

Abstract
LC3-associated phagocytosis (LAP) is a novel form of non-canonical autophagy where LC3 (microtubule-associated protein 1A/1B-light chain 3) is conjugated to phagosome membranes using a portion of the canonical autophagy machinery. The impact of LAP to immune regulation is best characterized in professional phagocytes, in particular macrophages, where LAP has instrumental roles in the clearance of extracellular particles including apoptotic cells and pathogens. Binding of dead cells via receptors present on the macrophage surface results in the translocation of the autophagy machinery to the phagosome and ultimately LC3 conjugation. These events promote a rapid form of phagocytosis that produces an “immunologically silent” clearance of the apoptotic cells. Consequences of LAP deficiency include a decreased capacity to clear dying cells and the establishment of a lupus-like autoimmune disease in mice. The ability of LAP to attenuate autoimmunity likely occurs through the dampening of pro-inflammatory signals upon engulfment of dying cells and prevention of autoantigen presentation to other immune cells. However, it remains unclear how LAP shapes both the activation and outcome of the immune response at the molecular level. Herein, we provide a detailed review of LAP and its known roles in the immune response and provide further speculation on the putative mechanisms by which LAP may regulate immune function, perhaps through the metabolic reprogramming and polarization of macrophages.
Graphical Abstract
Introduction
Traditional macro-autophagy (autophagy hereafter) has long been implicated in regulating multiple facets of the immune response including pathogen capture, metabolic regulation, and cellular homeostasis. Autophagy is best described as “self-eating”, and is a critical mechanism that cells utilize under conditions of nutrient deprivation and stress to compensate and preserve homeostasis. Activation of the autophagic pathway leads to both the en masse and selective capture and degradation of cytosolic components in a signaling dependent fashion [1, 2], resulting in energy production derived from the recycling of organelles, proteins, amino acids and other macromolecules. The ability of the autophagic pathway to govern metabolic status is of particular interest for cells of the immune system. Immune cells including lymphocytes and phagocytic cells such as macrophages require a fluidity in their energy architecture as they respond to activation signals [3, 4]. While both quiescent and activated immune cells require ATP, the transition in response to immune perturbation necessitates the appropriation of nutrients into varying pathways to support or direct functional alterations [3]. As expected, major energetic pathways including glycolysis, the tricarboxylic acid (TCA) cycle, the pentose phosphate pathway, fatty acid oxidation, fatty acid synthesis, and amino acid metabolism play vital roles in sculpting the energetic profile of individual immune cells [5]. Autophagy also plays a prominent role in facilitating these primary energetic pathways through the production of metabolic substrates and/or the quality control of organelles. While the autophagy pathway is the only known mechanism to dispose of and recycle intracellular organelles, multiple unique forms of autophagy exist. These divergent forms, deemed non-canonical autophagy, differ in their requirements for specific autophagy proteins (ATGs) required for the canonical autophagy pathway. These studies reveal limitations in our knowledge of how individual components function in either canonical or non-canonical autophagy. Perhaps it should not be surprising that components of a molecular pathway that has existed for aeons might acquire new roles in related processes.
One such form of non-canonical autophagy is present in phagocytic cells, including macrophages, epithelial cells, and endothelial cells. This novel pathway of non-canonical autophagy utilizes components of the canonical autophagy machinery including a subset of the ATGs to conjugate the microtubule-associated protein 1A/1B-light chain 3 (LC3) to phagosome membranes [6]. As such, this process of non-canonical autophagy is called LC3-associated phagocytosis (LAP). Unlike other forms of non-canonical autophagy, LAP is unique in that it is an autophagosome-independent process that results in the formation of LC3-positive phagosomes called LAPosomes. Animals deficient in the LAP pathway have a propensity to develop autoimmunity, particularly systemic lupus erythrematosus (SLE) as discussed below [7]. Additionally, LAP has been shown to have an obligatory function in the efficient clearance of dying cells by macrophages, and likely serves in dampening pro-inflammatory responses. Importantly, this ability for LAP to quench inflammatory responses results in a shaping of the immune response towards anti-inflammation.
In this review we provide an overview of the LAP pathway, signaling to LAP, and the role of LAP in not only the clearance of dying cells and other phagocytic substrates, but further speculate on the potential for LAP to be a mechanism for the metabolic reprogramming of immune cells including macrophages in a variety of both physiological and pathological scenarios.
LAP bridges phagocytosis and autophagy
To understand LAP, we must first distinguish how cells, in particular phagocytes, identify potential cargo to be internalized. Surface receptors at the plasma membrane of phagocytic cells, including macrophages, can be engaged by ligands that are present on target particles. This binding event can result in the activation of the traditional phagocytic pathway, characterized by cytoskeletal rearrangements that mechanically pull the particle into the cell, leading to the formation of a phagosome enclosing the cargo for degradation [8]. In subsequent steps, the phagosome acquires hydrolytic capacity through fusion with lysosomes leading to the destruction of the phagocytosed material [9]. On their route to the lysosome, and depending on the engulfed material, certain phagosomes will recruit members of the autophagic machinery that are responsible for conjugating LC3-family proteins to lipids at the phagosome membrane, the process of LAP (Figure 1). A livery of plasma membrane receptors are involved in cargo sensing and particle uptake leading to LAP. Pattern recognition receptors (PRR) such as toll-like receptors (TLR, in particular, TLR1/2, TLR2/6, and TLR4) able to sense diverse pathogen-associated molecular patterns (PAMPs), immunoglobulin (Ig) receptors that recognize foreign particles opsonized by the immune system, and receptors mediating the clearance of cell corpses such as TIM4 that binds phosphatydilserine (PtdSer) scrambled to the outer leaflet in the plasma membrane upon cell death, are major cell surface receptors that have been shown to participate in cargo sensing during LAP [6, 7, 10–14]. A currently unexplored facet of the LAP mechanism is whether soluble signals, such as “find me” signals, released by dying cells [15] or soluble PAMP moieties from extracellular pathogens are required to prime LAP or to regulate LAP outcome prior to the cell encountering any potential extracellular phagocytic cargo.
Figure 1. Overview of LC3-associated phagocytosis.

Molecules on the surface of cargo are detected by receptors located in the plasma membrane of the phagocyte (such as TIM4 for PtdSer, FcR for Ig-opsonized particles, or TLRs and other PRR for PAMPs). The cytoskeleton rearranges and curves the plasma membrane to produce the phagosome cup. When the plasma membrane closes around the extracellular cargo (not depicted), the phagosome cup is sealed into an early phagosome produced in the cytosol of the phagocyte. Then, the early phagosome matures to become a late phagosome in a process involving membrane modifications and ROS generation. Lysosomes contain hydrolases and by fusion to the late phagosome, these are transferred to the phagosome lumen and the enclosed phagosome cargo is degraded producing nutrients and second messengers. The three panels in the inset depict the molecular machinery in the pathway: the PI3KC3 complex (upper panel) produces PI3P, the NOX2 complex (middle panel) is responsible of the oxidative burst. The two UB-like conjugation systems (lower panel) lipidate LC3 proteins on the phagosome membranes. See text for further details.
While receptors that engage LAP have been identified, it remains unclear how these signal to initiate LAP. In the case of TLRs, the signaling molecules MyD88 and TRIF are dispensable for LAP [10]. LAP induced by zymosan is dependent on TLR2 [10], but may also rely on Dectin-1 signaling to DAPK1 [16]. While LAP is induced by TLR 1/2, TLR2/6, and TLR4 [10], it is not induced by TLR9 ligation [13]. In the latter case, LAP was not required for delivery of IgG-DNA complexes to TLR9 in plasmacytoid dendritic cells, but FcR-mediated LAP was involved in the formation of the acidic TLR9 interferon-signaling compartment [13].
Phagosome maturation, mediated by the fusion of phagosomes to lysosomes, is facilitated by LAP following TLR stimulation [10, 17] and the uptake of dying cells [6, 7]. However, it appears to be dispensable for phagosome maturation following FcR-mediated phagocytosis [18].
PI3K and Rubicon are required for LAP
Following the recognition of phagocytic cargo and after phagosome formation, the phosphotidylinositol 3-kinase complex (PI3KC3) is the first multi-protein complex involved in LAP regulation (Figure 1, upper inset). As such, it is the most upstream complex shared between LAP and the canonical autophagic pathway. The core components of the PI3KC3 complex are VPS34, VPS15 and Beclin1 [19]. VPS34 is the catalytic subunit in the complex that phosphorylates the inositol ring of phosphotidylinositides (PtdIns) at the 3-position to produce phosphatidylinositol 3-phosphate (PtdIns3P) [20]. VPS15 (also known as p150 or PI3KR4) is a pseudokinase that is myrostylated at the amino-terminus and regulates VPS34 activity [21]. Beclin1 (ATG6/VPS30) is a coiled-coil and BH3 domain containing protein that regulates VPS34 lipid kinase function and is involved in autophagic regulation from yeast to mammals [22, 23]. Inhibitors of PI3KC3 function or genetic deficiency in the core components Beclin1 and VPS34 abrogate LAP [6, 10, 17]. In addition to Beclin 1 and VPS15/34, the PI3KC3 complex requires additional subunits to be fully functional. UVRAG and Rubicon are required for LAP whereas ATG14L and AMBRA1 (required for canonical autophagy) are dispensable, arguing that a specific Rubicon-containing complex initiates the LAP pathway [17]. Rubicon is a RUN domain-containing protein that has been previously reported to inhibit PI3P production and LC3-lipidation during canonical autophagy [24, 25]. Rubicon deficiency reduces PI3P generation by VPS34 on phagosome membranes upon LAP induction, influencing downstream events such as reactive oxygen species (ROS) production and LC3-lipidation [17]. Furthermore, Rubicon deficiency in mice phenocopies deficiencies in other proteins involved in LAP regulation, whereas ATG14L, involved in canonical autophagic regulation, does not [7].
PI3KC3 serves as a molecular signaling platform by generating PI3P within the phagosome membrane. However, the means by which the LAP signal is transduced from diverse receptors, such as TLRs, FcRs or TIM4 to a common hub remains elusive. The AMPK-mTOR-ULK1 axis is the primary regulator of PI3KC3 activity and autophagosome initiation during canonical autophagy induced by starvation [26–28]. Inhibition of mTOR using rapamycin or starvation does not impact LAP, and deficiency of either the serine/threonine kinase ULK1, or its accessory subunits FIP200 or ATG13 does not disrupt LAP function [6, 13, 17, 29]. The absence of mTOR-ULK1-ATG13 mediated regulation of PI3KC3 activity during LAP might be explained by the necessity for maintaining LAP function, which is important for pathogen clearance and elimination of cellular debris, irrespective of nutritional status within the phagocyte.
Some evidence suggests that signaling events related to phagosome cup formation, cargo engulfment, and early phagosome maturation might regulate PI3KC3 activation, and plausibly, LAP initiation. For instance, Dynamin is a GTPase involved in phagosome sealing, and is required for VPS34 recruitment to phagosome membranes in C. elegans in a process mediated by the small GTPase Rab5 [30–32]. Rab5 activity has long been correlated to VPS34 function and PI3P generation on phagosome membranes [33–36]. Additionally, upon engulfment of yeast particles by phagocytes or in response to A. fumigatus monocytic infection, the kinase Syk regulates downstream LAP signaling events such as ROS production and LC3-lipidation [12, 37] [11]. SRC family kinases are also activated early during phagosome maturation [38]. Like Syk, Src kinases are involved in ROS production and LC3-lipidation during phagocytosis [11]. Moreover, PI3KC3 subunits are post-translationally modified upon autophagic induction [39]. It is alluring to speculate that signal transduction from upstream receptors may involve protein kinases including Src and Syk, that in turn may regulate the PI3KC3 complex upon LAP initiation through specific post-translation modifications. Inositide signaling can regulate early phagocytic events [40, 41] and diacyl-glycerol, produced by phospholipase C (PLC) from PI(4,5)P2 after phagosome closure, regulates ROS production and phagosome LC3 conjugation in response to Listeria monocytogenes [42] and Legionella dumoffii [43] infections. Whether these signaling moieties participate in all LAP-mediated processes is currently unknown. Similarly, their functional relationships within the LAP machinery and the order of events in the putative signaling pathway remain unaddressed questions.
An often-overlooked disparity between LAP and canonical autophagy is the timing of PI3P generation. During LAP, VPS34 activity produces PI3P in the outer leaflet of the phagosome membrane once the cargo has been engulfed and the phagosome fully sealed [10, 13, 36, 44]. Conversely, upon autophagic stimulation, PI3P is generated in certain portions of the endoplasmic reticulum in one of the first steps of canonical autophagy [45, 46]. In LAP, the timing of PI3P generation suggests that early phagosome events such as cargo recognition, phagosome cup formation, membrane elongation and cargo engulfment are processes most likely independent of VPS34 activity or PI3P production. In this regard, during the phagocytic clearance of apoptotic bodies in C. elegans, an event that involves LAP [10], the overexpression of a PI3P phosphatase that dephosphorylates PI3P to PIP, abrogates phagosome maturation without affecting phagosome formation [32]. On the other hand, during canonical autophagy, the VPS34 activity helps in localizing the phagophore to the precise site where it will elongate [45]. Overexpression of PI3P phosphatases abolishes the complete assembly of the autophagosome in both mammalian cells [47, 48] and C. elegans [49], indicating that PI3P is important for autophagosome closure and elongation. Interestingly, despite PI3P being generated prior to autophagosome sealing and the observation that PI3P may freely diffuse between the inner and the outer membranes; the majority of PI3P resides in the outer leaflet of the autophagosome following closure [50]. This localization is consistent with what occurs during LAP, suggesting a common function for PI3P following vesicle closure.
PI3P serves as a platform for the recruitment of effector proteins into membranes where it is generated. While many effector proteins use PX or FYVE domains to bind to PI3P [51], the primary PI3P effectors during canonical autophagy are the WIPI proteins that utilize an alternative lipid binding domain, the WD β-propeller PROPIN [52]. In early autophagosome structures WIPI2 simultaneous binds to PI3P generated by VPS34 and to ATG16L1 [53]. Since ATG16L labels the membrane site where LC3-lipidation will take place [54], WIPI2 links two important signaling platforms: the PI3KC3 and the lipidation machinery. In contrast, WIPI2 is dispensable for LAP [17], while LC3-lipidation depends on PI3P generated on the phagosome [6, 10, 17]. Interestingly, EEA1, a FYVE domain containing protein that participates in phagosome maturation [55], and the FYVE-containing protein FYCO1 have been shown to negatively regulate ROS production during LAP [56]. Whether these or other PI3P-binding proteins involved in autophagy regulation bridge the two signaling platforms upon LAP induction is an idea that requires further attention.
NOX2, ROS and LC3-lipidation in LAP
In addition to PI3P, we likewise have eluded to the importance of ROS in LAP activation. The nicotamide adenine dinucleotide phosphate (NADPH) oxidase-2 (NOX2) generates ROS during phagosome maturation in macrophages [57, 58] and both NOX2 function and ROS are indispensable for LAP [17, 42, 59]. NOX2 is a multiprotein complex comprised of both membrane-bound and cytosolic subunits (Figure 1, middle inset). The membrane-bound cytochrome b-245 is a heterodimer of two transmembrane proteins: gp91phox that has electron transferase activity from NADPH to oxygen (O2 + e− -> O2−) and p22phox that facilitates the reaction and serves as a docking site for the cytosolic subunits. The regulatory subunits p67phox, p47phox and p40phox aid in complex assembly with the small GTPase Rac1 and are required to fully trigger the ROS generating activity of NOX2 [60, 61]. The p67phox subunit participates in the catalysis reaction by promoting electron transfer from NADPH and FAD. Interestingly, p40phox has a PX-domain with the ability to bind PI3P [62, 63], however this binding site is masked under resting conditions. In early phagocytic events, inositide signaling activates PKC that in turn phosphorylates p47hox, exposing the PX-domain in p40phox. The licensed p40phox protein is then able to interact with PI3P generated in phagosome membranes [64–66] and brings together the remaining regulatory subunits. After the translocation of the regulatory subunit, the complex is rearranged to achieve full oxidative capacity [60]. NOX2 produces a superoxide anion (O2−) that is rapidly dismutated to hydrogen peroxide in the phagosome lumen (O2− + e− + 2H+ -> H2O2), this and other forms of ROS can cause oxidative damage, particularly relevant to the killing of certain bacteria [67]. To offset this form of immune response, some pathogens have been shown to express proteins that abrogate NOX2 assembly or function [68]. In addition to ROS production, NOX2 contributes to the alkalinization of phagosome and phagolysosomal lumens through the consumption of protons to dismutate superoxide anions. This event limits the activity of acidic lysosomal proteases. This event promotes antigen presentation to adaptive immune cells [69]. Furthermore, concomitantly to ROS production, the electron transferase activity of NOX2 generates an electric potential across the phagosome membrane that is buffered through ion exchange [70].
As mentioned above, NOX2 activity and ROS production are important for LC3-lipidation during LAP [17, 42, 59]. However, the signaling events that link NOX2 activity to downstream events such as recruitment of the conjugation machinery or LC3-lipidation remains obscure. Oxidation of lipids at the phagosome membrane has been implicated as a putative signaling modality in LAP [17], but the molecular mechanism has yet to be characterized. Alternatively, ROS produced in the phagosome lumen can diffuse to the cytosol where it may activate cytosolic enzymes required for LAP. ROS are also generated during canonical autophagy following induction by different stimuli as reported previously [71], and interestingly, the delipidation activity of ATG4 is inhibited by ROS [72], extending the presence of LC3 proteins in the autophagosomal membrane and favoring autophagy.
LAP requires the autophagy conjugation system
While it remains unclear how the conjugation machinery is recruited to the phagosome membrane, the two ubiquitin-like (UB-like) conjugation systems required for LC3 conjugation during canonical autophagy [73] are also involved in LAP (Figure 1, lower inset) [6, 10, 13, 17]. Briefly, in the first UB-like conjugation system, the exposed terminal glycine in ATG12 is covalently linked to an internal lysine in ATG5 through isopeptide bonding [74]. ATG7 and ATG10 function as the activating (E1) and the conjugating enzymes (E2) respectively [75, 76]. Under physiological conditions, most ATG5 and ATG12 proteins within the cell are conjugated in the form of a stable heterodimer that cannot readily be de-conjugated [74]. Further, the majority of ATG5-ATG12 conjugates are bound to ATG16L to produce the ATG16L complex [77]. In the second UB-like system, a glycine in the carboxyl-terminal tail of the UB-like proteins from the LC3 family are covalently conjugated to the polar head of a phosphatidylethanolamine lipid molecule within an (auto)phagosome membrane [78, 79]. Using a similar molecular mechanism [73], ATG7 again possesses the E1 activity for LC3 whereas ATG3 participates as the E2 enzyme in this system [76, 80]. Finally, LC3 is transferred from the ATG3-LC3 heterodimer to the lipid, using the E3 ligase activity of the ATG16L complex [81]. An important prerequisite for LC3 conjugation is the exposure of a glycine residue in the carboxy-terminal tail of LC3 [82]. ATG4 comprises a family of cysteine proteases that cleave LC3 proteins releasing a small oligopeptide (or a single amino acid), thus exposing the glycine residue required for lipid conjugation [83].
It is noteworthy that in stark contrast to the first system, some functional redundancy occurs in the second UB-like system. In the mouse genome four ATG4 proteases are encoded (ATG4A-D) that can cleave with low specificity, and at least five LC3 paralogs (LC3A, LC3B, GABARAP, GABARAPL1 and GABARAPL2) have been identified. Additionally, the E3 ligase activity is encoded in two proteins (ATG16L1 and ATG16L2). Most of these proteins are ubiquitously expressed in different tissues and the function of this redundancy remains unclear. One can speculate that a different combination of proteins might be required for specific autophagic processes (basal autophagy, nutritional autophagy, mitophagy, xenophagy, LAP) or might be differentially localized within the cell, generating spatially separated mechanistic pools. Importantly, upon LAP induction, several LC3 paralogs have been identified in phagosome membranes, but deficiency in either ATG16L1 or ATG4B seems to completely block LAP [17].
The complicated multistep mechanism described above has the sole purpose of LC3-lipidation and despite it being well conserved from yeast to mammals, the function of lipidated LC3 proteins in canonical autophagy is far from fully understood. Three main functions have been proposed for LC3 proteins: these include mediating cargo selection, favoring autophagosome closure, and facilitating membrane fusion. LC3 proteins are involved in autophagic cargo selection as certain cargo adaptor proteins including p62, NRB1, NDP52 and OPTN can simultaneously bind to LC3 proteins and ubiquitinated cargoes during the elongation and closure of the autophagosome [84] which enables cargo encapsulation. Different pathogens have been reported to induce LC3-lipidation when the bacteria still reside in single membrane vesicles during LAP or processes resembling LAP. However, due to the fact that LC3 is lipidated at the phagosome membrane after phagosome closure [10] and that the bacteria enclosed in such phagosomes are typically ubiquitin-negative, it seems improbable that cargo adaptors function during LAP.
Timing is everything
It is important to bear in mind, as exemplified above, the differences in the timing of lipidated LC3 production between LAP and canonical autophagy, and the functions derived from this divergence. LC3 is lipidated when the phagosome is fully sealed [10], and the lipidation machinery is most likely attached to the membrane after phagosome closure. Conversely, LC3 is conjugated during autophagosome membrane elongation [78, 79] and the ATG16L complex is released from autophagosome precursors just after or at approximately the same time as autophagosome closure [77]. Interestingly, the overexpression of a non-functional ATG4 mutant protein lacking the ability to process LC3 proteins precludes autophagosome closure [85], suggesting that lipidated LC3 proteins might be involved in autophagosome membrane elongation and/or closure. Nevertheless, LC3 lingers in the outer autophagosome membrane after autophagosome sealing, at least until autophagosome-lysosome fusion [86], pointing to additional functions. Interestingly, lipidated LC3 proteins have also been shown to be involved in hemifusion between membranes in vitro [87]. Cytosolic expression of the Legionella effector protein RavZ that specifically cleaves LC3-II from membranes [88], delayed the acquisition of the lysosomal marker LAMP1 during LAP [17], arguing for a role of lipidated LC3 during phagosome-lysosome fusion.
LAP promotes differential outcomes during immune activation
Upon fusion of the phagosome with the lysosome, luminal acidic hydrolases degrade the engulfed material and transmembrane pumps in the lysosomal membranes aid in the recycling of sugars, amino acids, lipids and nucleotides to replenish cellular stores. These surplus molecules generated from extracellular material are reminiscent of amoeboid nutrition and can potentially be used as building blocks and energy sources within the phagocyte. Therefore, while LAP most likely impacts cellular metabolism, how the phagocyte responds to the extra nutrient supplies has usually been overlooked. Furthermore, the ultimate goal of phagocytosis is the degradation of extracellular particles, but the final outcome in the phagocyte greatly varies in a cargo-dependent manner. It is intriguing to speculate that this control of phagocytosis and degradation of pathogenic cargo generates metabolites or triggers signaling pathways that reshape metabolic architecture, and ultimately, regulate the immune response. LAP-deficient mice develop an auto-inflammatory, lupus-like syndrome with aging, which can be recapitulated by chronic exposure to apoptotic cells [7]. Also, LAP-deficient mice fail to efficiently clear Aspergillus fumigatus infection [17, 89]. Thus, it is clear that defects in the LAP mechanism can have a breadth of immunological consequences.
A role for LAP in antigen presentation and pathogen clearance
Activation of the C-type lectin receptor Dectin-1 or TLR2 leads to recruitment of LC3 to phagosomal membranes [37, 90, 91]. It has been well characterized that activation of Dectin-1 occurs in response to fungal infections and fungal-derived antigens. This mechanism of LAP activation promotes the sustained presentation of antigen by MHC-II [37]. Further evidence supporting a function for LAP in antigen presentation stems from work performed in human macrophages and dendritic cells, showing TLR2 activation leads to LAP engagement and preservation of antigen presentation through the sequestration and binding of “LAPosomes” to TLR containing endosomes [90, 92]. Additionally, this process was shown to be ROS and NOX2 dependent, further confirming LAP activation and not another form of non-canonical autophagy. While these studies were conducted in macrophages and bone-marrow derived dendritic cells, the findings are consistent with, and may in part explain why LAP-deficient mice have a reduced capacity for clearing fungal infections.
Moreover, it has long been established that selective autophagy can participate in the clearance of microbes through a process known as xenophagy [93]. Likewise, it has been proposed that LAP can similarly function in the clearance of pathogens not just through altering antigen presentation and immune activation as described above, but through direct engulfment and trafficking for lysosomal degradation and by virtue of ROS production [59, 94, 95]. As described previously, NOX2 and ROS are essential for LAP and are likewise central components for the ROS-dependent killing of phagocytosed pathogens [59]. Interestingly, other NOX enzymes can promote selective xenophagy and ROS mediated killing of pathogens such as Salmonella Typhimurium in non-phagocytic cell types, suggesting that the role and function of NOX2 in LAP is restricted to professional phagocytes, while other NOX/autophagy dependent mechanisms exist for the clearance of pathogens in divergent cell types [59].
LAP in efferocytosis
The connection of LAP with immune regulation is even more evident in the elimination of apoptotic cells by macrophages, a process known as efferocytosis. At any given time, professional phagocytes, specifically macrophages, efficiently patrol and eliminate dead cells to prevent the release of inflammatory signals by cell corpses, and thus protect tissue homeostasis. Although efferocytosis is generally defined as an ‘immunologically silent’ process, the engulfment of apoptotic cells orchestrates a potent regulatory mechanism that actively induces the production of anti-inflammatory cytokines (IL-10, TGF-β) and inhibits the production of inflammatory cytokines and chemokines such as IL-6, IL-1β, CCL-2, CCL-3, CCL-4 and CXCL-10 [96–100]. Ultimately, efficient engulfment and elimination of cellular debris prevents the development of autoimmune syndromes, as accumulation of dead cells in the circulation and in the lymph nodes as well as serum levels of antibodies generated against self-antigens are pathological signatures of patients with SLE [101]. LAP mediates immunoregulation in response to apoptotic cells, and mice that are deficient for LAP, but not canonical autophagy, accumulate apoptotic bodies in their tissues and within the cytosol of phagocytic cells. Importantly, a polymorphism in ATG5 was identified in lupus patients by GWAS, suggesting a putative role for LAP in preventing autoimmunity in humans [102, 103]. Additionally, patients with chronic granulomatosis, a syndrome caused by mutations in NOX2, a major component of LAP signaling cascade detailed above, have a high occurrence of lupus [104–106].
Metabolic reprograming in macrophage functional polarization
The link between LAP and clearance of apoptotic cells by efferocytosis hints at a deeper involvement for LAP in the regulation of macrophage metabolism and polarization. Macrophage functions are defined in response to microenvironmental cues, such as recognition of dying cells, that drive the acquisition of a spectrum of polarization states that find their extremes in either a pro-inflammatory, pathogen eliminating direction (M1, classically activated) or antiinflammatory, immunosuppressive and wound-healing response (M2, alternatively activated). M1 prototypic macrophages are induced by Th1 inflammatory cytokines (IFNγ, TNFα), damage-associated molecules (e.g., ATP, alarmins, HMGB1) and pathogen-associated molecular patterns (e.g., LPS), and are characterized by secretion of inflammatory cytokines including IL-1β, TNFα, IL-6, IL-12 and production of antimicrobial nitric oxide (NO). M1 macrophages also express higher levels of MHCII and co-stimulatory CD80 and CD86 on their surface. In contrast, Th2 cytokines (IL-4, IL-13) activate M2 macrophages, generally characterized by secretion of anti-inflammatory IL-10 and TGF-β, low expression of inflammatory cytokines, and high expression of mannose and galactose C-type lectins (dectin-1, CD206, CD301) and arginase-1 activity [107]. Since efferocytosis is typically immunologically silent, an anti-inflammatory M2-like response is observed in response to apoptotic cells. However, mice that are LAP-deficient tend to have a pro-inflammatory response when challenged with dead cells, leading to autoinflammation as detailed above.
To sustain a heterogeneous functional repertoire that encompasses various activation states, it is important that a metabolic plasticity exists and allows for the establishment of a renewed homeostatic state, providing energy and catabolites for macromolecule biosynthesis. Metabolic reprogramming not only supports the energetic demands of different macrophage polarization phenotypes, but also dictates the signaling pathways that confer functional specificity to these cells. For instance, the metabolism of arginine, a hallmark distinction between M1 and M2 macrophage subsets, is essentially a metabolic mechanism of functional polarization toward pathogen elimination. M1 macrophages convert arginine into microbicide NO and citrulline through induction of nitric oxide synthase (iNOS) while in M2 macrophages arginine is metabolized by arginase-1 (Arg-1) into ornithine and urea [108–110].
Overall, inflammatory macrophages are characterized by high glycolytic rates, an increased pentose phosphate pathway (PPP), disturbed TCA cycle and impaired mitochondrial oxidative phosphorylation (OXPHOS). Anti-inflammatory M2 macrophages are also energetically demanding cells, but require an efficient electron transport and oxidative phosphorylation to supply this demand and dictate M2 polarization [111]. Finally, metabolic circuits that control the utilization and synthesis of lipids and amino acids to sustain these metabolic pathways also participate in regulation of the immune response and macrophage function, as discussed below.
Implications of LAP in macrophage immunometabolism
Upon engulfment of apoptotic cells, macrophages upregulate fatty acid oxidation, raising the possibility that metabolic changes contribute to the immune response to efferocytosis described above [112]. Whether LAP-dependent immunoregulation in macrophages upon engulfment and degradation of apoptotic cells is ultimately driven by metabolic reprogramming is still a matter of speculation, but some interesting studies suggest a possible relationship. Efferocytosis induces IL-4 production in response to apoptotic cells and this pathway is defective in macrophages of NOX2 defective mice [113]. The tryptophan (Trp)-metabolizing enzyme indoleamine 2,3 dioxygenase 1 (IDO1) is rapidly induced by phagocytosis of apoptotic cells, leading to consumption of tryptophan and activation of general controlled non-repressed 2 (GCN2). The activity of GCN2 is important for the production of IL-10 by macrophages following engulfment of apoptotic cells, and lupus-prone mice lacking myeloid GCN2 function develop an age-related autoimmune syndrome [114].
Another possible association between LAP and macrophage activation upon engulfment of apoptotic cells is the role of members of the nuclear receptor superfamily of transcription factors that regulate lipid homeostasis, including peroxisome-proliferator-activated receptors (PPAR isoforms γ and β/δ) and liver X receptor (LXR). Binding of ligands such as fatty acids and oxysterols as well as recruitment of co-factors induce the expression of a plethora of genes involved in lipid metabolism [115–117]. Different studies have shown that PPAR and LXR are activated in macrophages in response to apoptotic cells and express genes such as the cholesterol transporter ABCA1 and phospholipid transporter, phospholipid transfer protein 1 (PLTP1). Moreover, PPAR and LXR activity upregulate the expression of phagocytic receptors (Mer, CD36 and Axl) and opsonins (C1qb, Gas6, MFG-E8,) that support phagocytic activity and continuous engulfment of apoptotic bodies [118, 119]. Perhaps the most interesting feature of PPAR and LXR activation in response to efferocytosis that points towards a putative role in macrophage function is the induction of anti-inflammatory IL-10, and prevention of inflammatory cytokine production [120–122]. A mechanistic link between nuclear receptor activation in response to apoptotic cells and cytokine production is has not been established, however. While PPAR and LXR heterodimers with RXR are described to cis-repress NF-κB transcription, no evidence of such mechanisms operating during efferocytosis have been described. Importantly, genetic ablation of PPARγ, PPARδ, or LXR impairs phagocytic clearance and renders mice susceptible to age-related development of autoimmune disease, in a manner similar to LAP-deficient mice [120–122].
The LAP-dependent engulfment and clearance of apoptotic bodies raises other important questions: how is macrophage homeostasis affected by the extra content of engulfed cells? Do derived metabolites from the degradation of dying cells alter metabolic pathways of the macrophages? And do these derived metabolites, such as lipids, directly regulate immune signaling pathways? These questions further lead to a broader theme regarding the putative role of macrophage LAP in response to other stimuli, not only apoptotic cells and pathogenic perturbations such as bacterial infection.
LAP as a potential regulator of vascular inflammation
It is well established that macrophages constitute a large portion of immune cells found in atherosclerotic plaques. Additionally, macrophage specific depletion of Atg5 in an atherosclerotic mouse model increases atherosclerotic lesion size, at least in part due to an increase in infammasome activation, a defect in cholesterol efflux, an increase in ER/oxidative stress and a defect in efferocytosis [123–125]. Since Atg5 is a known regulator of both LAP and canonical autophagy, we can speculate that LAP-deficiency may contribute to the increase in atherosclerotic lesions and associated symptoms. Moreover, non-resolving inflammation during atherosclerosis progression is partly attributed to a defect in the clearance of dead cells by efferocytosis [126], correlating to what is observed in the LAP-deficient mouse model that develops SLE, described above.
Consistent with this notion, patients with SLE are at a high risk for prematurely developing an accelerated form of atherosclerosis [127, 128]. Since defects in LAP are involved in the pathogenesis of an SLE-phenotype, we can hypothesize that LAP-deficiency might accelerate the progression of atherosclerosis at different stages of the process. Atherosclerosis is defined as a non-resolving chronic inflammatory disease that progressively leads to functional alterations in medium and large arteries. From the traditional view of a passive lipid accumulation in the arterial wall, atherosclerosis has emerged in the last two decades as a more complex process where both innate and adaptive immunity play key roles in the pathophysiology of the disease [129]. Most atherosclerotic lesions remain silent through life, but in some individuals an erosion or a rupture of the plaque occurs, leading to two acute thrombotic complications, coronary artery disease and stroke [130].
LAP may function in vascular macrophages to clear modified lipids and lipoproteins in an immunologically silent manner. During acute or chronic hyperlipidemia, cholesterol transported on low-density lipoproteins (LDL) infiltrate the arterial wall, are trapped in the intimal extracellular matrix, and become more susceptible to diverse modifications such as oxidation and proteolysis [131]. Retention of modified lipids and lipoproteins synergized by a disturbed blood flow in branching points of the arterial tree activates endothelial cells via an upregulation of leukocyte adhesion molecules such as E-selectin, VCAM-1 and chemokines such as CCL2 and CX3XCL1, which attract monocytes, T-cells, and dendritic cells to the sub-endothelial space of the arterial wall [132, 133]. LDL retention and leukocyte activation/transmigration are the first hallmarks of vascular inflammation. In a LAP-deficient setting, the clearance of modified lipids and lipoproteins could lead to the establishment of a pro-inflammatory response, much like what is observed when LAP-deficient macrophages are exposed to dying cells [6].
During early atherogenesis, bone marrow-derived monocytes are recruited and differentiate into macrophages in response to macrophage colony stimulating factor produced by endothelial cells or smooth muscle cells [134, 135]. Genetic lineage-tracing experiments demonstrated that the subsets of arterial macrophages are more heterogeneous than previously thought, both in terms of origin and activation. Embryonic CX3CR1+ precursors mainly from the yolk sac and to a less extent from the fetal liver flt3+ cells, give rise to resident arterial macrophages which proliferate postnatally, sustaining the homeostasis of arterial macrophage pools [136]. Using parabiosis techniques, accumulation of macrophages in the vascular wall during atherosclerotic development have been shown to primarily arise from macrophage proliferation [137]. Even though exogenous Th2-cytokines such as IL4 have been implicated in tissue resident macrophage self-renewal [138], the mechanisms by which resident foam cell macrophages proliferate is not fully understood. Deficiency in autophagy markers such as p62, Beclin-1 or Ambra-1 stimulates cell migration and proliferation in MEFs and in different human carcinoma cells [138–141]. However, it is largely unknown whether the autophagic machinery shared by LAP can also be involved in regulating differentiation and proliferation of resident macrophages.
Even though it is unclear if LAP is able to impact the expansion of resident macrophages, it has been well characterized that both resident and bone-marrow derived macrophages can ingest modified lipoproteins and lipids such as oxidized LDL and cholesterol crystals through binding to scavenger receptors and ultimately become foam cells [142, 143]. Whether arterial macrophage foam cells give rise to a more pro-inflammatory response which would be consistent with a deficiency in LAP, is still under debate, since genetic deletion of the scavenger receptors in atherosclerotic mice models showed conflicting results regarding the progression of the disease [144]. A dynamic view of vascular inflammation has been proposed to include a crosstalk between the hypoxic environment, macrophage foam cells, and T-cells (T-reg and Th17), which are known to mutually engage vascular macrophages with a high degree of plasticity [145]. LAP-deficiency is likely to be found in macrophages associated with advanced atherosclerotic plaques, which are characterized by the presence of a large necrotic core and an accumulation of dead cells in the vascular wall [130]. If LAP governs the clearance of these dead cells, then the loss of LAP may account for the finding that key markers of efferocytosis such as Tg2, MER-TK, MFG-E8, C1q, and Cdkn2b are decreased in a mouse model of atherosclerosis [146]. An increase in the size of the necrotic core has also been observed in atherosclerotic mice with macrophage-specific depletion of Atg5 [123]. Whether LAP, which again also requires ATG5, might represent a protective mechanism by which an escalation in vascular inflammation could be dampened, remains an open question.
With heightened risk for the development of atherosclerosis in SLE patients and the recent studies linking accumulation of apoptotic bodies as a contributing factor to atherosclerotic lesion establishment, it is tempting to further speculate on the clinical importance of LAP. While many unanswered questions remain regarding when, how, and why LAP is activated, it has now been established that LAP tends to promote an anti-inflammatory response following activation with a variety of stimuli. The SLE disease that develops in LAP-deficient mice as they age is clearly a result of apoptotic corpse accumulation and inflammatory activation. Moreover, this autoimmune phenotype has the potential to have global impact, as is observed in SLE patients with atherosclerosis. Interestingly, SLE patients are also at a 15% increased risk of developing cancer and an even higher incidence of type-II diabetes, possibly linked to the increased adiposity and adipose macrophage infiltration as described below. Even though many studies have evaluated the secondary risks stemming from SLE, studies directed at the underlying molecular mechanisms responsible for these correlations are less prevalent.
LAP in adipose tissue macrophages and insulin resistance
SLE patients in particular are at a high propensity to have increased visceral adiposity and epicardial fat accumulation [147]. Since LAP promotes anti-inflammation, it is possible that adipose-infiltrating macrophages, which display profound pro-inflammatory characteristics, have either a restriction or deficiency in LAP. As is the case with generalized obesity, establishment of this inflammatory milieu in the adipose tissue has been directly linked with insulin resistance and type-II diabetes [148]. This may in part explain the increased risk SLE patients face with regards to developing insulin resistance and cardiac dysfunction.
While the role of adipose infiltrating macrophages and the correlation to insulin resistance has been well studied over the past few years, the relationship is still inadequately understood. A primary function for adipose tissue macrophages, specifically in the context of fat expansion and obesity, is to promote the clearance of dead and dying adipocytes through efferocytosis. However, unlike the canonical efferocytosis of dying cells that is described previously as being LAP-dependent and immunologically silent, the process in adipose tissue is categorized by pro-inflammatory cytokine release [149], suggesting a possible impairment in LAP. It is generally accepted that the presence of these pro-inflammatory macrophages around dead adipocytes reflects the inability of the macrophages to efficiently clear the adipocyte corpses, producing a pro-inflammatory response similar to what is described for macrophages in atherosclerosis mentioned above [149, 150]. Likewise, it is tempting to speculate that a defect in the LAP pathway may be a causative factor in not only adipose tissue expansion, but also insulin resistance as a result of the inflammatory response that is observed. Therefore, it is not out of context to imagine that macrophages with an intact and functioning LAP pathway may be suppressive against adipose expansion and ultimately obesity. These however are hypotheses that will required further investigation to fully delineate, yet it may be prudent to simply evaluate LAP in adipose tissue macrophages from lean, obese, and SLE patients or animal models in an effort to identify any correlation that may exist between the LAP pathway and clinical metabolic pathology.
Conclusions and Outlook
The functions of LAP in inflammation and autoimmunity, taken in combination with the important role of LAP in mediating bacterial clearance and antifungal host defense, reinforce the broad importance of this form of non-canonical autophagy to not only cell, but organismal homeostasis. As shown in Figure 2, the capacity for cells to engage LAP is a critical determinant of autoimmunity in response to apoptotic cells. In this model, a perturbation of LAP is likely a contributing factor to the establishment of SLE, atherosclerosis, visceral adiposity, and insulin resistance as discussed above. More importantly, it is evident that the function of LAP can be both “good” or “bad” depending on the context in which it is activated. As observed in autoimmunity, LAP activation dampens the inflammatory response and is beneficial (Figure 2), yet we can hypothesize that LAP activation may promote the establishment of the tumor microenvironment, which is characterized by anti-inflammatory M2-like macrophages [151]. It is important to note that most of the current work has been directed at investigating LAP in murine macrophages. The hyperinflammatory response that occurs when LAP is abrogated could be unique to the loss of LAP in macrophages specifically. Modulation of LAP in other cell types may produce distinct outcomes. As detailed above, in human macrophages and both conventional and plasmocytoid dendritic cells, “LAPosomes” seem to be preferentially stabilized and can fuse to TLR containing endosomes [90, 152]. These differing fates for LAPosomes across cell types may impact physiological outcomes including inflammatory responses and antigen presentation as detailed previously. Additionally, it is important to bear in mind that activation of canonical autophagy can suppress inflammasome activation [153]. Therefore, future studies investigating LAP and the inflammatory response should take caution in demarcating the contributions of not only LAP versus canonical autophagy, but cell specificity as well.
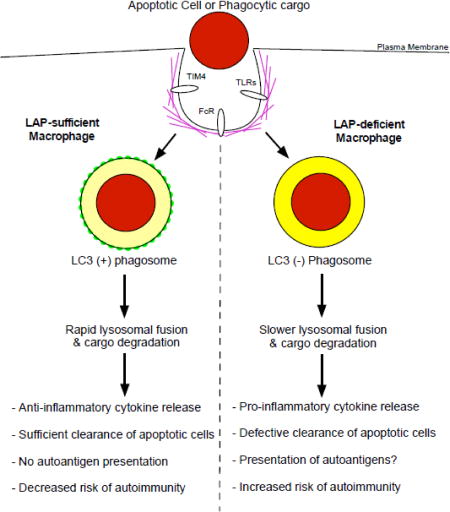
Figure 2. Physiological outcomes of LC3-associated phagocytosis in autoimmunity.

Macrophages that are sufficient in the LAP pathway are able to efficiently clear apoptotic cells and pathogens. There is a rapid lysosomal fusion and degradation of target cargo that occurs. This timing results in the production of anti-inflammatory cytokines and dampening of the immune response, leading to a decreased risk of autoimmunity. In contrast, macrophages deficient in LAP have an abrogation in the clearance of apoptotic cells. Moreover, the reduced rate of phagosome-lysosomal fusion results in the establishment of a more pro-inflammatory cytokine profile. Additionally, in the absence of LAP there is likely an increased amount of autoantigen presentation. Taken together, these factors greatly increase the risk for developing autoimmunity, as is evident in the establishment of SLE in LAP-deficient mice.
Due to the discrepancies in LAP function across tissues and settings, it will be of significant importance to identify the upstream signaling components that delineate activation of LAP versus traditional phagocytosis. Moreover, the differentiating signals between canonical autophagy and LAP will be of major interest, as the two pathways share a unique intimacy yet are distinct from one another. The primary void in the pathway to date is the absence of a factor controlling signal transduction following surface receptor engagement to Rubicon and the required PI3KC3 complex detailed herein. It would be fascinating if the identification of the signaling effector(s) in this pathway leads to the development of novel therapeutics whereby we can modulate LAP activity in a targeted fashion. Rubicon may prove a viable target for inhibition of LAP, as it is absolutely required for LAP function. However, Rubicon is not sufficient to induce the LAP pathway, and therefore would be an unlikely candidate to target for activing LAP.
As new aspects of both LAP signaling and mechanistic regulation are revealed, we believe that the importance of LAP to both normal physiological homeostasis as well as pathological insult will be further elucidated. The studies that detail the vital function of LAP in immune regulation provide a foundation upon which to continue investigation into the direct role of LAP in the modulation of immune metabolism, and how LAP is indeed a key player in shaping the immune response to infection, autoimmune regulation, and cancer.
Highlights.
-
-
Comprehensive overview of LC3-associated phagocytosis (LAP)
-
-
LAP bridges the phagocytic and autophagic pathways
-
-
Signaling to LAP and regulation of LAP activation by the canonical autophagy machinery
-
-
LAP quenches inflammation and shapes the immune response towards anti-inflammation
-
-
LAP promotes the immunosilent clearance of dying cells and prevents autoimmunity
Acknowledgments
EBR is a recipient of an EMBO Long-Term Fellowship (ALTF 1526 −2016). DRG is supported by funding from the NIH and the Lupus Research Alliance. We would also like to thank Dr. Bart Tummers for his input and helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157:65–75. doi: 10.1016/j.cell.2014.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martinez J, Verbist K, Wang R, Green DR. The relationship between metabolism and the autophagy machinery during the innate immune response. Cell Metab. 2013;17:895–900. doi: 10.1016/j.cmet.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38:633–43. doi: 10.1016/j.immuni.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang R, Green DR. Metabolic reprogramming and metabolic dependency in T cells. Immunol Rev. 2012;249:14–26. doi: 10.1111/j.1600-065X.2012.01155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Delmastro-Greenwood MM, Piganelli JD. Changing the energy of an immune response. Am J Clin Exp Immunol. 2013;2:30–54. [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A. 2011;108:17396–401. doi: 10.1073/pnas.1113421108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martinez J, Cunha LD, Park S, Yang M, Lu Q, Orchard R, et al. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature. 2016;533:115–9. doi: 10.1038/nature17950. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8.Freeman SA, Grinstein S. Phagocytosis: receptors, signal integration, and the cytoskeleton. Immunol Rev. 2014;262:193–215. doi: 10.1111/imr.12212. [DOI] [PubMed] [Google Scholar]
- 9.Kinchen JM, Ravichandran KS. Phagosome maturation: going through the acid test. Nat Rev Mol Cell Biol. 2008;9:781–95. doi: 10.1038/nrm2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–7. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 11.Kyrmizi I, Gresnigt MS, Akoumianaki T, Samonis G, Sidiropoulos P, Boumpas D, et al. Corticosteroids block autophagy protein recruitment in Aspergillus fumigatus phagosomes via targeting dectin-1/Syk kinase signaling. J Immunol. 2013;191:1287–99. doi: 10.4049/jimmunol.1300132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tam JM, Mansour MK, Khan NS, Seward M, Puranam S, Tanne A, et al. Dectin-1-dependent LC3 recruitment to phagosomes enhances fungicidal activity in macrophages. J Infect Dis. 2014;210:1844–54. doi: 10.1093/infdis/jiu290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henault J, Martinez J, Riggs JM, Tian J, Mehta P, Clarke L, et al. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity. 2012;37:986–97. doi: 10.1016/j.immuni.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Segawa K, Nagata S. An Apoptotic 'Eat Me' Signal: Phosphatidylserine Exposure. Trends Cell Biol. 2015;25:639–50. doi: 10.1016/j.tcb.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 15.Medina CB, Ravichandran KS. Do not let death do us part: 'find-me' signals in communication between dying cells and the phagocytes. Cell Death Differ. 2016;23:979–89. doi: 10.1038/cdd.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oikonomou V, Moretti S, Renga G, Galosi C, Borghi M, Pariano M, et al. Noncanonical Fungal Autophagy Inhibits Inflammation in Response to IFN-gamma via DAPK1. Cell Host Microbe. 2016;20:744–57. doi: 10.1016/j.chom.2016.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martinez J, Malireddi RK, Lu Q, Cunha LD, Pelletier S, Gingras S, et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol. 2015;17:893–906. doi: 10.1038/ncb3192. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Cemma M, Grinstein S, Brumell JH. Autophagy proteins are not universally required for phagosome maturation. Autophagy. 2016;12:1440–6. doi: 10.1080/15548627.2016.1191724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Backer JM. The intricate regulation and complex functions of the Class III phosphoinositide 3-kinase Vps34. Biochem J. 2016;473:2251–71. doi: 10.1042/BCJ20160170. [DOI] [PubMed] [Google Scholar]
- 20.Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3'-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem. 2000;275:992–8. doi: 10.1074/jbc.275.2.992. [DOI] [PubMed] [Google Scholar]
- 21.Volinia S, Dhand R, Vanhaesebroeck B, MacDougall LK, Stein R, Zvelebil MJ, et al. A human phosphatidylinositol 3-kinase complex related to the yeast Vps34p–Vps15p protein sorting system. EMBO J. 1995;14:3339–48. doi: 10.1002/j.1460-2075.1995.tb07340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kametaka S, Okano T, Ohsumi M, Ohsumi Y. Apg14p and Apg6/Vps30p form a protein complex essential for autophagy in the yeast, Saccharomyces cerevisiae. J Biol Chem. 1998;273:22284–91. doi: 10.1074/jbc.273.35.22284. [DOI] [PubMed] [Google Scholar]
- 23.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–6. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 24.Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009;11:385–96. doi: 10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- 25.Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 2009;11:468–76. doi: 10.1038/ncb1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284:12297–305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–91. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim JY, Zhao H, Martinez J, Doggett TA, Kolesnikov AV, Tang PH, et al. Noncanonical autophagy promotes the visual cycle. Cell. 2013;154:365–76. doi: 10.1016/j.cell.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gold ES, Underhill DM, Morrissette NS, Guo J, McNiven MA, Aderem A. Dynamin 2 is required for phagocytosis in macrophages. J Exp Med. 1999;190:1849–56. doi: 10.1084/jem.190.12.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kinchen JM, Doukoumetzidis K, Almendinger J, Stergiou L, Tosello-Trampont A, Sifri CD, et al. A pathway for phagosome maturation during engulfment of apoptotic cells. Nat Cell Biol. 2008;10:556–66. doi: 10.1038/ncb1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu N, Shen Q, Mahoney TR, Neukomm LJ, Wang Y, Zhou Z. Two PI 3-kinases and one PI 3-phosphatase together establish the cyclic waves of phagosomal PtdIns(3)P critical for the degradation of apoptotic cells. PLoS Biol. 2012;10:e1001245. doi: 10.1371/journal.pbio.1001245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Christoforidis S, Miaczynska M, Ashman K, Wilm M, Zhao L, Yip SC, et al. Phosphatidylinositol-3-OH kinases are Rab5 effectors. Nat Cell Biol. 1999;1:249–52. doi: 10.1038/12075. [DOI] [PubMed] [Google Scholar]
- 34.Mallo GV, Espina M, Smith AC, Terebiznik MR, Aleman A, Finlay BB, et al. SopB promotes phosphatidylinositol 3-phosphate formation on Salmonella vacuoles by recruiting Rab5 and Vps34. J Cell Biol. 2008;182:741–52. doi: 10.1083/jcb.200804131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shin HW, Hayashi M, Christoforidis S, Lacas-Gervais S, Hoepfner S, Wenk MR, et al. An enzymatic cascade of Rab5 effectors regulates phosphoinositide turnover in the endocytic pathway. J Cell Biol. 2005;170:607–18. doi: 10.1083/jcb.200505128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vieira OV, Botelho RJ, Rameh L, Brachmann SM, Matsuo T, Davidson HW, et al. Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. J Cell Biol. 2001;155:19–25. doi: 10.1083/jcb.200107069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma J, Becker C, Lowell CA, Underhill DM. Dectin-1-triggered recruitment of light chain 3 protein to phagosomes facilitates major histocompatibility complex class II presentation of fungal-derived antigens. J Biol Chem. 2012;287:34149–56. doi: 10.1074/jbc.M112.382812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berton G, Mocsai A, Lowell CA. Src and Syk kinases: key regulators of phagocytic cell activation. Trends Immunol. 2005;26:208–14. doi: 10.1016/j.it.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 39.Rostislavleva K, Soler N, Ohashi Y, Zhang L, Pardon E, Burke JE, et al. Structure and flexibility of the endosomal Vps34 complex reveals the basis of its function on membranes. Science. 2015;350:aac7365. doi: 10.1126/science.aac7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levin R, Grinstein S, Schlam D. Phosphoinositides in phagocytosis and macropinocytosis. Biochim Biophys Acta. 2015;1851:805–23. doi: 10.1016/j.bbalip.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 41.Swanson JA. Phosphoinositides and engulfment. Cell Microbiol. 2014;16:1473–83. doi: 10.1111/cmi.12334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lam GY, Cemma M, Muise AM, Higgins DE, Brumell JH. Host and bacterial factors that regulate LC3 recruitment to Listeria monocytogenes during the early stages of macrophage infection. Autophagy. 2013;9:985–95. doi: 10.4161/auto.24406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hubber A, Kubori T, Coban C, Matsuzawa T, Ogawa M, Kawabata T, et al. Bacterial secretion system skews the fate of Legionella-containing vacuoles towards LC3-associated phagocytosis. Sci Rep. 2017;7:44795. doi: 10.1038/srep44795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ellson CD, Anderson KE, Morgan G, Chilvers ER, Lipp P, Stephens LR, et al. Phosphatidylinositol 3-phosphate is generated in phagosomal membranes. Curr Biol. 2001;11:1631–5. doi: 10.1016/s0960-9822(01)00447-x. [DOI] [PubMed] [Google Scholar]
- 45.Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182:685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009;11:1433–7. doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- 47.Vergne I, Roberts E, Elmaoued RA, Tosch V, Delgado MA, Proikas-Cezanne T, et al. Control of autophagy initiation by phosphoinositide 3-phosphatase Jumpy. EMBO J. 2009;28:2244–58. doi: 10.1038/emboj.2009.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taguchi-Atarashi N, Hamasaki M, Matsunaga K, Omori H, Ktistakis NT, Yoshimori T, et al. Modulation of local PtdIns3P levels by the PI phosphatase MTMR3 regulates constitutive autophagy. Traffic. 2010;11:468–78. doi: 10.1111/j.1600-0854.2010.01034.x. [DOI] [PubMed] [Google Scholar]
- 49.Wu Y, Cheng S, Zhao H, Zou W, Yoshina S, Mitani S, et al. PI3P phosphatase activity is required for autophagosome maturation and autolysosome formation. EMBO Rep. 2014;15:973–81. doi: 10.15252/embr.201438618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng J, Fujita A, Yamamoto H, Tatematsu T, Kakuta S, Obara K, et al. Yeast and mammalian autophagosomes exhibit distinct phosphatidylinositol 3-phosphate asymmetries. Nat Commun. 2014;5:3207. doi: 10.1038/ncomms4207. [DOI] [PubMed] [Google Scholar]
- 51.Schink KO, Raiborg C, Stenmark H. Phosphatidylinositol 3-phosphate, a lipid that regulates membrane dynamics, protein sorting and cell signalling. Bioessays. 2013;35:900–12. doi: 10.1002/bies.201300064. [DOI] [PubMed] [Google Scholar]
- 52.Proikas-Cezanne T, Takacs Z, Donnes P, Kohlbacher O. WIPI proteins: essential PtdIns3P effectors at the nascent autophagosome. J Cell Sci. 2015;128:207–17. doi: 10.1242/jcs.146258. [DOI] [PubMed] [Google Scholar]
- 53.Dooley HC, Razi M, Polson HE, Girardin SE, Wilson MI, Tooze SA. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol Cell. 2014;55:238–52. doi: 10.1016/j.molcel.2014.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell. 2008;19:2092–100. doi: 10.1091/mbc.E07-12-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fratti RA, Backer JM, Gruenberg J, Corvera S, Deretic V. Role of phosphatidylinositol 3-kinase and Rab5 effectors in phagosomal biogenesis and mycobacterial phagosome maturation arrest. J Cell Biol. 2001;154:631–44. doi: 10.1083/jcb.200106049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ma J, Becker C, Reyes C, Underhill DM. Cutting edge: FYCO1 recruitment to dectin-1 phagosomes is accelerated by light chain 3 protein and regulates phagosome maturation and reactive oxygen production. J Immunol. 2014;192:1356–60. doi: 10.4049/jimmunol.1302835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 58.Lambeth JD, Neish AS. Nox enzymes and new thinking on reactive oxygen: a double-edged sword revisited. Annu Rev Pathol. 2014;9:119–45. doi: 10.1146/annurev-pathol-012513-104651. [DOI] [PubMed] [Google Scholar]
- 59.Huang J, Canadien V, Lam GY, Steinberg BE, Dinauer MC, Magalhaes MA, et al. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci U S A. 2009;106:6226–31. doi: 10.1073/pnas.0811045106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rastogi R, Geng X, Li F, Ding Y. NOX Activation by Subunit Interaction and Underlying Mechanisms in Disease. Front Cell Neurosci. 2016;10:301. doi: 10.3389/fncel.2016.00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Panday A, Sahoo MK, Osorio D, Batra S. NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell Mol Immunol. 2015;12:5–23. doi: 10.1038/cmi.2014.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ellson CD, Gobert-Gosse S, Anderson KE, Davidson K, Erdjument-Bromage H, Tempst P, et al. PtdIns(3)P regulates the neutrophil oxidase complex by binding to the PX domain of p40(phox) Nat Cell Biol. 2001;3:679–82. doi: 10.1038/35083076. [DOI] [PubMed] [Google Scholar]
- 63.Kanai F, Liu H, Field SJ, Akbary H, Matsuo T, Brown GE, et al. The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nat Cell Biol. 2001;3:675–8. doi: 10.1038/35083070. [DOI] [PubMed] [Google Scholar]
- 64.Ellson C, Davidson K, Anderson K, Stephens LR, Hawkins PT. PtdIns3P binding to the PX domain of p40phox is a physiological signal in NADPH oxidase activation. EMBO J. 2006;25:4468–78. doi: 10.1038/sj.emboj.7601346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bissonnette SA, Glazier CM, Stewart MQ, Brown GE, Ellson CD, Yaffe MB. Phosphatidylinositol 3-phosphate-dependent and -independent functions of p40phox in activation of the neutrophil NADPH oxidase. J Biol Chem. 2008;283:2108–19. doi: 10.1074/jbc.M706639200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Suh CI, Stull ND, Li XJ, Tian W, Price MO, Grinstein S, et al. The phosphoinositide-binding protein p40phox activates the NADPH oxidase during FcgammaIIA receptor-induced phagocytosis. J Exp Med. 2006;203:1915–25. doi: 10.1084/jem.20052085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Slauch JM. How does the oxidative burst of macrophages kill bacteria? Still an open question. Mol Microbiol. 2011;80:580–3. doi: 10.1111/j.1365-2958.2011.07612.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Flannagan RS, Cosio G, Grinstein S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol. 2009;7:355–66. doi: 10.1038/nrmicro2128. [DOI] [PubMed] [Google Scholar]
- 69.Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, et al. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell. 2006;126:205–18. doi: 10.1016/j.cell.2006.05.035. [DOI] [PubMed] [Google Scholar]
- 70.Nunes P, Demaurex N, Dinauer MC. Regulation of the NADPH oxidase and associated ion fluxes during phagocytosis. Traffic. 2013;14:1118–31. doi: 10.1111/tra.12115. [DOI] [PubMed] [Google Scholar]
- 71.Chen Y, Azad MB, Gibson SB. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009;16:1040–52. doi: 10.1038/cdd.2009.49. [DOI] [PubMed] [Google Scholar]
- 72.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–60. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Klionsky DJ, Schulman BA. Dynamic regulation of macroautophagy by distinctive ubiquitin-like proteins. Nat Struct Mol Biol. 2014;21:336–45. doi: 10.1038/nsmb.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152:657–68. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mizushima N, Yoshimori T, Ohsumi Y. Mouse Apg10 as an Apg12-conjugating enzyme: analysis by the conjugation-mediated yeast two-hybrid method. FEBS Lett. 2002;532:450–4. doi: 10.1016/s0014-5793(02)03739-0. [DOI] [PubMed] [Google Scholar]
- 76.Tanida I, Tanida-Miyake E, Ueno T, Kominami E. The human homolog of Saccharomyces cerevisiae Apg7p is a Protein-activating enzyme for multiple substrates including human Apg12p, GATE-16, GABARAP, and MAP-LC3. J Biol Chem. 2001;276:1701–6. doi: 10.1074/jbc.C000752200. [DOI] [PubMed] [Google Scholar]
- 77.Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, et al. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci. 2003;116:1679–88. doi: 10.1242/jcs.00381. [DOI] [PubMed] [Google Scholar]
- 78.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117:2805–12. doi: 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- 80.Tanida I, Tanida-Miyake E, Komatsu M, Ueno T, Kominami E. Human Apg3p/Aut1p homologue is an authentic E2 enzyme for multiple substrates, GATE-16, GABARAP, and MAP-LC3, and facilitates the conjugation of hApg12p to hApg5p. J Biol Chem. 2002;277:13739–44. doi: 10.1074/jbc.M200385200. [DOI] [PubMed] [Google Scholar]
- 81.Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282:37298–302. doi: 10.1074/jbc.C700195200. [DOI] [PubMed] [Google Scholar]
- 82.Kirisako T, Ichimura Y, Okada H, Kabeya Y, Mizushima N, Yoshimori T, et al. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J Cell Biol. 2000;151:263–76. doi: 10.1083/jcb.151.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fernandez AF, Lopez-Otin C. The functional and pathologic relevance of autophagy proteases. J Clin Invest. 2015;125:33–41. doi: 10.1172/JCI73940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014;16:495–501. doi: 10.1038/ncb2979. [DOI] [PubMed] [Google Scholar]
- 85.Fujita N, Hayashi-Nishino M, Fukumoto H, Omori H, Yamamoto A, Noda T, et al. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell. 2008;19:4651–9. doi: 10.1091/mbc.E08-03-0312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–60. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 87.Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 2007;130:165–78. doi: 10.1016/j.cell.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 88.Choy A, Dancourt J, Mugo B, O'Connor TJ, Isberg RR, Melia TJ, et al. The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science. 2012;338:1072–6. doi: 10.1126/science.1227026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sprenkeler EG, Gresnigt MS, van de Veerdonk FL. LC3-associated phagocytosis: a crucial mechanism for antifungal host defence against Aspergillus fumigatus. Cell Microbiol. 2016;18:1208–16. doi: 10.1111/cmi.12616. [DOI] [PubMed] [Google Scholar]
- 90.Romao S, Gasser N, Becker AC, Guhl B, Bajagic M, Vanoaica D, et al. Autophagy proteins stabilize pathogen-containing phagosomes for prolonged MHC II antigen processing. J Cell Biol. 2013;203:757–66. doi: 10.1083/jcb.201308173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Munz C. Of LAP, CUPS, and DRibbles - Unconventional Use of Autophagy Proteins for MHC Restricted Antigen Presentation. Front Immunol. 2015;6:200. doi: 10.3389/fimmu.2015.00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ligeon LA, Romao S, Munz C. Analysis of LC3-Associated Phagocytosis and Antigen Presentation. Methods Mol Biol. 2017;1519:145–68. doi: 10.1007/978-1-4939-6581-6_10. [DOI] [PubMed] [Google Scholar]
- 93.Bauckman KA, Owusu-Boaitey N, Mysorekar IU. Selective autophagy: xenophagy. Methods. 2015;75:120–7. doi: 10.1016/j.ymeth.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fang FC. Antimicrobial actions of reactive oxygen species. MBio. 2011:2. doi: 10.1128/mBio.00141-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lerena MC, Colombo MI. Mycobacterium marinum induces a marked LC3 recruitment to its containing phagosome that depends on a functional ESX-1 secretion system. Cell Microbiol. 2011;13:814–35. doi: 10.1111/j.1462-5822.2011.01581.x. [DOI] [PubMed] [Google Scholar]
- 96.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–8. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kim S, Elkon KB, Ma X. Transcriptional suppression of interleukin-12 gene expression following phagocytosis of apoptotic cells. Immunity. 2004;21:643–53. doi: 10.1016/j.immuni.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 98.Martin CJ, Peters KN, Behar SM. Macrophages clean up: efferocytosis and microbial control. Curr Opin Microbiol. 2014;17:17–23. doi: 10.1016/j.mib.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mukundan L, Odegaard JI, Morel CR, Heredia JE, Mwangi JW, Ricardo-Gonzalez RR, et al. PPAR-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat Med. 2009;15:1266–72. doi: 10.1038/nm.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Xiao YQ, Freire-de-Lima CG, Schiemann WP, Bratton DL, Vandivier RW, Henson PM. Transcriptional and translational regulation of TGF-beta production in response to apoptotic cells. J Immunol. 2008;181:3575–85. doi: 10.4049/jimmunol.181.5.3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Munoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol. 2010;6:280–9. doi: 10.1038/nrrheum.2010.46. [DOI] [PubMed] [Google Scholar]
- 102.International Consortium for Systemic Lupus. Erythematosus G, Harley JB, Alarcon-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008;40:204–10. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gateva V, Sandling JK, Hom G, Taylor KE, Chung SA, Sun X, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet. 2009;41:1228–33. doi: 10.1038/ng.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Brandrup F, Koch C, Petri M, Schiodt M, Johansen KS. Discoid lupus erythematosus-like lesions and stomatitis in female carriers of X-linked chronic granulomatous disease. Br J Dermatol. 1981;104:495–505. doi: 10.1111/j.1365-2133.1981.tb08163.x. [DOI] [PubMed] [Google Scholar]
- 105.Hafner J, Enderlin A, Seger RA, Wuthrich B, Bruckner-Tudermann L, Panizzoni P, et al. Discoid lupus erythematosus-like lesions in carriers of X-linked chronic granulomatous disease. Br J Dermatol. 1992;127:446–7. doi: 10.1111/j.1365-2133.1992.tb00471.x. [DOI] [PubMed] [Google Scholar]
- 106.Rupec RA, Petropoulou T, Belohradsky BH, Walchner M, Liese JG, Plewing G, et al. Lupus erythematosus tumidus and chronic discoid lupus erythematosus in carriers of X-linked chronic granulomatous disease. Eur J Dermatol. 2000;10:184–9. [PubMed] [Google Scholar]
- 107.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Munder M, Eichmann K, Modolell M. Alternative metabolic states in murine macrophages reflected by the nitric oxide synthase/arginase balance: competitive regulation by CD4+ T cells correlates with Th1/Th2 phenotype. J Immunol. 1998;160:5347–54. [PubMed] [Google Scholar]
- 109.Modolell M, Corraliza IM, Link F, Soler G, Eichmann K. Reciprocal regulation of the nitric oxide synthase/arginase balance in mouse bone marrow-derived macrophages by TH1 and TH2 cytokines. Eur J Immunol. 1995;25:1101–4. doi: 10.1002/eji.1830250436. [DOI] [PubMed] [Google Scholar]
- 110.Corraliza IM, Soler G, Eichmann K, Modolell M. Arginase induction by suppressors of nitric oxide synthesis (IL-4, IL-10 and PGE2) in murine bone-marrow-derived macrophages. Biochem Biophys Res Commun. 1995;206:667–73. doi: 10.1006/bbrc.1995.1094. [DOI] [PubMed] [Google Scholar]
- 111.Van den Bossche J, O'Neill LA, Menon D. Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol. 2017;38:395–406. doi: 10.1016/j.it.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 112.Park D, Han CZ, Elliott MR, Kinchen JM, Trampont PC, Das S, et al. Continued clearance of apoptotic cells critically depends on the phagocyte Ucp2 protein. Nature. 2011;477:220–4. doi: 10.1038/nature10340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zeng MY, Pham D, Bagaitkar J, Liu J, Otero K, Shan M, et al. An efferocytosis-induced, IL-4-dependent macrophage-iNKT cell circuit suppresses sterile inflammation and is defective in murine CGD. Blood. 2013;121:3473–83. doi: 10.1182/blood-2012-10-461913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ravishankar B, Liu H, Shinde R, Chaudhary K, Xiao W, Bradley J, et al. The amino acid sensor GCN2 inhibits inflammatory responses to apoptotic cells promoting tolerance and suppressing systemic autoimmunity. Proc Natl Acad Sci U S A. 2015;112:10774–9. doi: 10.1073/pnas.1504276112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nagy L, Szanto A, Szatmari I, Szeles L. Nuclear hormone receptors enable macrophages and dendritic cells to sense their lipid environment and shape their immune response. Physiol Rev. 2012;92:739–89. doi: 10.1152/physrev.00004.2011. [DOI] [PubMed] [Google Scholar]
- 116.Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, et al. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. 2000;14:2819–30. doi: 10.1101/gad.844900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Heckmann BL, Zhang X, Saarinen AM, Schoiswohl G, Kershaw EE, Zechner R, et al. Liver X receptor alpha mediates hepatic triglyceride accumulation through upregulation of G0/G1 Switch Gene 2 expression. JCI Insight. 2017;2:e88735. doi: 10.1172/jci.insight.88735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Roszer T. Transcriptional control of apoptotic cell clearance by macrophage nuclear receptors. Apoptosis. 2017;22:284–94. doi: 10.1007/s10495-016-1310-x. [DOI] [PubMed] [Google Scholar]
- 119.N AG, Hidalgo A. Nuclear Receptors and Clearance of Apoptotic Cells: Stimulating the Macrophage's Appetite. Front Immunol. 2014;5:211. doi: 10.3389/fimmu.2014.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lok AS, Liang RH, Chiu EK, Wong KL, Chan TK, Todd D. Reactivation of hepatitis B virus replication in patients receiving cytotoxic therapy. Report of a prospective study. Gastroenterology. 1991;100:182–8. doi: 10.1016/0016-5085(91)90599-g. [DOI] [PubMed] [Google Scholar]
- 121.N AG, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. 2009;31:245–58. doi: 10.1016/j.immuni.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Roszer T, Menendez-Gutierrez MP, Lefterova MI, Alameda D, Nunez V, Lazar MA, et al. Autoimmune kidney disease and impaired engulfment of apoptotic cells in mice with macrophage peroxisome proliferator-activated receptor gamma or retinoid X receptor alpha deficiency. J Immunol. 2011;186:621–31. doi: 10.4049/jimmunol.1002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, et al. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 2012;15:545–53. doi: 10.1016/j.cmet.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ouimet M, Franklin V, Mak E, Liao X, Tabas I, Marcel YL. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011;13:655–67. doi: 10.1016/j.cmet.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, et al. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012;15:534–44. doi: 10.1016/j.cmet.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. 2010;10:36–46. doi: 10.1038/nri2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Roman MJ, Shanker BA, Davis A, Lockshin MD, Sammaritano L, Simantov R, et al. Prevalence and correlates of accelerated atherosclerosis in systemic lupus erythematosus. The New England journal of medicine. 2003;349:2399–406. doi: 10.1056/NEJMoa035471. [DOI] [PubMed] [Google Scholar]
- 128.Wigren M, Nilsson J, Kaplan MJ. Pathogenic immunity in systemic lupus erythematosus and atherosclerosis: common mechanisms and possible targets for intervention. Journal of internal medicine. 2015;278:494–506. doi: 10.1111/joim.12357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Libby P, Lichtman AH, Hansson GK. Immune effector mechanisms implicated in atherosclerosis: from mice to humans. Immunity. 2013;38:1092–104. doi: 10.1016/j.immuni.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and rupture. Circulation research. 2014;114:1852–66. doi: 10.1161/CIRCRESAHA.114.302721. [DOI] [PubMed] [Google Scholar]
- 131.Skalen K, Gustafsson M, Rydberg EK, Hulten LM, Wiklund O, Innerarity TL, et al. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. 2002;417:750–4. doi: 10.1038/nature00804. [DOI] [PubMed] [Google Scholar]
- 132.Schnoor M, Alcaide P, Voisin MB, van Buul JD. Crossing the Vascular Wall: Common and Unique Mechanisms Exploited by Different Leukocyte Subsets during Extravasation. Mediators Inflamm. 2015;2015:946509. doi: 10.1155/2015/946509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gerhardt T, Ley K. Monocyte trafficking across the vessel wall. Cardiovascular research. 2015;107:321–30. doi: 10.1093/cvr/cvv147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Antonov AS, Munn DH, Kolodgie FD, Virmani R, Gerrity RG. Aortic endothelial cells regulate proliferation of human monocytes in vitro via a mechanism synergistic with macrophage colony-stimulating factor. Convergence at the cyclin E/p27(Kip1) regulatory checkpoint. J Clin Invest. 1997;99:2867–76. doi: 10.1172/JCI119480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ostriker A, Horita HN, Poczobutt J, Weiser-Evans MC, Nemenoff RA. Vascular smooth muscle cell-derived transforming growth factor-beta promotes maturation of activated, neointima lesion-like macrophages. Arterioscler Thromb Vasc Biol. 2014;34:877–86. doi: 10.1161/ATVBAHA.114.303214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ensan S, Li A, Besla R, Degousee N, Cosme J, Roufaiel M, et al. Self-renewing resident arterial macrophages arise from embryonic CX3CR1(+) precursors and circulating monocytes immediately after birth. Nat Immunol. 2016;17:159–68. doi: 10.1038/ni.3343. [DOI] [PubMed] [Google Scholar]
- 137.Robbins CS, Hilgendorf I, Weber GF, Theurl I, Iwamoto Y, Figueiredo JL, et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med. 2013;19:1166–72. doi: 10.1038/nm.3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, van Rooijen N, et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science. 2011;332:1284–8. doi: 10.1126/science.1204351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cianfanelli V, D'Orazio M, Cecconi F. AMBRA1 and BECLIN 1 interplay in the crosstalk between autophagy and cell proliferation. Cell cycle (Georgetown, Tex) 2015;14:959–63. doi: 10.1080/15384101.2015.1021526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cianfanelli V, Fuoco C, Lorente M, Salazar M, Quondamatteo F, Gherardini PF, et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat Cell Biol. 2015;17:706. doi: 10.1038/ncb3171. [DOI] [PubMed] [Google Scholar]
- 141.Qiang L, Zhao B, Ming M, Wang N, He TC, Hwang S, et al. Regulation of cell proliferation and migration by p62 through stabilization of Twist1. Proc Natl Acad Sci U S A. 2014;111:9241–6. doi: 10.1073/pnas.1322913111. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 142.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13:709–21. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11:155–61. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cole JE, Kassiteridi C, Monaco C. Toll-like receptors in atherosclerosis: a 'Pandora's box' of advances and controversies. Trends Pharmacol Sci. 2013;34:629–36. doi: 10.1016/j.tips.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 145.Tabas I, Bornfeldt KE. Macrophage Phenotype and Function in Different Stages of Atherosclerosis. Circulation research. 2016;118:653–67. doi: 10.1161/CIRCRESAHA.115.306256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kojima Y, Weissman IL, Leeper NJ. The Role of Efferocytosis in Atherosclerosis. Circulation. 2017;135:476–89. doi: 10.1161/CIRCULATIONAHA.116.025684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Versini M, Jeandel PY, Rosenthal E, Shoenfeld Y. Obesity in autoimmune diseases: not a passive bystander. Autoimmun Rev. 2014;13:981–1000. doi: 10.1016/j.autrev.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 148.Shah A, Mehta N, Reilly MP. Adipose inflammation, insulin resistance, and cardiovascular disease. JPEN J Parenter Enteral Nutr. 2008;32:638–44. doi: 10.1177/0148607108325251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Boutens L, Stienstra R. Adipose tissue macrophages: going off track during obesity. Diabetologia. 2016;59:879–94. doi: 10.1007/s00125-016-3904-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Surmi BK, Hasty AH. Macrophage infiltration into adipose tissue: initiation, propagation and remodeling. Future Lipidol. 2008;3:545–56. doi: 10.2217/17460875.3.5.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel) 2014;6:1670–90. doi: 10.3390/cancers6031670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Henault J, Martinez J, Riggs JM, Tian J, Mehta P, Clarke L, et al. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity. 2012;37:986–97. doi: 10.1016/j.immuni.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Han J, Bae J, Choi CY, Choi SP, Kang HS, Jo EK, et al. Autophagy induced by AXL receptor tyrosine kinase alleviates acute liver injury via inhibition of NLRP3 inflammasome activation in mice. Autophagy. 2016;12:2326–43. doi: 10.1080/15548627.2016.1235124. [DOI] [PMC free article] [PubMed] [Google Scholar]