

Abstract
TP53 is the most frequently mutated gene in human cancer. Functionally, p53 is activated by a host of stress stimuli and, in turn, governs an exquisitely complex anti-proliferative transcriptional program that touches upon a bewildering array of biological responses. Despite the many unveiled facets of the p53 network, a clear appreciation of how and in what contexts p53 exerts its diverse effects remains unclear. How can we interpret p53’s disparate activities and the consequences of its dysfunction to understand how cell type, mutation profile, and epigenetic cell state dictate outcomes, and how might we restore its tumor suppressive activities in cancer?
p53: the textbook view
p53 was discovered during the peak of tumor virus research as a 53 kD host protein bound to simian virus 40 large T antigen in virally-transformed cells (Lane and Crawford, 1979; Linzer and Levine, 1979). First classified as an oncogene, subsequent work established that wild-type p53, encoded by the TP53 gene, suppresses growth and oncogenic transformation in cell culture (Finlay et al., 1989), that inactivating TP53 mutations are common in human tumors (Baker et al., 1990), and in many cancers linked to poor patient prognosis (Olivier et al., 2010). Consistent with its action as a tumor suppressor, TP53 mutations are a hallmark of a hereditary cancer predisposition disorder known as Li-Fraumeni Syndrome (Malkin et al., 1990), and Trp53 knockout mice develop tumors at high penetrance (Donehower et al., 1992).
p53 is a sequence-specific DNA binding protein that regulates transcription (reviewed in Laptenko and Prives, 2006). The p53 protein consists of two N-terminal transactivation domains followed by a conserved proline rich domain, a central DNA binding domain, and a C-terminus encoding its nuclear localization signals and an oligomerization domain needed for transcriptional activity. Consistent with the importance of p53-mediated transcription in tumor suppression, the vast majority of tumor-derived TP53 mutations occur in the region encoding p53’s DNA binding domain. In normal cells, p53 protein is maintained at low levels by a series of regulators including MDM2, which functions as a p53 ubiquitin ligase to facilitate its degradation (Haupt et al., 1997; Honda et al., 1997; Kubbutat et al., 1997). However, p53 is stabilized in response to various cellular stresses, including DNA damage and replication stress produced by deregulated oncogenes. Mechanisms leading to p53 activation can be stimulus-dependent: for example, DNA damage promotes p53 phosphorylation, blocking MDM2-mediated degradation (Shieh et al., 1997), whereas oncogenic signaling induces the ARF tumor suppressor to inhibit MDM2 (Pomerantz et al., 1998; Quelle et al., 1995; Zhang et al., 1998).
The best understood functions of p53 focus on its ability to promote cell cycle arrest and apoptosis. Indeed, seminal studies from the early 1990s showed that p53 is crucial for a reversible DNA damage-induced G1 phase checkpoint (Kastan et al., 1991) that is mediated, in part, by its ability to transcriptionally activate the p21 cyclin-dependent kinase inhibitor gene (el-Deiry et al., 1993; Harper et al., 1993), presumably facilitating DNA repair prior to further cell division. In some circumstances, p53 induces cellular senescence, a stable if not permanent cell cycle arrest program that also involves the retinoblastoma (RB) gene product (Serrano et al., 1997; Shay et al., 1991). p53 can also promote apoptosis (Clarke et al., 1993; Lowe et al., 1993; Yonish-Rouach et al., 1991), relying on the induction of pro-apoptotic BCL-2 family members whose action facilitates caspase activation and cell death (Miyashita et al., 1994). Why p53 promotes cell cycle arrest in some cell types and apoptosis in others is incompletely understood (see below).
The settings in which p53 can be activated to arrest or eliminate pre-malignant cells have guided current thinking as to why p53 is such a potent tumor suppressor. On one hand, its ability to arrest or eliminate cells following DNA damage suggests that it might prevent cancer by preventing the accumulation of oncogenic mutations (Livingstone et al., 1992; Yin et al., 1992). In this model, p53 loss indirectly promotes cancer by increasing the number of mutations in surviving daughter cells. On the other hand, the ability of p53 to halt the proliferation in response to aberrant oncogene expression suggests a role in limiting the consequences of oncogenic mutations. Here, p53 loss directly enables cancer development by allowing oncogene-expressing cells to proliferate unabated, explaining why TP53 mutations cooperate with oncogenes in transformation (Lowe et al., 1994; Serrano et al., 1997). In both models, p53 acts as the “guardian of the genome” to limit the deleterious consequences of mutation (Lane, 1992). Although this historic view provides a basic conceptual framework as to why TP53 mutations are so common in human tumors, more recent work paints a much more nuanced picture of p53 action that highlights its context-dependent regulation and the broadly diverse consequences of its activation.
Revisiting the guardian of the genome
Upon DNA damage, p53 is activated to either promote the elimination or repair of damaged cells, ultimately reducing their risk of propagating mutations. DNA damage response (DDR) kinases phosphorylate p53, driving cell-cycle arrest, senescence, or apoptosis (reviewed in Williams and Schumacher, 2016). Additionally, p53 stimulates DNA repair by activating target genes that encode components of the DNA repair machinery, and p53-null cells are defective in certain DNA repair activities in vitro (Williams and Schumacher, 2016).
While TP53 mutation can correlate with patterns of single nucleotide variants and specific co-mutated genes, what is striking is that the association between TP53 mutation and copy number variation (CNVs) is strong and universal in a pan-cancer analysis (Ciriello et al., 2013). Also, cancers harboring TP53 mutations are typically aneuploid, with gross changes in numbers of whole chromosomes (Ciriello et al., 2013). Various biological explanations for this association have been proposed, but one mechanism contributing to this relationship is the ability of p53 to regulate processes in G2/M transitions (reviewed in Vitre and Cleveland, 2012). For example, p53 loss dysregulates the spindle assembly checkpoint by derepressing MAD2, leading to an increased rate of chromosome missegregation and tetraploidization (Schvartzman et al., 2011). In the context of tetraploid cells, p53 loss leads to an increased rate of multipolar mitoses and subsequent chromosome missegregation (Vitale et al., 2010).
In an alternative but non-mutually exclusive explanation, p53 can restrict chromosomal instability through its ability to cull cells at risk of aberrant mitoses, particularly following centrosome amplification and/or telomere dysfunction (Dewhurst et al., 2014; Eischen, 2016; Lanni and Jacks, 1998). Extra centrosomes lead to Hippo pathway up-regulation that, in turn, activates p53 by inhibiting MDM2 (Aylon et al., 2006; Ganem et al., 2014). Accordingly, TP53 mutations are also associated with whole genome doubling events in human tumors (Cancer Genome Atlas Research, 2013). Additional studies suggest that p53-deficient cells are better at tolerating proteomic stress produced by aberrant gene dosage (Tang et al., 2011), yet others suggest that p53-mediated culling of aneuploid cells is more efficient against structural aneuploidy than whole chromosome imbalances, implicating the role of DDR in response to chromosome shearing (Soto et al., 2017). Hence, it appears that the absence of p53 both facilitates the accumulation and permits the survival of aneuploid cells.
p53 also appears to suppress a particular type of chromosome shattering and rearrangement event known as chromothripsis. Cells that bypass replicative senescence after p53 and RB inactivation can proliferate despite telomere erosion (Hayashi et al., 2012). Failing this checkpoint, telomere dysfunction initiates chromosome breakage-fusion-bridge cycles that contribute to chromothripsis (Maciejowski et al., 2015). Although the extent to which chromothripsis fosters tumorigenesis remains an open question, the phenomenon is significantly more prevalent in tumors harboring TP53 mutations (Rausch et al., 2012).
An unanticipated way in which p53 helps maintain genomic integrity is by suppressing retrotransposons, which are latent virus-derived genetic elements whose aberrant expression can lead to mutagenesis through their mobilization and re-insertion throughout the genome (reviewed in Levine et al., 2016). Experimental activation of mobile elements in drosophila induces DNA double strand breaks and p53-mediated apoptosis (Wylie et al., 2014) that could, in principle, reduce their mutagenic effects. However, recent evidence demonstrates that the association between p53 mutation and retrotransposon expression is more than simply a culling effect: indeed, p53 binding to target sites within LINE elements and other transposon sequences are associated with their downregulation (Chang et al., 2007). p53-mediated repression is dependent on epigenetic silencing of retrotransposon loci and not apoptosis, and derepressed retrotransposons are competent for reintegration into the genome (Leonova et al., 2013; Wylie et al., 2016), promoting mutagenesis (Tubio et al., 2014). Genomic analyses have revealed that retrotransposon mobilization is common in human cancers (Ting et al., 2011; Tubio et al., 2014). While the precise impact remains to be determined, there is a significant association between repetitive element expression and p53 status in mouse and human tumors (Wylie et al., 2016).
The immediacy with which p53 loss cooperates with oncogenes to transform cells indicates that genomic instability is not absolutely required for tumor initiation (Lowe et al., 1994; Serrano et al., 1997). Still, the genomic instability fueled by p53 loss enables acquisition of additional driver events with the potential to accelerate transformation, metastasis and drug resistance (reviewed in McGranahan and Swanton, 2017). Just as species diversity in an ecosystem is associated with its robustness, subclonal diversity, not the total number of mutations in a tumor, dictates the resilience of a cancer cell population to changing conditions and challenges. In this regard, p53 inactivation may be unique in its ability to both promote genomic instability (by increasing the rate of new variants) and permit the survival of a wider pool of genetic configurations (decreasing the likelihood of extinction of variants). Together, these observations raise the possibility that p53 inactivation contributes to intratumoral heterogeneity.
p53 controls a broad and flexible network
As if regulating genome integrity, cell cycle arrest, and apoptosis were not enough functions for a single gene, an ever-growing body of work suggests that p53 also controls additional “non–canonical” programs that contribute to its effects (Figure 1). As examples, p53 can modulate autophagy, alter metabolism, repress pluripotency and cellular plasticity, and facilitate an iron-dependent form of cell death known as ferroptosis (reviewed in Aylon and Oren, 2016). Even basal levels of p53 can reinforce multiple other tumor suppressive networks (Pappas et al., 2017). Given extensive past research, it is surprising that there is no clear and simple answer to the question of what exactly p53 does and how. Nevertheless, a take-home message is that the p53 response is remarkably flexible and depends on the cell type, its differentiation state, stress conditions, and collaborating environmental signals.
Figure 1. The p53 network.
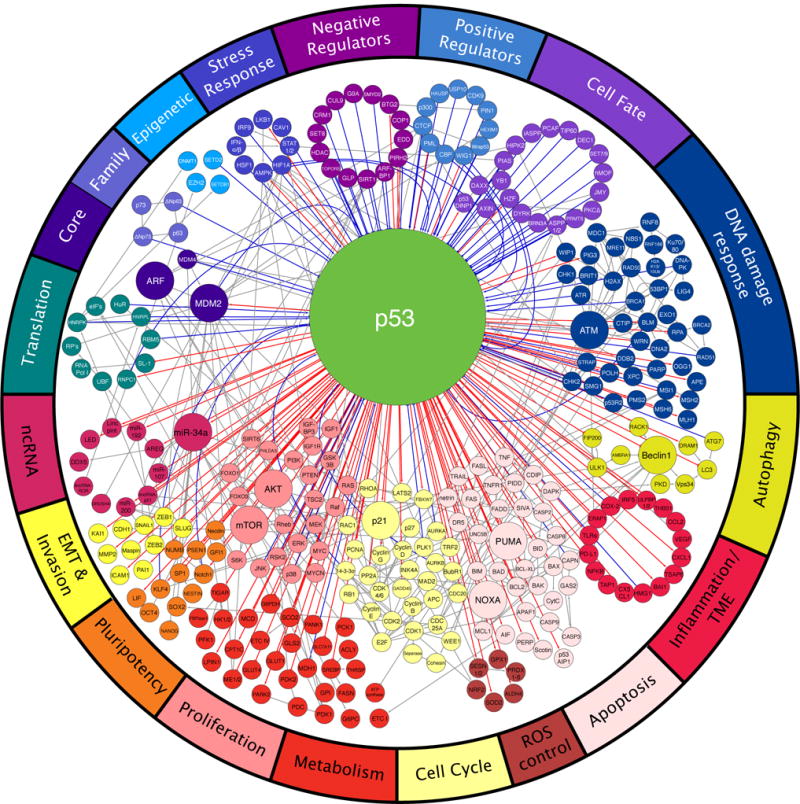
A wide variety of regulators govern the activity of p53 (top), which, in turn, controls many distinct biological processes (bottom). Each node represents a gene and each line represents an interaction. Direct p53 inputs are indicated as blue lines and direct p53 outputs are indicated as red lines. Noticeably, p53 controls effector processes by activating multiple target genes. Downstream pathways are highly interconnected (gray lines). Interactions are annotated as positive (arrow), negative (T-bar), or modifying (solid circle).
The varied functions of p53 are anchored in its ability to control distinct sets of its many target genes (Figure 1). For example, observations that cell cycle arrest and apoptosis are associated with upregulation of p21 or pro-apoptotic Bcl-2 proteins, respectively, obscure the fact that the global transcriptional response to p53 activation includes many other potential modifiers of outcome. Historically, genes have been implicated as p53 targets if p53 binds the locus and the mRNA is induced. More recently, Global Run-On Sequencing has improved specificity by enabling detection of nascent transcripts induced upon p53 activation (Allen et al., 2014). The nature of p53 targets identified in this analysis provides strong confirmation that non-canonical processes including ROS control, tissue remodeling, autophagy, and metabolism are bona fide processes controlled by p53 (Figure 1).
Efforts to identify a universal set of p53 target genes have invariably failed. Meta-analyses from 16 genome-wide datasets revealed that only about 60 genes were implicated as common targets (Fischer, 2017). It is noteworthy that these surveys involved a restricted number of different cell types and employed distinct methods for p53 induction. However, a central theme is that cellular context and various stimuli incite transcription of qualitatively different sets of genes, not just different levels of the same set of genes. It seems naive to expect that oncogene activation in different tissues (for example, KRAS activation in colon, pancreas, and lung) would precipitate an identical p53 transcriptional response. Moreover, one would not presume a priori that the p53 output generated by DNA damage would exactly mirror the gene expression signature elicited by oncogene activation, even in a single cell type. Despite data indicating that p53 can, in principle, control a wide variety of biological processes (reviewed in Olivos and Mayo, 2016), the physiological settings in which one or more processes predominate are incompletely understood and deserve more systematic study.
Cellular metabolism is one non-canonical p53-controlled process that has received much attention (reviewed in Kruiswijk et al., 2015). The collection of metabolic target genes controlled by p53 affect many individual processes: p53 is reported to increase glutamine catabolism, support anti-oxidant activity, downregulate lipid synthesis, increase fatty acid oxidation, or stimulate gluconeogenesis (Kruiswijk et al., 2015). Depending on the cell type, p53 can also have opposing effects on the same metabolic processes. For example, in breast and lung cancer cells, p53 inhibits glycolysis by attenuating glucose uptake (Zhang et al., 2013) or repressing the expression of glycolytic enzymes (Kim et al., 2013). By contrast, in muscle cells, p53 induces glycolytic enzymes (Kruiswijk et al., 2015). Likewise, p53 typically increases (Stambolsky et al., 2006) but can also restrict flux through the tricarboxylic acid (TCA) cycle (Jiang et al., 2013; Wang et al., 2013). Taken at face value, these results imply that p53 can regulate different aspects of metabolism that produce distinct, or even opposite, biochemical and phenotypic outcomes. Here again, precise contextual factors have yet to be identified.
While it is often assumed that each p53 effector function is a standalone process, there is increasing evidence that cross-talk between separate input and output pathways is more important than previously recognized (Figure 1, gray lines). For example, p53-driven cellular senescence may be supported by activation of autophagy (Young et al., 2009). Alterations in p53 control of metabolism undoubtedly contribute to apoptosis, autophagy, and ferroptosis (Gao et al., 2016). In some settings, p53-mediated processes can apparently be antagonistic: autophagy has the potential to delay apoptosis by reducing levels of PUMA (Thorburn et al., 2014). However, in contexts where p53 fails to repress glycolysis, autophagy is not efficiently engaged and apoptosis is favored (Duan et al., 2015). In these examples, interaction between distinct biochemical processes controlled by p53 elicits different biological outcomes.
The mechanistic basis underlying the ability of p53 to induce different biological outputs remains unclear. On one hand, p53 can induce qualitatively different programs that produce different biological outcomes depending on cell type and stimulus. One proposed mechanism for qualitatively modulating biological p53’s effects involves stimulus-dependent post-translational modifications (PTMs) that can alter p53 affinity for different target genes; for example, phospho-p53 (S46) or acetyl-p53 (K120) stimulates apoptosis, whereas PRMT5-methylated p53 activates p21 more readily than apoptotic genes (reviewed in Kumari et al., 2014). A broad array of other PTMs at many different sites in the p53 protein have been described to not only modify protein stability, but also influence target gene bias, such as SUMOylation, glycosylation, and prolyl isomerization (Kumari et al., 2014). Moreover, one post-translational modification may enhance acquisition of another, unlocking additional layers of regulation of protein stability, protein-protein interaction, and biasing DNA-binding toward select target genes.
p53 induction can yield either a steady signaling output or one that can oscillate in discrete waves; remarkably, the kinetics of its expression, independent of maximal p53 protein levels, can determine cell fate in response to genotoxic stress (reviewed in Stewart-Ornstein and Lahav, 2017). p53 activation kinetics can be translated into target gene bias owing to differences in p53 binding and dissociation rates at distinct target loci. Here, the p21 promoter is sensitive to short pulses of p53 activity whereas the pro-apoptotic p53 target FAS is not; consequently, a short pulse drives proliferative arrest but a sustained signal induces apoptosis (Espinosa et al., 2003; Gomes and Espinosa, 2010a; Morachis et al., 2010). Perhaps certain p53-driven stress responses instigate a short-term repair and salvage program that, if necessary, reaches a tipping point that progresses to cellular self-destruction.
On the other hand, several factors influence how the cell interprets p53 activation. For instance, cell lineage may be a large determinant in the nature of a hypothetical tipping point between alternative cell fates. First, cell type- and state-specific chromatin modifications may make particular genes more or less accessible to p53 transactivation (Su et al., 2015). For instance, CTCF insulates the PUMA locus from repressive histone modifications under certain conditions, governing whether PUMA is expressed and apoptosis occurs (Gomes and Espinosa, 2010b). In embryonic stem cells (ESCs), p53 can be induced to bind to the p21 promoter, but p21 is not efficiently activated, dependent on cell-type-specific repressive histone H3K27me3 marks at the locus (Itahana et al., 2016). Second, the p53 target spectrum can be altered by cooperation or antagonism with other transcription factors, such as FOXO and NF-κB, whose levels and occupancy are also context-dependent (Cooks et al., 2014; Eijkelenboom and Burgering, 2013). Finally, the same transcriptional output may have different effects depending on the state of the cell. ATM signaling protects cells from p53-mediated apoptosis, not by changing p53-driven transcriptional output, but by blocking autophagy, thus maintaining mitochondrial homeostasis and suppressing ROS levels (Sullivan et al., 2015).
Collectively, these observations imply that p53 response is not merely an “on-off” switch; to the contrary, cell fate is a result of a rich palette of p53-driven stress responses. Clearly, p53 is embedded in a densely populated and interconnected network of regulators and effectors (Figure 1) that permit a flexible p53 response coordinated to fit cell type and conditions at the time of activation. In short, cellular context (cell type, epigenetic state, tissue microenvironment, activating signal) is central to both the biochemical aspects of p53 activity as well as the biological outcome of a p53 response.
Putting Tumor Suppression in Context
By definition, tumor suppressor genes regulate processes that limit inappropriate cell expansion and whose inactivation facilitates tumor initiation or progression. Given the many processes that p53 controls, which of its effector functions are critical for tumor suppression has been the topic of much debate. Senescence and apoptosis can clearly be detected in tumors and when these processes are activated, they are certainly tumor suppressive. Still, a recent body of work suggests that apoptosis and senescence can be dispensable for tumor suppression and that, in some settings, other non-canonical p53 functions may be more critical (Valente et al., 2013). There is no consensus view on which p53-dependent process is most important.
The only relevant metric of “tumor suppression” is whether a gene impairs the onset or progression of tumors arising in vivo. In this regard, the p53 knockout mouse is a powerful model that develops thymic lymphoma (and sometimes sarcoma) at complete penetrance (Donehower et al., 1992). To address which p53 function(s) is crucial for tumor suppression, mutant strains have been produced in an attempt to isolate specific p53 functions, and the resulting animal cohorts monitored for tumors over time. If the ablation of a p53-driven function allows for tumorigenesis, the underlying process is crucial for the tumor suppressive activity of p53. If it does not, it is deemed dispensable. However, it bears consideration that thymic lymphoma rarely occurs in people, including Li-Fraumeni patients, so the requirements for suppressing this unusual cancer do not necessarily extend to other systems.
One line of investigation has compared tumor onset and pathology between mice harboring knockouts of p53 target genes versus p53 itself (Figure 2A). For example, mice deficient for p21, Puma, and Noxa do not develop thymic lymphoma, hinting that p53-mediated cell cycle arrest and apoptosis might be dispensable for tumor suppression (Valente et al., 2013). Still, p53 target genes may already expressed at basal levels so, for example, p53-null cells are by no means p21-null. Consequently, this approach could overestimate the contribution of a particular p53 effector to the null phenotype. Conversely, since multiple effectors mediate most p53 outputs, mouse strains deficient for individual p53 effector genes do not fully disable the associated p53 effector program (e.g. p21 loss does not completely disable p53-mediated cell cycle arrest). Hence, this approach may underestimate the contribution of the targeted process to tumor suppression. Changes in feedback loops and compensatory mechanisms arising as a consequence of manipulating the pathway may further complicate the interpretation of such studies (Sullivan et al., 2012).
Figure 2. Investigating mechanisms of tumor suppression.
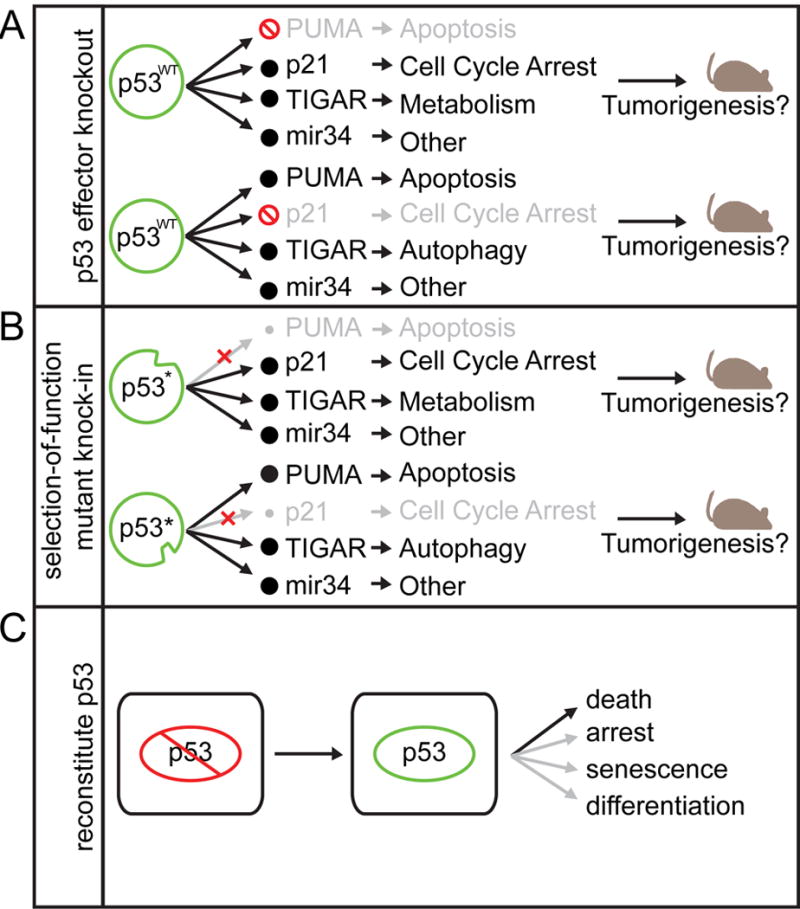
Defining the mechanism of p53-mediated tumor suppression has been interrogated in several ways: (A) knocking out p53 target genes and assessing tumor formation, (B) mutating p53 itself, such that it can activate some targets but not others, and (C) reconstituting p53 in p53 deficient cancer and determining the cell fate.
Another approach isolates p53 effects through separation-of-function mutants that selectively retain or lose the ability to regulate certain subsets of p53 target genes and activities (Figure 2B). For example, the tumor-derived p53R175P and p53E180R alleles show defects in apoptosis while retaining the capability to provoke cell cycle arrest, so that mice harboring the equivalent mutations display extended tumor-free survival compared to p53-null animals (Liu et al., 2004). Furthermore, the tumors that do arise in these mice exhibit far less CIN than p53-null tumors, indicating that different p53 mutants may impinge selectively on downstream effector pathways (Liu et al., 2004). Alternatively, engineered structure-function mutants that disrupt p53 transcriptional domains or are defective in being acetylated can separate key p53 functions, at least in vitro (Jiang et al., 2015). While these studies reinforce the importance of p53-mediated transcription for tumor suppression (Brady et al., 2011; Jiang et al., 2011), they do not pinpoint a single key process (Jiang et al., 2015).
Although such structure-function approaches are compelling, they also have caveats. Mutant p53 proteins can be more or less stable than the wild-type protein (Brady et al., 2011) and thus differential phenotypes may reflect quantitative as well as qualitative effects. Most structure-function mutants have only been characterized in a limited number of cell types, and given context dependencies, it cannot be assumed that these results extrapolate to tumorigenesis in all tissues. Perhaps these caveats explain why technically sound studies have failed to converge on a common mechanism or theme.
Several studies have circumvented the issues surrounding the manipulation of individual functions peripheral to p53: rather than measuring tumor onset upon p53 loss, they instead take advantage of mouse strains harboring “switchable” p53 alleles to reawaken endogenous p53 in established tumors (Figure 2C). In all situations examined, p53 restoration produces a marked anti-tumor response, the nature of which depends on the model employed (Martins et al., 2006; Ventura et al., 2007; Xue et al., 2007). In Myc-expressing B cell lymphomas, this response is massive apoptosis; in liver carcinomas and sarcomas, the response is senescence. In other contexts, p53 reactivation can trigger cellular differentiation and a loss of self-renewal (Messina et al., 2012). Although the consequences of p53 reactivation in an established cancer may not reflect the same processes lost during tumorigenesis, these studies reinforce the notion that the p53 response varies depending on context.
So then, what are the most important p53 activities needed for tumor suppression? Certainly, the above caveats preclude generalities without considering context-specificity. Indeed, the importance of context is readily observed in mouse studies demonstrating that Puma suppression approaches p53 loss in driving Myc-induced lymphomagenesis but not in promoting thymic lymphoma (Garrison et al., 2008; Hemann et al., 2004). Embracing this notion should enable the identification of tumor-specific modes of tumor suppression and pave the way for restoring the most relevant p53 functions in individual tumors.
The Origins of p53
How and why did the p53 network evolve? Most tumors arise after reproductive age, implying that the TP53 gene did not evolve to prevent cancer. Moreover, given the diverse outputs of the p53 network, it seems surprising that neonatal p53-null mice seem initially normal. Genes that resemble TP53 by sequence similarity and induction by DNA damage are found in simple invertebrates (including choanoflagellates, sea anemone and worms) that are not susceptible to cancer (Lane et al., 2010a; Lane et al., 2010b; Pearson and Sanchez Alvarado, 2010). Like mammalian p53, these genes induce apoptosis in response to stress but, in contrast, are expressed principally in germline stem cells. Perhaps protection of the germline is central and evolved further to suppress tumors in the soma at advanced age (Wylie et al., 2014).
Beyond the germline, a closer look at p53 and the consequences of its disruption indicate it has important roles in embryonic development. Fundamentally, multicellularity is a compromise among the cells of complex organisms. The most proliferative individuals outcompete populations of single cell organisms, while multicellular organisms require cellular cooperation, at the expense of competition, to maintain coordinated, specialized functions. The need for cooperation starts in embryonic development, where p53 restricts expansion of individual “cheater” cells, observed in chimeric blastocysts upon p53 knockdown (Dejosez et al., 2013) and following positive selection of spontaneous TP53 mutations detected in commonly used human ES lines (Merkle et al., 2017). Tight regulation of DNA methylation by DNMT and TET family enzymes requires p53 and it appears that epigenetic disorder contributes to this clonal heterogeneity in p53-deficient ESC colonies (Tovy et al., 2017). Inactivation of p53 rescues cultured cells from apoptosis caused by DMNT1 deficiency and subsequent genomic demethylation, supporting the notion of p53 can sense and respond to perturbations in the epigenome (Jackson-Grusby et al., 2001).
Other than its familiar role in restricting inappropriate clonal outgrowth, p53 also regulates target genes that fulfill specific biological requirements in development, such as LIF, which is required for efficient mammalian embryo implantation (Feng et al., 2011). Trp53 knockout mice exhibit a variety of low-penetrance tissue-specific developmental abnormalities in the neural tube, eyes and testes (Danilova et al., 2008). p53 orthologs in more primitive species can also exhibit conserved non-canonical p53 functions, such as promoting redox control and survival (reviewed in Aylon and Oren, 2016). Moreover, the ortholog Lvp53 is expressed in the soma in shrimp, where cross-talk with NF-kB eliminates virally infected cells and activates innate immunity (Li et al., 2017). Such observations are consistent with a role for the p53 family in promoting cell survival or fighting infection. Hence, the p53 network evolved diverse physiological roles prior to its implementation for tumor suppression.
TP53 is a member of a broader gene family that includes the TP63 and TP73 genes that have diverse and complementary roles. TP53 of higher eukaryotes diverged from TP63/TP73 sometime before the appearance of sharks (Lane et al., 2011). Since splitting from its homologs, TP53 and its network have acquired tumor suppressive capabilities not shared by TP63/TP73, which display even clearer ties to embryonic development (reviewed in Belyi et al., 2010). Triple p53/p63/p73 knockout mice demonstrate that the p53 family is required for mesendodermal differentiation (Wang et al., 2017), exemplifying how p53 can interact with p63/p73 in redundant or cooperative ways. It seems likely that compensation between p53 family members has masked other roles for p53 during development.
Although the p53 protein sequence itself is relatively conserved in higher eukaryotes, domains involved in p53 regulation on the N- and C-termini (Lane et al., 2011) as well as the downstream p53 response are under continued evolutionary pressure. Indeed, many p53 response elements exhibit surprisingly low conservation with respect to other transcription factor recognition sites (Horvath et al., 2007; Su et al., 2015). Another way in which the p53 network has evolved is by increasing gene dosage. That elephants have acquired up to 20 TP53 retrogenes may explain, at least in part, how an animal with such a large body size and relative longevity is not subject to high cancer risk (Abegglen et al., 2015; Sulak et al., 2016). A more detailed exploration of the factors selected and counter-selected in p53 biology over evolutionary time has the potential to provide insight into the biological processes critical for tumor suppression.
There is substantial evidence that p53 has additional functions in non-pathological tissue homeostasis. One illustrative example is that p53 function appears to be intertwined with stem cell biology and differentiation in the soma of higher organisms. In stem and progenitor cells of the hematopoietic system, liver, brain, and breast, p53 restricts cellular self-renewal (Friedmann-Morvinski et al., 2012; Tosoni et al., 2015; Tschaharganeh et al., 2014). Trp53-null mice consequently have expanded numbers of tissue specific stem cells, highlighting its importance in maintaining tissue homeostasis (Olivos and Mayo, 2016). p53 limits cellular plasticity (governing transition between cell states) and, at its extreme, the ability of somatic cells to undergo epigenetic reprogramming into induced pluripotent stem cells (Olivos and Mayo, 2016). The iPS-promoting factors KLF4 and Oct4 repress p53, and conversely, p53 activity antagonizes the efficiency of iPS cell reprogramming (Menendez et al., 2010).
An application of the above principles can be seen in tissue regeneration and the wound-healing response, which is a complex process involving waves of inflammation, angiogenesis, tissue regeneration, extracellular matrix (ECM) remodeling, and fibrosis to prevent infection and resolve tissue damage. During an initial proliferative phase of regeneration, mitogens are activated and p53 must be suppressed to allow tissue remodeling (Charni et al., 2017). By triggering cellular senescence, p53 promotes the release of secretory factors that allow resolution of fibrosis (Krizhanovsky et al., 2008) and coordinate ECM remodeling (Ritschka et al., 2017). Of note, the requirement for p53 to regulate plasticity appears to be evolutionarily conserved, which requires the coordinate suppression and derepression of p53 during salamander limb regeneration (Yun et al., 2013).
It is intriguing that the physiological and developmental functions of p53 are intertwined with the cancer-associated phenotype of p53 loss. Evading terminal differentiation is an essential step in malignant transformation and p53 loss may be one route to weaken this innate barrier to tumorigenesis. Consistent with this notion, an embryonic stem cell-like gene signature is observed in p53-mutant breast cancer (Mizuno et al., 2010). By affecting differentiation, incipient TP53 mutations facilitate the expansion of hematopoietic stem cell (HSC) clones in otherwise healthy individuals, occasionally overtaking the entire hematopoietic system (Steensma et al., 2015; Xie et al., 2014). The competitive expansion of pre-treatment p53 mutant HSC clones is accentuated by genotoxic chemotherapy, fostering therapy-related AML (t-AML) (Wong et al., 2015). At its extreme, p53 loss can even facilitate lineage switching as a mechanism of resistance to anti-androgen therapy in prostate cancer (Mu et al., 2017). Furthermore, p53 action in wound healing also shapes the tumor microenvironment. For example, the p53-driven senescence associated secretory phenotype (SASP) in tumor stroma can create a tumor suppressive immune milieu that influences the incidence of cancer (Lujambio et al., 2013; Xue et al., 2007). In other settings, the SASP can be tumor promoting, by inducing epithelial–mesenchymal transition (EMT) (Laberge et al., 2012; Ritschka et al., 2017).
It appears that evolution has selected for a delicate balance of p53 activity, since too little p53 leads to early onset cancer and too much p53 exacerbates aging. Regardless, the dangers of excess p53 are evident in pathologies beyond cancer, including aging, ischemic injury, and degeneration (reviewed in Gudkov and Komarova, 2010). As animals age, the cost of eliminating potentially dangerous cells is the attrition of stem cells required for tissue homeostasis. In an accelerated process, patients with the heritable DNA repair deficiency syndrome Fanconi anemia hyperactivate p53 in response to unresolved DNA damage and eventually experience bone marrow failure owing to progressive HSC loss (Ceccaldi et al., 2012). Excessive p53-dependent apoptosis can also drive developmental disorders of the brain (Houlihan and Feng, 2014) and aging-associated neurodegenerative diseases, namely Alzheimer’s and Parkinson’s diseases (reviewed in Checler and Alves da Costa, 2014). As a regulator of cell death, p53 has been implicated in the pathological response to cerebral and cardiac ischemia; p53 inhibition has been proposed as a protective strategy in the acute phase following injury (Gudkov and Komarova, 2010). Lastly, excessive p53-mediated ferroptosis can trigger lethal kidney ischemia (Friedmann Angeli et al., 2014). Collectively, the characteristics of p53 action in normal physiology and non-cancer pathologies shed light on additional regulatory mechanisms, downstream functions, and possible therapeutic targets.
The diversity of TP53 mutational events produces distinct functional consequences
Just as advances in our understanding of p53 biology have complicated, rather than simplified, our views on how TP53 mutations promote cancer, so has our appreciation of surprising range of ways in which the TP53 locus is altered in tumors. The most common and well-characterized TP53 mutations are missense mutations in the DNA binding domain, implying that this feature of p53 is crucial for tumor suppression. Current dogma tends to classify p53 as either wild-type or mutant, but TP53 mutations occur with different patterns, distinct co-mutations, and in many allelic configurations that lead to remarkably interesting functional and phenotypic ramifications.
Genome sequencing of thousands of tumors has confirmed that approximately half of all cancers harbor a TP53 mutation, though the frequency and the distribution of mutations can vary dramatically between tumor types (Figure 3A). Most of the single nucleotide variants (SNVs) observed across cancers are missense mutations, with 25% of those falling into 5 “hotspot” mutations (Shirole et al., 2016). Unexpectedly, nearly 25% of TP53 mutations are nonsense or frameshift mutations predicted to encode truncated proteins, whereas the remainder consists of splice site SNVs and in-frame indels of unclear biological significance (Shirole et al., 2016 ). While several modes to disable the second TP53 allele are possible, this typically occurs through “loss of heterozygosity” (LOH) by segmental deletion (Figure 3B). These deletions vary widely in size and occur at a frequency that is similar to p53 SNVs (Liu et al., 2016). Nearly all possible allelic combinations are observed such that, in reality, only ~25% of tumors harbor the canonical p53 missense mutation/deletion combination (Liu et al., 2016).
Figure 3. p53 alteration spectrum.
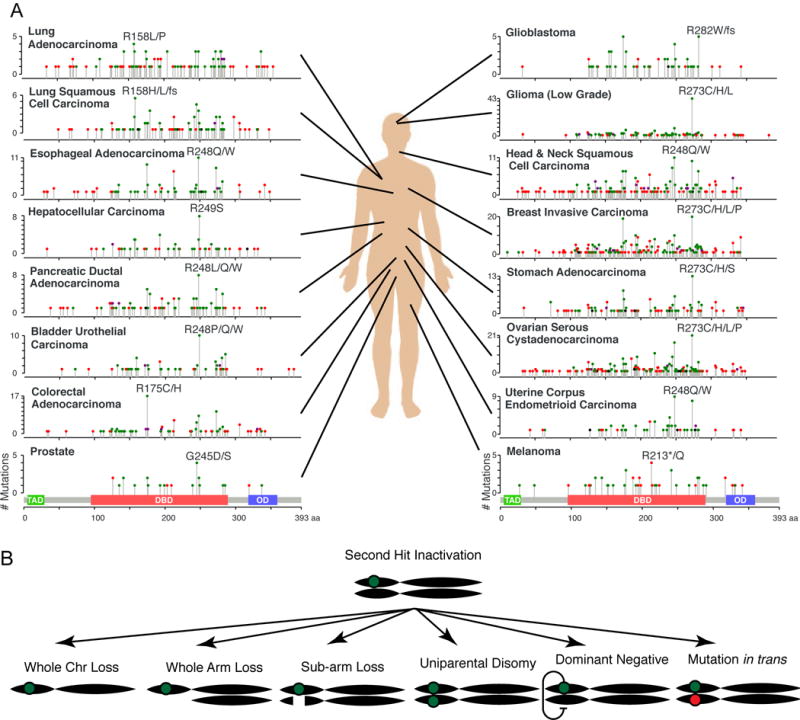
(A) The TP53 mutation distribution for 16 cancer types with sufficient available data and frequency of TP53 alteration. Each histogram depicts the number of mutations found at each position along the p53 protein coding sequence, with the transactivation domain (TAD), DNA-binding domain (DBD), and oligomerization domain (OD) illustrated below. Symbol color indicates mutation type, including missense (green), nonsense (red), inframe indels (black), or multiple mutation types (purple). Data source: MSKCC cbio portal (Gao, Schultz, Sci Signal, 2013). (B) Multiple avenues to inactivating the second allele of TP53.
Cancer genome projects have also produced interesting insights into the spectrum of TP53 mutation and its association with other somatic events. In some cancers, TP53 mutations often co-occur with activating KRAS mutations or MYC amplification, an observation reminiscent of age-old functional studies demonstrating the ability of p53 loss to cooperate with oncogenes to transform primary cells. And, as mentioned earlier, TP53 mutations are frequently associated with high rates of CNV, for example, as occurs in ovarian carcinoma and complex karyotype AML (Ciriello et al., 2013).
The extensive cataloging of TP53 alterations in different settings allows one to consider whether distinct alterations reflect functional selection or simply different mutagenic processes present during tumorigenesis. Distinct mutational signatures in TP53 and other genes can be attributed, in part, to the specific source of mutagenesis (Alexandrov et al., 2016). For instance, the R249S mutation prevalent in hepatocellular carcimoma arise from G-to-T transversions linked to aflatoxin exposure and R213* mutations in melanoma are associated with the C-to-T transition signature of UV mutagenesis (Alexandrov et al., 2016). Characterization of mutagenic signatures has revealed recurrent C-to-T mutation patterns attributed to cytidine deaminases, such as AID and APOBEC, which are an intrinsic source of mutagenesis with a physiological role in antibody diversification (Alexandrov et al., 2013). Curiously, the APOBECs can be induced by either wild type or mutant p53 (Menendez et al., 2017), though a clear link between p53 status and APOBEC-mediated mutagenesis has not yet been described (Burns et al., 2013; Shinmura et al., 2011). While epidemiology and genome sequencing can implicate environmental or endogenous mutagens as responsible for particular TP53 mutations, it is difficult, if not impossible, to assess individual alleles without direct functional studies.
In fact, experimental data emerging over the last 25 years have hinted that certain mutant TP53 alleles have “gain of function” properties that produce phenotypes distinct from the null. The most prominent phenotype produced by such mutant proteins is their ability to enhance invasion and metastasis, though in some settings particular mutants enhance drug resistance, epigenetic reprogramming or angiogenesis (reviewed in Aschauer and Muller, 2016). While proposed activities are diverse, an emerging “rule of thumb” is that tumor-derived p53 mutants oppose wild-type p53 functions or, more explicitly, exacerbate the consequences of p53 loss. In any case, the notion that not all p53 mutations are functionally equivalent is further supported by the fact that the onset and pathology of tumors in genetically engineered mouse models and in Li-Frameni patients varies by the type of mutant allele (Achatz and Zambetti, 2016; Olive et al., 2004; Xu et al., 2014).
A p53 mutant that has distinct phenotypes from the p53 null is not sufficient to define a mutant as “neomorphic.” Theoretically, p53 mutant alleles may reflect attenuation of function, separation of function, or neomorphic function. Attenuation of wild-type function (Figure 4A) can produce hypomorphs that can also yield produce unpredictable and qualitatively different phenotypes depending on the level of p53 suppression (Figure 4B). For instance, a series of p53-targeting shRNAs with varying knockdown efficiency display different abilities to disrupt p53 effector functions and drive lymphomagenesis in mice, with only complete p53 deletion capable of instigating chromosomal instability (Hemann et al., 2003). Loss of function is a common characteristic across all cancer-associated p53 mutants, given the failure of most mutants to induce apoptosis (Freed-Pastor and Prives, 2012). Separation of function – whereby a p53 mutant can retain some but not all interactions (reviewed in Muller and Vousden, 2014) – is also possible (Figure 4C), as exemplified by the aforementioned apoptosis-deficient p53R175P allele (Liu et al., 2004). Finally, neomorphic mutant activities (Figure 4D) have also been described (discussed below). In reality, the mutations encountered in cancer acquire some combination of these independent characteristics. Although p53 mutants are generally classified by their effect on structure – i.e. “contact” mutants that perturb DNA binding and “conformation” mutants that lose proper folding – it is currently not possible to predict precisely how a particular mutation impacts function.
Figure 4. Mutant p53 gain-of-function.
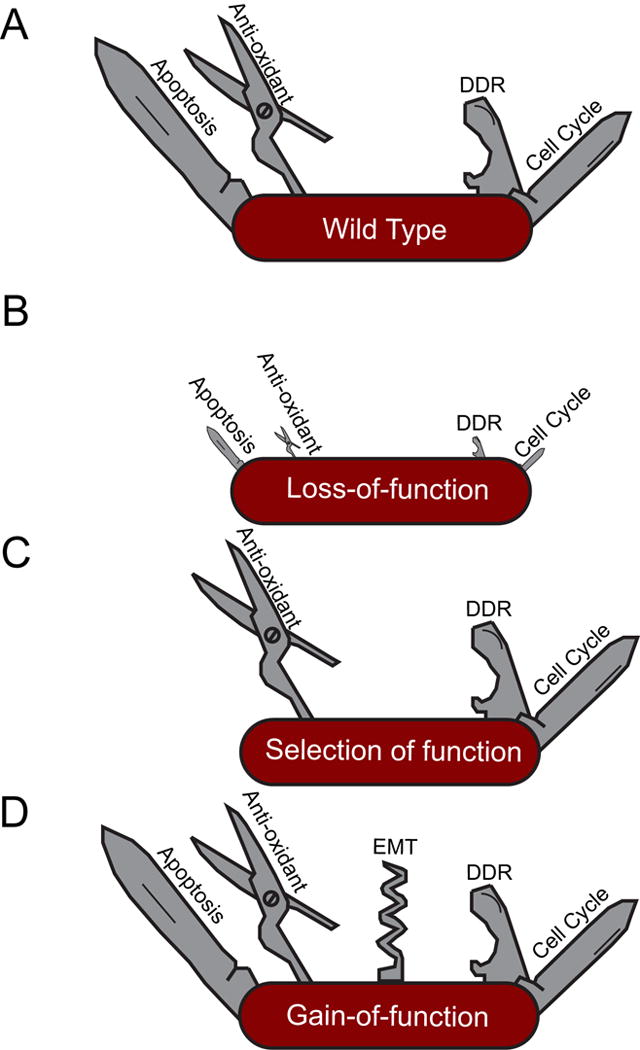
Several alternative mechanisms can lead to divergent phenotypes of p53 mutations. (A) wild-type, (B) loss or partial loss of function, (C) selection of function, or (D) neomorphic/gain-of-function.
The diversity of proposed mechanisms by which mutant p53 alleles elicit their pro-oncogenic effects are a source of much confusion in the field (Aschauer and Muller, 2016). First, some p53 mutant proteins retain residual transactivation activity and activate novel targets. For instance, mutant p53 is proposed to impact chromatin states by inducing MLL1/2 and MOZ (Zhu et al., 2015). Second, certain unstructured p53 mutants sequester other proteins that, in some settings, enable mutant p53 to bind p63 or p73, leading to changes in transcriptional profiles that alter receptor tyrosine kinase signaling to promote invasion and metastasis (Muller et al., 2013; Weissmueller et al., 2014). Finally, in an instance of gain-of-function protein-protein interaction, mutant p53 can cooperate with the SWI/SNF complex to upregulate the angiogenesis regulator VEGFR2 (Pfister et al., 2015). It remains difficult to reconcile how so many distinct yet selective protein-protein interactions can occur for disparate mutant proteins (reviewed in Freed-Pastor and Prives, 2012).
Although it is generally assumed that TP53 truncating mutations are null alleles, there are emerging data that even these alleles can have neomorphic activity. Implying some selective advantage, the frequency of TP53 nonsense mutations, particularly targeting exon 6, is greater than expected by chance (Shirole et al., 2016). At least some of these are not subject to nonsense-mediated decay, allowing certain truncated p53 mutants to promote invasion, metastasis, and sustain tumor maintenance in a manner that mirrors established gain of function missense mutants (Shirole et al., 2016). Provocatively, exon 6 truncated proteins mimic the structure and function of a naturally-occurring p53 splice variant (p53psi) that promotes cell invasion and is transiently expressed during certain wound healing responses (Senturk et al., 2014), suggesting that these mutants may represent “separation of function” alleles. Expression or mimicry of alternative splice variants may contribute to the phenotype of other common mutations as well (Candeias et al., 2016).
Beyond the heterogeneity produced by different p53 SNVs, the variable extent of human chromosome 17p deletions can produce heterogeneity in the nature and number of p53-linked genes subject to reduced dosage during tumorigenesis. Loss of these neighboring genes could well reflect a “passenger” event of no functional consequence; however, 17p deletions observed in human cancer often include other genes now functionally validated as tumor suppressors. Deletions engineered to be syntenic to 17p13 drive more aggressive cancers than simple p53 deficiency in mice by virtue of single copy loss of multiple haploinsufficient tumor suppressors, consistent with the negative prognostic association of 17p deletion independent of p53 mutation that is evident in AML (Liu et al., 2016). These observations and others underscore the unique biology underlying CNVs and highlight the importance of dissecting these understudied events (Tschaharganeh et al., 2016).
Collectively, our emerging understanding of the complexities of the gamut of TP53 alterations is changing our views on how “the most frequent event in human cancer” promotes tumorigenesis. While there is little doubt that the most substantial biological consequence results from inactivation of p53, it is now clear that both TP53 mutations and 17p deletions contribute phenotypes to cancer that go beyond p53 loss. Thus, as clinical decision-making in the future becomes increasingly based on genomic data, the current classification of tumors as simply “p53 wild-type” or “p53 mutant” must be replaced.
Harnessing the p53 Network
The potency of p53 in tumor suppression and the high rate of p53 alteration in cancers has spurred the development of strategies to target the p53 network in cancer therapy. Indeed, the potential value of engaging p53 in an anticancer response is clear from studies showing that, in some cases, robust responses to conventional chemotherapy can depend on p53, and studies in mice described above document massive tumor regressions in response to p53 reactivation in vivo. For instance, the dramatic cures achieved by retinoic acid and arsenic treatment of acute promyelocytic leukemia is dependent on p53-mediated senescence (Ablain et al., 2014). Since TP53 mutations inactivate wild-type p53 protein, they are widely considered undruggable and, consequently, efforts to rationally exploit p53 for therapeutic benefit have yet to reach fruition. Nonetheless, some strategies to target mutant p53 proteins, p53 regulators, or vulnerabilities created by TP53 mutation in cancer and other indications show promise.
One of the most advanced efforts to exploit our understanding of p53 biology for cancer therapy involves efforts to inhibit MDM2 in tumors harboring wild-type p53. Led by the development of Nutlin (Vassilev et al., 2004), a panoply of small molecule and peptide inhibitors of MDM2 and MDMX have been developed aimed at improving the properties of first generation inhibitors that generally act by targeting the p53 binding site in MDM2 (reviewed in Cheok and Lane, 2017). A number of phase I trials for MDM2 antagonists have been completed in leukemia and liposarcoma, with neutropenia and thrombocytopenia being prominent dose-limiting toxicities (Andreeff et al., 2016). While these dose-escalation studies preclude conclusions about drug efficacy, induction of p53 target gene expression was observed in most p53 wild-type samples. Moreover, a partial response occurred in 5–10% of patients, a promising result given that many were heavily pre-treated. Counterintuitively, only some of these MDM2 inhibitor clinical trials stratify patients by TP53 status (reviewed in Burgess et al., 2016; Wang et al., 2011).
Flipping the situation around, MDM2 inhibitors have also been used in efforts aimed at reducing the toxic side effects of chemotherapy. In a strategy termed cyclotherapy, these drugs are used to stabilize p53 and trigger a transient cell cycle arrest in normal cells, with the intention of having no effect on the cell cycle progression of p53 mutant tumor cells. As many cytotoxic drugs target actively cycling cells, this strategy is predicted to allow use of a higher tolerable dose of chemotherapy, enhancing efficacy against cancer cells that continue to cycle while reducing toxicity to arrested normal cells (Cheok and Lane, 2017). In preclinical studies, cyclotherapy protects mice treated with Polo kinase inhibitor from dose-limiting neutropenia (Sur et al., 2009).
One attractive therapeutic approach involves identifying agents that cause mutant p53 to regain sufficient wild-type p53 activity for tumor suppression. Although the thermodynamic requirements for achieving this seem daunting, structural studies and in silico predictions have propelled multiple strategies that supply proof-of-principle for this approach, including peptides and small molecules that stabilize unstructured mutants (Boeckler et al., 2008; Friedler et al., 2002; Yu et al., 2012). One drug, APR-246, which is purported to reactivate mutant p53 but also has off target effects, is currently in clinical trials (NCT03072043, NCT02999893)(Deneberg et al., 2016). Other agents that directly stabilize the p53 DNA binding domain show promise in preclinical studies (Cheok and Lane, 2017). Drugs known as metallochaperones can facilitate the reincorporation of zinc into unfolded p53 proteins leading to a more normal confirmation and an ability to bind DNA (reviewed in Blanden et al., 2015). Yet another approach exploits the unexpected observation that certain p53 mutant proteins have a penchant for aggregation into amyloid-like structures (reviewed in de Oliveira et al., 2015) that, when disrupted, restore p53 function and reportedly trigger tumor regression in xenograft models (Soragni et al., 2016).
While the above drugs all aim to coax native wild-type activity out of mutant proteins, instead disabling or suppressing mutant p53 represents an unexplored alternative direction that is justified by the observation that tumors can become “addicted” to mutant p53 (Alexandrova et al., 2015). Several indirect strategies have been proposed to destabilize mutant p53 protein including HSP90 inhibitors, HDAC inhibitors, or SIRT1 activators (reviewed in Parrales and Iwakuma, 2015). In the absence of readily available tools to directly inhibit mutant p53 function, the opportunity remains to apply existing drugs to target the underlying mechanism whereby mutant p53 promotes invasion, metastasis, and cellular survival (e.g. via HMG CoA reductase, EGFR, or PDGFRb inhibitors) (Aschauer and Muller, 2016; Weissmueller et al., 2014).
Another way in which to attack mutant p53 directly is to harness its potential to serve as a tumor-specific neoantigen. Mutant p53 proteins are typically expressed at high levels and can be antigenic (Crawford et al., 1982; DeLeo et al., 1979); furthermore, vaccination against mutant p53 can protect mice from cancer produced by transplanted tumors (Roth et al., 1996). Based on this premise, peptide vaccines (Zeestraten et al., 2013), viral vectors (van der Burg et al., 2002), and dendritic cell vaccines (Ellebaek et al., 2012) have entered Phase I/II clinical trials. Regardless of platform, immunotherapy has been able to induce p53-specific immune reactions in humans, though clinical responses have yet to be observed.
In theory, tumors that have escaped immunoediting are more likely to contain immunogenic neoantigens. Therefore, there is interest in combining p53 immunotherapy with so-called immune checkpoint blockade to enhance T cell reactivity, which may be able to translate previously observed generation of p53-specific T-cells into the desired cytotoxicity and clinical responses (Hardwick et al., 2014). Indeed, p53 loss can shield cancer cells from CD8+ T cells via PD-L1 derepression, an interaction that accelerates mouse models of cancer and is evident in human lung cancer (Cha et al., 2016; Cortez et al., 2016; Schuster et al., 2011), yet a positive association between p53 alteration and response to immunotherapy by PD-L1 inhibition has not been observed.
An attractive approach to targeting p53 mutant tumors is to exploit synthetic lethality, a term describing a situation in which gene mutation creates novel dependencies. Many previously characterized liabilities imposed by p53 mutation converge around the DNA damage response and metabolism. Although p53- deficient cells can evade apoptosis in the face of DNA damaging agents, further disabling the DDR leaves p53 mutant tumors hypersensitive to genotoxic damage (Ma et al., 2012). Accordingly, strategies combining DNA-damaging agents with inhibitors of DDR components ATM, CHK2, ATR, and CHK1 have been pursued (reviewed in Morandell and Yaffe, 2012). Supporting the potential of this approach, a WEE1 inhibitor that disables a G2 cell cycle checkpoint enhances the antitumor activity of genotoxic chemotherapy in previously refractory p53 mutant ovarian cancer patients (Leijen et al., 2016). Also, patients with TP53 mutations have been reported to have higher response rates to extended cycles of the demethylating agent decitabine (Welch et al., 2016). While the mechanistic basis for this observation is not know, one plausible explanation is that wild type cells arrest in G2/M upon drug treatment, whereas p53-deficient cells pass through the cell cycle checkpoint, resulting in severe chromosomal damage and death (Nieto et al., 2004). Although exacerbating instability may achieve therapeutic responses, the concern remains that mutagenesis associated with reducing the DDR likely fuels tumor evolution and perhaps even the emergence of treatment-associated cancers.
Additionally, the metabolic rewiring associated with p53 mutation also instills novel dependencies on druggable targets, including PIP4K2A/B, cholesterol biosynthesis, and IAPP (Emerling et al., 2013; Freed-Pastor et al., 2012; Venkatanarayan et al., 2015). Unlike synthetic lethal interactions related to p53 loss-of-function, a side-effect of single copy deletion of chromosome 17p deletions during LOH may be to expose cancers to heightened dependence on other essential genes in this region, such as POLR2A (Liu et al., 2015).
Beyond cancer, pharmacological modulation of p53 is a potentially useful and largely unexplored strategy to aid cell autonomous defense against infection. Pathogens evolved around mammalian cells, selected to keep the host alive despite the DNA damage, ROS induction, and activation of innate immunity through toll-like receptors that follows infection, all of which can be mediated by p53 (Shatz et al., 2012). Hence, p53 can act as a suppressor of bacterial infection, leading to the concept of pharmacological p53 activation to mitigate severe infections (Siegl et al., 2014). Some pathogens encode components that inhibit p53, and nutlin-based stabilization of p53 can hinder their propagation (Kaushansky et al., 2013; Siegl et al., 2014). Consequently, it may be worth considering use of MDM2 inhibitors in cases of life-threatening multi-drug resistant infections with no other treatment options. However, induction of p53 is not universally conducive to combating infection, and defining its disease-specific immune interactions will be a prerequisite for clinical relevance of p53 in infectious diseases. Trp53−/− mice are actually more capable of recovering from bacterial pneumonia than wild-type mice (Madenspacher et al., 2013).
Through restoring wild type function, inhibiting mutant function, or treating a dysregulated immune system, multiple avenues exist to target the p53 network in cancer. Given the obstacles that have been encountered using these strategies to date, further knowledge of basic p53 biology will be required for future successful clinical applications.
Concluding Remarks
p53 has captured the fascination of cancer biologists, and its detailed characterization has produced fundamental insights into mechanisms of gene regulation and nature’s safeguards against cancer. While the body of research on p53 is massive and sometimes contradictory, it is now abundantly clear that cellular responses to p53 activation involve a complex interplay between activation triggers, cell lineage, and cell state. While such context-dependent effects on p53 have stymied attempts to generalize the mechanism of p53-mediated tumor suppression, they provide opportunities to exploit the network in cancer cells, while avoiding deleterious consequences of manipulating p53 in all tissues.
Despite decades of intensive research and countless discoveries, there remains much to learn about the roles and regulation of p53. A challenge in the coming era of p53 research will be to distill convergent truths assembled from comprehensive studies, and to translate knowledge of p53 into clinical application. Indeed, the enormous challenge associated with exploiting p53 therapeutically does not mitigate the astounding morbidity associated with TP53 mutation. In the absence of new therapeutic innovations, TP53 mutant cancer will lead to the deaths of over 500 million people alive today. New technologies, together with our ever-increasing understanding of the complexity of p53 action and the diverse consequences of p53 mutation will hopefully set the stage for more robust clinical advances.
Figure 5. Harnessing p53.
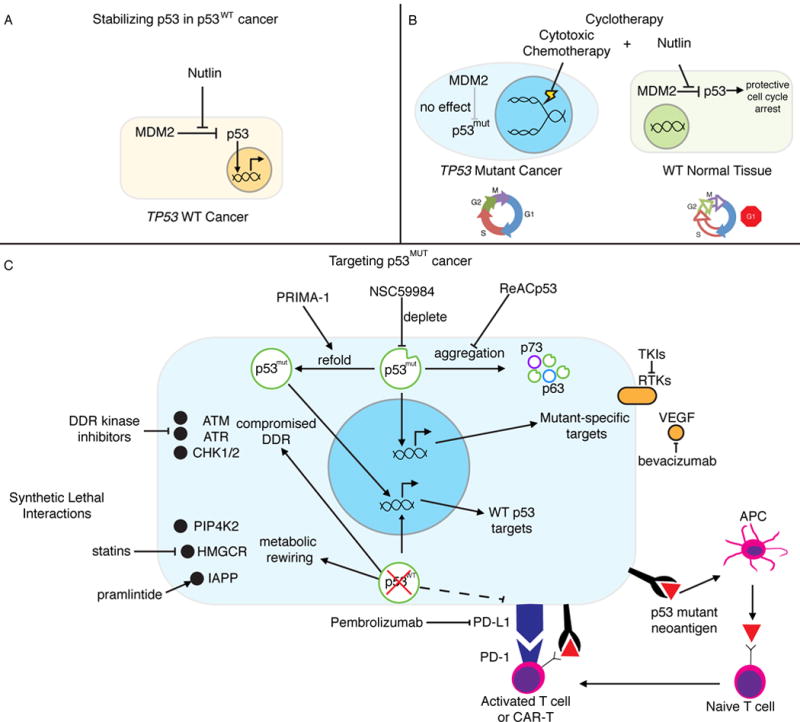
(A) Stabilizing p53 in p53WT cancer. Nutlin and other MDM2/MDMX inhibitors (RG7112, RO5503781, SAR405838, HDM201, MK4828, AMG232, and RG7388) allow for the accumulation and activity of p53 in cancer in which it is not mutated. (B) Cyclotherapy. Nutlin is used to transiently arrest p53WT normal cells, while p53MUT cancer cells continue to cycle and remain vulnerable to genotoxic chemotherapy. Sparing normal tissue allows for increased dosing and reduced toxicity. (C) Targeting p53MUT cancer. PRIMA-1 and other agents (APR-246, RITA, PK7088, p53R3, and ZMC1) are used to support proper folding of mutant p53 and restore wild type-like structure and activity. p53 mutant protein is depleted through a number of indirect mechanisms including inhibition of HSP90 (17-AAG), HDAC (SAHA), and SIRT1 (YK-3-237). The aggregation and inactivation of mutant p53 and its family members is inhibited by ReACp53. Synthetic lethal interactions are dependencies in p53 mutant cancer but not in p53WT cells. p53 deficient cells have a compromised DDR, leaving then vulnerable to even further genomic instability by inhibiting DDR-related kinases. Metabolic rewiring introduces druggable dependencies on PIP4K2, cholesterol biosynthesis/HMGCR (statins), and IAPP (pramlinitide). Some p53 mutations can result in recognizable neoantigens, which has led to the development of mutant p53-targeted immunotherapy. p53 ablation can also modify antigen presentation efficiency justifying the investigation of immune checkpoint inhibition, especially when combined with other strategies.
Acknowledgments
We would like to thank Charles Sherr of HHMI at St. Jude Children’s Research Hospital and all members of the Lowe lab for advice and critical discussions, especially Francisco Sanchez-Rivera, Shauna Houlihan, and John P Morris IV. E.R.K. is supported by an F31 NRSA predoctoral fellowship from the NCI/National Institutes of Health under award number F31CA192835. S.W.L. is an investigator of the Howard Hughes Medical Institute and the Geoffrey Beene Chair for Cancer Biology. We thank all of the investigators in the p53 field who have contributed to the p53 field and the many ideas presented herein. We apologize to the many investigators whose work could not be cited owing to space constraints.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abegglen LM, Caulin AF, Chan A, Lee K, Robinson R, Campbell MS, Kiso WK, Schmitt DL, Waddell PJ, Bhaskara S, et al. Potential Mechanisms for Cancer Resistance in Elephants and Comparative Cellular Response to DNA Damage in Humans. JAMA. 2015;314:1850–1860. doi: 10.1001/jama.2015.13134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ablain J, Rice K, Soilihi H, de Reynies A, Minucci S, de The H. Activation of a promyelocytic leukemia-tumor protein 53 axis underlies acute promyelocytic leukemia cure. Nat Med. 2014;20:167–174. doi: 10.1038/nm.3441. [DOI] [PubMed] [Google Scholar]
- Achatz MI, Zambetti GP. The Inherited p53 Mutation in the Brazilian Population. Cold Spring Harb Perspect Med. 2016;6 doi: 10.1101/cshperspect.a026195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik-Zainal S, Totoki Y, Fujimoto A, Nakagawa H, Shibata T, et al. Mutational signatures associated with tobacco smoking in human cancer. Science. 2016;354:618–622. doi: 10.1126/science.aag0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrova EM, Yallowitz AR, Li D, Xu S, Schulz R, Proia DA, Lozano G, Dobbelstein M, Moll UM. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature. 2015;523:352–356. doi: 10.1038/nature14430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen MA, Andrysik Z, Dengler VL, Mellert HS, Guarnieri A, Freeman JA, Sullivan KD, Galbraith MD, Luo X, Kraus WL, et al. Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. Elife. 2014;3:e02200. doi: 10.7554/eLife.02200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreeff M, Kelly KR, Yee K, Assouline S, Strair R, Popplewell L, Bowen D, Martinelli G, Drummond MW, Vyas P, et al. Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clin Cancer Res. 2016;22:868–876. doi: 10.1158/1078-0432.CCR-15-0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschauer L, Muller PA. Novel targets and interaction partners of mutant p53 Gain-Of-Function. Biochem Soc Trans. 2016;44:460–466. doi: 10.1042/BST20150261. [DOI] [PubMed] [Google Scholar]
- Aylon Y, Michael D, Shmueli A, Yabuta N, Nojima H, Oren M. A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes Dev. 2006;20:2687–2700. doi: 10.1101/gad.1447006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylon Y, Oren M. The Paradox of p53: What, How, and Why? Cold Spring Harb Perspect Med. 2016;6 doi: 10.1101/cshperspect.a026328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SJ, Preisinger AC, Jessup JM, Paraskeva C, Markowitz S, Willson JK, Hamilton S, Vogelstein B. p53 gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis. Cancer Res. 1990;50:7717–7722. [PubMed] [Google Scholar]
- Belyi VA, Ak P, Markert E, Wang H, Hu W, Puzio-Kuter A, Levine AJ. The origins and evolution of the p53 family of genes. Cold Spring Harb Perspect Biol. 2010;2:a001198. doi: 10.1101/cshperspect.a001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanden AR, Yu X, Loh SN, Levine AJ, Carpizo DR. Reactivating mutant p53 using small molecules as zinc metallochaperones: awakening a sleeping giant in cancer. Drug Discov Today. 2015;20:1391–1397. doi: 10.1016/j.drudis.2015.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeckler FM, Joerger AC, Jaggi G, Rutherford TJ, Veprintsev DB, Fersht AR. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:10360–10365. doi: 10.1073/pnas.0805326105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, Kenzelmann Broz D, Basak S, Park EJ, McLaughlin ME, et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell. 2011;145:571–583. doi: 10.1016/j.cell.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess A, Chia KM, Haupt S, Thomas D, Haupt Y, Lim E. Clinical Overview of MDM2/X-Targeted Therapies. Front Oncol. 2016;6:7. doi: 10.3389/fonc.2016.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns MB, Temiz NA, Harris RS. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nature genetics. 2013;45:977–983. doi: 10.1038/ng.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candeias MM, Hagiwara M, Matsuda M. Cancer-specific mutations in p53 induce the translation of Delta160p53 promoting tumorigenesis. EMBO Rep. 2016;17:1542–1551. doi: 10.15252/embr.201541956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Parmar K, Mouly E, Delord M, Kim JM, Regairaz M, Pla M, Vasquez N, Zhang QS, Pondarre C, et al. Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell. 2012;11:36–49. doi: 10.1016/j.stem.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha YJ, Kim HR, Lee CY, Cho BC, Shim HS. Clinicopathological and prognostic significance of programmed cell death ligand-1 expression in lung adenocarcinoma and its relationship with p53 status. Lung Cancer. 2016;97:73–80. doi: 10.1016/j.lungcan.2016.05.001. [DOI] [PubMed] [Google Scholar]
- Chang NT, Yang WK, Huang HC, Yeh KW, Wu CW. The transcriptional activity of HERV-I LTR is negatively regulated by its cis-elements and wild type p53 tumor suppressor protein. J Biomed Sci. 2007;14:211–222. doi: 10.1007/s11373-006-9126-2. [DOI] [PubMed] [Google Scholar]
- Charni M, Aloni-Grinstein R, Molchadsky A, Rotter V. p53 on the crossroad between regeneration and cancer. Cell Death Differ. 2017;24:8–14. doi: 10.1038/cdd.2016.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Checler F, Alves da Costa C. p53 in neurodegenerative diseases and brain cancers. Pharmacol Ther. 2014;142:99–113. doi: 10.1016/j.pharmthera.2013.11.009. [DOI] [PubMed] [Google Scholar]
- Cheok CF, Lane DP. Exploiting the p53 Pathway for Therapy. Cold Spring Harb Perspect Med. 2017;7 doi: 10.1101/cshperspect.a026310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45:1127–1133. doi: 10.1038/ng.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke AR, Purdie CA, Harrison DJ, Morris RG, Bird CC, Hooper ML, Wyllie AH. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature. 1993;362:849–852. doi: 10.1038/362849a0. [DOI] [PubMed] [Google Scholar]
- Cooks T, Harris CC, Oren M. Caught in the cross fire: p53 in inflammation. Carcinogenesis. 2014;35:1680–1690. doi: 10.1093/carcin/bgu134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez MA, Ivan C, Valdecanas D, Wang X, Peltier HJ, Ye Y, Araujo L, Carbone DP, Shilo K, Giri DK, et al. PDL1 Regulation by p53 via miR-34. J Natl Cancer Inst. 2016;108 doi: 10.1093/jnci/djv303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford LV, Pim DC, Bulbrook RD. Detection of antibodies against the cellular protein p53 in sera from patients with breast cancer. Int J Cancer. 1982;30:403–408. doi: 10.1002/ijc.2910300404. [DOI] [PubMed] [Google Scholar]
- Danilova N, Sakamoto KM, Lin S. p53 family in development. Mech Dev. 2008;125:919–931. doi: 10.1016/j.mod.2008.09.003. [DOI] [PubMed] [Google Scholar]
- de Oliveira GA, Rangel LP, Costa DC, Silva JL. Misfolding, Aggregation, and Disordered Segments in c-Abl and p53 in Human Cancer. Front Oncol. 2015;5:97. doi: 10.3389/fonc.2015.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejosez M, Ura H, Brandt VL, Zwaka TP. Safeguards for cell cooperation in mouse embryogenesis shown by genome-wide cheater screen. Science. 2013;341:1511–1514. doi: 10.1126/science.1241628. [DOI] [PubMed] [Google Scholar]
- DeLeo AB, Jay G, Appella E, Dubois GC, Law LW, Old LJ. Detection of a transformation-related antigen in chemically induced sarcomas and other transformed cells of the mouse. Proc Natl Acad Sci U S A. 1979;76:2420–2424. doi: 10.1073/pnas.76.5.2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deneberg S, Cherif H, Lazarevic V, Andersson PO, von Euler M, Juliusson G, Lehmann S. An open-label phase I dose-finding study of APR-246 in hematological malignancies. Blood Cancer J. 2016;6:e447. doi: 10.1038/bcj.2016.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewhurst SM, McGranahan N, Burrell RA, Rowan AJ, Gronroos E, Endesfelder D, Joshi T, Mouradov D, Gibbs P, Ward RL, et al. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov. 2014;4:175–185. doi: 10.1158/2159-8290.CD-13-0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Duan L, Perez RE, Davaadelger B, Dedkova EN, Blatter LA, Maki CG. p53-regulated autophagy is controlled by glycolysis and determines cell fate. Oncotarget. 2015;6:23135–23156. doi: 10.18632/oncotarget.5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eijkelenboom A, Burgering BM. FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol. 2013;14:83–97. doi: 10.1038/nrm3507. [DOI] [PubMed] [Google Scholar]
- Eischen CM. Genome Stability Requires p53. Cold Spring Harb Perspect Med. 2016;6 doi: 10.1101/cshperspect.a026096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- Ellebaek E, Engell-Noerregaard L, Iversen TZ, Froesig TM, Munir S, Hadrup SR, Andersen MH, Svane IM. Metastatic melanoma patients treated with dendritic cell vaccination, Interleukin-2 and metronomic cyclophosphamide: results from a phase II trial. Cancer Immunol Immunother. 2012;61:1791–1804. doi: 10.1007/s00262-012-1242-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerling BM, Hurov JB, Poulogiannis G, Tsukazawa KS, Choo-Wing R, Wulf GM, Bell EL, Shim HS, Lamia KA, Rameh LE, et al. Depletion of a putatively druggable class of phosphatidylinositol kinases inhibits growth of p53-null tumors. Cell. 2013;155:844–857. doi: 10.1016/j.cell.2013.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa JM, Verdun RE, Emerson BM. p53 functions through stress- and promoter-specific recruitment of transcription initiation components before and after DNA damage. Mol Cell. 2003;12:1015–1027. doi: 10.1016/s1097-2765(03)00359-9. [DOI] [PubMed] [Google Scholar]
- Feng Z, Zhang C, Kang HJ, Sun Y, Wang H, Naqvi A, Frank AK, Rosenwaks Z, Murphy ME, Levine AJ, et al. Regulation of female reproduction by p53 and its family members. FASEB J. 2011;25:2245–2255. doi: 10.1096/fj.10-180166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay CA, Hinds PW, Levine AJ. The p53 proto-oncogene can act as a suppressor of transformation. Cell. 1989;57:1083–1093. doi: 10.1016/0092-8674(89)90045-7. [DOI] [PubMed] [Google Scholar]
- Fischer M. Census and evaluation of p53 target genes. Oncogene. 2017 doi: 10.1038/onc.2016.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, Barsotti A, Chicas A, Li W, Polotskaia A, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 2012;148:244–258. doi: 10.1016/j.cell.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev. 2012;26:1268–1286. doi: 10.1101/gad.190678.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedler A, Hansson LO, Veprintsev DB, Freund SM, Rippin TM, Nikolova PV, Proctor MR, Rudiger S, Fersht AR. A peptide that binds and stabilizes p53 core domain: chaperone strategy for rescue of oncogenic mutants. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:937–942. doi: 10.1073/pnas.241629998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180–1191. doi: 10.1038/ncb3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann-Morvinski D, Bushong EA, Ke E, Soda Y, Marumoto T, Singer O, Ellisman MH, Verma IM. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science. 2012;338:1080–1084. doi: 10.1126/science.1226929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Cornils H, Chiu SY, O’Rourke KP, Arnaud J, Yimlamai D, Thery M, Camargo FD, Pellman D. Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell. 2014;158:833–848. doi: 10.1016/j.cell.2014.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26:1021–1032. doi: 10.1038/cr.2016.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison SP, Jeffers JR, Yang C, Nilsson JA, Hall MA, Rehg JE, Yue W, Yu J, Zhang L, Onciu M, et al. Selection against PUMA gene expression in Myc-driven B-cell lymphomagenesis. Mol Cell Biol. 2008;28:5391–5402. doi: 10.1128/MCB.00907-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes NP, Espinosa JM. Disparate chromatin landscapes and kinetics of inactivation impact differential regulation of p53 target genes. Cell Cycle. 2010a;9:3428–3437. doi: 10.4161/cc.9.17.12998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes NP, Espinosa JM. Gene-specific repression of the p53 target gene PUMA via intragenic CTCF-Cohesin binding. Genes Dev. 2010b;24:1022–1034. doi: 10.1101/gad.1881010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudkov AV, Komarova EA. Pathologies associated with the p53 response. Cold Spring Harb Perspect Biol. 2010;2:a001180. doi: 10.1101/cshperspect.a001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick N, Chung V, Cristea M, Ellenhorn JD, Diamond DJ. Overcoming immunosuppression to enhance a p53MVA vaccine. Oncoimmunology. 2014;3:e958949. doi: 10.4161/21624011.2014.958949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- Hayashi MT, Cesare AJ, Fitzpatrick JA, Lazzerini-Denchi E, Karlseder J. A telomere-dependent DNA damage checkpoint induced by prolonged mitotic arrest. Nat Struct Mol Biol. 2012;19:387–394. doi: 10.1038/nsmb.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemann MT, Fridman JS, Zilfou JT, Hernando E, Paddison PJ, Cordon-Cardo C, Hannon GJ, Lowe SW. An epi-allelic series of p53 hypomorphs created by stable RNAi produces distinct tumor phenotypes in vivo. Nat Genet. 2003;33:396–400. doi: 10.1038/ng1091. [DOI] [PubMed] [Google Scholar]
- Hemann MT, Zilfou JT, Zhao Z, Burgess DJ, Hannon GJ, Lowe SW. Suppression of tumorigenesis by the p53 target PUMA. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:9333–9338. doi: 10.1073/pnas.0403286101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–27. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- Horvath MM, Wang X, Resnick MA, Bell DA. Divergent evolution of human p53 binding sites: cell cycle versus apoptosis. PLoS Genet. 2007;3:e127. doi: 10.1371/journal.pgen.0030127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houlihan SL, Feng Y. The scaffold protein Nde1 safeguards the brain genome during S phase of early neural progenitor differentiation. Elife. 2014;3:e03297. doi: 10.7554/eLife.03297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itahana Y, Zhang J, Goke J, Vardy LA, Han R, Iwamoto K, Cukuroglu E, Robson P, Pouladi MA, Colman A, et al. Histone modifications and p53 binding poise the p21 promoter for activation in human embryonic stem cells. Sci Rep. 2016;6:28112. doi: 10.1038/srep28112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson-Grusby L, Beard C, Possemato R, Tudor M, Fambrough D, Csankovszki G, Dausman J, Lee P, Wilson C, Lander E, et al. Loss of genomic methylation causes p53-dependent apoptosis and epigenetic deregulation. Nature genetics. 2001;27:31–39. doi: 10.1038/83730. [DOI] [PubMed] [Google Scholar]
- Jiang D, Brady CA, Johnson TM, Lee EY, Park EJ, Scott MP, Attardi LD. Full p53 transcriptional activation potential is dispensable for tumor suppression in diverse lineages. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:17123–17128. doi: 10.1073/pnas.1111245108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Du W, Mancuso A, Wellen KE, Yang X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature. 2013;493:689–693. doi: 10.1038/nature11776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer research. 1991;51:6304–6311. [PubMed] [Google Scholar]
- Kaushansky A, Ye AS, Austin LS, Mikolajczak SA, Vaughan AM, Camargo N, Metzger PG, Douglass AN, MacBeath G, Kappe SH. Suppression of host p53 is critical for Plasmodium liver-stage infection. Cell Rep. 2013;3:630–637. doi: 10.1016/j.celrep.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HR, Roe JS, Lee JE, Cho EJ, Youn HD. p53 regulates glucose metabolism by miR-34a. Biochem Biophys Res Commun. 2013;437:225–231. doi: 10.1016/j.bbrc.2013.06.043. [DOI] [PubMed] [Google Scholar]
- Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657–667. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol. 2015;16:393–405. doi: 10.1038/nrm4007. [DOI] [PubMed] [Google Scholar]
- Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- Kumari R, Kohli S, Das S. p53 regulation upon genotoxic stress: intricacies and complexities. Mol Cell Oncol. 2014;1:e969653. doi: 10.4161/23723548.2014.969653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laberge RM, Awad P, Campisi J, Desprez PY. Epithelial-mesenchymal transition induced by senescent fibroblasts. Cancer Microenviron. 2012;5:39–44. doi: 10.1007/s12307-011-0069-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- Lane DP, Cheok CF, Brown CJ, Madhumalar A, Ghadessy FJ, Verma C. The Mdm2 and p53 genes are conserved in the Arachnids. Cell Cycle. 2010a;9:748–754. doi: 10.4161/cc.9.4.10616. [DOI] [PubMed] [Google Scholar]
- Lane DP, Crawford LV. T antigen is bound to a host protein in SV40-transformed cells. Nature. 1979;278:261–263. doi: 10.1038/278261a0. [DOI] [PubMed] [Google Scholar]
- Lane DP, Madhumalar A, Lee AP, Tay BH, Verma C, Brenner S, Venkatesh B. Conservation of all three p53 family members and Mdm2 and Mdm4 in the cartilaginous fish. Cell Cycle. 2011;10:4272–4279. doi: 10.4161/cc.10.24.18567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane DP, Verma C, Fang CC. The p53 inducing drug dosage may determine quiescence or senescence. Aging (Albany NY) 2010b;2:748. doi: 10.18632/aging.100229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanni JS, Jacks T. Characterization of the p53-dependent postmitotic checkpoint following spindle disruption. Mol Cell Biol. 1998;18:1055–1064. doi: 10.1128/mcb.18.2.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laptenko O, Prives C. Transcriptional regulation by p53: one protein, many possibilities. Cell Death Differ. 2006;13:951–961. doi: 10.1038/sj.cdd.4401916. [DOI] [PubMed] [Google Scholar]
- Leijen S, van Geel RM, Sonke GS, de Jong D, Rosenberg EH, Marchetti S, Pluim D, van Werkhoven E, Rose S, Lee MA, et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients With TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy Within 3 Months. J Clin Oncol. 2016;34:4354–4361. doi: 10.1200/JCO.2016.67.5942. [DOI] [PubMed] [Google Scholar]
- Leonova KI, Brodsky L, Lipchick B, Pal M, Novototskaya L, Chenchik AA, Sen GC, Komarova EA, Gudkov AV. p53 cooperates with DNA methylation and a suicidal interferon response to maintain epigenetic silencing of repeats and noncoding RNAs. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E89–98. doi: 10.1073/pnas.1216922110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ, Ting DT, Greenbaum BD. P53 and the defenses against genome instability caused by transposons and repetitive elements. Bioessays. 2016;38:508–513. doi: 10.1002/bies.201600031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wang S, Chen Y, Lu K, Yin B, Li S, He J, Li C. Identification of two p53 isoforms from Litopenaeus vannamei and their interaction with NF-kappaB to induce distinct immune response. Sci Rep. 2017;7:45821. doi: 10.1038/srep45821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linzer DI, Levine AJ. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell. 1979;17:43–52. doi: 10.1016/0092-8674(79)90293-9. [DOI] [PubMed] [Google Scholar]
- Liu G, Parant JM, Lang G, Chau P, Chavez-Reyes A, El-Naggar AK, Multani A, Chang S, Lozano G. Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat Genet. 2004;36:63–68. doi: 10.1038/ng1282. [DOI] [PubMed] [Google Scholar]
- Liu Y, Chen C, Xu Z, Scuoppo C, Rillahan CD, Gao J, Spitzer B, Bosbach B, Kastenhuber ER, Baslan T, et al. Deletions linked to TP53 loss drive cancer through p53-independent mechanisms. Nature. 2016;531:471–475. doi: 10.1038/nature17157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhang X, Han C, Wan G, Huang X, Ivan C, Jiang D, Rodriguez-Aguayo C, Lopez-Berestein G, Rao PH, et al. TP53 loss creates therapeutic vulnerability in colorectal cancer. Nature. 2015;520:697–701. doi: 10.1038/nature14418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingstone LR, White A, Sprouse J, Livanos E, Jacks T, Tlsty TD. Altered cell cycle arrest and gene amplification potential accompany loss of wild-type p53. Cell. 1992;70:923–935. doi: 10.1016/0092-8674(92)90243-6. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Jacks T, Housman DE, Ruley HE. Abrogation of oncogene-associated apoptosis allows transformation of p53-deficient cells. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:2026–2030. doi: 10.1073/pnas.91.6.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature. 1993;362:847–849. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, Zhao Z, Thapar V, Joyce JA, Krizhanovsky V, et al. Non-cell-autonomous tumor suppression by p53. Cell. 2013;153:449–460. doi: 10.1016/j.cell.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma CX, Cai S, Li S, Ryan CE, Guo Z, Schaiff WT, Lin L, Hoog J, Goiffon RJ, Prat A, et al. Targeting Chk1 in p53-deficient triple-negative breast cancer is therapeutically beneficial in human-in-mouse tumor models. J Clin Invest. 2012;122:1541–1552. doi: 10.1172/JCI58765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejowski J, Li Y, Bosco N, Campbell PJ, de Lange T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell. 2015;163:1641–1654. doi: 10.1016/j.cell.2015.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madenspacher JH, Azzam KM, Gowdy KM, Malcolm KC, Nick JA, Dixon D, Aloor JJ, Draper DW, Guardiola JJ, Shatz M, et al. p53 Integrates host defense and cell fate during bacterial pneumonia. J Exp Med. 2013;210:891–904. doi: 10.1084/jem.20121674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkin D, Li FP, Strong LC, Fraumeni JF, Jr, Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233–1238. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell. 2006;127:1323–1334. doi: 10.1016/j.cell.2006.12.007. [DOI] [PubMed] [Google Scholar]
- McGranahan N, Swanton C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell. 2017;168:613–628. doi: 10.1016/j.cell.2017.01.018. [DOI] [PubMed] [Google Scholar]
- Menendez D, Nguyen TA, Snipe J, Resnick MA. The Cytidine Deaminase APOBEC3 Family Is Subject to Transcriptional Regulation by p53. Mol Cancer Res. 2017;15:735–743. doi: 10.1158/1541-7786.MCR-17-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez S, Camus S, Izpisua Belmonte JC. p53: guardian of reprogramming. Cell Cycle. 2010;9:3887–3891. doi: 10.4161/cc.9.19.13301. [DOI] [PubMed] [Google Scholar]
- Merkle FT, Ghosh S, Kamitaki N, Mitchell J, Avior Y, Mello C, Kashin S, Mekhoubad S, Ilic D, Charlton M, et al. Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature. 2017;545:229–233. doi: 10.1038/nature22312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina RL, Sanfilippo M, Vella V, Pandini G, Vigneri P, Nicolosi ML, Giani F, Vigneri R, Frasca F. Reactivation of p53 mutants by prima-1 [corrected] in thyroid cancer cells. Int J Cancer. 2012;130:2259–2270. doi: 10.1002/ijc.26228. [DOI] [PubMed] [Google Scholar]
- Miyashita T, Krajewski S, Krajewska M, Wang HG, Lin HK, Liebermann DA, Hoffman B, Reed JC. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene. 1994;9:1799–1805. [PubMed] [Google Scholar]
- Mizuno H, Spike BT, Wahl GM, Levine AJ. Inactivation of p53 in breast cancers correlates with stem cell transcriptional signatures. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:22745–22750. doi: 10.1073/pnas.1017001108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morachis JM, Murawsky CM, Emerson BM. Regulation of the p53 transcriptional response by structurally diverse core promoters. Genes Dev. 2010;24:135–147. doi: 10.1101/gad.1856710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morandell S, Yaffe MB. Exploiting synthetic lethal interactions between DNA damage signaling, checkpoint control, and p53 for targeted cancer therapy. Prog Mol Biol Transl Sci. 2012;110:289–314. doi: 10.1016/B978-0-12-387665-2.00011-0. [DOI] [PubMed] [Google Scholar]
- Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, Wongvipat J, Ku SY, Gao D, Cao Z, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science. 2017;355:84–88. doi: 10.1126/science.aah4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller PA, Trinidad AG, Timpson P, Morton JP, Zanivan S, van den Berghe PV, Nixon C, Karim SA, Caswell PT, Noll JE, et al. Mutant p53 enhances MET trafficking and signalling to drive cell scattering and invasion. Oncogene. 2013;32:1252–1265. doi: 10.1038/onc.2012.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell. 2014;25:304–317. doi: 10.1016/j.ccr.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto M, Samper E, Fraga MF, Gonzalez de Buitrago G, Esteller M, Serrano M. The absence of p53 is critical for the induction of apoptosis by 5-aza-2′-deoxycytidine. Oncogene. 2004;23:735–743. doi: 10.1038/sj.onc.1207175. [DOI] [PubMed] [Google Scholar]
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, Jacks T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–860. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008. doi: 10.1101/cshperspect.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivos DJ, Mayo LD. Emerging Non-Canonical Functions and Regulation by p53: p53 and Stemness. Int J Mol Sci. 2016;17 doi: 10.3390/ijms17121982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappas K, Xu J, Zairis S, Resnick-Silverman L, Abate F, Steinbach N, Ozturk S, Saal LH, Su T, Cheung P, et al. p53 Maintains Baseline Expression of Multiple Tumor Suppressor Genes. Mol Cancer Res. 2017;15:1051–1062. doi: 10.1158/1541-7786.MCR-17-0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrales A, Iwakuma T. Targeting Oncogenic Mutant p53 for Cancer Therapy. Front Oncol. 2015;5:288. doi: 10.3389/fonc.2015.00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson BJ, Sanchez Alvarado A. A planarian p53 homolog regulates proliferation and self-renewal in adult stem cell lineages. Development. 2010;137:213–221. doi: 10.1242/dev.044297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister NT, Fomin V, Regunath K, Zhou JY, Zhou W, Silwal-Pandit L, Freed-Pastor WA, Laptenko O, Neo SP, Bargonetti J, et al. Mutant p53 cooperates with the SWI/SNF chromatin remodeling complex to regulate VEGFR2 in breast cancer cells. Genes Dev. 2015;29:1298–1315. doi: 10.1101/gad.263202.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz J, Schreiber-Agus N, Liegeois NJ, Silverman A, Alland L, Chin L, Potes J, Chen K, Orlow I, Lee HW, et al. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell. 1998;92:713–723. doi: 10.1016/s0092-8674(00)81400-2. [DOI] [PubMed] [Google Scholar]
- Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83:993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- Rausch T, Jones DT, Zapatka M, Stutz AM, Zichner T, Weischenfeldt J, Jager N, Remke M, Shih D, Northcott PA, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell. 2012;148:59–71. doi: 10.1016/j.cell.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, Morton JP, Sansom OJ, Zender L, Keyes WM. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017;31:172–183. doi: 10.1101/gad.290635.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth J, Dittmer D, Rea D, Tartaglia J, Paoletti E, Levine AJ. p53 as a target for cancer vaccines: recombinant canarypox virus vectors expressing p53 protect mice against lethal tumor cell challenge. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:4781–4786. doi: 10.1073/pnas.93.10.4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster C, Berger A, Hoelzl MA, Putz EM, Frenzel A, Simma O, Moritz N, Hoelbl A, Kovacic B, Freissmuth M, et al. The cooperating mutation or “second hit” determines the immunologic visibility toward MYC-induced murine lymphomas. Blood. 2011;118:4635–4645. doi: 10.1182/blood-2010-10-313098. [DOI] [PubMed] [Google Scholar]
- Schvartzman JM, Duijf PH, Sotillo R, Coker C, Benezra R. Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell. 2011;19:701–714. doi: 10.1016/j.ccr.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senturk S, Yao Z, Camiolo M, Stiles B, Rathod T, Walsh AM, Nemajerova A, Lazzara MJ, Altorki NK, Krainer A, et al. p53Psi is a transcriptionally inactive p53 isoform able to reprogram cells toward a metastatic-like state. Proc Natl Acad Sci U S A. 2014;111:E3287–3296. doi: 10.1073/pnas.1321640111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Shatz M, Menendez D, Resnick MA. The human TLR innate immune gene family is differentially influenced by DNA stress and p53 status in cancer cells. Cancer research. 2012;72:3948–3957. doi: 10.1158/0008-5472.CAN-11-4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shay JW, Pereira-Smith OM, Wright WE. A role for both RB and p53 in the regulation of human cellular senescence. Exp Cell Res. 1991;196:33–39. doi: 10.1016/0014-4827(91)90453-2. [DOI] [PubMed] [Google Scholar]
- Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91:325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- Shinmura K, Igarashi H, Goto M, Tao H, Yamada H, Matsuura S, Tajima M, Matsuda T, Yamane A, Funai K, et al. Aberrant expression and mutation-inducing activity of AID in human lung cancer. Ann Surg Oncol. 2011;18:2084–2092. doi: 10.1245/s10434-011-1568-8. [DOI] [PubMed] [Google Scholar]
- Shirole NH, Pal D, Kastenhuber ER, Senturk S, Boroda J, Pisterzi P, Miller M, Munoz G, Anderluh M, Ladanyi M, et al. TP53 exon-6 truncating mutations produce separation of function isoforms with pro-tumorigenic functions. Elife. 2016;5 doi: 10.7554/eLife.17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegl C, Prusty BK, Karunakaran K, Wischhusen J, Rudel T. Tumor suppressor p53 alters host cell metabolism to limit Chlamydia trachomatis infection. Cell Rep. 2014;9:918–929. doi: 10.1016/j.celrep.2014.10.004. [DOI] [PubMed] [Google Scholar]
- Soragni A, Janzen DM, Johnson LM, Lindgren AG, Thai-Quynh Nguyen A, Tiourin E, Soriaga AB, Lu J, Jiang L, Faull KF, et al. A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer Cell. 2016;29:90–103. doi: 10.1016/j.ccell.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto M, Raaijmakers JA, Bakker B, Spierings DCJ, Lansdorp PM, Foijer F, Medema RH. p53 Prohibits Propagation of Chromosome Segregation Errors that Produce Structural Aneuploidies. Cell Rep. 2017;19:2423–2431. doi: 10.1016/j.celrep.2017.05.055. [DOI] [PubMed] [Google Scholar]
- Stambolsky P, Weisz L, Shats I, Klein Y, Goldfinger N, Oren M, Rotter V. Regulation of AIF expression by p53. Cell Death Differ. 2006;13:2140–2149. doi: 10.1038/sj.cdd.4401965. [DOI] [PubMed] [Google Scholar]
- Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, Ebert BL. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126:9–16. doi: 10.1182/blood-2015-03-631747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart-Ornstein J, Lahav G. p53 dynamics in response to DNA damage vary across cell lines and are shaped by efficiency of DNA repair and activity of the kinase ATM. Sci Signal. 2017;10 doi: 10.1126/scisignal.aah6671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su D, Wang X, Campbell MR, Song L, Safi A, Crawford GE, Bell DA. Interactions of chromatin context, binding site sequence content, and sequence evolution in stress-induced p53 occupancy and transactivation. PLoS Genet. 2015;11:e1004885. doi: 10.1371/journal.pgen.1004885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulak M, Fong L, Mika K, Chigurupati S, Yon L, Mongan NP, Emes RD, Lynch VJ. TP53 copy number expansion is associated with the evolution of increased body size and an enhanced DNA damage response in elephants. Elife. 2016;5 doi: 10.7554/eLife.11994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan KD, Gallant-Behm CL, Henry RE, Fraikin JL, Espinosa JM. The p53 circuit board. Biochim Biophys Acta. 2012;1825:229–244. doi: 10.1016/j.bbcan.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan KD, Palaniappan VV, Espinosa JM. ATM regulates cell fate choice upon p53 activation by modulating mitochondrial turnover and ROS levels. Cell Cycle. 2015;14:56–63. doi: 10.4161/15384101.2014.973330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sur S, Pagliarini R, Bunz F, Rago C, Diaz LA, Jr, Kinzler KW, Vogelstein B, Papadopoulos N. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc Natl Acad Sci U S A. 2009;106:3964–3969. doi: 10.1073/pnas.0813333106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang YC, Williams BR, Siegel JJ, Amon A. Identification of aneuploidy-selective antiproliferation compounds. Cell. 2011;144:499–512. doi: 10.1016/j.cell.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorburn J, Andrysik Z, Staskiewicz L, Gump J, Maycotte P, Oberst A, Green DR, Espinosa JM, Thorburn A. Autophagy controls the kinetics and extent of mitochondrial apoptosis by regulating PUMA levels. Cell Rep. 2014;7:45–52. doi: 10.1016/j.celrep.2014.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting DT, Lipson D, Paul S, Brannigan BW, Akhavanfard S, Coffman EJ, Contino G, Deshpande V, Iafrate AJ, Letovsky S, et al. Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science. 2011;331:593–596. doi: 10.1126/science.1200801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosoni D, Zecchini S, Coazzoli M, Colaluca I, Mazzarol G, Rubio A, Caccia M, Villa E, Zilian O, Di Fiore PP, et al. The Numb/p53 circuitry couples replicative self-renewal and tumor suppression in mammary epithelial cells. J Cell Biol. 2015;211:845–862. doi: 10.1083/jcb.201505037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovy A, Spiro A, McCarthy R, Shipony Z, Aylon Y, Allton K, Ainbinder E, Furth N, Tanay A, Barton M, et al. p53 is essential for DNA methylation homeostasis in naive embryonic stem cells, and its loss promotes clonal heterogeneity. Genes Dev. 2017;31:959–972. doi: 10.1101/gad.299198.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschaharganeh DF, Bosbach B, Lowe SW. Coordinated Tumor Suppression by Chromosome 8p. Cancer Cell. 2016;29:617–619. doi: 10.1016/j.ccell.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschaharganeh DF, Xue W, Calvisi DF, Evert M, Michurina TV, Dow LE, Banito A, Katz SF, Kastenhuber ER, Weissmueller S, et al. p53-dependent Nestin regulation links tumor suppression to cellular plasticity in liver cancer. Cell. 2014;158:579–592. doi: 10.1016/j.cell.2014.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubio JM, Li Y, Ju YS, Martincorena I, Cooke SL, Tojo M, Gundem G, Pipinikas CP, Zamora J, Raine K, et al. Mobile DNA in cancer. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science. 2014;345:1251343. doi: 10.1126/science.1251343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente LJ, Gray DH, Michalak EM, Pinon-Hofbauer J, Egle A, Scott CL, Janic A, Strasser A. p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Cell Rep. 2013;3:1339–1345. doi: 10.1016/j.celrep.2013.04.012. [DOI] [PubMed] [Google Scholar]
- van der Burg SH, Menon AG, Redeker A, Bonnet MC, Drijfhout JW, Tollenaar RA, van de Velde CJ, Moingeon P, Kuppen PJ, Offringa R, et al. Induction of p53-specific immune responses in colorectal cancer patients receiving a recombinant ALVAC-p53 candidate vaccine. Clin Cancer Res. 2002;8:1019–1027. [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- Venkatanarayan A, Raulji P, Norton W, Chakravarti D, Coarfa C, Su X, Sandur SK, Ramirez MS, Lee J, Kingsley CV, et al. IAPP-driven metabolic reprogramming induces regression of p53-deficient tumours in vivo. Nature. 2015;517:626–630. doi: 10.1038/nature13910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- Vitale I, Senovilla L, Jemaa M, Michaud M, Galluzzi L, Kepp O, Nanty L, Criollo A, Rello-Varona S, Manic G, et al. Multipolar mitosis of tetraploid cells: inhibition by p53 and dependency on Mos. EMBO J. 2010;29:1272–1284. doi: 10.1038/emboj.2010.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitre BD, Cleveland DW. Centrosomes, chromosome instability (CIN) and aneuploidy. Curr Opin Cell Biol. 2012;24:809–815. doi: 10.1016/j.ceb.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Min S, Ma C, Liu Z, Zhang W, Wang Q, Li D, Li Y, Turner S, Han Y, et al. Synthesis of single-crystal-like nanoporous carbon membranes and their application in overall water splitting. Nat Commun. 2017;8:13592. doi: 10.1038/ncomms13592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang PY, Ma W, Park JY, Celi FS, Arena R, Choi JW, Ali QA, Tripodi DJ, Zhuang J, Lago CU, et al. Increased oxidative metabolism in the Li-Fraumeni syndrome. The New England journal of medicine. 2013;368:1027–1032. doi: 10.1056/NEJMoa1214091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Suh YA, Fuller MY, Jackson JG, Xiong S, Terzian T, Quintas-Cardama A, Bankson JA, El-Naggar AK, Lozano G. Restoring expression of wild-type p53 suppresses tumor growth but does not cause tumor regression in mice with a p53 missense mutation. J Clin Invest. 2011;121:893–904. doi: 10.1172/JCI44504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissmueller S, Manchado E, Saborowski M, Morris JPt, Wagenblast E, Davis CA, Moon SH, Pfister NT, Tschaharganeh DF, Kitzing T, et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell. 2014;157:382–394. doi: 10.1016/j.cell.2014.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JS, Petti AA, Miller CA, Fronick CC, O’Laughlin M, Fulton RS, Wilson RK, Baty JD, Duncavage EJ, Tandon B, et al. TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N Engl J Med. 2016;375:2023–2036. doi: 10.1056/NEJMoa1605949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AB, Schumacher B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb Perspect Med. 2016;6 doi: 10.1101/cshperspect.a026070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong TN, Ramsingh G, Young AL, Miller CA, Touma W, Welch JS, Lamprecht TL, Shen D, Hundal J, Fulton RS, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518:552–555. doi: 10.1038/nature13968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylie A, Jones AE, D’Brot A, Lu WJ, Kurtz P, Moran JV, Rakheja D, Chen KS, Hammer RE, Comerford SA, et al. p53 genes function to restrain mobile elements. Genes Dev. 2016;30:64–77. doi: 10.1101/gad.266098.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylie A, Lu WJ, D’Brot A, Buszczak M, Abrams JM. p53 activity is selectively licensed in the Drosophila stem cell compartment. Elife. 2014;3:e01530. doi: 10.7554/eLife.01530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, McMichael JF, Schmidt HK, Yellapantula V, Miller CA, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20:1472–1478. doi: 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Qian J, Hu Y, Wang J, Zhou X, Chen H, Fang JY. Heterogeneity of Li-Fraumeni syndrome links to unequal gain-of-function effects of p53 mutations. Sci Rep. 2014;4:4223. doi: 10.1038/srep04223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Tainsky MA, Bischoff FZ, Strong LC, Wahl GM. Wild-type p53 restores cell cycle control and inhibits gene amplification in cells with mutant p53 alleles. Cell. 1992;70:937–948. doi: 10.1016/0092-8674(92)90244-7. [DOI] [PubMed] [Google Scholar]
- Yonish-Rouach E, Resnitzky D, Lotem J, Sachs L, Kimchi A, Oren M. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature. 1991;352:345–347. doi: 10.1038/352345a0. [DOI] [PubMed] [Google Scholar]
- Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, Tavare S, Arakawa S, Shimizu S, Watt FM, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Vazquez A, Levine AJ, Carpizo DR. Allele-specific p53 mutant reactivation. Cancer Cell. 2012;21:614–625. doi: 10.1016/j.ccr.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun MH, Gates PB, Brockes JP. Regulation of p53 is critical for vertebrate limb regeneration. Proc Natl Acad Sci U S A. 2013;110:17392–17397. doi: 10.1073/pnas.1310519110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeestraten EC, Speetjens FM, Welters MJ, Saadatmand S, Stynenbosch LF, Jongen R, Kapiteijn E, Gelderblom H, Nijman HW, Valentijn AR, et al. Addition of interferon-alpha to the p53-SLP(R) vaccine results in increased production of interferon-gamma in vaccinated colorectal cancer patients: a phase I/II clinical trial. Int J Cancer. 2013;132:1581–1591. doi: 10.1002/ijc.27819. [DOI] [PubMed] [Google Scholar]
- Zhang C, Liu J, Liang Y, Wu R, Zhao Y, Hong X, Lin M, Yu H, Liu L, Levine AJ, et al. Tumour-associated mutant p53 drives the Warburg effect. Nat Commun. 2013;4:2935. doi: 10.1038/ncomms3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725–734. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- Zhu J, Sammons MA, Donahue G, Dou Z, Vedadi M, Getlik M, Barsyte-Lovejoy D, Al-awar R, Katona BW, Shilatifard A, et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature. 2015;525:206–211. doi: 10.1038/nature15251. [DOI] [PMC free article] [PubMed] [Google Scholar]