molecular dynamics (cMD) simulations performed for 30 ns are poorly converged, GaMD simulations of the same length yield free energy profiles that agree quantitatively with those of 1000 ns cMD simulation. Further GaMD simulations have captured folding of the chignolin and binding of the acetylcholine (ACh) endogenous agonist to the M3 muscarinic receptor. The reweighted free energy profiles are used to characterize the protein folding and ligand binding pathways quantitatively. GaMD implemented in the scalable NAMD is widely applicable to enhanced sampling and free energy calculations of large biomolecules.
Introduction
Accelerated molecular dynamics (aMD) is an enhanced sampling technique that works by smoothing the potential energy surface to lower energy barriers and thus accelerate conformational transitions of biomolecules.1 Without the need to set predefined collective variables (CVs), aMD allows “unconstrained” enhanced sampling of many complex biomolecules.2 For example, aMD simulations provide significant speed-up of peptide and protein conformational transitions,1,3 lipid diffusion and mixing,4 protein folding,5 and protein–ligand binding.6 Hundreds-of-nanosecond aMD simulations are able to capture millisecond time scale events in both globular and membrane proteins.7
While aMD is powerful for enhanced conformational sampling, its accuracy for free energy calculations has attracted lots of attention.8 In theory, frames of aMD simulations can be reweighted by the Boltzmann factors of the corresponding boost potential (i.e., eΔV/kBT) and averaged over each bin of selected CV(s) to obtain the canonical ensemble. However, the exponential reweighting is known to suffer from large statistical noise in practical calculations2,7c,8,9 because the Boltzmann reweighting factors are often dominated by a very few frames with high boost potential. The boost potential in aMD simulations of proteins is typically on the order of tens to hundreds of kilocalories per mole, which is much greater in magnitude and wider in distribution than that of CV-biasing simulations (e.g., several kilocalories per mole in metadynamics). It has been a long-standing problem to accurately reweight aMD simulations and recover the original free energy landscapes, especially for large proteins.6,7c Notably, when the boost potential follows near-Gaussian distribution, cumulant expansion to the second order provides improved reweighting of aMD simulations compared with the previously used exponential average and Maclaurin series expansion reweighting methods.10 The reweighted free energy profiles are in good agreement with the long-time scale conventional molecular dynamics (cMD) simulations as demonstrated on alanine dipeptide and fast-folding proteins.5b However, such improvement is limited to rather small systems (e.g., proteins with less than ∼35 amino acid residues).5b In simulations of larger systems, the boost potential exhibits significantly wider distribution and does not allow for accurate reweighting.
In order to achieve both unconstrained enhanced sampling and accurate energetic reweighting for free energy calculations of large biomolecules like proteins, Gaussian accelerated molecular dynamics (GaMD) has been developed by applying a harmonic boost potential to smooth the biomolecular potential energy surface.11 GaMD greatly reduces the energy barriers and accelerates conformational transitions and ligand binding by orders of magnitude.11 Moreover, because the boost potential follows Gaussian distribution, the original free energy profiles of biomolecules can be recovered through cumulant expansion to the second order.11 GaMD solves the long-standing energetic reweighting problem as encountered in the previous aMD method8 and allows us to characterize complex biomolecular conformational changes quantitatively.11 Compared with other enhanced sampling methods such as the metadynamics12 and adaptive biasing force (ABF),13 GaMD does not require predefined CVs, which is advantageous for studying “free” protein folding and ligand binding processes11 and efficient free energy calculations of large biomolecules.11
Here, we have implemented GaMD in the NAMD package on top of its existing aMD feature. The potential statistics of simulated biomolecules are collected from short cMD and used to construct the harmonic boost potential. The implemented GaMD in NAMD will be demonstrated on three model systems that have been extensively studied earlier: alanine dipeptide,10,11,14 the chignolin fast-folding protein,5b,11 and the M3 muscarinic G protein-coupled receptor (GPCR) bound by the acetylcholine (ACh) endogenous agonist.6,15
Methods
Theory
GaMD enhances the conformational sampling of
biomolecules by adding a harmonic boost potential to smooth the system
potential energy surface when the system potential 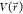 is lower than a reference energy E:11
is lower than a reference energy E:11
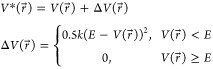 |
1 |
where k is
the harmonic force constant. The two adjustable parameters E and k are automatically determined by
applying the following three criteria. First, for any two arbitrary
potential values 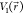 and
and 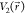 found on the original energy surface, if
found on the original energy surface, if 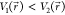 , ΔV should be a
monotonic function that does not change the relative order of the
biased potential values, i.e.,
, ΔV should be a
monotonic function that does not change the relative order of the
biased potential values, i.e., 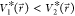 . Second,
if
. Second,
if 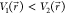 , the potential difference
observed on the
smoothed energy surface should be smaller than that of the original,
i.e.,
, the potential difference
observed on the
smoothed energy surface should be smaller than that of the original,
i.e., 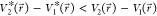 . By combining
the first two criteria and
plugging in the formula of
. By combining
the first two criteria and
plugging in the formula of 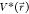 and ΔV, we obtain
and ΔV, we obtain
| 2 |
where Vmin and Vmax are the system minimum
and maximum potential energies. To ensure that eq 2 is valid, k has to satisfy: 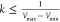 . Let us define
. Let us define 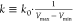 , then 0 < k0 ≤ 1. Third, the
standard deviation of ΔV needs to be small
enough (i.e., narrow distribution) to ensure accurate
reweighting using cumulant expansion to the second order:10 σΔV = k(E – Vavg)σV ≤ σ0, where Vavg and σV are the average and standard deviation of the system
potential energies, σΔV is
the standard deviation of ΔV with σ0 as a user-specified upper limit (e.g., 10kBT) for accurate reweighting. When E is set to the lower bound E = Vmax according to eq 2, k0 can be calculated
as
, then 0 < k0 ≤ 1. Third, the
standard deviation of ΔV needs to be small
enough (i.e., narrow distribution) to ensure accurate
reweighting using cumulant expansion to the second order:10 σΔV = k(E – Vavg)σV ≤ σ0, where Vavg and σV are the average and standard deviation of the system
potential energies, σΔV is
the standard deviation of ΔV with σ0 as a user-specified upper limit (e.g., 10kBT) for accurate reweighting. When E is set to the lower bound E = Vmax according to eq 2, k0 can be calculated
as
| 3 |
Alternatively, when the threshold energy E is set to its upper bound E = Vmin + 1/k, k0 is set to
| 4 |
if k0″ is found to be between 0 and 1. Otherwise, k0 is calculated using eq 3.
For energetic reweighting of GaMD simulations, the probability distribution along a selected reaction coordinate A(r) is written as p*(A), where r denotes the atomic positions {r1, ..., rN}.Given the boost potential ΔV(r) of each frame, p*(A) can be reweighted to recover the canonical ensemble distribution, p(A), as
| 5 |
where M is the number of bins, β = 1/kBT and ⟨eβΔV(r)⟩j is the ensemble-averaged Boltzmann factor of ΔV(r) for simulation frames found in the jth bin. In order to reduce the energetic noise, the ensemble-averaged reweighting factor can be approximated using cumulant expansion:16
| 6 |
where the first three cumulants are given by
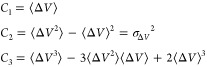 |
7 |
As shown earlier, when the boost potential
follows near-Gaussian distribution, cumulant expansion to the second
order provides the more accurate reweighting than the exponential
average and Maclaurin series expansion methods.10 Finally, the reweighted free energy is calculated as 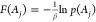 .
.
Implementation
Similar to the aMD implemented in NAMD,14 three modes are available for applying boost potential to biomolecules in GaMD: (1) boosting the dihedral energetic term only, (2) boosting the total potential energy only, and (3) boosting both the dihedral and total potential energetic terms (i.e., “dual-boost”). The major code modification is to extend the aMD function in NAMD 2.11 to include the boost potential calculation used in GaMD (Appendix A). As described in the previous section, GaMD boost potential is computed based on statistics of the system potential such as the minimum, maximum, average and standard deviation. Therefore, three stages of simulation are needed to collect the potential statistics. They include the (i) cMD, (ii) equilibration, and (iii) production stages. The program first collects potential statistics from a short cMD run. Subsequently, a boost potential is added to the system in the equilibration stage while update of the potential statistics continues. During this stage, the boost potential applied in each step is computed based on the energetic statistics collected up to that particular step. After the equilibration stage, the statistics collected is assumed to be sufficient to represent the potential energy landscape of interest. Hence, the potential statistics are fixed to calculate the boost potential for running the production simulation. Note that in both the cMD and equilibration stages, there are a small number of steps at the beginning of each stage during which we do not collect statistics. These steps, named preparation steps, are performed to allow the system to adapt to the simulation environment. The program starts collecting statistics of the potential energies after the preparation steps.
Simulation Protocols and Benchmarks
For alanine dipeptide and chignolin, the AMBER ff99SB force field was used and the simulation systems were built using the Xleap module in the AMBER package17 as described previously.9,11 By solvating the structures in a TIP3P18 water box that extends 8–10 Å from the solute surface, the alanine dipeptide system contained 630 water molecules and 2211 waters for chignolin. The total number of atoms in the two systems is 1912 and 6773 for alanine dipeptide and chignolin, respectively. Periodic boundary conditions were applied for the simulation systems. Bonds containing hydrogen atoms were restrained with the SHAKE algorithm19 and a 2 fs time step was used. Weak coupling to an external temperature and pressure bath was used to control both temperature and pressure.20 The electrostatic interactions were calculated using the particle mesh Ewald (PME) summation21 with a cutoff of 8.0 Å for long-range interactions. After the initial energy minimization and thermalization as described earlier,11 dual-boost GaMD was applied to simulate the two systems. The system threshold energy E for applying the boost potential was set to Vmax. The default parameter values were used for the GaMD simulations except stated otherwise. For alanine dipeptide, statistics of the system potential were first collected from an initial 2 ns cMD run, followed by a 6 ns equilibration run. Based on the statistics collected, the threshold energy E and harmonic force constant k were computed automatically according to eq 3. Finally, three independent 30 ns production runs were performed with different randomized initial atomic velocities. For chignolin, the dual-boost GaMD simulation includes 2 ns cMD, 50 ns equilibration after adding the boost potential, and then three independent 300 ns production runs.
For the M3 muscarinic GPCR, the system was set up following the same protocol as used previously.6,7d The tiotropium (TTP)-bound X-ray structure (PDB: 4DAJ) of the M3 receptor22 was used. After removal of TTP, the M3 receptor was inserted into a palmitoyl-oleoyl-phosphatidyl-choline (POPC) bilayer with all overlapping lipid molecules removed using the Membrane plugin and solvated in a water box using the Solvate plugin in VMD.23 Four ACh ligand molecules were placed at least 40 Å away from the receptor orthosteric site in the bulk solvent. The system charges were then neutralized with 18 Cl– ions. The simulation systems of the M3 receptor initially measured about 80 Å × 87 Å × 97 Å with 130 lipid molecules, ∼11 200 water molecules and a total of ∼55 500 atoms. Initial energy minimization and thermalization of the M3 receptor system follow the same protocol as used in the previous study.6 GaMD simulation was then performed using the dual-boost scheme with the threshold energy E set to Vmax. The simulation included 2 ns cMD, 50 ns equilibration after adding the boost potential and then three independent production runs with randomized atomic velocities (one for 400 ns and another two for 300 ns).
The GaMD simulations were carried out using NAMD 2.11 on the Gordon supercomputer at the San Diego Supercomputing Center. Excellent scalability was obtained as shown in Figure S1. For the M3 muscarinic GPCR system, benchmark simulations showed that GaMD ran at ∼10 ns/day with 64 CPUs and up to ∼61 ns/day with 640 CPUs, which were ∼8–11% slower than the corresponding cMD runs. This performance is very similar to that of the conventional aMD implemented in NAMD.14 GaMD production frames were saved every 0.1 ps. The VMD23 and CPPTRAJ24 tools were used for trajectory analysis. For chignolin, the root-mean square deviations (RMSD) of the Cα atoms relative to the native NMR structure (PDB: 1UAO) and the protein radius of gyration, Rg, were calculated. For the M3 muscarnic receptor, the density based spatial clustering of applications with noise (DBSCAN) algorithm25 was applied to cluster the diffusing ligand molecules for identifying the highly populated binding sites. Finally, the PyReweighting toolkit10 was applied to compute the potential of mean force (PMF) profiles of the backbone dihedrals Φ and Ψ in the alanine dipeptide, the (RMSD, Rg) of chignolin and structural clusters of the diffusing ACh in the M3 receptor system.
Results
Free Energy Profiles of the Alanine Dipeptide
With the three independent 30 ns GaMD production trajectories of the alanine dipeptide (Figure 1A), energetic reweighting was applied to calculate the free energy profiles of the backbone dihedrals Φ and Ψ. Analysis of the system boost potential showed that it followed Gaussian distribution with low anharmonicity (7.18 × 10–3; Figure 1B). The boost potential average is 6.93 and 1.87 kcal/mol for the standard deviation. With this, the cumulant expansion to the second order was applied for the reweighting.
Figure 1.

(A) Schematic representation of backbone dihedrals Φ and Ψ in alanine dipeptide. (B) Distribution of the boost potential ΔV applied in the GaMD simulations with anharmonicity equal to 7.18 × 10–3. (C–D) Potential of mean force (PMF) profiles of the (C) Φ and (D) Ψ dihedrals calculated from three 30 ns GaMD simulations combined using cumulant expansion to the second order. (E) 2D PMF profile of backbone dihedrals (Φ, Ψ). The low energy wells are labeled corresponding to the right-handed α helix (αR), left-handed α helix (αL), β-sheet (β), and polyproline II (PII) conformations. (F) Distribution anharmonicity of ΔV of frames found in each bin of the PMF profile.
In comparison, the reweighted PMF profiles obtained from 30 ns GaMD trajectories agree quantitatively with the original profiles from much longer 1000 ns cMD simulation. Although the GaMD derived PMF profile of Φ exhibits moderate fluctuations near the energy barrier at 0° and slightly elevated free energy well centered at ∼50° (Figure 1C), it essentially overlaps with the original profile in the other regions, similar for the entire PMF profile of Ψ (Figure 1D). In contrast, three cMD simulations did not properly sample the energy barriers of Φ at 0° and Ψ at 120° as shown in Figures S2A and S2B, respectively. For Φ, the energy well centered at 60° obtained from the 30 ns cMD simulations was higher than that from 1000 ns cMD simulation. Therefore, while cMD simulations performed for 30 ns are poorly converged for alanine dipeptide, GaMD simulations of the same length yielded significantly improved free energy profiles that agree quantitatively with those of the 1000 ns cMD simulation.
In addition, we calculated a 2D PMF of (Φ, Ψ) in alanine dipeptide by reweighting the three 30 ns GaMD trajectories combined. As shown in Figure 1E, five free energy wells were identified in the reweighted PMF profile of (Φ, Ψ), which are centered around (−144°, 0°) and (−72°, −18°) for the right-handed α helix (αR), (48°, −6°) for the left-handed α helix (αL), (−150°, 156°) for the β-sheet, and (−72°, 162°) for the polyproline II (PII) conformation (Figure 1E). Their corresponding minimum free energies are estimated as 0, 0.47, 1.82, 1.44, and 2.35 kcal/mol, respectively. In addition, the distribution anharmonicity of ΔV of frames clustered in each bin of the 2D PMF is smaller than 0.10 in all low-energy regions (Figure 1F), suggesting that reweighting using second order cumulant expansion is a reasonable approximation. Indeed, the reweighted 2D PMF profile obtained from three 30 ns GaMD trajectories (Figure 1E) is very similar to that obtained from 1000 ns cMD (Figure S2D), but not for the 30 ns cMD simulations (Figure S2C). Therefore, GaMD accurately samples the free energy profiles of alanine dipeptide within much shorter simulation time compared with cMD, demonstrating the efficiency and accuracy of GaMD for the biomolecular free energy calculations.
Folding of Chignolin
Started from an extended conformation of chignolin, simulations using GaMD implemented in NAMD 2.11 were able to capture complete folding of the protein into its native structure within 300 ns. The RMSD obtained between the simulation-folded chignolin and NMR experimental native structure (PDB: 1UAO) reaches a minimum of 0.2 Å (Figure 2A). The system boost potential applied in the GaMD simulations followed Gaussian distribution with the anharmonicity equal to 9.66 × 10–3 (Figure 2B). The average and standard deviation of the boost potential are 11.2 and 2.8 kcal/mol, respectively. During the three independent 300 ns GaMD simulations, chignolin folded into the native conformational state with RMSD < 2 Å and unfolded repeatedly in two of the simulations. It remained in the folded state after rapid folding within ∼20 ns in the third simulation (Figure 2C). Upon folding, the chignolin showed decrease of the radius of gyration, Rg, to 4.2 Å (Figure 2D). The average folding time of chignolin obtained from the GaMD simulation is ∼28 ns, which is significantly shorter than the 600 ns folding time obtained from the previous long-time scale cMD simulations26 (i.e., ∼30 times speedup).
Figure 2.

Folding of chignolin captured in the GaMD simulations: (A) comparison of simulation-folded chignolin (blue) with the PDB (1UAO) native structure (red) that exhibits 0.2 Å RMSD, (B) distribution of the boost potential ΔV with anharmonicity equal to 9.66 × 10–3, (C) the RMSD of chignolin between simulation snapshots and the PDB native structure, and (D) the radius of gyration, Rg, of chignolin calculated from the three independent 300 ns GaMD simulations. (E) 2D (RMSD, Rg) PMF calculated by reweighting the three 300 ns GaMD simulations combined. (F) Distribution anharmonicity of ΔV of frames found in each bin of the PMF profile.
Based on Gaussian distribution of the boost potential, cumulant expansion to the second order was applied to reweight the three 300 ns GaMD simulations of chignolin combined. A 2D PMF profile was calculated for the protein RMSD relative to the PDB native structure and the radius of gyration, (RMSD, Rg) as shown in Figure 2E. The reweighted PMF allowed us to identify the folded (“F”) and intermediate (“I”) conformational states, which correspond to the global energy minimum at (1.0 and 4.0 Å) and a low-energy well centered at (4.5 and 5.5 Å), respectively. Figure 2F plots the distribution anharmonicity of ΔV for frames found in each bin of the 2D PMF as shown in Figure 2E. The anharmonicity exhibits values smaller than 0.05 in the simulation sampled conformational space, suggesting that the boost potential follows Gaussian distribution for proper reweighting using cumulant expansion to the second order. In summary, GaMD enables efficient enhanced sampling and free energy calculations of protein folding as demonstrated on the chignolin.
Ligand Binding to a Muscarinic G Protein-Coupled Receptor
Finally, the GaMD implemented in NAMD 2.11 was demonstrated on binding of the ACh endogenous agonist to the M3 muscarinic GPCR (Figure 3A). The M3 muscarinic receptor is widely expressed in human tissues and a key seven-transmembrane (TM) GPCR that has been targeted for treating various human diseases, including cancer,27 diabetes,28 and obesity.29 Three independent GaMD simulations were performed on the M3 receptor, one for 400 ns and another two for 300 ns. As shown in Figure 3B, the system boost potential follows Gaussian distribution with anharmonicity equal to 1.33 × 10–2. The average and standard deviation of the boost potential are 10.9 and 3.0 kcal/mol, respectively. Such narrow distribution will ensure accurate reweighting for free energy calculation using cumulant expansion to the second order.
Figure 3.

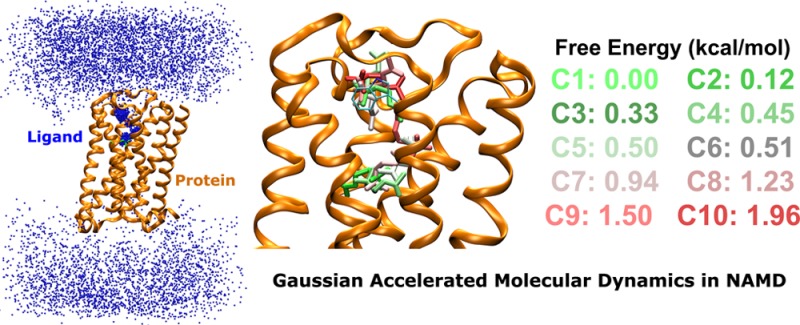
Binding of the acetylcholine (ACh) endogenous agonist to the M3 muscarinic G-protein-coupled receptor simulated via GaMD: (A) Schematic representation of the computational model, in which the receptor is shown in ribbons (orange), lipid in sticks, ions in small spheres, and four ligand molecules in large spheres. (B) Distribution of the boost potential ΔV with anharmonicity equal to 1.33 × 10–2. (C) Probability distribution of the ACh (the N atom in blue dots) diffusing in the bulk solvent and bound to the M3 receptor (orange ribbons), in which the Glide docking pose of ACh is shown in green sticks. (D) RMSD of the diffusing ACh relative to the Glide docking pose calculated from the 400 ns GaMD simulation. (E) Ten lowest energy structural clusters of ACh that are labeled and colored in a green–white–red (GWR) scale according to the PMF values obtained from reweighting of the GaMD simulation.
During the 400 ns GaMD simulation of the M3 muscarinic receptor, ACh was observed to enter the receptor and then bind to the receptor endogenous ligand-binding (“orthosteric”) site (Figure 3C). Highly populated clusters were identified for the ligand in the extracellular vestibule and orthosteric site of the receptor, while the ligand diffuses nearly homogeneously in the bulk solvent. Note that periodic boundary conditions were applied on the simulation system and thus ACh diffused to the cytoplasmic side of the lipid membrane, which may not occur in the real cells. Nonetheless, ACh entered the receptor from only the extracellular side, recapitulating the first step of GPCR-mediated cellular signaling machinery. Figure 3D plots the RMSD of the four diffusing ACh molecules relative to the ligand binding pose predicted from Glide docking30 in the orthosteric site. The ACh-3 molecule was observed to bind the extracellular vestibule with ∼10 Å RMSD, dissociate completely from the receptor, rebind to the extracellular vestibule at ∼200 ns and then enter the receptor to the orthosteric pocket at ∼270 ns. It finally rearranged its conformation in the orthosteric pocket, reached a minimum RMSD of 2.0 Å at ∼340 ns, and stayed bound in the orthosteric site until the end of the 400 ns GaMD simulation. Moreover, during the dissociation of the ACh-3, another ligand molecule (ACh-2) bound briefly to the receptor extracellular vestibule during ∼125–180 ns. Similar observations were obtained in the other 300 ns GaMD simulations of the M3 receptor as shown in Figures S3 and S4, during which different ACh molecules were able to bind the extracellular vestibule but could not reach the orthosteric site within the limited simulation time.
In order to obtain a quantitative picture of the ligand binding pathway, the DBSCAN algorithm25 was applied to cluster trajectory snapshots of four diffusing ligand molecules from the 400 ns GaMD simulation. Energetic reweighting10,11 was then applied to each of the ligand structural clusters to recover the original free energy. Ten structural clusters with the lowest free energies are shown in Figure 3E. Global free energy minimum (0.0 kcal/mol) was found for cluster “C1” in the orthosteric site. The second lowest energy minimum was identified for cluster “C2” (0.12 kcal/mol) located in the extracellular vestibule formed between ECL2/ECL3. Moreover, cluster “C3” that exhibits a different conformation compared with cluster “C1” and higher free energy (0.33 kcal/mol) was also identified in the orthosteric pocket. In addition to cluster “C2”, clusters “C4” with 0.45 kcal/mol, “C6” with 0.51 kcal/mol, “C8” with 1.23 kcal/mol, and “C10” with 1.96 kcal/mol were also identified in the extracellular vestibule, in which the positively charged N atom of the ligand interacts with residue Trp5257.35 through cation−π interactions. The residue superscripts denote the Ballesteros–Weinstein (BW) numbering of GPCRs.31 Three clusters of higher free energies, “C5” with 0.50 kcal/mol, “C7” with 0.94 kcal/mol, and “C9” with 1.50 kcal/mol, appear to connect “C1” in the orthosteric pocket and “C2” in the extracellular vestibule. Therefore, structural clusters “C1”, “C3” ↔ “C7”, “C5”, “C9” ↔ “C2”, “C4”, “C6”, “C8”, and “C10” appear to represent an energetically preferred pathway for the endogenous agonist binding to the M3 muscarinic receptor.
Discussion
By adding a harmonic boost potential to the potential energy surface, GaMD provides both unconstrained enhanced sampling and free energy calculation of biomolecules. Important statistical properties of the system potential, such as the average, maximum, minimum, and standard deviation values, are used to calculate the simulation acceleration parameters, particularly the threshold energy E and force constant k for applying the boost potential. In this study, we have implemented GaMD in the NAMD software version 2.11. GaMD computes the potential boost according to statistics of the system potential collected during the cMD and equilibration stages. It is worth noting that the statistics collection in GaMD-NAMD slightly differs from the previous version of GaMD implemented in AMBER.11 In the AMBER version, the average and standard deviation of potential energies are calculated in every “ntave” steps, a native function available in the AMBER program.32 In the NAMD version of GaMD, the average and standard deviation of the potential are calculated using the potential values collected up to the current step (see details in Appendix A).33 The two variables are updated every step until the end of the cMD and equilibration stages. Moreover, similar to the implementation of conventional aMD in NAMD, the LJ correction term is not included in the total potential energy calculation in GaMD.
With the implementation of GaMD in NAMD, we have demonstrated the code on three model systems: the alanine dipeptide, chignolin (a globular protein) and the M3 muscarinic GPCR (a membrane protein). For the alanine dipeptide biomolecular model system, short GaMD simulations performed for only 30 ns were able to reproduce highly accurate free energy profiles of the backbone dihedrals that may need as long as 1000 ns cMD simulation to converge. The free energy errors were almost negligible except the elevated free energy well of Φ near 50° by ∼0.5 kcal/mol and slight fluctuations in the energy barriers (particularly Φ at 0° and Ψ at −120°; Figure 2). In contrast, cMD simulations lasting 30 ns hardly sample these free energy barriers and exhibit poor convergence. Therefore, GaMD-NAMD greatly accelerates the conformational sampling and accurate free energy calculation of the alanine dipeptide.
For the chignolin, the GaMD-NAMD simulations were able to fold the protein rapidly. In two of the three 300 ns GaMD simulations, chignolin undergoes both folding and unfolding repeatedly (Figure 2C). Compared with the average folding time obtained from long-time scale cMD simulations (600 ns),26 GaMD folds the protein within ∼28 ns, i.e., ∼30 times faster. Unlike the previous GaMD-AMBER simulations,11 the fully unfolded state of chignolin does not appear as a low-energy well in the reweighted free energy profile obtained from the present GaMD-NAMD simulations. This behavior will be subject to further investigation in future GaMD studies. Nonetheless, in addition to sampling the folded state in the global free energy minimum, the GaMD-NAMD simulations also captured the intermediate state during the folding of the protein. This is consistent with the previous long-time scale cMD26 and aMD5b simulations.
Finally, we have demonstrated GaMD-NAMD on ligand binding to the M3 muscarinic GPCR as a model membrane protein system. While the ACh endogenous agonist binds only transiently to the receptor extracellular vestibule in two 300 ns GaMD simulations, the ligand enters the receptor and binds to the target orthosteric site in a 400 ns GaMD simulation. Although in principle multiple binding and unbinding events may be needed in order to compute converged ligand binding free energy, structural clustering and reweighting of the GaMD simulation allows us to identify energetically preferred binding sites and pathway of the diffusing ligand. Particularly, the lowest energy cluster of ACh is identified in the orthosteric site, in excellent agreement with the Glide docking pose. The second lowest energy cluster is located in the extracellular vestibule, with the positively charged N atom of ACh forming cation−π interaction with the receptor residue Trp5257.35. This is consistent with previous extensive experimental and computational studies that the extracellular vestibule of class A GPCRs acts as a metastable intermediate site during binding of orthosteric ligands.6,15 The energetically preferred pathway of agonist binding to the M3 receptor identified from the current GaMD-NAMD simulation is similar to that found in previous long-time scale cMD,34 aMD,6 and GaMD simulations35 of class A GPCRs. GaMD is thus promising to predict low-energy conformations of ligand binding and serve as a useful tool in structural biology and drug discovery. Moreover, GaMD mainly accelerates transitions across enthalpic energy barriers because it is based on boosting the system potential energy surface. However, GaMD can be potentially combined with the parallel tempering36 and replica exchange37 algorithms for further enhanced sampling over entropic barriers as discussed earlier.11
In summary, we have implemented GaMD in NAMD 2.11 that shows excellent scalability for supercomputer simulations of large biomolecules.38 The updated source code files of NAMD 2.11 for implementing GaMD are now publicly available at http://gamd.ucsd.edu. The implementation shall be released in the upcoming version 2.12 of NAMD. It is complementary to the original implementation of GaMD in the graphics processing unit (GPU) version of the AMBER software11 that runs extremely fast simulations with one or a small number of GPU cards.17c,32 As demonstrated on selected model systems, results of the current work shall facilitate the applications of GaMD in enhanced sampling and free energy calculations of a wide range of large biomolecules, such as proteins, lipid membrane, nucleic acids, virus particles, and cellular structures.
Acknowledgments
Computing time was provided on the Gordon and Comet supercomputers at the San Diego Supercomputer Center through award TG-MCA93S013. Y.M. and J.A.M. acknowledge support by NSF (grant MCB1020765), NIH (grant GM31749), Howard Hughes Medical Institute and National Biomedical Computation Resource (NBCR). Y.W. acknowledges support of project 409813 from University Grants Committee as well as project 4053165 from the Chinese University of Hong Kong. Finally, we would like to dedicate this work to Prof. Klaus Schulten for his remarkable contributions in computational biophysics, in particular development of the highly popular NAMD.
Appendix A: Implementation of Gaussian Accelerated Molecular Dynamics in NAMD
The Gaussian Accelerated Molecular
Dynamics (GaMD) algorithm is
implemented in NAMD 2.1138 as the following:
The following is a list of the input parameters for GaMD simulation in NAMD:
-
accelMDG < Is Gaussian accelerated MD on? >
Acceptable Values: on or off
Default Value: off
Description: Specifies whether Gaussian accelerated MD (GaMD) is on.
-
accelMDGiE < Flag to set the threshold energy E for adding boost potential >
Acceptable Values: 1, 2
Default Value: 1
Description: Specifies how the threshold energy E is set in GaMD. A value of 1 indicates that the threshold energy E is set to its lower bound E = Vmax. A value of 2 indicates that the threshold energy E is set to its upper bound E = Vmin + (Vmax – Vmin)/k0.
-
accelMDGcMDPrepSteps < no. of preparatory cMD steps >
Acceptable Values: Zero or positive integer
Default Values: 200 000
Description: The number of preparatory conventional MD (cMD) steps in GaMD. This value should be smaller than accelMDGcMDSteps (see below). Potential energies are not collected for calculating the values of Vmax, Vmin, Vavg, σV during the first accelMDGcMDPrepSteps.
-
accelMDGcMDSteps < no. of total cMD steps >
Acceptable Values: Positive integer
Default Value: 1 000 000
Description: The number of total cMD steps in GaMD. With accelMDGcMDPrepSteps < t < accelMDGcMDSteps, Vmax, Vmin, Vavg, σV are collected and at t = accelMDGcMDSteps, E, and k0 are computed.
-
accelMDGEquiPrepSteps < no. of preparatory equilibration steps in GaMD >
Acceptable Values: Zero or positive integer
Default Value: 200 000
Description: The number of preparatory equilibration steps in GaMD. This value should be smaller than accelMDGEquiSteps (see below). With accelMDGcMDSteps < t < accelMDGEquiPrepSteps + accelMDGcMDSteps, GaMD boost potential is applied according to E and k0 obtained at t = accelMDGcMDSteps.
-
accelMDGEquiSteps < no. of total equilibration steps in GaMD >
Acceptable Values: Zero or positive integer
Default Value: 1 000 000
Description: The number of total equilibration steps in GaMD. With accelMDGEquiPrepSteps + accelMDGcMDSteps < t < accelMDGEquiSteps + accelMDGcMDSteps, GaMD boost potential is applied, and E and k0 are updated every step.
-
accelMDGSigma0P < upper limit of the standard deviation of the total boost potential in GaMD >
Acceptable Values: Positive real number
Default Value: 6.0 (kcal/mol)
Description: Specifies the upper limit of the standard deviation of the total boost potential. This option is only available when accelMDdihe is off or when accelMDdual is on.
-
accelMDGSigma0D < upper limit of SD of the dihedral potential boost in GaMD >
Acceptable Values: Positive real number
Default Value: 6.0 (kcal/mol)
Description: Specifies the upper limit of the standard deviation of the dihedral boost potential. This option is only available when accelMDdihe or accelMDdual is on.
-
accelMDGRestart < Flag to restart GaMD simulation >
Acceptable Values: on or off
Default Value: off
Description: Specifies whether the current GaMD simulation is the continuation of a previous run. If this option is turned on, the GaMD restart file specified by accelMDGRestartFile (see below) will be read.
-
accelMDGRestartFile < Name of GaMD restart file >
Acceptable Values: UNIX filename
Description: A GaMD restart file that stores the current number of steps, maximum, minimum, average, standard deviation of the dihedral and/or total potential energies (depending on the accelMDdihe and accelMDdual parameters), and the current time step settings. This file is saved automatically every restartfreq steps. If accelMDGRestart is turned on, this file will be read and the simulation will restart from the point where the file was written.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jctc.6b00931.
Four supplementary figures (S1–S4; PDF)
Author Contributions
⊥ Y.T.P. and Y.M. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Hamelberg D.; Mongan J.; McCammon J. A. Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J. Chem. Phys. 2004, 120, 11919–11929. 10.1063/1.1755656. [DOI] [PubMed] [Google Scholar]
- Markwick P. R. L.; McCammon J. A. Studying functional dynamics in bio-molecules using accelerated molecular dynamics. Phys. Chem. Chem. Phys. 2011, 13, 20053–20065. 10.1039/c1cp22100k. [DOI] [PubMed] [Google Scholar]
- a Hamelberg D.; Shen T.; McCammon J. A. Phosphorylation effects on cis/trans isomerization and the backbone conformation of serine-proline motifs: Accelerated molecular dynamics analysis. J. Am. Chem. Soc. 2005, 127, 1969–1974. 10.1021/ja0446707. [DOI] [PubMed] [Google Scholar]; b De Oliveira C. A. F.; Hamelberg D.; McCammon J. A. Estimating kinetic rates from accelerated molecular dynamics simulations: Alanine dipeptide in explicit solvent as a case study. J. Chem. Phys. 2007, 127, 175105. 10.1063/1.2794763. [DOI] [PubMed] [Google Scholar]; c Baron R.; Wong S. E.; de Oliveira C. A. F.; McCammon J. A. E9-Im9 Colicin DNase-Immunity Protein Biomolecular Association in Water: A Multiple-Copy and Accelerated Molecular Dynamics Simulation Study. J. Phys. Chem. B 2008, 112, 16802–16814. 10.1021/jp8061543. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Grant B. J.; Gorfe A. A.; McCammon J. A. Ras Conformational Switching: Simulating Nucleotide-Dependent Conformational Transitions with Accelerated Molecular Dynamics. PLoS Comput. Biol. 2009, 5, e1000325. 10.1371/journal.pcbi.1000325. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Bucher D.; Grant B. J.; Markwick P. R.; McCammon J. A. Accessing a Hidden Conformation of the Maltose Binding Protein Using Accelerated Molecular Dynamics. PLoS Comput. Biol. 2011, 7, e1002034. 10.1371/journal.pcbi.1002034. [DOI] [PMC free article] [PubMed] [Google Scholar]; f de Oliveira C. A. F.; Grant B. J.; Zhou M.; McCammon J. A. Large-Scale Conformational Changes of Trypanosoma cruzi Proline Racemase Predicted by Accelerated Molecular Dynamics Simulation. PLoS Comput. Biol. 2011, 7, e1002178. 10.1371/journal.pcbi.1002178. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Markwick P. R. L.; Pierce L. C. T.; Goodin D. B.; McCammon J. A. Adaptive Accelerated Molecular Dynamics (Ad-AMD) Revealing the Molecular Plasticity of P450cam. J. Phys. Chem. Lett. 2011, 2, 158–164. 10.1021/jz101462n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Markwick P. R. L.; de Oliveira C. A. F.; McCammon J. A. Enhanced Lipid Diffusion and Mixing in Accelerated Molecular Dynamics. J. Chem. Theory Comput. 2011, 7, 3199–3207. 10.1021/ct200430c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Doshi U.; Hamelberg D. Achieving Rigorous Accelerated Conformational Sampling in Explicit Solvent. J. Phys. Chem. Lett. 2014, 5, 1217–1224. 10.1021/jz500179a. [DOI] [PubMed] [Google Scholar]; b Miao Y.; Feixas F.; Eun C.; McCammon J. A. Accelerated molecular dynamics simulations of protein folding. J. Comput. Chem. 2015, 36, 1536–49. 10.1002/jcc.23964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappel K.; Miao Y.; McCammon J. A. Accelerated Molecular Dynamics Simulations of Ligand Binding to a Muscarinic G-protein Coupled Receptor. Q. Rev. Biophys. 2015, 48, 479–487. 10.1017/S0033583515000153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Pierce L. C. T.; Salomon-Ferrer R.; de Oliveira C. A. F.; McCammon J. A.; Walker R. C. Routine Access to Millisecond Time Scale Events with Accelerated Molecular Dynamics. J. Chem. Theory Comput. 2012, 8, 2997–3002. 10.1021/ct300284c. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Miao Y.; Nichols S. E.; Gasper P. M.; Metzger V. T.; McCammon J. A. Activation and dynamic network of the M2 muscarinic receptor. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 10982–10987. 10.1073/pnas.1309755110. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Miao Y.; Nichols S. E.; McCammon J. A. Free Energy Landscape of G-Protein Coupled Receptors, Explored by Accelerated Molecular Dynamics. Phys. Chem. Chem. Phys. 2014, 16, 6398–6406. 10.1039/c3cp53962h. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Miao Y.; Caliman A. D.; McCammon J. A. Allosteric Effects of Sodium Ion Binding on Activation of the M3 Muscarinic G-Protein Coupled Receptor. Biophys. J. 2015, 108, 1796–1806. 10.1016/j.bpj.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen T. Y.; Hamelberg D. A statistical analysis of the precision of reweighting-based simulations. J. Chem. Phys. 2008, 129, 034103. 10.1063/1.2944250. [DOI] [PubMed] [Google Scholar]
- Sinko W.; Miao Y.; de Oliveira C. A. F.; McCammon J. A. Population Based Reweighting of Scaled Molecular Dynamics. J. Phys. Chem. B 2013, 117, 12759–12768. 10.1021/jp401587e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y.; Sinko W.; Pierce L.; Bucher D.; McCammon J. A.; Walker R. C. Improved reweighting of accelerated molecular dynamics simulations for free energy calculation. J. Chem. Theory Comput. 2014, 10, 2677–2689. 10.1021/ct500090q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y.; Feher V. A.; McCammon J. A. Gaussian Accelerated Molecular Dynamics: Unconstrained Enhanced Sampling and Free Energy Calculation. J. Chem. Theory Comput. 2015, 11, 3584–3595. 10.1021/acs.jctc.5b00436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laio A.; Gervasio F. L. Metadynamics: a method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Rep. Prog. Phys. 2008, 71, 126601. 10.1088/0034-4885/71/12/126601. [DOI] [Google Scholar]
- Darve E.; Rodriguez-Gomez D.; Pohorille A. Adaptive biasing force method for scalar and vector free energy calculations. J. Chem. Phys. 2008, 128, 144120. 10.1063/1.2829861. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Harrison C. B.; Schulten K.; McCammon J. A. Implementation of Accelerated Molecular Dynamics in NAMD. Comput. Sci. Discovery 2011, 4, 015002. 10.1088/1749-4699/4/1/015002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse A. C.; Hu J.; Pan A. C.; Arlow D. H.; Rosenbaum D. M.; Rosemond E.; Green H. F.; Liu T.; Chae P. S.; Dror R. O.; Shaw D. E.; Weis W. I.; Wess J.; Kobilka B. K. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 2012, 482, 552–556. 10.1038/nature10867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hummer G. Fast-growth thermodynamic integration: Error and efficiency analysis. J. Chem. Phys. 2001, 114, 7330–7337. 10.1063/1.1363668. [DOI] [Google Scholar]; b Eastwood M. P.; Hardin C.; Luthey-Schulten Z.; Wolynes P. G. Statistical mechanical refinement of protein structure prediction schemes: Cumulant expansion approach. J. Chem. Phys. 2002, 117, 4602–4615. 10.1063/1.1494417. [DOI] [Google Scholar]
- a Case D. A.; Darden T. A., Cheatham T.E. III; Simmerling C. L.; Wang J.; Duke R. E.; Luo R.; Walker R. C.; Zhang W.; Merz K. M.; Roberts B.; Hayik S.; Roitberg A.; Seabra G.; Swails J.; Goetz A. W.; Kolossváry I.; Wong K. F.; Paesani F.; Vanicek J.; Wolf R. M.; Liu J.; Wu X.; Brozell S. R.; Steinbrecher T.; Gohlke H.; Cai Q.; Ye X.; Wang J.; Hsieh M.-J.; Cui G.; Roe D. R.; Mathews D. H.; Seetin M. G.; Salomon-Ferrer R.; Sagui C.; Babin V.; Luchko T.; Gusarov S.; Kovalenko A.; Kollman P. A.. AMBER 12; University of California, San Francisco 2012.; b Gotz A. W.; Williamson M. J.; Xu D.; Poole D.; Le Grand S.; Walker R. C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. 10.1021/ct200909j. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Salomon-Ferrer R.; Götz A. W.; Poole D.; Le Grand S.; Walker R. C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]; d Salomon-Ferrer R.; Case D. A.; Walker R. C. An overview of the Amber biomolecular simulation package. Wiley Interdisciplinary Reviews-Computational Molecular Science 2013, 3, 198–210. 10.1002/wcms.1121. [DOI] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Ryckaert J.-P.; Ciccotti G.; Berendsen H. J. C. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; Vangunsteren W. F.; Dinola A.; Haak J. R. Molecular-Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. 10.1063/1.448118. [DOI] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. 10.1063/1.470117. [DOI] [Google Scholar]
- Haga K.; Kruse A. C.; Asada H.; Yurugi-Kobayashi T.; Shiroishi M.; Zhang C.; Weis W. I.; Okada T.; Kobilka B. K.; Haga T.; Kobayashi T. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 2012, 482, 547–551. 10.1038/nature10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Roe D. R.; Cheatham T. E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Ester M.; Kriegel H.-P.; Sander J.; Xu X. A density-based algorithm for discovering clusters in large spatial databases with noise. Knowledge Discov. Data Min. 1996, 96, 226–231. [Google Scholar]
- Lindorff-Larsen K.; Piana S.; Dror R. O.; Shaw D. E. How fast-folding proteins fold. Science 2011, 334, 517–520. 10.1126/science.1208351. [DOI] [PubMed] [Google Scholar]
- Spindel E. R. Muscarinic receptor agonists and antagonists: effects on cancer. Handb. Exp. Pharmacol. 2012, 208, 451–68. 10.1007/978-3-642-23274-9_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ruiz de Azua I.; Scarselli M.; Rosemond E.; Gautam D.; Jou W.; Gavrilova O.; Ebert P. J.; Levitt P.; Wess J. RGS4 is a negative regulator of insulin release from pancreatic beta-cells in vitro and in vivo. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 7999–8004. 10.1073/pnas.1003655107. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gregory K. J.; Sexton P. M.; Christopoulos A. Allosteric modulation of muscarinic acetylcholine receptors. Curr. Neuropharmacol. 2007, 5, 157–67. 10.2174/157015907781695946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston-Green K.; Huang X. F.; Lian J. M.; Deng C. Effects of olanzapine on muscarinic M3 receptor binding density in the brain relates to weight gain, plasma insulin and metabolic hormone levels. Eur. Neuropsychopharmacol. 2012, 22, 364–373. 10.1016/j.euroneuro.2011.09.003. [DOI] [PubMed] [Google Scholar]
- a Halgren T. A.; Murphy R. B.; Friesner R. A.; Beard H. S.; Frye L. L.; Pollard W. T.; Banks J. L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]; b Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Ballesteros J. A.; Weinstein H.. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In Methods in Neuroscience; Stuart C. S., Ed.; Academic Press: New York, 1995; Vol. 25, pp 366–428. [Google Scholar]
- Case D.; Babin V.; Berryman J.; Betz R.; Cai Q.; Cerutti D.; Cheatham T. III; Darden T.; Duke R.; Gohlke H.. Amber 14; 2014.
- Knuth D. E. Artistic Programming - a Citation-Classic Commentary on the Art of Computer-Programming, Vol 1, Fundamental Algorithms, Vol 2, Seminumerical Algorithms, Vol 3, Sorting and Searching by Knuth, D. E. Current Contents/Engineering Technology & Applied Sciences 1993, 8-8. [Google Scholar]
- Dror R. O.; Pan A. C.; Arlow D. H.; Borhani D. W.; Maragakis P.; Shan Y.; Xu H.; Shaw D. E. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 13118–23. 10.1073/pnas.1104614108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y.; McCammon J. A. Graded activation and free energy landscapes of a muscarinic G-protein–coupled receptor. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 12162–12167. 10.1073/pnas.1614538113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansmann U. H. E. Parallel tempering algorithm for conformational studies of biological molecules. Chem. Phys. Lett. 1997, 281, 140–150. 10.1016/S0009-2614(97)01198-6. [DOI] [Google Scholar]
- a Sugita Y.; Okamoto Y. Replica-exchange multicanonical algorithm and multicanonical replica-exchange method for simulating systems with rough energy landscape. Chem. Phys. Lett. 2000, 329, 261–270. 10.1016/S0009-2614(00)00999-4. [DOI] [Google Scholar]; b Fajer M.; Hamelberg D.; McCammon J. A. Replica-Exchange Accelerated Molecular Dynamics (REXAMD) Applied to Thermodynamic Integration. J. Chem. Theory Comput. 2008, 4, 1565–1569. 10.1021/ct800250m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kale L.; Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.